

ABSTRACT
Epstein-Barr virus (EBV) attachment to human CD21 on the B-cell surface initiates infection. Whether CD21 is a simple tether or conveys vital information to the cell interior for production of host factors that promote infection of primary B cells is controversial, as the cytoplasmic fragment of CD21 is short, though highly conserved. The ubiquity of CD21 on normal B cells, the diversity of this population, and the well-known resistance of primary B cells to gene transfer technologies have all impeded resolution of this question. To uncover the role(s) of the CD21 cytoplasmic domain during infection initiation, the full-length receptor (CD21 = CR), a mutant lacking the entire cytoplasmic tail (CT), and a control vector (NEO) were stably expressed in two pre-B-cell lines that lack endogenous receptor. Genome-wide transcriptional analysis demonstrated that stable CD21 surface expression alone (either CR or CT) produced multiple independent changes in gene expression, though both dramatically decreased class I melanoma-associated antigen (MAGE) family RNAs and upregulated genes associated with B-cell differentiation (e.g., C2TA, HLA-II, IL21R, MIC2, CD48, and PTPRCAP/CD45-associated protein). Temporal analysis spanning 72 h revealed that not only CR- but also CT-expressing lines initiated latency. In spite of this, the number and spectrum of transcripts altered in CR- compared with CT-bearing lines at 1 h after infection further diverged. Differential modulation of immediate early cellular transcripts (e.g., c-Jun and multiple histones), both novel and previously linked to CD21-initiated signaling, as well as distinct results from pathway analyses support a separate role for the cytoplasmic domain in initiation of intracellular signals.
IMPORTANCE Membrane proteins that mediate virus attachment tether virus particles to the cell surface, initiating infection. In addition, upon virus interaction such proteins may transmit signals to the interior of the cell that support subsequent steps in the infection process. Here we show that expression of the Epstein-Barr virus B-cell attachment receptor, CD21, in B cells that lack this receptor results in significant changes in gene expression, both before and rapidly following EBV-CD21 interaction. These changes translate into major signaling pathway alterations that are predicted to support stable infection.
INTRODUCTION
The human herpesvirus Epstein-Barr virus (EBV) is implicated in the pathogenesis of infectious mononucleosis and several hematologic and epithelial cancers, and in promotion of certain autoimmune diseases (1). EBV is so efficiently transmitted that by adulthood most (>95%) of the world's population has been infected (2). In vivo, virus persists for life through establishment of a latent B-cell reservoir. In vitro, normal resting B cells alone are actively infected and immortalized by EBV. A high-affinity protein-protein interaction (3) between the virion envelope glycoprotein gp350/220 and human CD21 (4–6) can initiate virus attachment to B-cell surfaces, culminating in EBV gene expression that promotes latent, although in some cases lytic or abortive, infection (reviewed in reference 7). Persistent infection of primary B cells in vitro and in vivo begins with a robust viral expression pattern known as latency type III (EBNAs 1, 2, and 3A, B, and C, LP, LMP 1, 2A, and 2B, and noncoding RNAs, including EBERs 1/2 and viral microRNAs). However, in vivo and in most EBV-positive tumor lines, progressive silencing of viral gene expression is the norm (8).
For many viruses, contact with a specific attachment receptor is a decisive event that not only tethers the virion to the plasma membrane but also delivers intracellular signals that support establishment of infection. Whether and how interaction with CD21 initiates changes in host gene expression that alter the efficiency, pattern, or stability of primary EBV infection is not well understood. CD21 is uniformly expressed on mature B cells and follicular dendritic cells although it can be variably detected on certain other cell types. A type 1 membrane glycoprotein, CD21, consists of an extracellular domain (ED) of 15 or 16 short consensus repeat (SCR) sequences, a transmembrane (TM) region, and a short 34-amino-acid intracellular peptide. In addition to EBV, the two N-terminal CD21 SCRs bind the C3d fragment of complement C3, the activation antigen CD23, and alpha interferon (reviewed in reference 9). CD21 is transiently phosphorylated by exposure to phorbol myristate acetate (PMA). When physically cross-linked to the B-cell receptor (BCR) or triggered immediately prior to BCR cross-linking, toggling of CD21 (a surrogate for immune complex interaction) amplifies mIg signaling, B-cell proliferation, antibody production, and antigen presentation (reviewed in reference 10). CD21-CD23 (low-affinity IgE receptor) interaction blocks B-cell apoptosis (11). These findings highlight CD21's role in the integration of innate and adaptive immune responses. However, the cytoplasmic domain is short, and thus it has been suggested that intracellular signals are transduced not directly by CD21 but rather through B-cell surface proteins, such as CD19, that complex with CD21 (12). Downstream phosphorylation upon ligation of CD21 has been described in CD19-negative cell lines (13), and interactions with the cytoplasmic domain of CD21 have been detected in two-hybrid assays (14, 15), even though the intervening molecules that link the intracellular domain of CD21 with signaling pathways are not well characterized. To identify whether and which cell regulatory pathways are activated upon EBV ligation of CD21 and to precisely uncover how the intracellular fragment of CD21 contributes to these events, we developed a strategy based on comparative transcriptional analysis. Because (i) virtually all normal B cells express endogenous CD21, as well as the CD21-associated proteins CD19 and CD35, (ii) normal B cells circulating in peripheral and cord blood, as well as those localized to lymphoid organs, are extremely diverse, consisting of mixtures of cells expressing different BCRs suspended at distinct stages of B-cell differentiation and cycling at different rates, (iii) primary human B cells are altered by positive selection procedures, e.g., with CD19, and (iv) primary B cells are very difficult to transfect and are modified by transfection-infection procedures, we developed a cell line-based approach. We stably introduced two unrelated human pre-B-cell lines (Nalm6 and Laz221) that lack CD21 (and also CD35), established clones, and engineered the respective cloned lines to express either full-length CD21 (referred to as CR), CD21 devoid of the cytoplasmic domain (referred to as CT), or a vector control (NEO). Clones derived from the six sublines were synchronized to eliminate cell cycle effects, simultaneously infected with purified EBV, and analyzed for viral gene expression over 72 h. The expression pattern of EBV RNAs (by Northern blotting) was consistent with the pattern observed after primary infection of B cells. Cellular RNAs harvested at 1 min, at 1 h (this study), and at time points throughout the first week were compared. In this report, we identify the major changes in cellular gene expression just 1 h after EBV-CD21 interaction. We show that a subset of transcripts altered following contact of EBV with full-length CD21 (CR) are distinct from those that contact the truncated receptor (CT). This reveals modulation of downstream signaling networks by the cytoplasmic tail of CD21 and identifies pathways that may be essential for infection of primary B cells (though not tumor lines). Unexpectedly, the transcriptome of cells constitutively expressing full-length compared with truncated CD21 were more diverged from one another than predicted, based on an established literature describing the intracellular fragment as being predominantly silent. Nevertheless, among cells that stably expressed CR or CT compared with NEO, conserved patterns of stable gene expression were evident. Among the most notable changes, class I melanoma-associated antigen (MAGE) family gene expression was dramatically reduced, whereas expression of multiple genes (e.g., HLA class II) associated with B-cell maturation increased, consistent with a key role for interactions mediated solely through association with the extracytoplasmic domains of CD21 in B-cell differentiation.
MATERIALS AND METHODS
Cell lines and cultivation.
The human pre-B-cell (acute lymphoblastic leukemia) lines Nalm6 and Laz221(LAZ 221) were obtained from Margaret Shipp, Dana-Farber Cancer Institute, Boston, MA. The Nalm6 cell line has been extensively characterized, lacks a c-myc translocation, and contains wild-type p53 (16). Less is known about Laz221 (17). The EBV-positive Burkitt lymphoma line Akata was provided by Kenzo Takada (Nihon University, Tokyo, Japan). The EBV-positive marmoset producer B-lymphoblastoid cell line (LCL) B95-8 (American Type Culture Collection [ATCC]), EBV-positive B LCLs, and the human T-cell line HPB-MLT (Jack Strominger, Dana-Farber Cancer Institute, Boston, MA) were provided as indicated. All cell lines were maintained in a humidified 5% CO2 incubator at 37°C in RPMI 1640 containing l-glutamine. Culture media were supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml) (HyClone), and 10% heat-inactivated calf serum (HyClone).
Plasmid preparation.
A full-length CD21 cDNA encoding 16 SCRs was cloned from a pH3M expression library derived from the CD21+ T-cell line HPB-ALL (18) using a cDNA probe. Mutant CD21 lacking the 35-amino-acid cytoplasmic tail (SKH…PAS) was generated by a subcloning strategy. A 923-bp PstI-PstI fragment encoding the transmembrane region, cytoplasmic tail, and 3′ untranslated region was removed from CD21pH3M and subcloned into PACYC177 (ATCC). The internal 105-bp EcoRV-PvuII fragment encoding the cytoplasmic tail was excised, and PACYC177 was then religated. The truncated PstI-PstI fragment was then excised from PACYC177 and recloned into pH3MCD21 in the correct orientation. The fidelity of full-length CD21 and CD21 deleted of its intracellular domain (CD21CT−) was confirmed by PCR and by DNA sequence analysis (not shown).
Electroporation.
Wild-type (CR)- and mutant (CT)-containing plasmids were individually transfected, together with pSV2neo (Addgene), into cells that were then selected with Geneticin (Gibco). PSV2neo was transfected alone as the vector control. Briefly, Nalm6 cells and Laz221 cells were grown to log phase, and 1 × 107 cells were washed twice in 10 ml serum-free RPMI and resuspended in 0.8 ml of 0.9% NaCl (Baxter) containing 10 mM HEPES buffer (BioWhittaker). Expression plasmid pH3M, pH3MCD21, or pH3MCD21− (15 μg) was mixed with pSV2neo (1.5 μg), ethanol precipitated, washed with 70% ethanol, and then air dried under sterile conditions. The DNA pellet was resuspended into the cells, and the mixture was transferred to a 0.4-cm-gap electroporation cuvette (Bio-Rad). Electroporation was performed at room temperature on a Bio-Rad gene pulser apparatus at 0.3 kV and 960 μF, with a resultant time constant of ∼11. Cells were maintained at room temperature for 10 min and then cultured in complete medium containing 20% heat-inactivated calf serum that was mixed 1:1 with filtered (0.22 μm) (Easy Flow filter; Becton Dickinson) conditioned media from the respective lines. Forty-eight hours later, cells were washed once with complete medium and then transferred into complete medium supplemented with 720 μg/ml (active) Geneticin (Gibco/BRL). Seventy-two hours later, cells were transferred into complete medium containing 900 μg/ml (active) Geneticin, in which they were maintained permanently. After 2 weeks of growth in Geneticin, either transfectants were single-cell cloned and evaluated for CR2 expression by flow cytometry or the bulk population was sorted four or more times for 100% CR2 expression on a FACSVantage cell sorter (Becton Dickinson).
Antibodies.
