

ABSTRACT
Barrier-to-autointegration factor (BAF) is a DNA binding protein with multiple cellular functions, including the ability to act as a potent defense against vaccinia virus infection. This antiviral function involves BAF's ability to condense double-stranded DNA and subsequently prevent viral DNA replication. In recent years, it has become increasingly evident that dynamic phosphorylation involving the vaccinia virus B1 kinase and cellular enzymes is likely a key regulator of multiple BAF functions; however, the precise mechanisms are poorly understood. Here we analyzed how phosphorylation impacts BAF's DNA binding, subcellular localization, dimerization, and antipoxviral activity through the characterization of BAF phosphomimetic and unphosphorylatable mutants. Our studies demonstrate that increased phosphorylation enhances BAF's mobilization from the nucleus to the cytosol, while dephosphorylation restricts BAF to the nucleus. Phosphorylation also impairs both BAF's dimerization and its DNA binding activity. Furthermore, our studies of BAF's antiviral activity revealed that hyperphosphorylated BAF is unable to suppress viral DNA replication or virus production. Interestingly, the unphosphorylatable BAF mutant, which is capable of binding DNA but localizes predominantly to the nucleus, was also incapable of suppressing viral replication. Thus, both DNA binding and localization are important determinants of BAF's antiviral function. Finally, our examination of how phosphatases are involved in regulating BAF revealed that PP2A dephosphorylates BAF during vaccinia infection, thus counterbalancing the activity of the B1 kinase. Altogether, these data demonstrate that phosphoregulation of BAF by viral and cellular enzymes modulates this protein at multiple molecular levels, thus determining its effectiveness as an antiviral factor and likely other functions as well.
IMPORTANCE The barrier-to-autointegration factor (BAF) contributes to cellular genomic integrity in multiple ways, the best characterized of which are as a host defense against cytoplasmic DNA and as a regulator of mitotic nuclear reassembly. Although dynamic phosphorylation involving both viral and cellular enzymes is likely a key regulator of multiple BAF functions, the precise mechanisms involved are poorly understood. Here we demonstrate that phosphorylation coordinately regulates BAF's DNA binding, subcellular localization, dimerization, and antipoxviral activity. Overall, our findings provide new insights into how phosphoregulation of BAF modulates this protein at multiple levels and governs its effectiveness as an antiviral factor against foreign DNA.
INTRODUCTION
Barrier-to-autointegration factor (BAF/BANF1) is an essential, highly conserved metazoan protein with multiple functions linked to maintaining the integrity of the cellular genome. BAF can interact with double-stranded DNA in a sequence-independent manner, homodimerize to crossbridge DNA, and form higher-order nucleoprotein complexes (1–4). BAF also interacts with many cellular proteins, including LAP2/emerin/MAN1 (LEM) domain proteins that reside in the nuclear envelope, histones, lamins, transcription factors, and DNA damage response (DDR) proteins (5–10). Using these interactions, BAF is thought to act as a tethering protein to bring together chromatin DNA and LEM proteins during late stages of mitosis when the nuclear envelope (NE) is being reassembled. The importance of BAF's role during mitosis is underscored by evidence that misregulation of these BAF-dependent processes leads to chromosome segregation and NE defects, mislocalization of LEM proteins, and embryonic lethality in Caenorhabditis elegans and Drosophila melanogaster (11–14).
An additional mechanism through which BAF acts to protect genomic integrity is as a host defense factor against foreign DNA in the cytoplasm. BAF was first identified as a host protein that targets invading genomes when it was copurified as a component of the proviral preintegration complex (PIC). In those studies it was observed that BAF can prevent suicidal autointegration of retroviral DNA in vitro (15, 16). The ability of BAF to condense DNA contributes to the compaction of viral DNA within the preintegration complex, thus protecting viral DNA from autointegration in vitro (2, 16, 17). Interestingly, the DNA binding/compaction activity of BAF also allows it to be capable of potent antiviral activity against vaccinia virus.
