

Abstract
Defective interfering (DI) RNAs are highly deleted forms of the infectious genome that are made by most families of RNA viruses. DI RNAs retain replication and packaging signals, are synthesized preferentially over infectious genomes, and are packaged as DI virus particles which can be transmitted to susceptible cells. Their ability to interfere with the replication of infectious virus in cell culture and their potential as antivirals in the clinic have long been known. However, until now, no realistic formulation has been described. In this review, we consider the early evidence of antiviral activity by DI viruses and, using the example of DI influenza A virus, outline developments that have led to the production of a cloned DI RNA that is highly active in preclinical studies not only against different subtypes of influenza A virus but also against heterologous respiratory viruses. These data suggest the timeliness of reassessing the potential of DI viruses as a novel class of antivirals that may have general applicability.
INTRODUCTION
The generation of new or improved vaccines to prevent virus disease in people, domestic animals, and farmed stock remains a considerable challenge, as vaccines are not always effective and viruses have a disquieting ability to become resistant to the limited range of antivirals currently available. For example, influenza A virus mutants resistant to oseltamivir appeared very rapidly in the early stages of the 2009-2010 pandemic (1). There is therefore an urgent need for more effective measures to control the virus burden.
It has been suggested that small interfering RNAs (siRNAs), microRNAs (miRNAs), and synthetic interfering RNAs (RNAi) may have therapeutic potential (2, 3). The siRNAs are made by plant and invertebrate animal cells, while miRNAs are encoded by most eukaryotic cells and some DNA viruses. Both siRNAs and miRNAs are enzymically excised from larger precursors by cellular endonucleases and loaded into a RNA-induced silencing complex (RISC). The RISC with its RNA cargo binds pre-mRNA or mRNA, and either the complex is cleaved by RISC (siRNA) or translation is inhibited in some other way (miRNA). RNAi molecules act in a similar way when introduced into cells. Defective interfering (DI) RNAs differ from other interfering RNAs as they originate from a viral genome and act by competing with viral genomes for replication and/or packaging. Their capacity to be packaged provides specific and efficient targeting of DI RNAs, which cannot yet be achieved for the other inhibitory RNA approaches, although progress is being made (4). New developments suggest that a reevaluation of the antiviral potential of DI genomes would be timely.
DI genomes are made by nearly all viruses and are antiviral by definition in vitro. In the years following their initial characterization, DI viruses were considered to have exciting clinical potential, but translating in vitro promise into in vivo activity proved problematic (5). Now, after decades of slow progress, the application of new techniques is moving the field rapidly forward. In particular, cloning technology has been applied to influenza virus to generate a homogeneous population of DI genomes that has afforded a previously lacking consistency and reproducibility in experimentation. This approach can now inform a search in other virus systems for DI viruses that may be therapeutically and prophylactically useful. Here, we review the evolution of the field to its present state, the directions in which it is heading with particular reference to DI influenza virus, and the implications in general for the use of DI viruses in the control of human and animal virus infections.
HISTORY
The interference phenomenon was first reported over a half century ago by Preben von Magnus in a series of experiments on the growth characteristics of influenza viruses. During successive high-multiplicity passages in embryonated chicken's eggs, a whole-animal system, von Magnus recognized a rise in the ratio of virus particle to infectivity that reflected a loss in the yield of infectious virus (6). He referred to the noninfectious particles generated as “incomplete” virus. This description was remarkably prescient in the absence of suitable analytical techniques with which to fully characterize the viruses. The term DI virus was coined later when molecular studies became possible (7).
DI viruses are virus particles that contain a heavily deleted version of the infectious genome (i.e., they are defective in one or more genes) and have the ability in vitro to interfere with, and diminish the production of, infectious progeny produced by the infectious virus from which that DI particle originated (7). DI genomes and DI particles are made by nearly all viruses and may represent an evolutionarily acceptable by-product of an inefficient replication process. Alternatively, they may be produced for evolutionary reasons, such as the stimulation of innate immunity (8–10), that could favor survival of the host species and hence perpetuation of the virus population itself. Because of its deleted genome, a DI virus is by definition noninfectious and is replicated only in a cell coinfected by an infectious “helper” virus, usually the virus from which the DI genome is derived. This provides the function(s) that the DI genome lacks and enables the DI genome to be packaged into virus particles. A high multiplicity of infection favors the propagation of DI viruses, as more cells can be infected simultaneously with DI and infectious viruses. It follows that all DI genomes retain the molecular signals that permit their replication and packaging into new DI particles and that the DI genome is packaged by proteins made by the helper virus. Hence, the progeny DI and helper viruses are antigenically identical.
It is believed that the ratio of DI genomes to infectious genomes within the cell determines the outcome of infection in the culture system as a whole. The level of DI RNA that is needed to establish protection from a particular dose of infectious virus has only been determined empirically. In one scenario, the DI genome enters some cells but others have no DI genome. Similarly, some cells will be infected and others not. Finally, some infected cells contain both a DI and an infectious genome. When the number of infectious particles is below the (unknown) threshold, the DI genome population inhibits the spread of infectious virus to a degree determined by many factors, including the ratio of DI genomes to virus genomes in the culture system as a whole and their respective rates of replication. At a low ratio of DI genomes to infectious genomes, there is replication of both genome types without interference and no reduction of specific infectivity or total particles produced. As the ratio of DI genomes to infectious genomes in the cell increases, more DI genomes than infectious genomes are produced, although the total number of virus particles produced is unaltered. However, here the majority of particles are DI virus. With an even greater increase in the ratio of DI genomes to infectious genomes, there is a reduction in replication of both DI and infectious genomes and a fall in the total number of particles (DI virus and infectious virus) produced. In essence, this is a race between the amplification of the DI RNA, which leads to the production of more DI virus and the establishment of more DI virus-protected cells, and the spread of infectious virus. When the DI RNA “wins,” clinical disease is inhibited and the animal is protected.
Analysis of DI genomes from single-stranded RNA viruses has allowed them to be classified into four main types (11). The “simplest” form of DI RNA comprises a deletion of the majority of the infectious genome (which can be over 90%) while retaining the original termini (Fig. 1, section i). A version of this has repeated, rearranged, or further deleted sequences flanked by the original termini. A second form (Fig. 1, section ii) comprises an authentic 5′ terminus and an inverted repeat of the same terminus at the other, while between the termini is a genome constituted as shown in Fig. 1, section i. A third form, known as a copyback DI RNA, contains a variable length of the infectious genome and an authentic 5′ terminus followed immediately by an inverted repeat of some of that sequence and the 5′ end (Fig. 1, section iii). Last, there is a snapback DI RNA comprising a single authentic terminus but with an inverted repeat of almost all the sequence (Fig. 1, section iv). The last two DI RNAs have the potential for a high degree of double strandedness when not complexed with nucleoprotein that would prevent base pairing.
FIG 1.
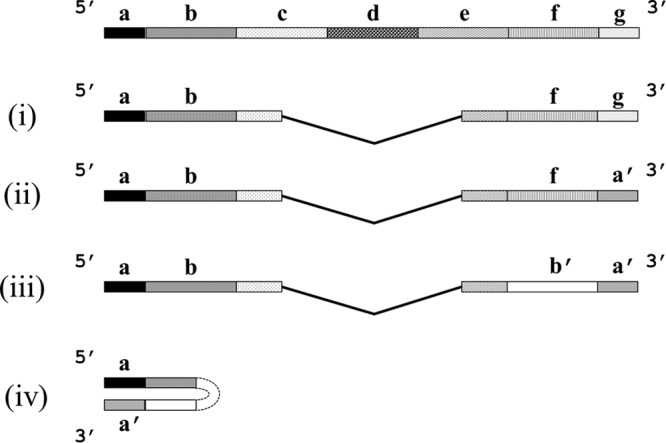
Schematic of the different types of DI RNA (not to scale). The virus genome is shown at the top with terminal sequences containing the replication signals labeled at the 5′ end (a) and at the 3′ end (g). The remainder of the genome is arbitrarily divided into sections to indicate the possible origin of some of the DI RNA sequences. See the text for explanations of sections i to iv.
A significant complexity for these early analyses was that DI viruses produced by high-multiplicity-of-infection passage varied from preparation to preparation and consisted of a complex mixture of different DI genome sequences (12, 13). These mixtures inevitably contained defective genomes that did not interfere efficiently (or indeed at all) with the replication of the helper parental virus and genomes that interfered extremely well (14–16). This uncontrollable heterogeneity frequently meant that promising evidence of in vivo antiviral activity could not be reproduced.
Early investigations failed to detect DI genomes in vivo, and it was commonly believed that they were solely artifacts of the laboratory. However, with the advent of genome amplification techniques, including most recently deep sequencing, putative defective and/or defective interfering genomes have been identified in a number of human infections, including those with dengue virus (17), hepatitis B virus (18), hepatitis C virus (19), hepatitis A virus (20), and influenza A virus (21). Defective genomes have also been isolated from hepatitis A virus-infected marmosets (20) and from infectious hematopoietic necrosis virus-infected trout (22). In addition, it appears likely that DI influenza A virus and DI dengue virus can be transmitted from person to person (21, 23). It was further argued that infectious dengue virus mixed with DI dengue virus has a higher transmission potential than that of infectious virus alone. An understanding of the role(s) of DI genomes in the evolution of viruses and their infections has attracted much speculation over the years and remains very much a work in progress.