Murine monoclonal antibodies (MAbs) were used for cytometric analysis of cell lines: HB-5 anti-CD21 (ATCC), OKB7 anti-CD21 (generously provided by Ortho Corporation), B-C3 anti-CD19 (Biosource), 5A6 anti-CD81 (Shoshana Levy, Stanford University School of Medicine, Stanford, CA), 44D anti-CD35, anti-CD48, and anti-CD59 (Becton Dickinson), anti-Leu 13 (Sharon Evans, Roswell Park Cancer Institute, Buffalo, NY), LB3.1 anti-HLA-DR (Jack Strominger), anti-HLA-DP and anti-HLA-DQ (Biodesign International, Saco, ME), HB168 (also known as 72A1) anti-gp350 (ATCC), 2L10 anti-gp350 (Gary R. Pearson, Georgetown University, Washington, DC, retired), CS1-4 anti LMP-1 (Abcam), anti-UPC10 control IgG2a (ICN), and MOPC 21 control IgG1 (ICN). Antibodies were used at a concentration of 10 to 40 μg/ml, which was determined to be approximately 10-fold in excess of saturation based on cytometric analyses of standard B-cell lines. For detection, fluorescein isothiocyanate (FITC)-labeled goat F(ab′)2 anti-mouse IgG (Biosource) at a concentration of 10 μg/ml was used. Monospecific rabbit anti-CD21/CR2 antibody, prepared as described previously (19, 20), was used to detect human CD21 protein by immunoblotting. Anti-CD21 antibody was detected with 125I-goat F(ab′)2 anti-rabbit IgG (New England Nuclear). Rabbit anti-human CD21 and normal rabbit serum (control) were used for some cytometric analyses, followed by detection with FITC-goat F(ab′)2 anti-rabbit IgG (Biosource).
Flow cytometry.
Cells (1 × 106/sample) were washed in RPMI containing 2% heat-inactivated fetal calf serum (FCS) and 0.04% sodium azide. Cells were then incubated with first antibody (either a specific murine MAb or an isotype-matched control or, alternatively, rabbit anti-CR2 or normal rabbit serum as a control) for 30 min on ice. The antibody concentrations that were used were 10-fold in excess of saturation based on prior titration by cytometry on appropriate positive- and negative-control LCLs (commonly 10 to 40 μg/ml for MAbs). Cells were then washed twice and further incubated with FITC–goat F(ab′)2 anti-mouse IgG (murine MAbs) absorbed for cross-reactivity to human and rabbit IgG or FITC–goat anti-rabbit IgG (rabbit sera) for 30 min on ice. The cells were washed two additional times. In some experiments, propidium iodide was added to live cells (final concentration, 5 μg/ml). Cells were analyzed by flow cytometry on a FACScan instrument (Becton, Dickinson).
Immunoblotting.
Cells (2 × 107/sample) were washed twice in phosphate-buffered saline (PBS) and then solubilized in 1 ml of lysis buffer consisting of 1% NP-40 and 0.02% NaN3 to which 20 μl of 0.4 M iodoacetamide and 10 μl of 0.2 M phenylmethylsulfonyl fluoride (PMSF) in methanol were added. The lysate was incubated for 30 min on ice and then centrifuged for 10 min at 1,000 × g to remove insoluble material. Fifty microliters of soluble cell extract was boiled for 5 min with sample buffer under nonreducing conditions, and individual proteins were separated on an SDS-10% polyacrylamide gel (21). The proteins were transferred to nitrocellulose. CD21 was detected with rabbit anti-human CR2 followed by 125I-labeled goat F(ab′)2 anti-rabbit IgG (New England Nuclear) and analyzed by autoradiography using X-OMAT XAR film (Eastman Kodak) or with goat anti-rabbit horseradish peroxidase (A5420; Sigma) and developed with a chemiluminescent substrate mixture (Amersham ECL, GE Life Sciences) according to the directions of the manufacturer.
Virus preparation and infection.
B95-8 cells were seeded at a density of 5 × 105 cells/ml in 8 liters of complete medium and cultured in a spinner flask with constant stirring for 1 week. The contents of the flask were poured over coarse-porosity filters (grade 802; Whatman) to remove cells. An additional filtration step was performed over fine-porosity filters (0.8 μm) (Nalge). The resulting filtrate was 40-fold concentrated on a Pelicon filtration system (Millipore). An additional centrifugation at 6,000 rpm in a JA-10 rotor (Beckman) for 10 min at 4°C was performed to remove any remaining insoluble material. The supernatant was then centrifuged at 20,000 rpm in an SS-27 rotor (Sorvall) for 90 min at 4°C, and the EBV-containing pellets were pooled, resuspended in 50 ml of complete medium, and stored in 1-ml aliquots at −70°C until use.
Duplicate samples of 2 × 107 Nalm6 or Laz221 transfectant cells were removed from log-phase cultures and washed once in complete medium. Cells were centrifuged at 800 × g for 10 min at 4°C, and the supernatants were removed. Cell pellets were either processed immediately for preparation of total RNA (see below) or resuspended in 4 ml of complete medium containing EBV at a 1:2 dilution with complete medium. EBV incubation was continued for 4 h under cell culture conditions as described above. After incubation, cells were washed twice by dilution in 50 ml complete medium and separated by centrifugation as described above. Cells were resuspended at a concentration of 4 × 105/ml in complete medium and cultured in T75 vented-cap tissue culture flasks (Costar) as described above. At 24, 48, and 72 h, 1 × 107 cells were removed from the appropriate tissue culture flasks and separated by centrifugation, and total RNA was prepared.
RNA blot hybridization.
RNA preparation and blot hybridization were performed as previously described (22). cDNAs utilized as probes were as previously described (22) with the exception of the probe for BHRF1. This probe consisted of an ∼1,100-bp BclI-BamHI subfragment of the BamHI H genomic fragment (23).
RNA preparation and reverse transcription-PCR (RT-PCR).
Cells (107/sample) were harvested, and total RNA was prepared using the RNeasy total RNA kit (Qiagen) according to the directions provided by the manufacturer. All resultant RNAs were treated with DNase (Promega) and further purified on RNeasy total RNA spin columns as directed. One microgram of each sample of total RNA was reverse transcribed according to directions provided in the GeneAmp PCR kit (Perkin-Elmer), using a PTC-100 programmable thermal cycler (MJ Research). PCR amplification primers spanning the cytoplasmic domain of CD21 were 5′-GTGCCAATCGGATCACC-3′ (sense) and 5′-TCCGCTGAATTCCAAGC-3′ (antisense). PCR amplification primers for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and amplification conditions were as described previously (24).
Validation of differentially expressed genes and genes belonging to specific pathways and functions was performed by quantitative RT-PCR (qRT-PCR). Two hundred nanograms of high-quality RNA samples was reverse transcribed to first-strand cDNA, and 1 μl cDNA was used for each well RT-PCR. Reactions were performed in triplicate. SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA) was used for two-step real-time RT-PCR analysis on an Applied Biosystems StepOnePlus real-time PCR instrument. Primer sequences were designed using the rpimer3 tool (http://bioinfo.ut.ee/primer3-0.4.0/primer3/). The expression value of the targeted gene in a given sample was normalized to the corresponding expression of GAPDH. The 2−ΔΔCT method was used to calculate relative expression of the targeted genes.
Microarray analysis.
Total RNA was isolated using the RNeasy kit (Qiagen). Transcriptional profiling of RNAs extracted from cell lines that express NEO, CR, or CT was performed using the Affymetrix oligonucleotide microarray. Five micrograms of fragmented, labeled cRNA from each sample was hybridized to the U133 Plus 2.0 chip, which has 54,675 probe sets, or to the U133A chip, which has 22,283 probe sets, using standard Affymetrix protocols. Chips were scanned using an Affymetrix microarray scanner (Hewlett-Packard), while fluorescence intensity values were captured using GeneChip software (Affymetrix). The quality of hybridized chips was assessed using Affymetrix guidelines on the basis of average background, scaling factor, number of genes called present, 3′-to-5′ ratios for beta-actin and GAPDH, and values for spike-in control transcripts. The quality of the chips was determined using the affyQCR package of BioConductor. Reproducibility of samples was checked using chip-to-chip correlation and signal-to-noise ratio (SNR) methods for replicate arrays. All of the high-quality arrays were included for low- and high-level bioinformatics analyses.
Statistical analysis.
To obtain the signal values, high-quality chips were analyzed by dChip that was applied independently to two different microarrays, which is more robust than MAS5.0 and robust multiarray average (RMA) in signal calculation. The raw probe-level data were normalized using a smoothing-spline invariant set method. The signal value for each transcript was summarized using a perfect-match–mismatch (PM-MM)-based signal-modeling algorithm as described in dChip. In this way, the signal value corresponds to the absolute level of expression of a transcript. To reduce false-positive results, transcripts that were scored as absent on the basis of MAS5 were removed from further analysis. MAS5-retained transcripts were then calculated using the Affy package in BioConductor. Normalized data from both microarrays were combined into one data set using transcripts represented on both arrays. To reduce the batch effect, the ComBat method (http://www.biostat.harvard.edu/wjohnson/ComBat/ComBat.html) was used to normalize gene expression values between data sets before single-gene analysis.
A hierarchical clustering technique was applied to construct an unweighted-pair group method using average linkages (UPMGA) tree using Pearson's correlation as the metric of similarity. Clustering was performed using the “Hclust” package in R language. Principal-component analysis (PCA) was performed by computation and then plotting of the two principal components using the R statistical software package with default settings. PCA projects multivariate data objects onto a lower-dimensional space while retaining as much of the original variance as possible. When comparing two groups of samples to identify genes enriched in a given group, we used the lower confidence bound (LCB) of the fold change (FC) between two groups (NEO versus CR, CR versus CT, and NEO versus CT) as the cutoff criterion. If the 90% LCB of the FC between the two groups was greater than 1.2, the corresponding gene was considered to be differentially expressed. LCB is a stringent estimate of the fold change and has been shown to be the better-ranking statistic (25). Recently, dChip's LCB method for assessing differentially expressed genes has been shown to be superior to other commonly used approaches, such as Microarray Analysis Suite 5.0 and robust multiarray average-based methods. By using custom arrays and quantitative reverse transcriptase real-time PCR, it has been suggested that Affymetrix chips might underestimate differences in gene expression. Based on previous studies, a criterion of selecting genes that have an LCB of greater than 1.2 most likely corresponds to genes with an “actual” fold change in gene expression of at least 3.
Pathway and functional analyses.
Pathway and functional analyses of the differentially expressed genes were performed using the commercial systems biology-oriented package Ingenuity Pathway Analysis (IPA) (Qiagen). The knowledge base contains information on human, mouse, and rat genes, including annotations, synonyms, and over 1.4 million published biological interactions. Thus, this knowledge base provides a framework by which the lists of genes identified by large microarray experiments can be annotated in terms of functional relationships to understand the underlying biological mechanisms. It calculates the P value using Fisher's exact test for each pathway and functions according to the fit of the user's data to IPA databases. The P value measures how likely the observed association between a specific pathway/function and the data set would be if it were due only to random chance, by also considering the total number of functions/pathways/lists of eligible genes in the data set and the reference set of genes (those which potentially could be significant in the data set). In case of interactive networks, all the identified genes were mapped to genetic networks available in the Ingenuity database and were ranked by the score. The score (−log P value) is calculated using Fisher's exact test and indicates the likelihood that a gene will be found in a network due to random chance. For example, if a network achieves a score of 2, it has at least 99% confidence of not being generated by chance alone.