Poxviruses are large and complex DNA viruses that are pathogenic to human and animals. Vaccinia virus, the prototypical member of this family, exhibits significant autonomy from the host cell, as demonstrated by its ability to complete its entire life cycle in the cytoplasm of infected cells. Such autonomy is attributed to the large poxviral genome, which encodes essential replication and transcription proteins (18, 19). One of these essential proteins is the B1 Ser/Thr kinase, which is expressed early in the viral life cycle and performs functions in viral DNA replication and intermediate gene expression (20–26). The temperature-sensitive mutant viruses Cts2 and Cts25 harbor point mutations within the B1 locus and serve as critical tools for gaining insights about B1's functions. For example, using the Cts2 virus, it was determined that a primary function of B1 is to phosphorylate BAF, thereby inactivating its DNA binding capability. Both in vitro and in vivo data support that B1 can directly phosphorylate BAF at Thr2, Thr3, and Ser4 at BAF's N terminus (27, 28). If not phosphorylated by B1, BAF localizes to the sites of viral DNA and interferes with genome replication (27, 29) and transcription (25), thereby leading to a sharp reduction in the number of viral progeny produced (29).
Interestingly, the cell also modulates BAF phosphorylation at its N terminus via both kinases and phosphatases of its own. For example, BAF is a substrate of the cellular vaccinia-related kinases (VRKs) (28, 30–33) and protein phosphatases PP2A and PP4 (32, 34). These enzymes are critical regulators of BAF's mitotic function, controlling its interaction with DNA and other proteins (28, 31, 32, 35). However, there are also data that VRK1 can inhibit BAF's association with retroviral PICs in vitro (36), suggesting that cellular enzymes may impact not only BAF's mitotic function but its effectiveness in binding to foreign DNA as well.
Here we further explored the impact of BAF phosphorylation on its antipoxviral activity. Specifically, we tested the hypothesis that multiple properties of BAF are impacted by its phosphorylation state, thereby regulating BAF's antiviral activity through interconnected mechanisms involving phosphorylation, DNA binding, and cellular localization. Through the studies of cells stably expressing BAF phosphorylation mutants, we demonstrate that BAF phosphomimetic mutants accumulate in the cytoplasm but lack DNA binding activity and thus cannot suppress viral DNA replication and production of the Cts2 B1-deficient virus. Strikingly, the absence of suppression is also observed in a nonphosphorylatable BAF mutant that is fully capable of binding DNA but adopts a predominantly nuclear localization. Furthermore, we confirm that PP2A is a BAF phosphatase, and we provide evidence that it and/or other phosphatases dephosphorylate BAF in the cytoplasm. Together, these data reveal that phosphoregulation of BAF by both viral and cellular enzymes modulates this protein at multiple molecular levels, thus determining its effectiveness as an antiviral effector and likely other functions as well.
MATERIALS AND METHODS
Cell culture.
African green monkey kidney CV-1 cells (Flp-In-CV-1 cells) were obtained from Life Technologies (catalog number R752-07) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals) and penicillin-streptomycin and incubated at 37°C in a 5% CO2 atmosphere.
Mutagenesis and cloning of BAF mutants.
To construct expression vectors for 1× FLAG-tagged BAF (short hairpin RNA [shRNA] resistant) and 1× FLAG-tagged MAAAQ (shRNA resistant), pcDNA5/FRT/TO/3×FLAG-BAF (27) and pcDNA5/FRT/TO/3×FLAG-MAAAQ (29) were used as templates for mutagenesis by overlapping PCR using outside primers Bam Kozak 1×Flag (5′-CTCGAGGGATCCGCCACCATGGATTACAAGGATGACGATGAC-3′) and BAF BamHI 3′ (5′-GCAGGATCCTCACAAGAAGGCGTCGCAC-3′) and internal mutagenesis primers BAF resistant F (5′-GACAAGGCTTATGTGGTCCTTGGCCAG-3′) and BAF resistant R (5′-CTGGCCAAGGACCACATAAGCCTTGTC-3′). PCR products were BamHI digested and ligated to BamHI-digested pHAGE-HYG-MCS (pHM) lentiviral vector (a generous gift from Paula Traktman, Medical College of Wisconsin) to generate pHM/HYG/1×FLAG-BAF and pHM/HYG/1×FLAG-MAAAQ.