ROLE OF THE HOST CELL IN THE DI VIRUS PHENOMENON
DI virus genomes are thought to arise from errors that occur during the replication process. This is principally a result of the replication complex containing the nascent RNA detaching from the template and reattaching at a different point on the same or a different template molecule one or more times. Alternatively, synthesis may continue using the nascent RNA as the template to produce an inverted repeat sequence (Fig. 1, sections ii to iv). However, the generation of DI viruses is not determined exclusively by the virus itself. Although most cell-virus combinations yield DI genomes, this is not always the case, and the cell can determine whether or not DI RNA arises de novo from a particular virus; e.g., DI RNAs were not generated by Semliki Forest virus (SFV) in some sublines of HeLa cells even after 200 high-multiplicity passages of infectious virus (24), by Sendai virus in chicken embryo lung cells (25), by vesicular stomatitis virus (VSV) in primary human skin fibroblast (26) and HeLa cells (27), or by measles virus in human diploid lung fibroblast (WI-38) cells (28). Generation of a DI genome may involve a host-coded function, as actinomycin D, which inhibits transcription by DNA-dependent RNA polymerases, abrogated the generation of DI VSV (29), and in human cells the generation of VSV DI genomes was mapped to chromosome 16 (26). To add to the complexity, there are examples of cells which do not generate DI RNA de novo but which with the aid of helper virus can propagate a DI RNA with which they have been inoculated (26). To explain these phenomena, it was postulated that the viral polymerase has a cellular constituent that contributes to its processing fidelity. Thus, a high-fidelity polymerase makes no errors and gives rise to no DI genomes, while a low-fidelity, error-prone enzyme generates a great many (30). However, once generated, a DI RNA can be replicated. The role of the host cell in vivo is also important as demonstrated by the failure of a DI SFV RNA to be replicated by infectious virus in mouse brain, although this happened perfectly well in cell culture (31).
ANTIVIRAL ACTIVITY
The antiviral interfering activity of DI viruses is easy to demonstrate in vitro and is a necessary requirement for distinguishing them from defective viruses which are not interfering. Demonstrating the presence of in vivo antiviral activity proved much more difficult, and this failure, coinciding with the availability of cloned interferon (IFN), may have contributed to the loss of interest in DI viruses as antivirals in the 1980s. Key in vivo studies are outlined below. DI viruses from a broad range of virus systems have been investigated for their potential as antivirals, but the best studied remain vesicular stomatitis virus, Semliki Forest virus, and influenza virus, which are considered in detail below. However, studies with other virus systems, such as lymphocytic choriomeningitis virus and Junin virus (32, 33), reovirus (34), and various herpesviruses (35–37), have provided encouraging results that demonstrate the potential of the DI virus antiviral approach. The precision of investigation has been helped by the production of cloned DI genomes and viruses, for example, Sendai virus (38), classical swine fever virus (39), mouse hepatitis virus (40), Semliki forest virus (31, 41), and influenza A virus (42, 43).
DI VESICULAR STOMATITIS VIRUS
The genome of vesicular stomatitis virus (VSV) is a single molecule of single-stranded, negative-sense RNA. DI VSV is an attractive experimental system, since in the Rhabdoviridae, genome size determines the length of the rod-shaped particle. Thus, a small DI RNA is encapsidated in a short particle and can be separated by velocity gradient centrifugation from longer infectious particles, but 4 successive gradients were required to completely remove infectivity (7). Infection with VSV by the intracerebral or intranasal route in adult mice causes death in 2 to 3 days. Administration by the same route of partially purified VSV DI particles containing some residual infectious virus only delayed death by 1 to 2 days, but complete protection was achieved by intracerebrally injecting a large dose of highly purified DI virus together with a low dose of infectious virus (44, 45). DI virus treatment also reduced the infectious virus load of challenge virus in the brain. This protection was not mediated through type I interferon and did not protect against another strain of VSV. DI virus injected intracerebrally on its own was immunizing (44). Protection was dependent on the ratio of DI virus to challenge virus, and lowering the dose of DI virus or increasing the amount of infectious virus rendered protection less effective, although animals survived until 6 to 9 days postinfection (44, 46). No DI virus could be detected in brains of DI virus-treated infected adult mice, raising doubts about the role of the DI virus in protection from disease (45). However, DI RNA was detectable in the brains of newborn mice inoculated intracerebrally, although protection was weak (3/24 animals protected). DI particles isolated from the brain were the same length as the inoculum DI particles and could therefore have been progeny particles rather de novo-generated particles (47). Similar data were obtained with DI rabies virus and infectious rabies virus challenge in mice (47–49). There appears to be a real difference in levels of replication of DI RNA in the brains of newborn and older mice, which as with other viruses may have its root in the age-related maturity of neurons (50). Later work using the intranasal route of inoculation and Northern blotting showed the presence of VSV RNA sequences in the brains of mice inoculated with a high dose of VSV plus a lower relative amount of DI VSV particles (51). However, the presence of DI RNA in the brain did not correlate strongly with protection (52). Intraperitoneal injection of Syrian hamsters with VSV resulted in a systemic infection with a lymphoreticular pathology and little or no involvement of the central nervous system (53). Intraperitoneal injection of DI VSV protected partially against a lethal dose of virus, and it was thought that in contrast to the situation in the brain, type I interferon was partly responsible for protection. Sequencing has shown that copyback and snapback DI VSV genomes occur most commonly, but centrally deleted genomes are also known (Fig. 1). In vivo work with cloned DI VSV has yet to be carried out.
DI SEMLIKI FOREST VIRUS
Semliki Forest virus (SFV; Togaviridae) has a genome comprising a single molecule of single-stranded, positive-sense RNA. Mice infected with a virulent strain of SFV contract a fatal encephalitis. The dose required to cause disease varies with the route of infection, with intranasal inoculation requiring one of the lowest doses. Initially, it was demonstrated that DI SFV produced by high-multiplicity passage and inoculated intranasally completely protected mice from disease (5, 54), although some DI SFV preparations did not protect (14). Subsequently, two DI RNAs were cloned and sequenced, and the cloned DI virus was produced in SFV-infected cells transfected with transcribed DI RNA (41). The two cloned DI RNA sequences were similar but differed in length (1,124 and 2,146 nt) and were of the type described in Fig. 1, section i, with discontinuous regions derived from the genome. However, although both interfered to the same extent in vitro, only DI virus reconstituted with the smaller DI RNA protected mice from a lethal intranasal SFV infection (31). It appeared that the nonprotecting larger DI RNA was not replicated in mouse brain, although this is the main target organ for infectious virus. This example explicitly demonstrates that in vitro interference is not necessarily synonymous with in vivo protection.
DI INFLUENZA VIRUSES
Influenza viruses A and B (Orthomyxoviridae) have genomes comprising 8 segments of single-stranded, negative-sense RNA. It has long been known that DI influenza A virus interferes with the multiplication in vivo of influenza A virus in embryonated chicken's eggs (6, 55–62). Early data provided evidence of protection from pneumonia in adult mice inoculated intranasally with virus preparations containing putative DI virus (55, 63–65) and from encephalitis in adult mice inoculated intracerebrally (55, 66). In another study of respiratory disease, there was no protection but the infectious virus load in the lung was reduced (45). However, in general, the level and consistency of protection were low, and controls to permit the conclusion that protection was due to the activity of the DI RNA, rather than some other part of the virus particle, were lacking. Indeed, one study concluded that DI virus-mediated protection of mice from pneumonia resulted from stimulation of humoral immunity (46).
Unlike the case with VSV, the problem of helper influenza virus infectivity contaminating the DI virus preparations could not be solved by purification as the two types of particle differ only in having a different amount of one RNA segment. However, it was circumvented by inactivating the infectivity of the helper virus with a low dose of UV irradiation. This does not appreciably affect the antiviral activity of the DI virus. This works because a single UV-induced lesion anywhere in the infectious genome of approximately 13,600 nucleotides kills the infectivity, while the DI virus RNA target is typically only 400 to 500 nucleotides. Prolonged UV irradiation of DI virus or inactivation with β-propiolactone, each of which targets RNA and destroys interfering activity without affecting the integrity of the virus particle or its hemagglutinin or neuraminidase titers, provides a control for active DI virus. Such preparations provide an in vivo control for active DI virus, as they have the same antigens (and impurities) and stimulate the same host responses. Indeed, in a large number of experiments and in a variety of experimental settings, DI virus given by the intranasal route reproducibly protected the majority of mice (also intranasally infected), while inactivated DI virus gave no significant protection (67–74). Use of different strains of mice demonstrated that the protection phenomenon was not restricted to a particular genotype (72). Ferrets too were significantly protected from influenza by active but not inactivated DI virus (75). Thus, protection can be attributed solely to the properties of the DI RNA. However, there was still the problem of there being many different DI RNA sequences in each preparation (12).