Upstream regulator analysis.
Ingenuity's upstream regulator analysis is a tool that allows transcription regulator prediction based on the genome-wide differentially expressed gene signature. This tool predicts which transcriptional regulators are involved and whether they are likely activated or inhibited. The activation status of a given regulator is predicted through a calculated activation z score where z > 2 predicts activation and z < −2 predicts inhibition.
RESULTS
Stable transduction of the CD21-deficient pre-B-cell lines Nalm6 and Laz221 enables EBV binding.
Virtually all mature B cells and B-cell lines susceptible to EBV infection already express the EBV receptor CD21 (CR) (26). This has impeded analysis of whether the signals transduced upon attachment of EBV impact the outcome of infection and whether CR and/or a noncovalently associated partner is responsible for signal generation. The early pre-B lines Nalm6 and Laz221 lack CR (Fig. 1A). Therefore, we analyzed whether they express B-cell surface proteins known to form complexes with CR (complex 1, CD19/CD81/+/−Leu13/CD225, and complex 2, CD35) (27) in addition to the EBV coreceptor, HLA class II (DR, DP, DQ), required for B-cell fusion and entry (28). Although no CR was detected by cytometry, the coreceptor HLA-II as well as CD19/CD81 complex members were expressed, whereas CD35 was absent (Fig. 1A, left and middle). CD225 (Leu-13/IFITM1/CD225) and CD23 (not shown) were detected only on Laz221 cells, consistent with derivation of each line from separate patients. Staining of an EBV-positive LCL is displayed as a control (Fig. 1A, right).
FIG 1.

Pre-B-cell lines Nalm6 and Laz221, which lack endogenous CR and variably express CR complex members, can stably express recombinant CR and CT and bind EBV. (A) Flow cytometric analysis (FC) of CD21, CD35, CD19/CD81/Leu13(CD225), and HLA-II (DR, DP, and DQ) expression on Nalm6 and Laz221 cells. x axis, log fluorescence intensity; y axis, cell number. An LCL is displayed as a staining control. (B) Expression of CR on two Nalm6 clones (1 and 2) compared with a lymphoma line (BL41) and a LCL that express endogenous CR at different levels. (C) Verification of CR and CT expression by immunoblot (left) and by PCR (right). (D) Demonstration that expression of associated proteins is unmodified in Nalm6CR although coreceptor, HLA-II expression is increased ∼3-fold (left shift of log mean fluorescence intensity). (E) Top four histograms, demonstration that EBV attaches to cells expressing CR and also CT by FC. CR and CT were directly detected with MAb HB-5 anti-CD21 (black line) compared with an isotype control (block). MAb 2L10 anti-EBVgp350/220 was used for detection of bound EBV (black line). No virus served as the negative control (block). An LCL is displayed as a positive control. Bottom histogram, EBV blockade, confirms that rabbit anti-CR blocks EBV attachment to Laz221CR and CT (dark gray block), whereas normal rabbit serum does not (light gray block). See Materials and Methods for details. (F) Demonstration that CT is not cleaved by phosphatidylinositol-specific phospholipase C (PIPLC) (not PI linked) by FC. Top, the GPI-linked proteins CD48 and CD59 are cleaved by PIPLC in HPB-ALL, a control T-cell line, resulting in decreased surface expression. Neither HLA-I nor CR (endogenous), which lack PI linkages, is altered. Bottom, concurrent analysis of Nalm6CT showed no evidence of CT cleavage. CD48 and CD59 are absent and HLA-I uncleaved on Nalm6CT.
Receptor expression on Nalm6 (Fig. 1B) and Laz221 (not shown) cells at levels corresponding to those observed on normal resting B cells was achieved by sequential transfection, selection with Geneticin, and cell sorting (see Materials and Methods). A Burkitt lymphoma cell line, BL41, that expresses endogenous CR at approximately the same level as normal B cells, and an EBV-positive LCL that upregulates CR, a characteristic of many LCLs (29), are displayed for comparison. Expression was further documented by immunoblotting (Fig. 1C, left) and PCR (Fig. 1C, right). Surface expression of CR and of CT (not shown) did not significantly alter expression of the CR-associated cell surface proteins, as shown for Nalm6CR compared with Nalm6NEO (Fig. 1D), although coreceptor expression (HLA-II) clearly increased (∼3-fold) as indicated by a shift in log mean fluorescence intensity (Fig. 1D). While CR is the attachment protein for EBV on normal B cells, EBV initiates infection of certain epithelial cell types through alternate interactions (28, 30). Therefore, we confirmed that purified EBV bound to CR, but not to the NEO control line (Fig. 1E), and verified by flow cytometry that infection was blocked with rabbit anti-CD21 (AP12) directed to the binding site on SCRs 1 and 2 (20) (Fig. 1E, bottom), with the anti-CD21 MAb OKB7 likewise directed to this site (not shown) and the anti-gp350/220 neutralizing antibody 72A1 (not shown). Surprisingly, these results showed that in the complete absence of a cytoplasmic anchor, the ability of CT to support virion attachment to the B-cell surface was not demonstrably altered (Fig. 1E). Type Ia plasma membrane proteins with short intracellular domains of less than 10 amino acids sometimes become tethered to the cytoplasm by lipid, specifically glycosyl phosphatidylinositol (GPI) linkages. CR is a member of the regulators of complement activation (RCA) gene family. In fact, several RCA proteins (e.g., CD55 and CD59) can be GPI linked. To ensure that CT was not GPI linked in these cells, we treated Nalm6CT cells with phospholipase C, which releases GPI. As shown in Fig. 1F, although upon treatment with phospholipase C, PI-linked proteins CD48 and -59 (31) were released from the surface of the T-cell line HPB-ALL, surface expression of both endogenous (HPB-ALL also expresses CR) and Nalm6CT remained unaltered (Fig. 1F, bottom) (note that Nalm6 and Laz221 cells and many LCLs lack CD48 and -59 expression, and therefore HPB-ALL was used as the cleavage control).
EBV infects CR- but also CT-expressing cell lines and expresses latent cycle transcripts.
No viral gene expression was detected after incubation of Nalm6NEO or Laz221NEO (not shown) cells with EBV, as assessed by RNA blot hybridization. However, after introduction of CR into Nalm6 and Laz221 cells, EBV infection was manifest by initiation (24 to 72 h) of a type III latency transcript pattern (Fig. 2A and B). There was minimal evidence of lytic gene expression in these cells, consistent with absence of BHRF1, a highly expressed lytic transcript (Fig. 2A, left, right bottom segment). Preincubation of cells with OKB7, a MAb directed to the EBV binding site of CR, blocked infection, as shown by loss of LMP1 expression, whereas preincubation with an isotype-matched irrelevant MAb, UPC10, did not prevent infection (Fig. 2A, bottom panel). Latent viral RNAs, including LMP1 and EBERS (Fig. 2A) EBNA 2, LMP2A, and EBNA 1, were readily detected by 24 h (Fig. 2A and B and data not shown). Cellular transcripts, including Rag-1, TdT, and CD10, previously observed to be downmodulated upon EBV infection of normal pre-B cells (22), were also observed to decrease (Fig. 2C).
FIG 2.

EBV infection of CR-bearing pre-B-cell lines analyzed by RNA blot hybridization. (A) Top, expression of LMP1 and EBER in Nalm6CR and Laz221CR cells was detected 24 to 72 h after EBV incubation. Nalm6NEO cells served as a negative control. Expression of BHRF1, a highly expressed lytic RNA, was absent. Ak (Akata) cells served as the positive control. GAPDH or 28S RNA (arrow) provided loading controls. Bottom, preincubation of cells with OKB7, a MAb that blocks binding of EBV gp350/220 to CR, prevented infection of Nalm6CR, whereas UPC10, an isotype-matched control, did not. (B) EBNA-1 (top), EBNA-2 (middle), and LMP-2A (bottom) transcripts were also detected, as shown in Nalm6CR cells. (C) Cellular RNAs encoding RAG-1, TdT, and CD10 declined upon EBV infection, as previously observed (22). (D) Infection of CT-bearing cells induced LMP1 (Nalm6 and Laz221) and EBNA2 (Nalm6) expression with a time course similar to that of CR.
Significantly, not only was the intracellular domain of CD21 dispensable for stable virus attachment, it was also dispensable for subsequent infection, as the efficiency and pattern of infection among CT-bearing cells were similar to those with CR (Fig. 2D). No major differences in viral transcripts at 24 to 72 h (Fig. 2D and not shown) were observed between lines expressing wild-type and mutant receptors. A switch in viral promoter usage together with altered patterns of viral gene expression and hyperproliferation, events that typically begin ∼72 h after normal B-cell infection, were not examined here (32, 33).
Although major differences in the efficiency and pattern of infection in the respective CR- and CT-bearing lines were lacking, we reasoned that normal resting B cells could require signals that were no longer relevant for infection or immortalization of established tumor cell lines.
Gene expression profiles of the respective CR, CT and, NEO control lines form separate clusters.
To gain insight into events that immediately followed EBV attachment to its wild-type receptor, we compared the transcriptional responses of CR-, CT-, and NEO-expressing cells before and 1 h after incubation with EBV. Unsupervised clustering using all transcripts showed that the three coordinate cell line sets, Nalm6NEO/Laz221NEO, Nalm6CR/Laz221CR, and Nalm6CT/Laz221CT, formed separate clusters (Fig. 3A) at each time point investigated. The first principal component (PC1) captured 42.5% of the variance, and the second (PC2) captured 17.9% of the variance. The first PCA component revealed that CT-bearing cells exhibited a more distinct pattern of gene expression than cells expressing NEO or CR. The second PCA component, PC2, confirmed that cells expressing wild-type receptor, CR, varied significantly in gene expression patterns compared with those expressing CT and NEO (Fig. 3B). Although PCA and hierarchical clustering (Fig. 3C) uncovered some increased variance in the founder line Laz221NEO, this variation was small compared to that detected between different data sets defined by NEO, CT, and CR.
FIG 3.

Unsupervised clustering and principal-component analysis of the gene expression data. (A) An unsupervised Euclidean distance-based cluster of arrays after normalizing the data demonstrates that B-cell lines that express CT, CR, or NEO form separate clusters that correlate better with one another than with cells expressing an alternate receptor form. (B) PCA analysis demonstrates that CT, CR, and NEO cells have distinct gene expression patterns. (C) Heat map of the differentially expressed genes by comparing NEO, CT, and CR at 1 min and 60 min demonstrates that NEO, CR, and CT cells have a set of unique differentially expressed genes. Rows represent genes, and columns represent samples from CT, CR, and NEO groups. Genes are clustered using row-normalized signals and mapped to the [−1,1] interval (shown in scales beneath each heat map). Red and green represent high and low expression values, respectively.
Stable expression of CR and CT alters the transcriptome of pre-B cells.