To construct expression vectors introducing the amino acid mutations MTTDQ (S4D) and MDDDQ (T2D/T3D/S4D), pHM/HYG/1×FLAG-BAF was used as a template for overlapping PCR using outside primers Bam Kozak 1×Flag and BAF BamHI 3′ and internal mutagenesis primers pair Flag-MTTDQ-F (5′-GACAAGCTCATGACAACCGACCAAAAGCACCGAGAC-3′) and Flag-MTTDQ-R (5′-GGTCGGTTGTCATGAGCTTGTCATCGTCATCCTTGTAATCG-3′) or Flag-MDDDQ-F (5′-GACAAGCTCATGGACGACGACCAAAAGCACCGAGAC-3′) and Flag-MDDDQ-R (5′-GGTCGTCGTCCATGAGCTTGTCATCGTCATCCTTGTAATCG-3′). PCR products were BamHI digested and ligated to BamHI-digested pHAGE-HYG-MCS (pHM) vector. Each clone was verified by restriction digestion and DNA sequencing.
Production of stably expressing cells.
Each lentiviral vector carrying BAF and BAF mutants was used to generate lentivirus as previously described (29). The stable overexpression of BAF in CV1 cells was then performed by transducing cells using these lentivirus preparations expressing 1× FLAG-BAF or BAF mutants. For transduction, CV-1 cells were seeded in 35-mm dishes at ∼0.4 × 106 per well. The next day, medium was replaced with 1 ml of lentivirus supernatant and left for 24 h. Medium was then replaced with fresh medium and left for an additional 24 h. Cells were then passaged in medium containing 200 μg/ml of hygromycin to select for stable lentiviral integration.
The stable depletion of BAF was performed using a BAF shRNA or scrambled shRNA (control) as previously described in (29). Cells were then passaged in medium containing 10 μg/ml of puromycin to select for stable lentiviral integration. For some experiments, the stable overexpression of BAF and its mutants in shBAF cells was performed by further transducing shBAF cells with lentivirus expressing 1× FLAG-BAF or its mutants while shControl cells were further transduced with empty vector pHM-HYG-MCS as a control. Transduced cells were selected with both 200 μg/ml of hygromycin and 10 μg/ml of puromycin.
Virus infections and viral yield assay.
The wild-type (WT) vaccinia virus (WR strain) and the B1-deficient Cts2 virus (20) were used. For viral DNA replication, viral titer determination, and immunoblotting, CV1 cells (1 × 106) were infected with WT or Cts2 virus at a multiplicity of infection (MOI) of 5 at 40°C for 24 h. The next day, cells were harvested onto 1 ml phosphate-buffered saline (PBS) (10 mM Na2HPO4 · 7H2O, 1 mM KH2PO4, 2 mM KCl, 140 mM NaCl, pH 7.4). Cells were aliquoted (200 μl for viral DNA replication, 400 μl for viral titer determination, and 400 μl for immunoblotting) prior to the appropriate analyses performed on each aliquot. For immunofluorescence, CV1 cells were infected with Cts2 vaccinia virus at an MOI of 5 at 32°C for 16 h, followed by a shift to 40°C for 3 h.
For viral yield assays, following cell harvest, cells were pelleted and cell pellets were resuspended in 10 mM Tris (pH 9). Samples were freeze-thawed three times prior to titration on BSC40 cells at 32°C.
Immunoprecipitation analysis.