CLONED DI INFLUENZA A VIRUS
The 8 RNA segments that comprise the influenza genome are believed to replicate independently of each other but are packaged in a coordinated fashion so that each virus particle contains a single copy of each of the 8 segments (76, 77). In agreement with the earlier finding that DI influenza virus had a lower RNA content than infectious virus (78), it was apparent that preparations of DI influenza virus are deficient in one or more genome segments, usually segment(s) 1, 2, or 3, and these are replaced by a cognate DI RNA (12, 13, 79–86). Thus, an influenza virus particle can have a full-length virion RNA segment or a DI RNA derived from that segment, but not both. It is not known if a DI particle can have more than one genome segment replaced with a cognate DI RNA. Influenza DI RNAs are all simple deletions of the parental segment from which they are derived and are probably formed as a result of a faulty replication process which results in the loss of a central region of the genome segment and the linking together of the remaining terminal portions by read-through (Fig. 1, section i). The position of the deletion is highly variable; hence, there can be a large number of different DI RNA sequences derived from just one virion segment with a single DI influenza virus preparation containing over 50 different DI RNA sequences (12). The mean and median for influenza DI RNAs derived from segment 1, 2, or 3 was 400 to 500 nucleotides, so these will have lost approximately 80% of their sequence.
Reverse genetics plasmids encoding individual influenza virus enabled DI RNAs and infectious helper virus to be “rescued” into virus particles, producing stocks of cloned DI virus (and helper virus) containing a single DI RNA sequence (87, 88). After amplification in eggs and purification, the cloned DI virus preparation was UV irradiated as described above to inactivate helper virus infectivity. It was administered intranasally to mice together with a lethal dose of infectious virus. However, the first cloned DI virus succeeded only in delaying disease and death (43).
Consistent with data from noncloned DI virus stocks, it was found that different cloned DI RNA sequences protect mice to differing extents (42). One of these (244 DI RNA) provided strong in vivo prophylaxis and therapy (42, 89–91). 244 DI RNA was generated spontaneously from segment 1 of A/Puerto Rico [PR]/8/34 virus (PR8; H1N1) during a reverse genetics experiment to produce infectious virus. It was subsequently cloned and expressed as a DI PR8 virus. 244 DI RNA is a classical influenza virus DI RNA of 395 nucleotides, being formed by a single deletion and having the original terminal sequences (Fig. 1, section i, and Fig. 2). Expression by reverse genetics results in a mixture in which 244 DI virus predominates over infectious virus. The strong interfering properties of 244 DI virus were seen when transfection of increasing amounts of 244 DI plasmid relative to plasmids encoding infectious virus abrogated virus production in an MDCK cell-based interference assay titration (42) and reduced virus yield by >99% in embryonated eggs inoculated with increasing amounts of 244 DI virus (our unpublished data).
FIG 2.

Schematic diagram of the structure of influenza 244 DI RNA. This 395-nucleotide RNA was derived by a single central deletion from the full-length genomic segment 1 (2,341 nucleotides) of A/PR/8/34 (H1N1) (42). Numbers indicate the nucleotide positions of the breakpoints in the genomic segment 1 RNA (negative sense, 3′ to 5′). The sequence is in GenBank under accession number FB718012.
244 DI and infectious particles appear identical in morphology and protein content as determined by transmission electron microscopy and polyacrylamide gel electrophoresis, respectively (our unpublished data). The use of cloned DI RNA has allowed the biological properties of an influenza virus DI RNA to be related to its sequence for the first time, making 244 DI RNA the most extensively characterized of the influenza A virus DI RNAs.
PROTECTION OF ANIMALS FROM TYPE A INFLUENZA VIRUSES BY CLONED INFLUENZA A VIRUS DI RNA (HOMOLOGOUS PROTECTION)
The targeted delivery of a DI influenza virus RNA packaged by an influenza virus particle to cells that are susceptible to infectious virus is an essential requirement for successful treatment and explains how a small dose of DI RNA can be effective as an antiviral. DI influenza RNA, delivered by nasal administration to the respiratory tract, reaches precisely the cells that are, or potentially can be, infected by an invading pathogenic influenza virus, because it is carried there and introduced into the cell by a virus particle that retains cell targeting and entry functions. Influenza A viruses bind to one of two isomeric forms of sialic acid on the surface of target cells through their hemagglutinin major surface glycoprotein. This is often oversimplified to avian influenza viruses using the N-acetylneuraminic acid (NANA) α2,3Gal form of the receptor and mammalian viruses the NANA α2,6Gal form. However, the actual situation is more complex, with both receptor types being present in both avians and mammals, although in differing proportions in the various parts of the respiratory tract. For example, the human upper respiratory tract has a high ratio of NANA α2,6Gal to NANA α2,3Gal, while this is reversed in the lower respiratory tract (92). For optimum protection, it is therefore important that DI virus delivers its DI RNA to cells bearing either type of receptor. To this end, a substrain of PR8 was identified that is dual receptor specific (93). Its hemagglutinin and neuraminidase proteins were cloned and used to create a delivery particle that recognizes both types of receptor. This was effective in delivering DI RNA to the respiratory tracts of mice and ferrets, in which NANA α2,3Gal and NANA α2,6Gal predominate, respectively.
For preclinical investigations, inbred strains of mice are infected with virulent influenza viruses under light anesthesia. Such mice develop an acute respiratory disease of approximately 3 to 7 days' duration which progresses to pneumonia and death through a readily recognizable sequence of clinical events. This is accompanied by weight loss which can exceed 25%. A single dose of 244 DI virus (1.2 μg DI virus protein containing 8 pg 244 DI RNA), given intranasally to mice before or at the same time as infection by the same route prevented nearly all clinical signs of infection (Fig. 3a), while a dose of 0.12 μg DI virus (0.8 pg DI RNA) protected nearly as well (42). A single dose of cloned 244 DI virus protected several different inbred mouse strains from clinical influenza, indicating that host genetics is not an issue.
FIG 3.

Comparison of prophylactic (a, b) and therapeutic (c, d) protection of mice mediated by a single dose of intranasal 244 DI virus. (a, c) Homologous protection from clinical disease caused by intranasal influenza A virus (A/WSN, H1N1); (b, d) heterologous protection from clinical disease caused by intranasal pneumonia virus of mice (PVM). Mice were 5-week-old C3H/He-mg. The filled and open arrows indicate the delivery of the DI virus and the challenge virus (10 50% lethal doses [LD50]), respectively. Mice were anesthetized before inoculation. DI virus was administered at a time that gives optimum protection in each virus system. On the y axis, a score of 1 indicates a normal healthy mouse and 5 is a dead mouse; a score of 2 to 4 refers to increasingly severe clinical disease estimated on a formal scale (42). In parentheses is the percentage survival at the end of the study. Helper virus infectivity was removed from the DI virus preparation by UV irradiation for 40 s. For the prophylaxis studies depicted in panels a and b, the symbols indicate: ◼, 1.2 μg DI virus protein; ◆, 0.12 μg DI virus; ▲, 0.012 μg DI virus (PVM only); ◻, 1.2 μg DI virus inactivated by extensive UV irradiation (8 min) for influenza and PVM challenges, respectively; ○, a control group inoculated with diluent—these all had a value of 1. For the therapeutic studies (panels c and d), influenza and PVM infections were treated with optimally effective amounts of DI virus (12 μg and 1.2 μg protein, respectively). Mice were treated with DI virus after infection at the following times: 1 day (◼), 2 days (◆), or 3 days (▲; PVM only); with inactivated DI virus at 1 day (◻); or with diluent (○)—these all had a value of 1. Data are from references 42 and 96.
Therapeutic administration of 244 DI virus to mice previously given a lethal dose of infectious virus is also protective (Fig. 3c) (42). Treatment at 1 day after infection with 12 μg DI virus protein per mouse was completely effective, and treatment at 2 days after infection delayed the appearance of clinical signs and reduced mortality. Treatment at 3 days after infection (not shown) was ineffective. The ratio of DI genomes to infectious genomes is key to understanding this situation, since as the infection takes hold, the number of infectious genomes increases to a point where the amount of DI RNA administered is insufficient to influence the course of disease.
The ferret does not usually develop pneumonia and thus is a better model of human influenza than the mouse and is the gold standard for agencies that regulate human medicines. A single dose of DI RNA (2 μg RNA in 300 μg virus protein) given 2 h ahead of infection protected ferrets from infection with 2009 H1N1 pandemic virus, significantly reducing the severity of all clinical disease parameters. This protection was statistically better than that achieved with oseltamivir, the current major influenza antiviral, which was given as 10 doses over 5 days (25 mg/kg) (as prescribed for humans) (89, 90). The 50% protective dose of DI virus in ferrets was not determined. Amplified DI virus and progeny infectious virus, which both carry the surface antigens of the challenge virus, were cleared from the respiratory tract at the same time as the virus in infected, untreated control animals. It is thought that clearance is effected by the action of the innate and adaptive immune systems destroying infected cells that express budding DI virus or infectious virus on their surface and mopping up free DI virus and infectious virus.