A relevant question when recombinant receptors are expressed in cells prior to introduction of a specific ligand(s) is whether and how the receptor or mutant forms of the receptor alter baseline transcription. CR, a developmentally expressed protein in the B-cell lineage, forms cell surface complexes that have the potential to constitutively modify the composition of the transcriptome independent of changes induced by binding of an exogenous ligand(s). Therefore, before investigating the effects of EBV binding, we analyzed whether CR or CT synthesis expression altered constitutive gene expression (time = 1 min). As revealed by hierarchical clustering (Fig. 3C), numerous alterations in gene expression were observed in CR and CT compared with NEO control cells and between CR and CT (top ca. two-thirds of heat map) (Fig. 4A and B). Nevertheless, closer inspection of individual genes with the highest fold change (Tables 1 and 2) uncovered several changes common to CR and CT. Among the most prominent findings was profound downregulation of multiple class I melanoma-associated antigen (MAGE) family member mRNAs encoding the cancer testis antigens MAGEA3, MAGEC1, MAGEA2, MAGEA5, and MAGEA6 (LCBs for CR/CT of −8/−20, −6/−5, −3/−4, −2/−2, and −11/−39, respectively). These transcripts, encoded by genes on chromosome X, are highly expressed during embryogenesis and in many tumors (e.g., malignant melanomas and carcinomas of the lung, head, and neck, as well as urinary bladder, colorectal, prostatic, and renal cell carcinomas and others) but are suppressed in virtually all normal cells (except male germ cells). On the basis of these properties, MAGE family members have been targeted for tumor vaccine development, and some are currently undergoing clinical trials (e.g., trial NCT00944580, to determine the immunologic effects of a MAGE-A1, MAGE-A3, and NY-ESO-1 vaccine in patients with high-risk neuroblastoma, osteogenic sarcoma, and rhabdomyosarcoma). MAGED2, a class II MAGE, is ubiquitously expressed (34) and, unlike the other MAGEs, was not downregulated in CR- or CT-expressing cells. Additional transcripts strongly downregulated in both CR and CT included vesicle-associated membrane protein 8/endobrevin (VAMP8) (−4.1/−7.2), leukocyte receptor tyrosine kinase (LTK and TYK1) (−5.3/−2.3), dopamine decarboxylase (DDC) (−6.8/−1.3), and ZNF32, a zinc finger protein that declined more in CR than CT. Enolase 2 (ENO2) increased most in both CR- and CT-expressing cells (LCBs of 4.55 and 3.13, respectively) and then declined in both following infection (Table 2), as did transcripts encoding the intracellular proteins programmed cell death 4 (PCD4) (LCBs of 2.63/2.78), alpha interferon-inducible gene 6-16 (G1P3) (1.65/2.51), and formin binding protein 2 (FNBP2) (1.77/2.48). Transcripts encoding several immune cell surface antigens, in particular members of the HLA class II family (HLA-DM, HLA-DP, HLA-DR, and HLA-DQ) were upregulated (CT > CR) as were MIC2, CDW52, IL-21R, and CD48 (CR > CT). Notably, fluorescence-activated cell sorter (FACS) analysis (Fig. 1D) confirmed that HLA-DR protein expression was increased as predicted. Based on the comprehensive nature of the microarray analysis, not surprisingly, the majority of gene expression changes observed in CR and CT at 1 min persisted at 1 h despite exposure to virus (Fig. 3C). The upregulated genes that displayed the highest constitutive (one-minute) fold change (LCB scores in Table 2 and heat maps in Fig. 4B) in CR compared with CT included BHLHB2, CASP1, CEBPB, HIG2, IRF1, JUN, MDM2, N33, PLAC8, PLXNB2, PP1R15A, RLF, and RRAD, whereas E2F2, HEY1, ID4, and FASN were most downregulated. Based on these candidates, upstream regulator analysis predicted significantly increased activation of pathways in CR compared with CT related to tumor necrosis factor (TNF) signaling (P value, 1.42E−07), lipopolysaccharide (LPS) signaling (P value, 3.08E−06), and TP53 (P value, 4.49E−06) as well as others discussed below.
FIG 4.

Differentially expressed genes in NEO, CR, and CT cells at 1 min (constitutive changes). (A) Venn diagrams of differentially expressed (downregulated and upregulated) genes in NEO, CT, and CR cells. (B) Heat map of selected top genes constitutively altered (1 min) after recombinant receptor expression. Left, NEO versus CT; middle, NEO versus CR; right, CT versus CR. Genes are clustered using row-normalized signals and mapped to the [−1,1] interval (shown in scales beneath each heat map). Red and blue represent high and low expression values, respectively.
TABLE 1.
Identification of top downregulated genes based on hierarchical clustering of all differentially expressed genes in NEO, CR, and CT
| Gene | Fold change |
|||||
|---|---|---|---|---|---|---|
| 1 min |
60 min |
|||||
| NEO-CR | NEO-CT | CT-CR | NEO-CR | NEO-CT | CT-CR | |
| MAGEA6 | −11.4 | −39.2 | −0.21 | 0 | 0 | −0.21 |
| MAGEA3 | −8.31 | −19.59 | −0.26 | 0 | 0 | −0.12 |
| DDC | −6.77 | −1.26 | −2.05 | 0 | 0 | −1.09 |
| MAGEC1 | −6.12 | −5.11 | 0 | 0 | 0 | 0 |
| ZNF32 | −5.62 | −1.52 | −1.71 | −2.57 | −0.3 | −3.04 |
| VAMP8 | −4.1 | −7.2 | 1.04 | 0 | 0 | 1.73 |
| HSPA1A | −0.54 | −1.71 | 0.65 | −0.37 | −1.35 | 0.67 |
| BHLHB2 | 0.74 | −2.74 | 4.58 | 0.69 | −1.95 | 2.83 |
| HSPA1B | −0.59 | −2.1 | 0.68 | −0.59 | −1.88 | 0.98 |
| LYZ | −0.59 | −3.32 | 2.05 | 0.5 | −2.14 | 1.96 |
| GPRC5B | −0.92 | −4.04 | 1.64 | 0.35 | 0 | 1.53 |
| LAMA3 | −1.83 | −2.86 | 0.82 | −0.21 | −0.33 | 0.87 |
| MYB | −1.02 | −1.9 | 0.96 | −0.97 | −1.64 | 0.98 |
| E2F2 | −1.17 | 2.02 | −3.49 | −0.89 | 1 | −1.1 |
| HEY1 | −0.94 | 1.92 | −3.22 | −0.9 | 1.6 | −1.86 |
| DDC | −4.62 | −0.83 | −2.54 | 0 | 0.4 | −2.13 |
| ID4 | −1.98 | 0.75 | −2.52 | −1.1 | 0.8 | −1.56 |
| FASN | −0.66 | 1.38 | −2.39 | −1.15 | 1.37 | −2.49 |
| HLA-DQA1 | 0.19 | 1.81 | −2.37 | 0.5 | 2.14 | −2.07 |
| LTK | −5.3 | −2.31 | −0.87 | −3.69 | −3.78 | −0.54 |
| TPM2 | −3.61 | −1.62 | −1.05 | −3.25 | −1.25 | −0.5 |
| AGTRL2 | 0.29 | −0.19 | 0.3 | −2.64 | −0.89 | −2.04 |
| ANXA2 | −2.94 | −1.25 | −0.38 | −2.49 | −0.86 | −0.11 |
| G6PD | −1.22 | −0.6 | −0.92 | −2.46 | −0.81 | −1.46 |
| TXNIP | −0.95 | −1.54 | 0.67 | −1.01 | −2.75 | 1.19 |
| C20orf97 | 1.21 | −1.55 | 3.01 | 0.92 | −2.72 | 3.06 |
| GADD45B | 0.9 | −1.74 | 1.91 | −0.72 | −2.63 | 1.86 |
| PPP1R15A | 0.93 | −4.14 | 4.48 | 0.96 | −2.63 | 2.77 |
| ZNF198 | −0.55 | −1.59 | 1.02 | −1.1 | −2.53 | 1.63 |
| PSCD3 | −1.99 | −2.26 | 0.89 | −0.74 | −2.52 | 0.53 |
| HLA-DQA1 | 0.83 | 2.31 | −1.68 | 0.6 | 3.67 | −2.49 |
| GNMT | 0.75 | 1.04 | −1.07 | −1.79 | 1.18 | −2.48 |
| CCNE2 | 0.25 | 1.47 | −0.89 | −0.98 | 1.35 | −2.47 |
| GPRK6 | −1.42 | 1.15 | −1.74 | −0.6 | 1.37 | −2.32 |
| DLAT | −1.49 | −0.94 | −1.54 | −2.07 | 1.01 | −2.31 |
| MAP2K6 | 0.88 | 3.13 | −1.48 | −1.7 | 1.13 | −2.3 |
TABLE 2.