CV1 cells (1 × 106) were harvested and pelleted at 8,000 × g for 10 min at 4°C. Cells were then lysed in 1 ml of cell lysis buffer (50 mM Tris-HCl [pH 6.8], 2 mM MgCl2, 200 mM NaCl, and 0.2% Triton X-100, supplemented with Complete protease inhibitor [Roche], PhosSTOP phosphatase inhibitor [Roche], and 10 units benzonase) on ice for 30 min, followed by centrifugation at 8,000 × g for 10 min at 4°C. The supernatant was transferred to a new tube, followed by another centrifugation at 10,000 × g for 10 min at 4°C. Next, this supernatant was incubated with 2.5 μg of mouse anti-FLAG M2 antibody on an end-over-end rotator for 2 h at 4°C, at which point 10 μl of magnetic Dynabeads protein G (Life Technologies) equivalent to a 5-μl bead volume was added and rotated overnight at 4°C. The next day, beads were washed 4 times in Tris-buffered saline (TBS) (20 mM Tris-HCl [pH 7.4], 150 mM NaCl). Bound proteins were eluted by the addition of 120 μl of 2× SDS sample buffer (100 mM Tris [pH 6.8], 2% β-mercaptoethanol, 2% SDS, 32.5% glycerol, bromophenol blue).
Subcellular fractionation.
Subcellular fractionations were performed as described previously (37) with modifications. Cell pellets obtained from CV-1 cells (1.2 × 106) upon harvest and centrifugation at 500 × g for 10 min at 4°C were treated with cell lysis buffer (50 mM Tris-HCl [pH 6.8], 2 mM MgCl2, 150 mM NaCl, and 0.5% saponin, supplemented with Complete protease inhibitor [Roche] and PhosSTOP phosphatase inhibitor [Roche]) on ice for 10 min. Soluble cytoplasmic fractions were collected following centrifugation at 500 × g at 4°C for 10 min. The insoluble pellets were further treated with Triton X-100 lysis buffer (50 mM Tris-HCl [pH 6.8], 2 mM MgCl2, 75 mM NaCl, 0.2% Triton X-100, 0.1% SDS, 1× Complete protease inhibitor [Roche], PhosSTOP phosphatase inhibitor [Roche]) on ice for 10 min. Soluble nuclear fractions were collected following centrifugation at 10,000 × g at 4°C for 10 min. To each fraction, 2× SDS sample buffer was added, followed by loading for SDS-PAGE.
Phosphatase inhibitor treatment.
Calyculin A (Sigma C5552 or Santa Cruz 24000) and okadaic acid (SantaCruz 3513) stock and serial dilution solutions were prepared in dimethyl sulfoxide (DMSO). CV-1 cells (1.2 × 106) were treated with phosphatase inhibitor diluted in DMEM-FBS (final DMSO concentration of 1%) at 37°C for 2 h. Cells were then harvested by scraping, washed in PBS, and lysed in 2× SDS sample buffer prior to loading for SDS-PAGE.
Plasmid transfection.
For transfection experiments, pG8-luciferase plasmid (a generous gift from B. Moss, NIAID, Bethesda, MD) and pUC19 plasmid were used. For chromatin immunoprecipitation (ChIP) experiments, CV-1 cells (6 × 106) were transfected with 150 ng pG8-Luc plasmid for 24 h by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. For immunofluorescence, CV-1 cells were transfected with 1 μg of pUC19 plasmid for 24 h.
Immunofluorescence.
Cells were fixed in 4% paraformaldehyde in 1× PBS at room temperature for 20 min. Following fixation, cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature. Cells were then incubated with mouse anti-FLAG M2 antibody (Sigma) and/or rabbit anti-I3 (1:500) for 1 h at room temperature. Following washing with PBS, cells were then incubated with Alexa Fluor 488-conjugated goat anti-mouse-488 (Invitrogen) in PBS and/or Alexa Fluor 594-conjugated goat anti-rabbit-594 (Invitrogen) in PBS for 1 h at room temperature. Cells were then washed in PBS, and DNA was stained with DAPI (4′,6′-diamidino-2-phenylindole). Fluorescence images were taken by indirect fluorescence on an inverted (Olympus IX 81) confocal microscope, and final images were obtained by pseudocoloring using ImageJ software.
Immunoblotting analysis.