An important clinical consequence of 244 DI virus administration is that replication of infectious challenge virus was reduced but not completely inhibited. The reduced amount of infectious virus was compensated for by production of DI virus of identical antigenic composition, and together these stimulate adaptive immunity (including hemagglutination-inhibiting and neutralizing antibody) and render animals (mice and ferrets) immune to reinfection with a high dose of the same virus (42, 89, 90). However, a very high dose of 244 DI virus inhibited this immunity, by presumably reducing antigen production to a subimmunogenic level (42). In addition to protecting young adult animals, 244 DI virus protected elderly, obese mice from acute infection, and these mice also developed solid immunity to the same infectious virus in a subsequent challenge (94). Thus, 244 DI RNA converts a potentially lethal disease into an avirulent infection with no overt symptoms of disease—effectively, a live vaccine. Finally, it is worth noting how much more effective cloned 244 DI virus is than noncloned DI virus at protecting mice from influenza in vivo. It reproducibly provides a longer window for prophylaxis and therapy, is effective at a lower concentration, and requires only a single dose.
INFLUENZA A DI VIRUS ALSO PROTECTS IN VIVO AGAINST HETEROLOGOUS VIRUSES
Although it is generally held that a DI virus interferes only with the virus from which it was derived or one to which it is closely related, 244 DI virus protected mice from clinical disease caused by pneumonia virus of mice (PVM; Paramyxoviridae, genus Pneumovirus) and an influenza B virus (95, 96). Members of the Influenzavirus A and Influenzavirus B genera of the Orthomyxoviridae are deemed heterologous, as they are genetically distinct, do not undergo RNA segment reassortment, and do not support each other's replication. DI virus-mediated prophylaxis and therapy in the heterologous systems were both highly effective. Prophylaxis was optimal when given 1 day ahead of infection with PVM, and 1.2 μg 244 DI virus protein (8 pg 244 DI RNA) per mouse gave complete protection (Fig. 3b). In a dilution series, 0.12 μg DI virus protein (0.8 pg DI RNA) gave significant protection, while 12 pg DI virus (80 fg DI RNA) merely delayed the infection by 1 day. A comparison of heterologous protection with PVM with homologous protection (Fig. 3a and b) suggests that the latter is approximately 5-fold more effective. Twelve and 1.2 μg of DI virus protein given 3 to 4 days before heterologous challenge gave significant protection, but homologous protection was effective for a much longer time (our unpublished data). This difference presumably reflects the different mechanisms that operate between heterologous protection (type I interferon) and homologous protection (direct interference) and the intrinsic inability of heterologous viruses to amplify the influenza A virus DI RNA (see below).
Therapy with 1.2 μg DI virus protein per mouse at 1 day after infection with PVM greatly reduced clinical disease and abrogated mortality completely (Fig. 3d). Therapy at 2 days after infection halved the mortality, but therapy at 3 days after infection was ineffective. However, heterologous therapy was achieved with approximately 10% of the dose required for homologous protection (Fig. 3c and d). This result might reflect the relative sensitivities of PVM and influenza A/WSN virus to type I interferon, but that was not tested. Extensive UV irradiation of DI virus, described above, abrogated heterologous protection, suggesting that the 244 DI RNA was the entity responsible (95, 96).
HOW DOES INFLUENZA 244 DI VIRUS RNA MEDIATE PROTECTION IN VIVO?
In general terms, DI RNA-mediated protection can be viewed as an alteration in the virus-host relationship in favor of the host. Interference with the replication of infectious virus delays the progress of infection and reduces the virus load, allowing host defenses sufficient time to mount effective antiviral responses that clear infectious and progeny DI virus and go on to establish protective immunity. Analysis of how such protection is achieved indicates that there are at least two distinct mechanisms at play, one relevant to influenza A viruses and a second that acts against other respiratory viruses.
PROTECTION AGAINST DISEASE MEDIATED BY INFLUENZA A (HOMOLOGOUS) VIRUSES
The current view of the protection scenario is that DI virus follows the route of infection taken by infectious virus. It binds via its hemagglutinin to target cells and introduces its DI RNA, as a ribonucleoprotein, into the cell. DI RNA then moves to the nucleus, and is transcribed by its associated RNA-dependent RNA polymerase into positive-sense antigenome DI RNA and DI mRNA. No replication is possible as this DI virus lacks a complete segment 1 RNA and cannot synthesize PB2 protein, an essential component of the de novo RNA-dependent RNA polymerase required to transcribe newly synthesized antigenome positive sense DI and full-length RNA into their negative sense complements (B. Meng, N. J. Dimmock and A. J. Easton, unpublished data). If no infectious influenza A virus enters that cell, the DI RNA eventually decays. The dynamics of nonreplicating DI RNA in mouse lung after intranasal instillation of DI virus was followed by quantitative PCR. Figure 4 shows that the majority of DI RNA is lost in the first 2 h, possibly by the loss of excess volume from the respiratory tract due to the relatively large amount inoculated (20 μl per nare), with much of the virus failing to reach the host cells. Nevertheless, based on quantitative data (97), at 2 h postinoculation there were approximately 2.3 copies of DI RNA per lung epithelial cell. The loss of DI RNA was higher over the first week than that over the succeeding 6 weeks, and the amount of DI RNA was extrapolated to zero after 6 months. This gives an indication of the protective amount, as the dose of 244 DI virus routinely employed would result in approximately 1 DI RNA copy per 5 respiratory tract epithelial cells and protected mice for 1 to 2 weeks. A higher dose of DI RNA provides longer lasting protection, as it takes longer for the DI RNA to degrade to the 50% protecting dose. However, PCR may exaggerate the stability of intact DI RNA as it records the presence of only a fragment of the DI RNA. Nonetheless, these data are in broad agreement with an earlier report in which MDCK cells were inoculated with noncloned, noninfectious DI influenza virus. Cells showed no pathology and grew at a normal rate, and cultures were subdivided every few days as usual. Initially, these cells completely resisted challenge with homologous virus. The half-life for resistance was 3 to 4 weeks (98). It was inferred that cells received multiple DI genomes and that these segregated to daughter cells at mitosis. Loss of resistance was presumed to result from dilution of DI genomes as cells divided and from degradation of the DI RNA.
FIG 4.
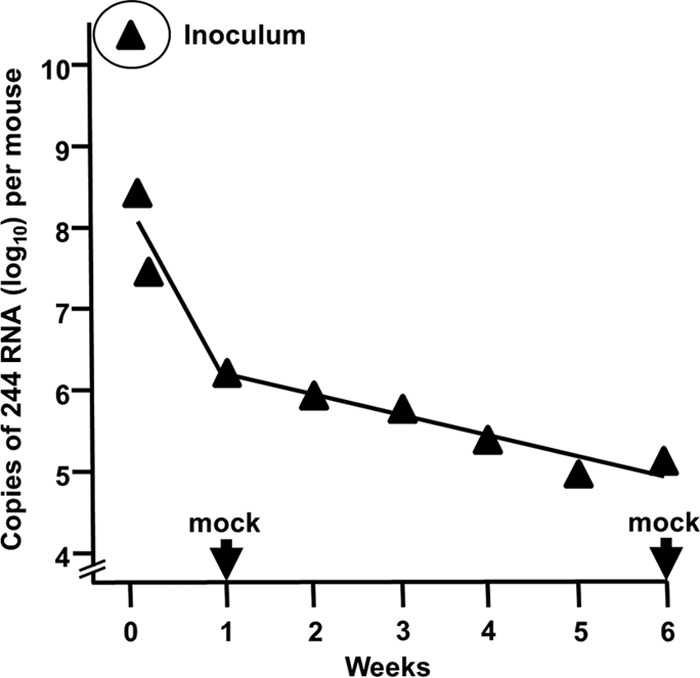
Decay of 244 DI RNA in the lungs of mice inoculated with 12 μg 244 DI virus protein. The amount of 244 RNA associated with the lung was determined by quantitative PCR (89). Also shown is the amount of 244 RNA administered initially (inoculum) and the lack of signal at 1 and 6 weeks in the lungs of mice inoculated with diluent (mock) (B. Meng, A. J. Easton, and N. J. Dimmock, unpublished data).
Table 1 indicates the amount of 244 DI RNA that protected mice against a lethal dose of influenza virus as shown in Fig. 3a. This is the amount contained in 1.2 μg 244 DI virus protein. There is an initial loss of DI RNA in the first 2 h after infection (Fig. 4), which results in a maximum total of 1 DI RNA per 5 respiratory tract cells. The ratio of DI RNA copies to infectious virus units inoculated is estimated as a million to one, and this indicates in approximate terms the amount of DI RNA that is needed for protection. However, Fig. 3a shows that mice were also protected using 10-fold less DI virus.
TABLE 1.
Calculation of the number of copies of 244 DI RNA required for protection of a mouse from 10 LD50 of influenza A/WSN virusa
| Total no. of cells covering the surface of the RTb | No. of DI RNA copies inoculated per RTc | No. of DI RNA copies remaining in the RT after 2 hd | No. of DI RNA copies per cell in the RT after 2 h |
|---|---|---|---|
| 1.36 × 108 | 1.32 × 108 | 2.6 × 107 | 0.19 |
Delivered intranasally as 100 50% tissue culture infective doses (TCID50) of 244 DI virus; the infectious virus was also delivered intranasally.
Based on a previous study (97) which determined the areas of the various parts of the mouse respiratory tract (RT); assumes that the area of a respiratory tract epithelial cell is 1.6 × 10−3 mm2.