Identification of top upregulated genes based on hierarchical clustering of all differentially expressed genes in NEO, CR, and CT
| Gene | Fold change |
|||||
|---|---|---|---|---|---|---|
| 1 min |
60 min |
|||||
| NEO-CR | NEO-CT | CT-CR | NEO-CR | NEO-CT | CT-CR | |
| AK1 | 1.91 | 0.86 | 1.54 | 2.08 | −0.69 | 1.67 |
| ARHGEF9 | 0.86 | 1.34 | −1.18 | 1.73 | 2.4 | −1.15 |
| ATF3 | 0.92 | −0.96 | 1.99 | 0.95 | −0.61 | 3.46 |
| ATRN | 0.94 | 2.41 | −0.99 | 0.6 | 1.24 | −1.32 |
| BHLHB2 | 0.74 | −2.74 | 4.58 | 0.69 | −1.95 | 2.83 |
| C14orf1 | −0.97 | 1.97 | −2.15 | −0.82 | 2.18 | −1.96 |
| C20orf97 | 1.21 | −1.55 | 3.01 | 0.92 | −2.72 | 3.06 |
| CASP1 | 2.04 | −0.92 | 2.19 | 2.84 | −0.67 | 2.14 |
| CD48 | 2.41 | 1.2 | 1.68 | 2.1 | 1.03 | 1.9 |
| CDKN1C | 3.49 | 0 | 0.54 | 0 | 0 | 0 |
| CDW52 | 3.77 | 1.7 | 1.42 | 4.42 | 2.45 | 1.39 |
| CEBPB | 1.28 | −1.38 | 2.99 | 1.05 | −2.19 | 2.94 |
| COL5A1 | 2.6 | 0.74 | 1.04 | 2.98 | 0.51 | 1.09 |
| COL6A3 | 0.81 | 1.6 | −1.25 | 0.9 | 2.02 | −1.66 |
| CPM | 1.37 | 3.48 | −1.37 | −0.01 | 0.81 | −1.75 |
| CYBA | 1.01 | 1.62 | −1.08 | 1.39 | 1.92 | −1.1 |
| DEPP | 1.13 | −0.41 | 1.65 | 2.14 | −0.6 | 3.33 |
| ENO2 | 4.55 | 3.13 | 1.35 | 0.95 | 0.78 | 0.92 |
| FADS2 | −0.68 | 2.31 | −2.01 | 0.45 | 2.2 | −1.76 |
| FLJ11126 | 0 | 2.91 | −1.01 | −0.2 | 0.98 | −2.23 |
| FLJ12595 | −1.73 | −1.92 | 0.7 | 0.77 | −3.45 | 5.43 |
| FLJ12895 | 4.61 | 0 | 3.97 | 1.96 | −0.94 | 5.94 |
| FLJ20452 | 0.84 | 1.73 | −1.06 | 0.89 | 2.16 | −1.37 |
| FNBP2 | 1.77 | 2.48 | −1.24 | 1.02 | 1.11 | −0.75 |
| FOSB | 0.7 | −0.53 | 0.64 | 1.08 | −0.7 | 2.73 |
| G1P3 | 1.65 | 2.51 | −0.66 | 0.57 | 0.93 | −0.94 |
| GALNAC4S-6ST | 3.02 | 1.26 | 1.56 | 2.82 | 0.92 | 2.67 |
| H1F3 | 0.98 | −1.09 | 1.77 | 1.42 | −1.7 | 4.24 |
| H1F4 | 0.67 | −0.68 | 1.63 | 0.83 | −2.08 | 3.91 |
| H2AFC | 0.57 | −0.99 | 1.4 | 0.93 | −2.06 | 2.79 |
| H2BFA | 1.2 | −1.39 | 1.94 | 2.11 | −1.05 | 2.83 |
| H3FB | −0.81 | −2.28 | 1.75 | 0.94 | −2.3 | 2.86 |
| H3FL | −0.57 | −1.68 | 1.15 | 0.81 | −1.56 | 2.99 |
| H4F2 | 0.61 | −1.06 | 0.71 | 1.02 | −0.83 | 3.05 |
| H4FI | 1.02 | −0.71 | 1.08 | 0.96 | −0.76 | 4.21 |
| H4FK | 1.04 | −0.65 | 1.05 | 0.7 | −1.52 | 5.78 |
| HLA-DMA | 1.18 | 2.08 | −1.2 | 0.89 | 2.08 | −1.16 |
| HLA-DPA1 | 1.36 | 1.99 | −1.28 | 1.56 | 2.22 | −1.21 |
| HLA-DRB3 | 1.32 | 2.31 | −0.78 | 1.14 | 1.79 | −0.79 |
| HLA-DRB4 | 1.27 | 2.32 | −0.81 | 1.23 | 1.8 | −0.83 |
| HLA-DRB6 | 1.24 | 2.39 | −0.77 | 1.03 | 1.76 | −0.84 |
| IL21R | 2.01 | 1.19 | 0.95 | 2.04 | 1.24 | 0.99 |
| IRF1 | 0.97 | −1.71 | 2.42 | 1.2 | −0.97 | 2.07 |
| JUN | 1.07 | −1.45 | 2.68 | 1.79 | −1.04 | 2.24 |
| KIAA1332 | 1.32 | −0.88 | 2.25 | 1.89 | −0.51 | 2.7 |
| MAF | 0 | 1.7 | −1.31 | 0 | 2.01 | −0.9 |
| MAGED2 | 0.71 | 1.81 | −1 | 0.94 | 2.15 | −1.88 |
| MDM2 | 1.32 | −1.95 | 2.9 | 1.06 | −1.45 | 2.01 |
| MGC861 | 0.58 | 1.47 | −1.25 | 0.37 | 2.06 | −0.91 |
| MIC2 | 3.72 | 0.84 | 1.07 | 1.16 | 0.32 | 0.85 |
| MICA | 1.31 | −1.25 | 2.34 | −0.7 | −1.42 | 1.63 |
| N33 | 3.86 | −0.6 | 3.28 | 4.11 | 0.67 | 3.64 |
| PALM | 2.6 | 0.76 | 1.58 | 2.89 | −0.79 | 2.7 |
| PDCD4 | 2.63 | 2.78 | −1.03 | 1.16 | 1.05 | 0.86 |
| PFKL | 1.59 | 1.52 | 0.92 | 2.05 | 1.53 | 1.3 |
| PLAC8 | 1.72 | −0.92 | 2.34 | 1.77 | 0.76 | 1.62 |
| PLXNB2 | 2.31 | −0.49 | 2.21 | 1.65 | −0.45 | 3.77 |
| PPP1R15A | 0.93 | −4.14 | 4.48 | 0.96 | −2.63 | 2.77 |
| PTPRCAP | 1.56 | 2.5 | −1.17 | 1.28 | 2.08 | −0.98 |
| RLF | 1.06 | −2.26 | 2.49 | 0.99 | −1.3 | 1.89 |
| RNU2 | −0.28 | −0.78 | 0.77 | 0.77 | −2.5 | 3.18 |
| RRAD | 1.6 | −0.72 | 2.61 | 1.45 | −0.58 | 2.72 |
| SCGF | 1.68 | 0.62 | 1.48 | 2.05 | −0.88 | 1.98 |
| SERPINE1 | 0 | −0.41 | 0 | 1.06 | −0.43 | 3.21 |
| SIAH1 | 0.98 | 1.87 | −1.1 | 0.84 | 1.92 | −1.12 |
| SNX17 | 1.02 | 2.31 | −1.37 | 0.54 | 2.04 | −1.26 |
| SPINK4 | 1.47 | 1.38 | 0.51 | 1.59 | 2 | 0.55 |
| TCL6 | 0.77 | 2.45 | −1.2 | 0.82 | 0.9 | −0.66 |
| TNS | 4.66 | 0.91 | 3.44 | 5.5 | 0.78 | 4.53 |
| TRIM44 | 2.76 | 1.8 | 1.45 | 3.13 | 1.39 | 1.85 |
The B-cell transcriptome is minimally altered by EBV incubation in the absence of receptor expression.
Gp350/220, the EBV glycoprotein ligand that binds CR, abundantly decorates the virion surface. Gp350/220, or possibly other envelope proteins, might contact cells through low-affinity interactions that cross-link cellular pattern recognition proteins (e.g., Toll-like receptors [TLRs]) or other cell surface innate immune proteins. Such interactions would not result in infection but could alter the cellular transcriptome, inducing signals that are distinct from or, more likely, would overlap those delivered through CT or CR, itself an innate immune system protein.
The observation that the NEO control cell lines in the absence of infection displayed some consistent changes among transcripts after 60 min (Fig. 3) suggested that responses to EBV might occur even virus was present only in the surrounding cellular milieu. Therefore, we performed a class comparison of NEO lines to identify transcripts that were differentially expressed after 1 h. We detected 253 upregulated (215 unique to NEO) and 260 downregulated (167 unique to NEO) transcripts with an LCB of >1.2 (Fig. 5A). However, ∼90% of these differentially expressed mRNAs had an LCB of <1.5, indicating that changes were modest. A heat map of the top differentially expressed transcripts in NEO control cells at 1 and 60 min is displayed in Fig. 5B (left). We also analyzed whether the differentially expressed genes in NEO lines (1 versus 60 min) were retained in CR and CT as a vestige of receptor-independent low-affinity interactions between EBV and the cell. Common to all samples, 5 transcripts were upregulated and 31 were downregulated (Fig. 5B) To address whether specific biological effects could be attributed to these shared genes, we performed separate function and pathways analyses on the transcripts that populated the overlapping NEO compartments of the Venn diagrams. Canonical pathway analysis, using the Ingenuity Pathway Analysis package on overlapping subsets of transcripts, did not detect significant enrichment in any pathway. Functional and network-based analysis of NEO-expressing cells uncovered only minor changes in cellular assembly, regulation of cellular morphology, development, and proliferation. No major changes in interferon response pathways/factors or TLR family-related downstream signaling effector transcripts were identified in the NEO control cells after EBV incubation. Overall, in the absence of an attachment protein, incubation with EBV did not appear to have major effects on cellular gene expression beyond limited modulation of acute-phase response-related transcripts that were largely negative and potentially reflect nonspecific plasma membrane perturbation.
FIG 5.

Comparison of genes differentially altered in NEO, CT, and CR at 1 versus 60 min after incubation with EBV. (A) Venn diagrams of differentially expressed (downregulated and upregulated) genes in NEO, CT, and CR cells. (B) Heat map of selected top genes changing from 1 to 60 min within the cell lines NEO, CT, and CR. (C) Heat map depicting selected top genes changing at 60 min in NEO versus CT, NEO versus CR, and CT versus CR. Genes are clustered using row-normalized signals and mapped to the [−1,1] interval (shown in scales beneath each heat map). Red and blue represent high and low expression values, respectively.
Cellular genes representing distinct functional categories are differentially expressed in CR- and CT-expressing cells at 60 min after exposure to EBV.
Although cells stably expressing CR and CT constitutively expressed several transcripts in common compared with NEO-bearing cells, the baseline transcriptome was more widely divergent than had been predicted. Following EBV incubation (60 min), specific differences in the respective transcript profiles of CR and CT could be clearly identified (Tables 1 and 2; Fig. 5A to C). Two hundred fifty-eight transcripts were uniquely upregulated and 425 were downregulated upon EBV binding to CR-bearing cells, compared with 269 upregulated and 219 downregulated in CT-bearing cells (Fig. 5A). Thus, almost twice as many transcripts were downregulated in CR- compared with CT-expressing cells. The heat map displayed in Fig. 5B shows selected top changes in gene expression within each receptor grouping after 60 min, whereas the heat map displayed in Fig. 5C reveals major differences between these receptor groupings at 60 min after receptor contact. Histones, particularly those belonging to histone cluster 1, were a frequent target of altered gene regulation, particularly in CR. The transcription factor c-JUN, first identified as a proto-oncogene and active in many cellular processes, was among the genes whose expression was clearly increased in CR-bearing cells (60 min), but this was accompanied by minimal changes of FOS and FOSB transcripts compared with those in CT-bearing cells. The FOS family members are encoded by leucine zipper proteins that can dimerize with Jun to form the AP1 transcription factor complex, which is implicated in the regulation of cell proliferation, differentiation, and transformation. c-Fos transcription was previously found to be minimally activated within the first hour after cross-linking of CR (35) on primary B cells, whereas c-Myc was unaltered, a point we will return to. Though approximately twice as many genes were downregulated in CR- compared with CT-expressing cells after EBV exposure (Fig. 5A), Tables 1 and 2 indicate that the degree of down- compared with upregulation (based on LCB values) of altered transcripts was less. Interestingly, among the major downregulated genes, several were involved in metabolism, particularly lipid metabolism, and therefore may reflect transient growth inhibition (SPHK2, HMGCR, MRPL12, INSIG1, and FASN). The top modified genes for CT versus CR at 60 min (Fig. 5C) included MDM2, GADD45B, UBQLN3, IER3, TIEG, PIM1, and SMAD7 (up) and TNFRSF21, ARHGEF12, PIK3R3, and CCNE2 (down). These and others were subjected to a more comprehensive functional and pathway analyses, as discussed below. In many cases there was already evidence of altered constitutive signaling through full-length CR that was then amplified by EBV interaction.
Functional and pathway analyses of differentially expressed genes implicate the cytoplasmic domain in gene regulation.
The Ingenuity Pathway Analysis program provides a framework by which lists of genes identified by large microarray experiments can be annotated in terms of functional relationships to provide new understanding of fundamental biological mechanisms. For interactive networks, all identified genes are mapped to genetic networks available in the Ingenuity database and ranked by a score (−log P value) that indicates the likelihood that a gene will be found in a network by random chance. Table 3 displays the significantly ranked functional categories derived from the entire data sets for CR and CT (and compared with NEO) at 60 min after EBV incubation. For each of the categories cell growth and proliferation, hematological system development and function, cellular development, cellular movement, cell death, cell-mediated immune response, and cell cycle, the P value of CR exceeded those of CT and NEO. This finding is consistent with an augmented role of the wild-type receptor in relation to these functions at 1 h after infection. In contrast, the functional category related to antigen presentation ranked in CT alone. Although these data reveal that specific functional categories are impacted, they do not uncover predicted outcomes, as that requires inspection of the individual components of each functional category.