CV-1 cells (1.2 × 106) were freshly collected, pelleted, and resuspended in 300 μl of SDS sample buffer supplemented with 10 units of benzonase as described previously (25). Lysate volumes equivalent to 105 cells were resolved by 18% SDS-PAGE followed by protein transfer to polyvinylidene difluoride (PVDF) membranes overnight. Blots were then incubated with the appropriate antibodies prior to signal development with chemiluminescent reagents. Quantifications of the chemiluminescence signal were performed by using a Bio-Rad ChemiDoc XRS instrument and Bio-Rad ImageLab software. The primary antibodies used were as follows: GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 1:200 (Santa Cruz Biotechnology sc-25778); β-tubulin, 1:20,000 (Sigma T7816); FLAG M2 monoclonal, 1:8,000 (Sigma F1804); BAF, 1:5,000 (custom antibody); phospho-BAF (Ser4) antibody, 1:1500 (custom antibody); PP1 (E9) monoclonal, 1:200 (Santa Cruz Biotechnology sc-7482); PP1β monoclonal, 1:5,000 (Epitomics 2029-1); PP2A monoclonal, 1:1,000 (Millipore 05-421); PPP4C, 1:500 (Bethyl Laboratories A300-835A); and PPP6C, 1:1,000 (Bethyl Laboratories A300-844A).
siRNA depletion.
Small interfering RNAs (siRNAs) against PP1, PP2A, PP4, and PP6 and nontargeting siRNAs were designed and ordered from Dharmacon. The siRNA sense sequences were as follows: PP1α, 5′-CCGCAUCUACGGUUUCUACUU-3′; PP1β, 5′-UUAUGAGACCUACUGAUGUUU-3′; PP1γ, 5′-GCAUGAUUUGGAUCUUAUAUU-3′; PP2Aα, 5′-CGUGCAAGAGGUUCGAUGUUU-3′; PP2Aβ/PPP2CB, 5′-GCGAGAAGGCAAAGGAAAUUU-3′; PPP4C, 5′-GACAAUUGACCGAAAGCAAUU-3′; PPP6C, 5′-GCACGAAGGCUAUAAAUUUUU-3′; and scrambled siRNA (siControl), 5′-CAGUCGCGUUUGCGACUGGUU-3′. CV-1 cells were transfected with 50 nM siRNAs using RNAimax (Life Technologies) as per the manufacturer's protocol. Depletion was measured by both quantitative PCR (qPCR) and immunoblotting. For qPCR, RNA was extracted at 48 h posttransfection. For immunoblotting, protein depletions were analyzed at 72 h posttransfection. All infection experiments were performed following 72 h of siRNA treatment.
ChIP Assays.
Chromatin immunoprecipitation was performed as described previously (25) with the following slight modification. After overnight incubation of lysates and antibodies and 2 h of incubation of Dynabeads, beads were washed 3 times with final wash buffer (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 2 mM EDTA [pH 8], 1% Triton X-100, and 0.1% SDS), followed by elution of bound complexes. Immunoprecipitated and input materials were analyzed by quantitative PCR (StepOne Plus real-time PCR; Applied Biosystems).
DNA/RNA purification and qPCR.
Viral DNA was extracted using the GeneJET whole-blood genomic DNA purification minikit (Thermo Scientific K0782) or QIAamp DNA blood minikit (Qiagen 51106). For ChIP analysis, DNA was purified by glass bead purification using agarose dissolving buffer (Zymo Research D4001-1-50).
qPCR was performed using SYBR green PCR Master Mix (Applied Biosystems 4309155) or Bio-Rad iTaq Universal SYBR green Supermix (172-5121). Serial dilutions were included in each qPCR run to develop a standard curve and determine the PCR efficiency of the primer sets in that experiment set. qPCR analyses were performed using 1 μl of purified DNA and 1 μM each primer as follows: vaccinia HA F (5′-CATCATCTGGAATTGTCACTACTAAA-3′) and R (5′-ACGGCCGACAATATAATTAATGC-3′), human GAPDH F (5′-GCAAATTCCATGGCACCGT-3′) and R (5′-TCGCCCCACTTGATTTTGG-3′), and G8Pro ChIP Fwd (5′-CTTCGTGGATCCTGTAGAACG-3′) and G8Pro ChIP Rev (5′-CCATCTTCCAGCGGATAGAATG-3′).