B. Meng, A. J. Easton, and N. J. Dimmock, unpublished data.
See Fig. 4 for the loss of DI RNA from the mouse RT (approximately 50-fold) that occurs in the first 2 h after infection.
When infectious virus infects a cell in which the DI RNA is present, the proteins it provides in trans direct replication of full-length and DI RNAs, with DI RNA amplified up to 10,000-fold (89–91). The UV irradiation that inactivates helper virus has the additional benefit of inactivating full-length RNAs present in the DI particle. Hence, these are not replicated. Observation shows that DI RNA is made preferentially over its cognate full-length RNA and that packaging into progeny particles, which appears to be a stochastic process, ensures that DI particles predominate (>99%). The overall result is a preponderance of DI virus progeny over infectious virus progeny. De novo-synthesized DI RNA is packaged by the proteins of the infecting virus and thus is antigenically identical to the infecting virus. In a cycle of events that continues until the infection is resolved, progeny DI virus delivers newly synthesized DI RNA to other cells of the respiratory tract. Empirical evidence suggests that the ability of a DI RNA sequence to protect is specific to that sequence, although that relationship is not understood (42). Having the infecting virus amplify the very antiviral that will lead to its own demise is a satisfying twist to the DI virus story and makes DI RNA unique among prodrugs.
The activity of the 244 DI RNA is based on a commonality of genome replication which has been retained by all influenza A viruses irrespective of origin. As a result, DI RNA interference/protection is likely to be universal among members of the Influenzavirus A genus, as their common genetic system means that any influenza A virus can replicate any influenza A DI RNA and, hence, be inhibited by it. Indeed, 244 DI virus protects against virus strains from a number of different influenza A subtypes (42). 244 DI virus completely suppressed clinical disease in mice with severe combined immunodeficiency (SCID), but after a further week, these mice developed disease of the normal pattern and succumbed. Thus, adaptive immunity is not needed for DI RNA to control the acute phase of disease. Further investigation showed that the amount of DI RNA in the lung had not declined and that infectious virus had not mutated to escape the DI RNA. It was concluded that adaptive immunity was required for clearance of infectivity (91), although the facet of the adaptive immune system responsible was not established.
PROTECTION AGAINST DISEASE MEDIATED BY NON-INFLUENZA A (HETEROLOGOUS) VIRUSES
Heterologous and homologous interference and protection differ intrinsically, as by definition heterologous viruses cannot replicate and amplify the DI genome or be subject to DI virus-mediated genome competition. Use of type I interferon receptor-null mice and direct assay for interferon in the lungs of mice inoculated with 244 DI virus (in the absence of infectious virus) demonstrated that heterologous prophylaxis was mediated through the type I interferon system (95, 96). There were also indications that 244 DI RNA may stimulate factors in addition to type I interferon that contribute to protection, as there was a consistent 4-day delay in the onset of disease compared with that for the normal mouse infected controls (95, 96). A possible candidate is type III lambda interferon (IFN-λ), shown recently to be important in protection against influenza A and B viruses in vivo (99–101). It is possible that the interferon stimulated by influenza DI virus acts by enhancing the maturation of dendritic cells (8, 9). The inference from these studies is that 244 DI RNA may be able to protect against all type I interferon-sensitive respiratory viruses. Unlike the systemic administration of a large dose of pure interferon, intranasal 244 DI virus caused no observable toxic effects (fever, weight loss, or other clinical signs) in mice or ferrets, probably because it generated relatively small amounts of interferon, locally and over a period of time.
DI RNAs from the Rhabdoviridae and Paramyxoviridae, which are known to stimulate interferon, are all extensively double stranded and are formed by partial or complete self-copying (copyback and snapback RNAs, respectively) (Fig. 1, sections iii and iv) (102–105). However, their ability to protect against virus infections in vivo has not been tested. As influenza A DI RNAs in general and 244 DI RNA in particular have no extensive double-stranded regions, it is surprising that an influenza 244 DI virus can protect mice in a type I interferon-dependent manner from heterologous respiratory viruses.
Animals treated with 244 DI virus and challenged with heterologous viruses generated solid protective immunity to those challenge viruses, indicating that the DI RNA reduced but did not eliminate replication of the infectious virus, a scenario which parallels that seen with influenza A virus (95, 96).
RESISTANCE TO INFLUENZA VIRUS DI RNA IS HIGHLY UNLIKELY
Resistance to antivirals is well known in the virus world, with mutations arising at a rate of approximately 10−5 nucleotide changes per round of replication. Resistance of influenza viruses circulating in the human population to the antiviral oseltamivir phosphate (Tamiflu) has been widely reported (1, 106, 107). To overcome such resistance, combinations of drugs, each with a different target, can be used to reduce the likelihood of escape mutants arising. It was only when 3 antivirals, acting on different elements of human immunodeficiency virus, were given simultaneously that infections were brought under control. In so doing, the chance of a mutant virus arising that was resistant to all 3 antivirals was reduced from 10−5 to 10−15. Resistance of infectious viruses to interference by a cognate DI genome is theoretically possible only if the replicase and its recognition sequence mutate simultaneously so that they can no longer efficiently recognize the DI genome. The only recorded example occurred when a persistent VSV infection was established with the aid of DI VSV in a continuous tissue culture system (108–110). Because influenza A virus DI RNA-mediated protection against influenza A virus infection operates at the genetic level and the infectious genome is composed of 8 separate RNAs, a resistant mutant can arise only if there is simultaneous mutation of each of the 8 genome segments. With a mutation rate of 10−5 for each genome segment, the chance of an escape mutant arising is 1 in 1040. This is 1025-fold less likely than resistance to the triple HIV-1 therapy, of which there has been none since its inception in clinical practice some 30 years ago. In addition, there would also have to be a mutation(s) of the influenza RNA-dependent RNA polymerase to match the mutation in the 8 genome segments. The 10−40 figure is thus a low estimate of the chance of infectious influenza A virus escaping interference by an influenza virus DI RNA.
CHOICE OF DELIVERY VEHICLE
The human respiratory tract contains cells bearing the NANA α2,6Gal and NANA α2,3Gal receptors for influenza viruses, with the former found predominantly in the upper respiratory tract and the latter in the lower respiratory tract. Consequently, a helper-delivery system is required to have dual receptor specificity as discussed above. While it is possible to treat immunologically naive mice 10 times (at 1 dose per week) with a protective dose of DI virus with little loss of protective effect (P. D. Scott and N. J. Dimmock, unpublished data), the prevailing host immunity needs to be considered, as a DI virus can be neutralized by the same antibodies as those that neutralize infectious virus. Choice of the delivery system would therefore depend inter alia on the immune status of the population designated for treatment. In principle, any influenza A virus can package DI RNA and serve as the helper-delivery virus, providing it meets with other criteria, such as seroprevalence and growth requirements. The current choice is A/PR/8/34 (H1N1), a human virus isolated in 1934. The seroprevalence of neutralizing antibodies to the glycoproteins of PR8 is likely to currently be extremely low, as even in 1975 there was no difficulty in finding antibody-free individuals (111). Further, the average life expectancy for those born in the United States in 1934 was only 58 years (112) or less in many other parts of the world. Thus, only a small proportion of people alive today (80 years later) will have been infected with PR8. PR8 is safe to use as it has negligible infectivity for people (111) and has served as the backbone for the recombinant viruses that are used for making human vaccines since their inception.
CONCLUDING REMARKS
DI RNA is an entirely novel type of antiviral that has been designed by nature. DI influenza RNA could potentially be delivered by any influenza A virus. Thus, cells that are susceptible to influenza virus are specifically targeted, and a miniscule amount of DI RNA is clinically effective. DI RNA is amplified over 10,000-fold by influenza A viruses, and nascent progeny DI particles are spread throughout the respiratory system, making DI virus a unique type of prodrug. Without influenza A virus infection, DI RNA naturally decays, but with infection virus-specific immunity, which clears free DI and infectious virus and infected cells, it is stimulated. Finally, DI RNA-treated individuals infected with a homologous influenza A virus or a heterologous virus develop solid immunity to the challenge virus but not the DI virus. Thus, in addition to being a “prodrug,” DI virus is also a “provaccine.” The net result is that influenza 244 DI RNA is highly protective in vivo against influenza A viruses and other respiratory viruses in terms of prophylaxis, therapy, and postinfection immunity. Identification of the infectious agent is not required, as the DI virus is predicted to be universally active against all respiratory viruses. Unlike a vaccine, which takes several weeks to stimulate full immunity, DI virus-mediated protection is effective immediately. It inspires confidence in the fight against the continuously evolving influenza viruses that 244 DI virus, formulated in 2006, protected against a pandemic influenza A virus that did not exist until 2009 (89, 90). The likelihood of resistance arising is extremely low. The preclinical success of the DI virus approach as an antiviral against influenza A and other respiratory viruses argues for considering the development of a family of antivirals based on DI nucleic acids that can be targeted to a range of important human and animal diseases. However, the transfer of this technology to people now requires validation in clinical trials.
ACKNOWLEDGMENTS
We express our appreciation for support from the Wellcome Trust and the Medical Research Council.