TABLE 3.
Significantly affected functional categoriesa
| Functional category |
P valueb (1–60 min) |
||
|---|---|---|---|
| CR | CT | NEO | |
| Cellular growth and proliferation | 1.35E−05–6.15E−02 | 1.28E−03–1.27E−01 | 1.26E−04–1.21E−01 |
| Hematological system development and function | 4.99E−05–7.25E−02 | 1.28E−03–1.27E−01 | 6.74E−03–1.21E−01 |
| Cellular development | 4.41E−05–7.25E−02 | 4.05E−03–1.27E−01 | 9.43E−03–1.21E−01 |
| Cellular movement | 4.43E−05–7.25E−02 | 2.52E−02–1.27E−01 | 9.58E−03–1.21E−01 |
| Cell death | 1.01E−04–7.25E−02 | 6.54E−03–1.27E−01 | 1.58E−02–1.21E−01 |
| Cell-mediated immune response | 5.25E−04–7.25E−02 | 1.28E−03–1.27E−01 | 2.34E−02–1.21E−01 |
| Cell cycle | 5.46E−04–6.52E−02 | 6.81E−02–1.27E−01 | 2.34E−02–1.21E−01 |
| Lipid metabolism | 3.12E−02–5.96E−02 | 4.19E−02–1.27E−01 | 3.66E−02–1.21E−01 |
| Antigen presentation | 9.96E−03–1.27E−01 | 6.51E−02–1.21E−0 | |
| Immune cell trafficking | 5.96E−02–5.96E−02 | 1.28E−03–1.27E−01 | 6.51E−02–1.21E−01 |
Significant perturbations in biological function categories were identified using the Ingenuity Pathway Analysis biological function analysis tool. Differentially expressed genes in each cell type were uploaded into IPA for analysis.
The P value is corrected for multiple tests.
Identification of the top pathways affected at 60 min after EBV infection in CR- compared with CT-bearing cells (Fig. 6) was based upon the pooled analysis of individual genes identified in the original heat map. Of note, several findings were consistent with results from certain early studies that had interrogated CR signaling upon EBV contact. Pathways significantly activated by CR interaction and that supported some of the prior studies included NfkB activation by viruses (36), interleukin-6 (IL-6) signaling (37, 38), RAR activation (suppresses the EBV lytic cycle factor Z) (39), and phosphatidylinositol 3-kinase (PI3K)/Akt signaling (implicated in a prior study of a CD19-negative cell line) (40). Insulin-like growth factor 1 (IGF-1) signaling and molecular mechanisms of cancer signaling may reflect increased expression of immediate early transcripts that become increasingly relevant as latent infection progresses. Previously unrecognized functions include upregulation of several potentially interrelated pathways such as IL-15 signaling, IL-2 signaling, CTLA4 signaling in cytotoxic T lymphocytes, and ceramide signaling. Upregulation of ephrin receptor signaling, platelet-derived growth factor (PDGF) signaling, and CML signaling were novel, and their relevance is currently undefined. In contrast to CR-bearing cells, CT-bearing cells showed activation of several proapoptotic pathways, including death receptor signaling, apoptosis signaling, lymphocyte-mediated apoptosis signaling, and interferon signaling. The absence of CR activation of these same proapoptotic pathways following infection is notable.
FIG 6.

Comparison of top signaling pathways affected in different receptor-bearing cell types. (A) CT; (B) CR. Sets of differentially expressed genes (LCB of >1.2) between cell lines were uploaded onto Ingenuity Pathway Analysis and corresponding signaling pathways predicted. Statistical significance was set at −log P = 2.
In addition to direct pathway analysis, we also probed the database to identify predicted upstream regulators of functional pathway components. This analysis documented an overall increased number and diversity of activated upstream regulators in CR compared with CT and NEO after EBV incubation, highlighting the increased modulation of cellular gene expression following virus contact with CR (Fig. 7).
FIG 7.

Comparison of predicted upstream regulators of NEO, CT, and CR transcripts at 60 min after EBV infection. Differentially expressed genes were processed through Ingenuity Pathway Analysis to predict the downstream regulators whose activation status was affected following EBV interaction. No regulators reached the significant activation z score of −2 or +2 in NEO (A), whereas several regulators were predicted to be activated (z-score of >2) in CT (B) and a significantly larger number of regulators was either activated or inhibited (z score of <−2) in CR (C).
Validation by RT-PCR of selected differentially expressed transcripts.
To validate the results of Affymetrix analysis and assess whether modulation of gene expression was consistent with older findings, some based on interrogation of primary B cells, we performed RT-PCR on selected transcripts from the Nalm6 line, as Nalm6 is known to express normal p53 and lacks a c-myc translocation. Analyzed transcripts included the tumor-associated class I MAGE transcripts A3 and C1 (downregulated) contrasted with the ubiquitously expressed class II MAGE D2 (unchanged), the immediate early gene Jun (upregulated) contrasted with c-fos and FosB (upregulated but minimally changed upon infection in CR) and c-myc (unchanged), selected histone proteins (more variable than predicted by microarray analysis, suggesting that the Laz221 line may have contributed to detected changes), and other genes that predict novel CR functions (e.g., PIM1 and ATF3) (Fig. 8).
FIG 8.

RT-PCR validation of selected differentially expressed genes, including MAGE family, immediate early proto-oncogenes, histones, and novel candidates.
DISCUSSION
For many viruses, the first encounter with a surface receptor not only tethers the virion to the cell but also activates signal transduction pathways that help create a receptive intracellular environment. Downstream signaling events emanating from this interaction can be critical for establishing successful infection, and their investigation may identify potential cellular targets for antiviral therapy.
Attachment of EBV gp350/220 to CD21 (CR) initiates efficient latent infection of primary B cells, CR-positive B-cell lines, and certain other cell lines transfected with CR and HLA-II (the coreceptor) (28). The observation that the cytoplasmic domain of CD21 is short (34 amino acids) has led to speculation that it does not signal (12). In support of this proposal and in contrast to an earlier report, expression of recombinant receptor lacking its intracellular domain (CT) in an epithelial cell line (41) and, as we now show, in two B-cell lines initiates latent infection (42). Nevertheless, normal resting B cells, the physiologic target, differ from established immortal lines that proliferate constitutively and are protected from apoptosis by genetic alterations. Therefore, primary B cells may be dependent on CD21-mediated signaling events.
Here we investigated the changes in cellular transcripts that occur 60 min after EBV attachment but before initiation of EBV-driven transcription (43, 44). Our hypothesis was that in the absence of a tractable system for analysis of normal primary B-cell infection, comparative study of clonally derived and synchronized pre-B cells expressing CR, compared with CT (illustrated in Fig. 9), would identify cellular pathways required for initial EBV infection in normal B cells, even though they were no longer essential in a tumor cell microenvironment. To that end, the cellular transcriptome was analyzed in Nalm6 and Laz221 sublines stably expressing a NEO control vector, CR, or CT at 60 min after infection, when viral transcripts could not yet be detected, although infection was documented by continued culture and analysis. Profiling of mRNAs from the respective sublines revealed cellular gene expression patterns within each of the respective CR, CT, and NEO subsets that diverged at 60 min after exposure to EBV. Two major sets of findings stem from these studies.
FIG 9.
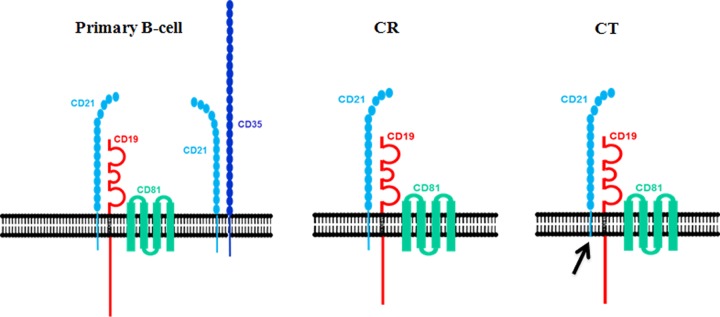
Schematic of CD21 complex formation on normal resting (primary) B cells compared with transfectant cell line models (CR and CT) used in this work.
First, although we did not set out to investigate the effects of constitutive recombinant receptor expression, upon analysis it became apparent that stable expression of CR compared with CT exerted very different effects on cellular gene expression, despite the fact that only the short intracellular fragment was removed. Nevertheless many of the top changes were shared and fell into categories related to the state of B-cell differentiation. In stable CR- and CT-bearing lines (pre-EBV exposure), multiple class I MAGE family transcripts dramatically decreased, whereas diverse mRNAs associated with B-cell maturation increased. The MAGE family consists of more than 60 genes that share a conserved MAGE homology domain (45). Class I MAGEs are highly expressed tumor antigens that are silent in all normal adult cells except germ line cells, which lack HLA and, as a result, are under intense study as targets for cancer immunotherapy (46). Some MAGE proteins interact with RING domain proteins such as E3 ubiquitin ligases, potentially modulating their activity (34). Although little is currently known about class I MAGE function(s), accumulating evidence suggests a role for the class I MAGE A and C proteins, identified here, in blockade of apoptosis through suppression of p53 activity (47) as well as in prevention of senescence (MAGE A2) (48). Additional genes coordinately downregulated that have also been directly or indirectly linked to suppression of differentiation include MYB, LTK, and TXNIP.
In contrast to the MAGE family, diverse mRNAs associated with B-cell maturation constitutively increased in CR- and/or CT-bearing cells, including C2TA, HLA-II family genes (DR, DP, DQ, and DM), other immune system genes, and genes more generally linked to differentiation (IL21R, MIC2, CD48, CDw52, CPM, SNX17, SCGF, TNS, ENO2, FNBP2, and PTPRCAP). Moreover, HLA-II was clearly increased on the plasma membrane of Nalm6CR cells (Fig. 1). As noted, on resting B cells CR is associated primarily with either CD19/CD81 or CD35. However, established B-cell lines (including Nalm6 and Laz221) typically lack CD35, though it is not well understood why (49). Because stable, synchronized CR- and CT-bearing lines, compared with NEO controls, displayed multiple changes in gene expression patterns associated with B-cell maturation, the observed changes could not be attributed to the cytoplasmic domain of CR, nor was CD35 required. Rather, previously described interactions between the extracellular and transmembrane regions of CD21 and CD19/CD81 (50), perhaps constitutively activating CD19, a molecule with a long signal-rich intracellular domain that is expressed earlier during B-cell differentiation than CD21 (51), may, upon constitutive interaction with CD21, transmit information that promotes B-cell maturation. The results of this analysis should serve as a useful resource for further studies.