To analyze mRNA transcript levels following siRNA depletion, RNA was isolated from cells by using RiboPure kit (Ambion, Life Technologies, AM1924). cDNA was then synthesized by using a high-capacity cDNA reverse transcription kit (Invitrogen, Life Technologies, 4368814). qPCR analyses were further performed using 2 μl of cDNA (40 to 50 ng) and 1 μM each primer as follows: PPP1CA (PP1α) F (5′-GAGACCATCTGCCTGCTGCT-3′) and R (5′-CAGTTTGATGTTGTAGCGTCTCTTG-3′), PPP1CB (PP1β) F (5′-CGAGTTTGATAATGCTGGTGGAATG-3′) and R (5′-GCTGTTCGAGTTGGAGTGAC-3′), PPP1CC (PP1γ) F (5′-AAAGAGGCAGTTGGTCACTCTG-3′) and R (5′-TTACAGGTCTCGTGGCATTTGG-3′), and PPP2CA (PP2Aα) F (5′-AGTTACACTGCTTGTAGCTCTTAAGGT-3′) and R (5′-GCTCTCATGATTCCCTCGAA-3′).
Statistics.
Error bars shown represent standard deviations from the mean. The P values indicated were calculated using the Student t test.
RESULTS
Stable expression of BAF phosphorylation mutants in CV1 cells.
We hypothesize that BAF's antiviral activity depends on the interwoven regulation of its phosphorylation, cellular localization, and other molecular properties. Here we examined the impact of BAF phosphorylation on its localization, DNA binding, dimerization, and antipoxviral activity using cells stably expressing BAF phosphorylation mutants. Specifically, we changed the amino acids at one or several known BAF phosphorylation sites (Thr2, Thr3, and Ser4) to either an alanine (A) (a nonpolar and uncharged amino acid) or an aspartic acid (D) (a negatively charged amino acid). We designated these mutants MAAAQ, MTTDQ, and MDDDQ (Fig. 1A). Each construct, including WT BAF, also contains a single FLAG epitope tag at its N terminus and was expressed using a lentiviral expression system which allows for stable integration into the host genome and hygromycin resistance. Following antibiotic selection of transduced cells, expression of endogenous BAF and each FLAG-BAF mutant was verified by immunoblot analysis with an anti-BAF antibody (Fig. 1B). As described in previous studies, endogenous BAF proteins can be separated into two or three distinct protein bands of around 10 kDa in size (27, 28, 36). Each distinct band corresponds to certain BAF phosphorylation states in which the protein migration decreases as BAF becomes more phosphorylated. Thus, the unphosphorylated form migrates the fastest, followed by the Ser4-phosphorylated form and finally the hyperphosphorylated (phosphorylated at all three residues) form. Consistent with these migration patterns, the 1× FLAG-tagged WT BAF and mutant proteins also displayed distinct electrophoretic mobilities, which demonstrated that tagging the protein with a single FLAG epitope still allows us to examine the BAF phosphorylation pattern via immunoblotting (Fig. 1B). All mutant and WT BAF proteins, averaging from three separate transductions, were expressed at 3.8- to 5.5-fold-higher levels than the endogenous BAF proteins (Fig. 1C). In addition, the growth rate of each of these CV1 cells was comparable to that of cells expressing an empty-vector control (apparent doubling times ranged between 30 and 31 h), with the exception of MAAAQ (doubling times of 34.8 h; ∼15% reduction compared to control). Overall, these data confirm the expression of our BAF phosphomutants.
FIG 1.