Footnotes
Published ahead of print 26 February 2014
REFERENCES
- 1.Hurt AC, Chotpitayasunondh T, Cox NJ, Daniels R, Fry AM, Gubareva LV, Hayden FG, Hui SD, Hungnes O, Lackenby A, Lim W, Meijer A, Penn C, Tashiro M, Uyeki TM, Zambon M, WHO Consultation on Pandemic Influenza A (H1N1) 2009 Virus Resistance to Antivirals 2012. Antiviral resistance during the 2009 influenza A H1N1 pandemic: public health, laboratory, and clinical perspectives. Lancet Infect. Dis. 12:240–248. 10.1016/S1473-3099(11)70318-8 [DOI] [PubMed] [Google Scholar]
- 2.Cullen BR. 2009. Viral RNAs: lessons from the enemy. Cell 136:592–597. 10.1016/j.cell.2009.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655. 10.1016/j.cell.2009.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid S, Zony LC, Tenoever BR. 2014. A versatile RNA vector for delivery of coding and noncoding RNAs. J. Virol. 88:2333–2336. 10.1128/JVI.03267-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett ADT, Dimmock NJ. 1986. Defective interfering viruses and infections of animals. Curr. Top. Microbiol. Immunol. 128:55–84. 10.1007/978-3-642-71272-2_2 [DOI] [PubMed] [Google Scholar]
- 6.von Magnus P. 1954. Incomplete forms of influenza virus. Adv. Virus Res. 21:59–79 [DOI] [PubMed] [Google Scholar]
- 7.Huang AS. 1973. Defective interfering viruses. Annu. Rev. Microbiol. 27:101–117. 10.1146/annurev.mi.27.100173.000533 [DOI] [PubMed] [Google Scholar]
- 8.Yount JS, Kraus TA, Horvath CM, Moran TM, Lopez CB. 2006. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J. Immunol. 177:4503–4513 [DOI] [PubMed] [Google Scholar]
- 9.Yount JS, Gitlin L, Moran TM, Lopez CB. 2008. MDA5 participates in the detection of paramyxovirus infection and is essential for early activation of dendritic cells in response to Sendai virus defective interfering particles. J. Immunol. 180:4910–4918 [DOI] [PubMed] [Google Scholar]
- 10.Killip MJ, Young DF, Gatherer D, Ross CS, Short JA, Davison AJ, Goodbourn S, Randall RE. 2013. Deep sequencing analysis of defective genomes of parainfluenza virus 5 and their role in interferon induction. J. Virol. 87:4798–4807. 10.1128/JVI.03383-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pringle CR. 1987. Rhabdovirus genetics, p 176–243 In Wagner RR. (ed), The rhabdoviruses. Plenum Press, New York, NY [Google Scholar]
- 12.Duhaut SD, Dimmock NJ. 1998. Heterologous protection against a lethal human H1N1 influenza virus infection of mice by a H3N8 equine defective interfering virus: comparison of defective RNA sequences isolated from the DI inoculum and mouse lung. Virology 248:241–253. 10.1006/viro.1998.9267 [DOI] [PubMed] [Google Scholar]
- 13.Jennings PA, Finch JT, Winter G, Robertson JS. 1983. Does the higher order of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 34:619–627. 10.1016/0092-8674(83)90394-X [DOI] [PubMed] [Google Scholar]
- 14.Barrett ADT, Dimmock NJ. 1984. Modulation of Semliki Forest virus-induced infection of mice by defective interfering virus. J. Infect. Dis. 150:98–104. 10.1093/infdis/150.1.98 [DOI] [PubMed] [Google Scholar]
- 15.Jones CL, Holland JJ. 1980. Requirements for DI particle prophylaxis against vesicular stomatitis virus infection in vivo. J. Gen. Virol. 49:215–220. 10.1099/0022-1317-49-1-215 [DOI] [PubMed] [Google Scholar]
- 16.Fultz P, Shadduck JA, Kang CY, Streilein JW. 1981. On the mechanism of DI particle protection against lethal VSV infection in hamsters, p 893–899 In Bishop DHL, Compans RW. (ed), The replication of negative strand viruses. Elsevier-North Holland, New York, NY [Google Scholar]
- 17.Li D, Lott WB, Lowry K, Jones A, Thu HM, Aaskov J. 2011. Defective interfering viral particles in acute dengue infections. PLoS One 6:e19447. 10.1371/journal.pone.0019447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan TT-T, Lin M-H, Chen D-S, Shih V. 1998. A defective interference-like phenomenon of human hepatitis B virus in chronic carriers. J. Virol. 72:578–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeh C-T, Lu SC, Chu CM, Liaw YF. 1997. Molecular cloning of a defective hepatitis C virus genome from the ascitic fluid of a patient with hepatocellular carcinoma. J. Gen. Virol. 78:2761–2770 [DOI] [PubMed] [Google Scholar]
- 20.Nuesch JPF, de Chastonay J, Siegl G. 1989. Detection of defective genomes in hepatitis A virus particles present in clinical specimens. J. Gen. Virol. 70:3475–3480. 10.1099/0022-1317-70-12-3475 [DOI] [PubMed] [Google Scholar]
- 21.Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V, Dwyer DE, Freiberg M, Horban A, Losso M, Lynfield R, Wentworth DN, Holmes EC, Davey R, Wentworth DE, Ghedin E, INSIGHT FLU002 Study Group Group IFS 2013. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J. Virol. 87:8064–8074. 10.1128/JVI.00240-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drolet BS, Chiou PP, Heidel J, Leong JA. 1995. Detection of truncated virus particles in a persistent RNA virus infection in vivo. J. Virol. 69:2140–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ke R, Aaskov J, Holmes EC, Lloyd-Smith JO. 2013. Phylodynamic analysis of the emergence and epidemiological impact of transmissible defective dengue viruses. PLoS Pathog. 9:e1003193. 10.1371/journal.ppat.1003193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark C, Kennedy SIT. 1978. The generation and propagation of defective-interfering particles of Semliki Forest virus in different cell types. Virology 89:285–299. 10.1016/0042-6822(78)90060-0 [DOI] [PubMed] [Google Scholar]
- 25.Kingsbury DW, Portner A. 1970. On the genesis of incomplete Sendai virions. Virology 42:872–879. 10.1016/0042-6822(70)90336-3 [DOI] [PubMed] [Google Scholar]
- 26.Kang CY, Weide LG, Tischfield JA. 1981. Suppression of vesicular stomatitis virus defective interfering particle generation by a function(s) associated with human chromosome 16. J. Virol. 40:946–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holland JJ, Villarreal LP, Breindel M. 1976. Factors involved in the generation and replication of rhabdovirus defective T particles. J. Virol. 17:805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whistler T, Bellini WJ, Rota PA. 1996. Generation of defective interfering particles from two vaccine strains of measles virus. Virology 220:480–484. 10.1006/viro.1996.0335 [DOI] [PubMed] [Google Scholar]
- 29.Kang CY, Allen R. 1978. Host function-dependent induction of defective-interfering particles of vesicular stomatitis virus. J. Virol. 25:202–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimmock NJ. 1996. Antiviral activity of defective interfering influenza virus in vivo, p 421–445 In Myint S, Taylor-Robinson D. (ed), Viral and other infections of the respiratory tract. Chapman and Hall, London, United Kingdom [Google Scholar]
- 31.Thomson M, White CL, Dimmock NJ. 1998. The genomic sequence of defective interfering Semliki Forest virus (SFV) determines its ability to be replicated in mouse brain and to protect against a lethal SFV infection in vivo. Virology 241:215–223. 10.1006/viro.1997.8975 [DOI] [PubMed] [Google Scholar]
- 32.Welsh RM, Lampert PW, Oldstone MBA. 1977. Prevention of virus-induced cerebellar disease by defective interfering lymphocytic choriomeningitis virus. J. Infect. Dis. 136:391–399. 10.1093/infdis/136.3.391 [DOI] [PubMed] [Google Scholar]
- 33.Help GI, Coto CE. 1980. Synthesis of interfering particles of Junin virus in baby mouse brain. Medicina (Buenos Aires) 40:531–536 [PubMed] [Google Scholar]
- 34.Spandidos DA, Graham AF. 1976. Generation of defective virus after infection of newborn rats with reovirus. J. Virol. 20:234–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frenkel N, Jacob RJ, Honess RW, Haywood GS, Locker H, Roizman B. 1975. Anatomy of herpes simplex virus DNA. III. Characterization of defective DNA molecules and biological properties of virus populations containing them. J. Virol. 16:153–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell DE, Kemp MC, Perdue ML, Randall CC, Gentry GA. 1976. Equine herpesvirus in vivo: cyclic production of a DNA density variant with repetitive sequences. Virology 69:737–750. 10.1016/0042-6822(76)90502-X [DOI] [PubMed] [Google Scholar]
- 37.Bronson DL, Dreesman GR, Biswal N, Benyesh-Melnick M. 1973. Defective virions of herpes simplex viruses. Intervirology 1:141–143. 10.1159/000148841 [DOI] [PubMed] [Google Scholar]
- 38.Calain P, Curran J, Kolakofsky D, Roux L. 1992. Molecular cloning of natural paramyxovirus copy-back defective interfering RNAs and their expression from DNA. Virology 191:62–71. 10.1016/0042-6822(92)90166-M [DOI] [PubMed] [Google Scholar]