Second, demonstration of differential gene expression in lines expressing CR versus CT versus NEO at 1 h after EBV incubation provided new evidence that the intracellular fragment of CD21 is functional and added support for some older findings that when directly toggled (antibody and recombinant ligand), CR can signal independently of CD19/CD81 and CD35 (i.e., in induction of c-fos but not c-myc transcripts [35] and activation of PI3K/Akt signaling [40]). As shown in Table 2 and the Fig. 6 heat maps, AP1 family members c-jun, and to a lesser extent fos (FOS) and FOSB, were among the transcripts upregulated at 1 h after EBV incubation in CR compared with CT. Pathway analysis provided indirect evidence of PI3K/AKT activation as previously detected in a study of a CD19- and CD35-negative myeloid cell line (40). Although pathway analysis also supported older studies indicating that NF-κB is rapidly activated upon contact with CD21, these data did not eliminate the possibility that in the earlier studies NF-κB activation resulted from CD35 binding and/or CD19 interaction (36, 38). Two previous reports (37, 52) described rapid elevation of IL-6 transcripts upon CR contact with either EBV or recombinant gp350/220. We did not detect an early increase in IL-6 transcripts in any of our comparisons (though downstream targets were consistent with emerging activation by IL-6 as well as other cytokines). One possibility is that the 1-h time point was too early to detect changes (in one study IL-6 transcripts were increasing at 1 h but peaked later) (52). Another possibility is that IL-6 signaling is altered in these tumor lines or that the signal in these earlier studies actually emanated from CD35 or CD19/CD81 (in both studies CD19 was used for positive selection and may have primed cells). Furthermore, as shown in Fig. 1E, there is some variation in the level of receptor expression on the respective lines that may have contributed to these and certain other differences in signaling (e.g., for Nalm6, CT > CR, whereas for Laz221, CR > CT). However, pooling of data sets as depicted in Fig. 3 indicates that most such variance is compensated.
It is well known that expression of EBV genes reprograms the cellular transcriptome. The ongoing convergence of cellular responses postinfection facilitated informative profiling of cellular responses in a mixed primary B-cell population (four donors) starting 3 days after infection and in a related elegant study documenting a cellular DNA repair response during the first week after infection (7, 33). But what of the earliest event: virion attachment to an extremely heterogeneous B-cell population before de novo expression of EBV genes? To circumvent obstacles and limit the amount of noise (though not without the caveat of interrogating tumor lines), we stably transfected clones from two B-cell lines that lack CD21 and CD35, replacing CD21 with either wild-type receptor, CR, or a receptor devoid of the cytoplasmic domain, CT (and documenting that it was not PI linked). Thus, we were able to maintain equivalent levels of surface receptor expression, mimic the expression level on normal B cells, and begin each experiment with synchronized, clonally derived cells. Because CD35 was absent and CR/CD21 is known to associate with CD19 only through the extracytoplasmic regions of the respective proteins, deletion of the intracellular domain was predicted to distinguish cellular changes in gene expression resulting from CD19 association from those requiring the cytoplasmic fragment of CR/CD21 (Fig. 9, diagram of wild-type and mutant receptors). Although differences in the levels of expression of individual genes within and between CR and CT were often small, as predicted at this very early time point, the functional categories and the pathway analyses which incorporated the total number of gene changes revealed many more changes in addition to activation of distinct pathways in CR. Functional categories significantly impacted by CR, compared with CT and/or NEO, included cell growth and proliferation/cell cycle, hematological system development and function/cellular development, cell death, lipid metabolism, and, interestingly, immune cell trafficking/cellular movement as well as cell-mediated immune response. The content of the functional categories was further revealed in a pathway analysis that independently confirmed activated categories, providing additional detail. Upon comparison of CR with CT, pathways with the highest comparative P values for CR included molecular mechanisms of cancer/NF-κB, activation by viruses/CML signaling/PI3K/Akt signaling, growth factor activation (IGF-1 and PDGF), cytokine activation (IL-2, IL-6, and IL-15 signaling), and, interestingly, CTLA-4 signaling in cytotoxic T lymphocytes (negative signaling). The modulation of functions/pathways typically associated with CD4 T-cell mobilization may support the previous observation that EBV-immortalized B cells behave as T-cell-activated B cells (8) and suggests that relevant signals may begin as soon as cells are contacted. In striking contrast, pathways with the highest P values in CT, which were nonsignificant in CR, included death receptor signaling/apoptosis signaling/lymphocyte-mediated apoptosis of cells, VDR/RXR activation (potentially antiproliferative), and interferon signaling. These observations raise the possibility that the latter pathways are, by comparison, repressed in CR-expressing cells both constitutively and more prominently upon multimerization following EBV engagement as a result of signals transduced through the cytoplasmic domain. This hypothesis is conversely supported by examination of the heat maps in Fig. 5 and 6, which show enhanced expression of MDM2, PIM1, and other antiapoptotic genes at 1 h after infection of CR but not CT cells. Although it was not formally tested, we did not note a comparative decrease in the number of infected CT cells over the subsequent week, perhaps because of compensatory mechanisms in the tumor lines.
Similar to the case for other herpesviruses, EBV DNA is introduced into the cell nucleus as a linear fragment essentially devoid of nucleosomes. Evidence suggests that it is several hours before covalently closed circular episomes form, eliminating exposure of the cell to double-strand breaks (53). Signals transduced through CR may increase expression of genes able to protect acutely infected cells from cell death until homologous recombination or another form of DNA repair is achieved. No functional category or pathway associated with a classic DNA repair program was highly activated at this early time point, in contrast to that reported for days 3 to 4 postinfection (7, 33). The alteration of mRNAs encoding multiple cluster 1 histones (chromosome 6) comprising all four of the core histones that make up the nucleosome (i.e., H2AFC, H2AFM, H2BF, H2BFG, H2BI, H3FA, H3FB, H3FL, H4FL, H4FK, H4FI, and H4FK) and also a single cluster 2 histone, H4F2 (note that nomenclature varies; the NCBI database and references 54 and 55 provide alternative names), as well as linker H1 histones (H1F3 and H1F4) that bind DNA between nucleosomes, was much more prominent in CR than CT at 1 h after EBV exposure and raises speculation that virus attachment may modulate specific histone clusters. One possibility is that some of these histones are rapidly regulated in order to provide a host cell-generated chromatin barrier that rapidly covers naked virus DNA that otherwise would be a nidus for onset of virus replication, leading to cell death. Modified expression of other histones may allow for activation of innate immune responses or alter cell cycle regulation, to name just a few alternatives. Although little has been reported about the precise functions of many of these histones, the conserved linker histones (H1F3 and H1F4, also known as H1D and H1E) were recently directly implicated in epigenetic silencing in mice by regulating both DNA methylation and posttranslational modification of histone H3 (56). Most current studies of histone-based regulation, particularly of herpesvirus DNA, have focused on epigenetic modifications. An intriguing question raised here is whether herpesvirus genomes become decorated with specific histone cluster members to support latency and/or whether following virus attachment, distinct histone proteins are specifically expressed or repressed.
Although we show in this report that the cytoplasmic domain of CD21 is not required for infection of immortalized B-cell lines, comparative gene expression profiling indicates that modifications induced at 1 h following EBV attachment to the wild-type receptor differ from those transduced through a receptor that lacks the cytoplasmic domain. Somewhat surprisingly, despite discernible similarities, the transcriptome of cells that stably expressed CT compared with CR was highly variant, suggesting that much of the signaling mediated by the CR cytoplasmic domain is constitutive and that this signaling is predominantly amplified rather than globally transformed upon EBV binding and receptor multimerization. Our findings provide insight into the specific pathways and genes altered upon virus contact with its high-affinity receptor CD21. Given the heterogeneity of normal B-cell populations at the time of primary infection, future studies can now focus on uncovering whether candidate genes identified here are key determinants of a successful primary infection and/or whether other attachment proteins, such as CD35, contribute major signals that determine the course of primary B-cell infection. Recent advances in technology, including better methods for transduction of primary cells and analysis of RNA presence through methods such as whole-transcriptome shotgun sequencing (RNA-seq), will refine future work.