- 39.Meyers G, Thiel H-J, Rumenapf T. 1996. Classical swine fever virus: recovery of infectious viruses from cDNA constructs and generation of recombinant cytopathic defective interfering particles. J. Virol. 70:1588–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Groot RJ, van der Most RG, Spaan WJM. 1992. The fitness of defective interfering murine coronavirus DI-a and its derivatives is decreased by nonsense and frameshift mutations. J. Virol. 66:5898–5905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomson M, Dimmock NJ. 1994. Common sequence elements in structurally unrelated genomes of defective interfering Semliki Forest virus. Virology 199:354–365 [DOI] [PubMed] [Google Scholar]
- 42.Dimmock NJ, Rainsford EW, Scott PD, Marriott AC. 2008. Influenza virus protecting RNA: an effective prophylactic and therapeutic antiviral. J. Virol. 82:8570–8578. 10.1128/JVI.00743-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duhaut SD, Dimmock NJ. 2003. Defective influenza A virus generated entirely from plasmids: its RNA is expressed in infected mouse lung and modulates disease. J. Virol. Methods 108:75–82. 10.1016/S0166-0934(02)00260-4 [DOI] [PubMed] [Google Scholar]
- 44.Doyle M, Holland JJ. 1973. Prophylaxis and immunisation in mice by use of virus-free defective T particles to protect against intracerebral infection by vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 70:2105–2108. 10.1073/pnas.70.7.2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holland JJ, Doyle M. 1973. Attempts to detect homologous autointerference in vivo with influenza virus and vesicular stomatitis virus. Infect. Immun. 7:526–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rabinowitz SG, Huprikar J. 1979. The influence of defective-interfering particles of the PR-8 strain of influenza A virus on the pathogenesis of pulmonary infection in mice. J. Infect. Dis. 140:305–315. 10.1093/infdis/140.3.305 [DOI] [PubMed] [Google Scholar]
- 47.Holland JJ, Villarreal LP. 1975. Purification of defective interfering T particles of vesicular stomatitis and rabies viruses generated in vivo in brains of newborn mice. Virology 67:438–449. 10.1016/0042-6822(75)90445-6 [DOI] [PubMed] [Google Scholar]
- 48.Koprowski H. 1954. Biological modification of rabies virus as a result of its adaptation to chicks and developing chick embryos. Bull. World Health Organ. 10:709–724 [PMC free article] [PubMed] [Google Scholar]
- 49.Wiktor TJ, Dietzschold B, Leamnson RN, Koprowski H. 1977. Induction and biological properties of defective interfering particles of rabies virus. J. Virol. 21:626–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oliver KR, Fazakerley JK. 1998. Transneuronal spread of Semliki Forest virus in the developing mouse olfactory system is determined by neuronal maturity. Neuroscience 82:867–877 [DOI] [PubMed] [Google Scholar]
- 51.Cave DR, Hagen TS, Palma EL, Huang AS. 1984. Detection of vesicular stomatitis virus RNA and its defective interfering particles in individual mouse brains. J. Virol. 50:86–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cave DR, Hendrickson FM, Huang AS. 1985. Defective interfering virus particles modulate virulence. J. Virol. 55:366–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fultz PN, Shadduck JA, Kang CY, Streilein JW. 1982. Mediators of protection against lethal systemic vesicular stomatitis virus infection in hamsters: defective interfering particles, polyinosinate-polycytidylate, and interferon. Infect. Immun. 37:679–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dimmock NJ, Kennedy SIT. 1978. Prevention of death in Semliki Forest virus-infected mice by administration of defective interfering Semliki Forest virus. J. Gen. Virol. 39:231–242. 10.1099/0022-1317-39-2-231 [DOI] [PubMed] [Google Scholar]
- 55.Bernkopf H. 1950. Study of infectivity and hemagglutination of influenza virus in de-embryonated eggs. J. Immunol. 65:571–583 [PubMed] [Google Scholar]
- 56.Fazekas de St Groth S, Graham DM. 1954. Artificial production of incomplete influenza virus. Nature 173:637–638. 10.1038/173637b0 [DOI] [PubMed] [Google Scholar]
- 57.Fazekas de St Groth S, Graham DM. 1954. The production of incomplete influenza virus particles among influenza strains. Experiments in eggs. Br. J. Exp. Pathol. 35:60–74 [PMC free article] [PubMed] [Google Scholar]
- 58.von Magnus P. 1947. Studies on interference in experimental influenza. I. Biological observations. Ark. Kemi Mineral. Geol. 24b:1–6 [Google Scholar]
- 59.von Magnus P. 1951. Propagation of the PR 8 strain of influenza virus in chick embryos. III. Properties of the incomplete virus produced in serial passages of undiluted virus. Acta Pathol. Microbiol. Scand. 29:157–181 [DOI] [PubMed] [Google Scholar]
- 60.von Magnus P. 1951. Propagation of the PR 8 strain of influenza virus in chick embryos. II. The formation of “incomplete” virus following the inoculation of large doses of seed virus. Acta Pathol. Microbiol. Scand. 28:278–293 [DOI] [PubMed] [Google Scholar]
- 61.von Magnus P. 1952. Propagation of the PR8 strain of influenza virus in chick embryos. IV. Studies on the factors involved in the formation of incomplete virus upon serial passage of undiluted virus. Acta Pathol. Microbiol. Scand. 30:311–335 [PubMed] [Google Scholar]
- 62.Werner GH. 1956. Quantitative studies on influenza virus infection of the chick embryo by the amniotic route. J. Bacteriol. 71:505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ginsberg HS. 1954. Formation of non-infectious influenza virus in mouse lungs: its dependence upon extensive pulmonary consolidation initiated by the viral inoculum. J. Exp. Med. 100:581–603. 10.1084/jem.100.6.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horsfall FL. 1954. On the reproduction of influenza virus. Quantitative studies with procedures which enumerate infective and hemagglutinating virus particles. J. Exp. Med. 100:135–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horsfall FL. 1955. Reproduction of influenza viruses. Quantitative investigations with particle enumeration procedures on the dynamics of influenza A and B virus reproduction. J. Exp. Med. 102:441–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gamboa ET, Harter DH, Duffy PE, Hsu KC. 1976. Murine influenza virus encephalomyelitis. III. Effect of defective interfering virus particles. Acta Neuropathol. (Berlin) 34:157–169 [DOI] [PubMed] [Google Scholar]
- 67.Dimmock NJ, Beck S, McLain L. 1986. Protection of mice from lethal influenza: evidence that defective interfering virus modulates the immune response and not virus multiplication. J. Gen. Virol. 67:839–850. 10.1099/0022-1317-67-5-839 [DOI] [PubMed] [Google Scholar]
- 68.McLain L, Dimmock NJ. 1989. Protection of mice from lethal influenza by adoptive transfer of non-neutralizing haemagglutination-inhibiting IgG obtained from the lungs of infected animals treated with defective-interfering virus. J. Gen. Virol. 70:2615–2624. 10.1099/0022-1317-70-10-2615 [DOI] [PubMed] [Google Scholar]
- 69.McLain L, Dimmock NJ. 1991. An influenza haemagglutinin-specific IgG enhances class I MHC-restricted CTL killing in vitro. Immunology 73:12–18 [PMC free article] [PubMed] [Google Scholar]
- 70.McLain L, Morgan DJ, Dimmock NJ. 1992. Protection of mice from lethal influenza by defective interfering virus: T cell responses. J. Gen. Virol. 73:375–381. 10.1099/0022-1317-73-2-375 [DOI] [PubMed] [Google Scholar]
- 71.Morgan DJ, Dimmock NJ. 1992. Defective interfering virus inhibits immunopathological effects of infectious virus in the mouse. J. Virol. 66:1188–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morgan DJ, McLain L, Dimmock NJ. 1993. Protection of three strains of mice against lethal influenza in vivo by defective interfering virus. Virus Res. 29:179–193. 10.1016/0168-1702(93)90058-U [DOI] [PubMed] [Google Scholar]
- 73.Noble S, Dimmock NJ. 1994. Defective interfering type A equine influenza virus (H3N8) protects mice from morbidity and mortality caused by homologous and heterologous subtypes of type A influenza virus. J. Gen. Virol. 75:3485–3491. 10.1099/0022-1317-75-12-3485 [DOI] [PubMed] [Google Scholar]
- 74.Noble S, McLain L, Dimmock NJ. 2004. Interfering vaccine: a novel antiviral that converts a potentially virulent infection into one that is subclinical and immunizing. Vaccine 22:3018–3025. 10.1016/j.vaccine.2004.02.013 [DOI] [PubMed] [Google Scholar]
- 75.Mann A, Marriott AC, Balasingam S, Lambkin R, Oxford JS, Dimmock NJ. 2006. Interfering vaccine (defective interfering influenza A virus) protects ferrets from influenza, and allows them to develop solid immunity to reinfection. Vaccine 24:4290–4296. 10.1016/j.vaccine.2006.03.004 [DOI] [PubMed] [Google Scholar]
- 76.Noda T, Kawaoka Y. 2010. Structure of influenza virus ribonucleoprotein complexes and their packaging into virions. Rev. Med. Virol. 20:380–391. 10.1002/rmv.666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hutchinson EC, von Kirchbach J, Gog J, Digard P. 2010. Genome packaging in influenza A virus. J. Gen. Virol. 91:313–328. 10.1099/vir.0.017608-0 [DOI] [PubMed] [Google Scholar]
- 78.Ada GL, Perry BT. 1955. Infectivity and nucleic acid content of influenza virus. Nature 175:209–210. 10.1038/175209a0 [DOI] [PubMed] [Google Scholar]
- 79.Crumpton WC, Dimmock NJ, Minor PD, Avery RJ. 1978. The RNAs of defective-interfering influenza virus. Virol. 90:370–378. 10.1016/0042-6822(78)90322-7 [DOI] [PubMed] [Google Scholar]
- 80.Davis AR, Nayak DP. 1979. Sequence relationships among defective interfering influenza viral RNAs. Proc. Natl. Acad. Sci. U. S. A. 76:3092–3096. 10.1073/pnas.76.7.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davis AR, Hiti AL, Nayak DP. 1980. Influenza defective interfering viral RNA is formed by internal deletion of genomic RNA. Proc. Natl. Acad. Sci. U. S. A. 77:215–219. 10.1073/pnas.77.1.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nayak DP, Sivasubramanian N, Davis AR, Cortini R, Sung J. 1982. Complete sequence analyses show that two defective interfering influenza viral RNAs contain a single internal deletion of a polymerase gene. Proc. Natl. Acad. Sci. U. S. A. 79:2216–2220. 10.1073/pnas.79.7.2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakajima K, Ueda M, Suguira A. 1979. Origin of small RNA in von Magnus virus particles of influenza virus. J. Virol. 29:1142–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moss BA, Brownlee GG. 1981. Sequence of DNA complementary to a small segment of influenza virus A/NT/60/68. Nucleic Acids Res. 9:1941–1947. 10.1093/nar/9.8.1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Crumpton WC, Clewley JP, Dimmock NJ, Avery RJ. 1979. Origin of the subgenomic RNAs in defective interfering influenza virus. FEMS Microbiol. Letts. 6:431–434. 10.1111/j.1574-6968.1979.tb03758.x [DOI] [Google Scholar]
- 86.Frensing T, Heldt FS, Pflugmacher A, Behrendt I, Jordan I, Flockerzi D, Genzel Y, Reichl U. 2013. Continuous influenza virus production in cell culture shows a periodic accumulation of defective interfering particles. PLoS One 8:e72288. 10.1371/journal.pone.0072288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Duhaut S, Dimmock NJ. 2000. Approximately 150 nt from the 5′ end of an influenza A virus segment 1 defective virion RNA are needed for genome stability during passage of defective virus in infected cells. Virology 275:278–285. 10.1006/viro.2000.0502 [DOI] [PubMed] [Google Scholar]
- 88.Duhaut SD, Dimmock NJ. 2002. Defective segment 1 RNAs that interfere with the production of infectious influenza virus require at least 150 nucleotides of 5′ sequence: evidence from a plasmid-driven system. J. Gen. Virol. 83:403–411 [DOI] [PubMed] [Google Scholar]
- 89.Dimmock NJ, Dove BK, Scott PD, Meng B, Taylor I, Cheung L, Hallis B, Marriott AC, Carroll M, Easton AJ. 2012. Cloned DI influenza virus protects ferrets from pandemic 2009 influenza A virus (A/Cal/04/09, H1N1) and allows protective immunity to A/Cal/04/09 to be established. PLoS One 7:e49394. 10.1371/journal.pone.0049394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dimmock NJ, Dove BK, Meng B, Scott PD, Taylor I, Cheung L, Hallis B, Marriott AC, Carroll MW, Easton AJ. 2012. Comparison of the protection of ferrets against pandemic 2009 influenza A virus (H1N1) by 244 DI virus and oseltamivir. Antiviral Res. 96:376–385. 10.1016/j.antiviral.2012.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. 2011. Defective interfering influenza virus confers only short-lived protection against influenza virus disease: evidence for a role for adaptive immunity in DI virus-mediated protection in vivo. Vaccine 29:6584–6591. 10.1016/j.vaccine.2011.06.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Belser JA, Rota PA, Tumpey TM. 2013. Ocular tropism of respiratory viruses Microbiol. Mol. Biol. Rev. 77:144–156. 10.1128/MMBR.00058-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meng B, Marriott AC, Dimmock NJ. 2010. The receptor preferences of influenza viruses. Influenza Other Respir. Viruses 4:147–153. 10.1111/j.1750-2659.2010.00130.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. 2011. Defective interfering virus protects elderly mice from influenza. Virology J. 8:212. 10.1186/1743-422X-8-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. 2011. DI influenza A virus protects in vivo against disease caused by a heterologous influenza B virus. J. Gen. Virol. 92:2122–2132. 10.1099/vir.0.034132-0 [DOI] [PubMed] [Google Scholar]
- 96.Easton AJ, Scott PD, Edworthy NL, Meng B, Marriott AC, Dimmock NJ. 2011. A novel broad-spectrum treatment for respiratory virus infections: influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine 29:2777–2784. 10.1016/j.vaccine.2011.01.102 [DOI] [PubMed] [Google Scholar]
- 97.Ito R, Ozaki YA, Yoshikawa T, Hasegawa H, Sato Y, Suzuki Y, Inoue R, Morishima T, Kondo N, Sata T, Kurata T, Tamura S-I. 2003. Roles of anti-hemagglutinin IgA and IgG antibodies in different sites of the respiratory tract of vaccinated mice in preventing lethal influenza pneumonia. Vaccine 21:2362–2371. 10.1016/S0264-410X(03)00078-1 [DOI] [PubMed] [Google Scholar]
- 98.Cane C, McLain L, Dimmock NJ. 1987. Intracellular stability of the interfering activity of a defective interfering influenza virus in the absence of virus multiplication. Virol. 159:259–264. 10.1016/0042-6822(87)90463-6 [DOI] [PubMed] [Google Scholar]
- 99.Jewell NA, Cline T, Mertz SE, Smirnov SV, Flaño E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, Durbin JE. 2010. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J. Virol. 84:11515–11522. 10.1128/JVI.01703-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, Staeheli P. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4:e1000151. 10.1371/journal.ppat.1000151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, Sorgeloos F, Ehl S, Mayer D, Kochs G, Schwemmle M, Günther S, Drosten C, Michiels T, Staeheli P. 2010. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 84:5670–5677. 10.1128/JVI.00272-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marcus PI, Gaccione C. 1989. Interferon induction by viruses. XIX. Vesicular stomatitis virus-New Jersey: high multiplicity passages generate interferon-inducing, defective-interfering particles. Virology 171:630–633 [DOI] [PubMed] [Google Scholar]
- 103.Marcus PI, Sekellick MJ. 1977. Defective interfering particles with covalently linked [±]RNA induce interferon. Nature 266:815–819. 10.1038/266815a0 [DOI] [PubMed] [Google Scholar]
- 104.Sekellick MJ, Marcus PI. 1982. Interferon induction by viruses. VIII. Vesicular stomatitis virus: [±]DI-011 particles induce interferon in the absence of standard virions. Virology 117:280–285 [DOI] [PubMed] [Google Scholar]
- 105.Strahle L, Garcin D, Kolakofsky D. 2006. Sendai defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111. 10.1016/j.virol.2006.03.022 [DOI] [PubMed] [Google Scholar]
- 106.Govorkova EA, Ilyushina NA, McClaren JL, Webster RG. 2009. Oseltamivir-resistant subpopulations of H5N1 influenza variants are genetically stable and virulent in ferrets. Antiviral Res. 82:A33. 10.1016/j.antiviral.2009.02.061 [DOI] [Google Scholar]
- 107.Hamelin M-E, Baz M, Abed Y, Couture C, Joubert P, Beaulieu E, Bellerose N, Plante M, Mallett C, Schumer G, Kobinger GP, Boivin G. 2010. Oseltamivir-resistant pandemic A/H1N1 virus is as virulent as its wild-type counterpart in mice and ferrets. PLoS Pathog. 6:e1001015. 10.1371/journal.ppat.1001015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.DePolo N, Giachetti C, Holland JJ. 1987. Continuing coevolution of virus and defective interfering particles and of viral genome sequences during undiluted passages: virus mutants exhibiting nearly complete resistance to formerly dominant defective interfering particles. J. Virol. 61:454–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giachetti C, Holland JJ. 1988. Altered replicase specificity is responsible for resistance to defective interfering particle interference of an Sdi− mutant of vesicular stomatitis virus. J. Virol. 62:3614–3621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Horodyski FM, Holland JJ. 1980. Viruses isolated from cells persistently infected with vesicular stomatitis virus show altered interactions with defective interfering particles. J. Virol. 36:627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Beare AS, Schild GC, Craig JW. 1975. Trials in man with live recombinants made from A/PR/8/34 (H0N1) and wild H3N2 influenza viruses. Lancet 306:729–732. 10.1016/S0140-6736(75)90720-5 [DOI] [PubMed] [Google Scholar]
- 112.Blacklow RS. 2007. Actuarially speaking: an overview of life expectancy. What can we anticipate? Am. J. Clin. Nutr. 86(Suppl):1560S–1562S [DOI] [PubMed] [Google Scholar]