Footnotes
Published ahead of print 5 March 2014
REFERENCES
- 1.Rickinson AB, Kieff E. 2007. Epstein-Barr Virus, p 2655–2700 In Knipe DM, Howley PM. (ed), Fields virology, vol 2 Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Luzuriaga K, Sullivan JL. 2010. Infectious mononucleosis. N. Engl. J. Med. 362:1993–2000. 10.1056/NEJMcp1001116 [DOI] [PubMed] [Google Scholar]
- 3.Tanner J, Weis J, Fearon D, Whang Y, Kieff E. 1987. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 50:203–213. 10.1016/0092-8674(87)90216-9 [DOI] [PubMed] [Google Scholar]
- 4.Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. 1984. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. U. S. A. 81:4510–4514. 10.1073/pnas.81.14.4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nemerow GR, Mold C, Schwend VK, Tollefson V, Cooper NR. 1987. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J. Virol. 61:1416–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frade R, Barel M, Ehlin-Henriksson B, Klein G. 1985. gp140, the C3d receptor of human B lymphocytes, is also the Epstein-Barr virus receptor. Proc. Natl. Acad. Sci. U. S. A. 82:1490–1493. 10.1073/pnas.82.5.1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikitin PA, Yan CM, Forte E, Bocedi A, Tourigny JP, White RE, Allday MJ, Patel A, Dave SS, Kim W, Hu K, Guo J, Tainter D, Rusyn E, Luftig MA. 2010. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8:510–522. 10.1016/j.chom.2010.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thorley-Lawson DA. 2001. Epstein-Barr virus: exploiting the immune system. Nat. Rev. Immunol. 1:75–82. 10.1038/35095584 [DOI] [PubMed] [Google Scholar]
- 9.Asokan R, Hua J, Young KA, Gould HJ, Hannan JP, Kraus DM, Szakonyi G, Grundy GJ, Chen XS, Crow MK, Holers VM. 2006. Characterization of human complement receptor type 2 (CR2/CD21) as a receptor for IFN-alpha: a potential role in systemic lupus erythematosus. J. Immunol. 177:383–394 http://www.jimmunol.org/content/177/1/383.long [DOI] [PubMed] [Google Scholar]
- 10.Roozendaal R, Carroll MC. 2007. Complement receptors CD21 and CD35 in humoral immunity. Immunol. Rev. 219:157–166. 10.1111/j.1600-065X.2007.00556.x [DOI] [PubMed] [Google Scholar]
- 11.Mongini PK, Jackson AE, Tolani S, Fattah RJ, Inman JK. 2003. Role of complement-binding CD21/CD19/CD81 in enhancing human B cell protection from Fas-mediated apoptosis. J. Immunol. 171:5244–5254 http://www.jimmunol.org/content/171/10/5244.long [DOI] [PubMed] [Google Scholar]
- 12.Tedder TF, Zhou LJ, Engel P. 1994. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol. Today 15:437–442. 10.1016/0167-5699(94)90274-7 [DOI] [PubMed] [Google Scholar]
- 13.Barel M, Balbo M, Le Romancer M, Frade R. 2003. Activation of Epstein-Barr virus/C3d receptor (gp140, CR2, CD21) on human cell surface triggers pp60src and Akt-GSK3 activities upstream and downstream to PI 3-kinase, respectively. Eur. J. Immunol. 33:2557–2566. 10.1002/eji.200324059 [DOI] [PubMed] [Google Scholar]
- 14.Gill MB, Roecklein-Canfield J, Sage DR, Zambela-Soediono M, Longtine N, Uknis M, Fingeroth JD. 2004. EBV attachment stimulates FHOS/FHOD1 redistribution and co-aggregation with CD21: formin interactions with the cytoplasmic domain of human CD21. J. Cell Sci. 117:2709–2720. 10.1242/jcs.01113 [DOI] [PubMed] [Google Scholar]
- 15.Tessier J, Cuvillier A, Glaudet F, Khamlichi AA. 2007. Internalization and molecular interactions of human CD21 receptor. Mol. Immunol. 44:2415–2425. 10.1016/j.molimm.2006.10.014 [DOI] [PubMed] [Google Scholar]
- 16.Filippini G, Griffin S, Uhr M, Eppenberger H, Bonilla J, Cavalli F, Soldati G. 1998. A novel flow cytometric method for the quantification of p53 gene expression. Cytometry 31:180–186. [DOI] [PubMed] [Google Scholar]
- 17.Lazarus H, Barell EF, Krishan A, Livingston DM, Harris K, Schlossman SF, Chess L. 1978. Characterization of a unique cell line (LAZ 221) from human acute lymphocytic (“null” cell) leukemia. Cancer Res. 38:1362–1367 [PubMed] [Google Scholar]
- 18.Simmons D, Makgoba MW, Seed B. 1988. ICAM, an adhesion ligand of LFA-1, is homologous to the neural cell adhesion molecule NCAM. Nature 331:624–627. 10.1038/331624a0 [DOI] [PubMed] [Google Scholar]
- 19.Fingeroth JD, Benedict MA, Levy DN, Strominger JL. 1989. Identification of murine complement receptor type 2. Proc. Natl. Acad. Sci. U. S. A. 86:242–246. 10.1073/pnas.86.1.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prota AE, Sage DR, Stehle T, Fingeroth JD. 2002. The crystal structure of human CD21: implications for Epstein-Barr virus and C3d binding. Proc. Natl. Acad. Sci. U. S. A. 99:10641–10646. 10.1073/pnas.162360499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shapiro AL, Vinuela E, Maizel JV., Jr 1967. Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels. Biochem. Biophys. Res. Commun. 28:815–820. 10.1016/0006-291X(67)90391-9 [DOI] [PubMed] [Google Scholar]
- 22.Kuhn-Hallek I, Sage DR, Stein L, Groelle H, Fingeroth JD. 1995. Expression of recombination activating genes (RAG-1 and RAG-2) in Epstein-Barr virus-bearing B cells. Blood 85:1289–1299 [PubMed] [Google Scholar]
- 23.Skare J, Strominger JL. 1980. Cloning and mapping of BamHI endonuclease fragments of DNA from the transforming B95-8 strain of Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 77:3860–3864. 10.1073/pnas.77.7.3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamin D, Sharma V, Knobloch TJ, Armitage RJ, Dayton MA, Goodwin RG. 1994. B cell IL-7. Human B cell lines constitutively secrete IL-7 and express IL-7 receptors. J. Immunol. 152:4749–4757 [PubMed] [Google Scholar]
- 25.Li C, Wong WH. 2001. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl. Acad. Sci. U. S. A. 98:31–36. 10.1073/pnas.98.1.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlossman SF, Boumsell L, Gilks W, Harlan JM, Kishimoto T, Morimoto C, Ritz J, Shaw S, Silverstrin R, Springer T, Tedder TF, Todd RF. 1995. Leukocyte typing V, vol 1 Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 27.Tuveson DA, Ahearn JM, Matsumoto AK, Fearon DT. 1991. Molecular interactions of complement receptors on B lymphocytes: a CR1/CR2 complex distinct from the CR2/CD19 complex. J. Exp. Med. 173:1083–1089. 10.1084/jem.173.5.1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutt-Fletcher LM. 2007. Epstein-Barr virus entry. J. Virol. 81:7825–7832. 10.1128/JVI.00445-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Gregory C, Sample C, Rowe M, Liebowitz D, Murray R, Rickinson A, Kieff E. 1990. Epstein-Barr virus latent membrane protein (LMP1) and nuclear proteins 2 and 3C are effectors of phenotypic changes in B lymphocytes: EBNA-2 and LMP1 cooperatively induce CD23. J. Virol. 64:2309–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutt-Fletcher LM, Chesnokova LS. 2010. Integrins as triggers of Epstein-Barr virus fusion and epithelial cell infection. Virulence 1:395–398. 10.4161/viru.1.5.12546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwong YL, Lee CP, Chan TK, Chan LC. 1994. Flow cytometric measurement of glycosylphosphatidyl-inositol-linked surface proteins on blood cells of patients with paroxysmal nocturnal hemoglobinuria. Am. J. Clin. Pathol. 102:30–35 [DOI] [PubMed] [Google Scholar]
- 32.Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2655 In Knipe DM, Howley PM. (ed), Fields virology, vol 2 Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 33.Price AM, Tourigny JP, Forte E, Salinas RE, Dave SS, Luftig MA. 2012. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-kappaB activation. J. Virol. 86:11096–11106. 10.1128/JVI.01069-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doyle JM, Gao J, Wang J, Yang M, Potts PR. 2010. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 39:963–974. 10.1016/j.molcel.2010.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luxembourg AT, Cooper NR. 1994. Modulation of signaling via the B cell antigen receptor by CD21, the receptor for C3dg and EBV. J. Immunol. 153:4448–4457 [PubMed] [Google Scholar]
- 36.Sugano N, Chen W, Roberts ML, Cooper NR. 1997. Epstein-Barr virus binding to CD21 activates the initial viral promoter via NF-kappaB induction. J. Exp. Med. 186:731–737. 10.1084/jem.186.5.731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanner JE, Alfieri C, Chatila TA, Diaz-Mitoma F. 1996. Induction of interleukin-6 after stimulation of human B-cell CD21 by Epstein-Barr virus glycoproteins gp350 and gp220. J. Virol. 70:570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D'Addario M, Libermann TA, Xu J, Ahmad A, Menezes J. 2001. Epstein-Barr virus and its glycoprotein-350 upregulate IL-6 in human B-lymphocytes via CD21, involving activation of NF-kappaB and different signaling pathways. J. Mol. Biol. 308:501–514. 10.1006/jmbi.2001.4589 [DOI] [PubMed] [Google Scholar]
- 39.Sista ND, Pagano JS, Liao W, Kenney S. 1993. Retinoic acid is a negative regulator of the Epstein-Barr virus protein (BZLF1) that mediates disruption of latent infection. Proc. Natl. Acad. Sci. U. S. A. 90:3894–3898. 10.1073/pnas.90.9.3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bouillie S, Barel M, Frade R. 1999. Signaling through the EBV/C3d receptor (CR2, CD21) in human B lymphocytes: activation of phosphatidylinositol 3-kinase via a CD19-independent pathway. J. Immunol. 162:136–143 [PubMed] [Google Scholar]
- 41.Valencia SM, Hutt-Fletcher LM. 2012. Important but differential roles for actin in trafficking of Epstein-Barr virus in B cells and epithelial cells. J. Virol. 86:2–10. 10.1128/JVI.05883-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carel JC, Myones BL, Frazier B, Holers VM. 1990. Structural requirements for C3d,g/Epstein-Barr virus receptor (CR2/CD21) ligand binding, internalization, and viral infection. J. Biol. Chem. 265:12293–12299 [PubMed] [Google Scholar]
- 43.Alfieri C, Birkenbach M, Kieff E. 1991. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology 181:595–608. 10.1016/0042-6822(91)90893-G [DOI] [PubMed] [Google Scholar]
- 44.Spender LC, Cannell EJ, Hollyoake M, Wensing B, Gawn JM, Brimmell M, Packham G, Farrell PJ. 1999. Control of cell cycle entry and apoptosis in B lymphocytes infected by Epstein-Barr virus. J. Virol. 73:4678–4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Y, Gao J, Yang M. 2011. When MAGE meets RING: insights into biological functions of MAGE proteins. Protein Cell 2:7–12. 10.1007/s13238-011-1002-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sang M, Wang L, Ding C, Zhou X, Wang B, Lian Y, Shan B. 2011. Melanoma-associated antigen genes—an update. Cancer Lett. 302:85–90. 10.1016/j.canlet.2010.10.021 [DOI] [PubMed] [Google Scholar]
- 47.Ladelfa MF, Peche LY, Toledo MF, Laiseca JE, Schneider C, Monte M. 2012. Tumor-specific MAGE proteins as regulators of p53 function. Cancer Lett. 325:11–17. 10.1016/j.canlet.2012.05.031 [DOI] [PubMed] [Google Scholar]
- 48.Peche LY, Scolz M, Ladelfa MF, Monte M, Schneider C. 2012. MageA2 restrains cellular senescence by targeting the function of PMLIV/p53 axis at the PML-NBs. Cell Death Differ. 19:926–936. 10.1038/cdd.2011.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ogembo JG, Kannan L, Ghiran I, Nicholson-Weller A, Finberg RW, Tsokos GC, Fingeroth JD. 2013. Human complement receptor type 1/CD35 is an Epstein-Barr virus receptor. Cell Rep. 3:371–385. 10.1016/j.celrep.2013.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsumoto AK, Martin DR, Carter RH, Klickstein LB, Ahearn JM, Fearon DT. 1993. Functional dissection of the CD21/CD19/TAPA-1/Leu-13 complex of B lymphocytes. J. Exp. Med. 178:1407–1417. 10.1084/jem.178.4.1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tedder TF, Inaoki M, Sato S. 1997. The CD19-CD21 complex regulates signal transduction thresholds governing humoral immunity and autoimmunity. Immunity 6:107–118. 10.1016/S1074-7613(00)80418-5 [DOI] [PubMed] [Google Scholar]
- 52.D'Addario M, Ahmad A, Morgan A, Menezes J. 2000. Binding of the Epstein-Barr virus major envelope glycoprotein gp350 results in the upregulation of the TNF-alpha gene expression in monocytic cells via NF-kappaB involving PKC, PI3-K and tyrosine kinases. J. Mol. Biol. 298:765–778. 10.1006/jmbi.2000.3717 [DOI] [PubMed] [Google Scholar]
- 53.Hurley EA, Thorley-Lawson DA. 1988. B cell activation and the establishment of Epstein-Barr virus latency. J. Exp. Med. 168:2059–2075. 10.1084/jem.168.6.2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ. 2002. The human and mouse replication-dependent histone genes. Genomics 80:487–498. 10.1006/geno.2002.6850 [DOI] [PubMed] [Google Scholar]
- 55.Izzo A, Kamieniarz K, Schneider R. 2008. The histone H1 family: specific members, specific functions? Biol. Chem. 389:333–343. 10.1515/BC.2008.037 [DOI] [PubMed] [Google Scholar]
- 56.Yang SM, Kim BJ, Norwood Toro L, Skoultchi AI. 2013. H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proc. Natl. Acad. Sci. U. S. A. 110:1708–1713. 10.1073/pnas.1213266110 [DOI] [PMC free article] [PubMed] [Google Scholar]