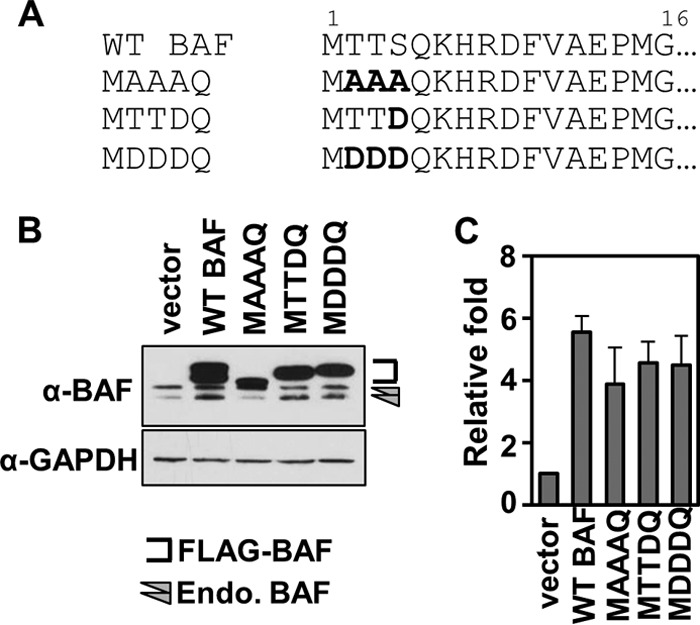
Stable expression of BAF phosphorylation mutants in CV1 cells. (A) Point mutations were introduced into BAF N-terminal residue Thr2, Thr3, or Ser4 as indicated in the amino acid alignment. The residues were mutated to A (alanine) or D (aspartic acid). (B) Representative Western blot analysis of whole-cell lysates from cells stably expressing WT BAF and BAF mutants. Anti-BAF antibody recognizes endogenous BAF protein (arrowheads) and FLAG-tagged proteins (bracketed). Anti-GAPDH antibody was used as a loading control. (C) Quantification of protein expression relative to empty-vector control cells. Data were obtained from three independent transduction experiments. Error bars represent standard deviations.
Unphosphorylated BAF is retained in the nucleus, while phosphorylated BAF is localized in the cytoplasm.
Previous studies have suggested that BAF's subcellular localization depends on its phosphorylation states. In those studies, transient coexpression of green fluorescent protein (GFP)-WT BAF and its kinase 3× FLAG-VRK1 led to relocalization of GFP-WT BAF to the cytosol in U2OS cells (28). In contrast, the nonphosphorylatable mutant GFP-MAAAQ remained nuclear despite VRK1 overexpression (28). To further investigate how posttranslational modification affects BAF's location, we began by analyzing the subcellular distribution of our stably expressed mutant proteins by immunofluorescence assay using an anti-FLAG antibody. Like endogenous BAF (14, 29, 38–40), WT BAF is expressed in the nucleus, nuclear envelope, and cytoplasm (Fig. 2B), and such localization is also consistent with the published results on GFP-tagged and 3× FLAG-tagged BAF (27, 28, 30, 41). With regard to the mutant BAF, we found that MAAAQ is found almost exclusively in the nucleus (Fig. 2C), while both our phosphomimetic mutants MTTDQ and MDDDQ displayed a cellular distribution similar to that of WT BAF (Fig. 2D and E). Next, to complement our immunofluorescence analyses of the subcellular distribution of these mutant proteins, we also performed subcellular fractionation assays followed by immunoblotting analyses. First, cells were lysed with the mild detergent saponin to release cytoplasmic proteins, followed by treatment of the saponin-insoluble fraction with Triton X-100 to release nuclear proteins (37). As expected, WT BAF was distributed in both the cytoplasm (42.4%) and the nucleus (57.6%) (Fig. 2F). In contrast, more than 90% of MAAAQ was distributed to the Triton X-100-soluble nuclear fraction (Fig. 2F), which is consistent with the immunofluorescence data above. Interestingly, >75% of MTTDQ and >85% of MDDDQ accumulated in the saponin-soluble fraction (Fig. 2F), which suggested a cytosolic accumulation of these mutants that could not be detected by immunofluorescence analysis. Thus, these results confirm and expand the evidence that BAF phosphorylation affects its subcellular distribution. Specifically, these data are consistent with the model that BAF phosphorylation facilitates its cytosolic accumulation, while complete BAF dephosphorylation leads to its nuclear retention.
FIG 2.
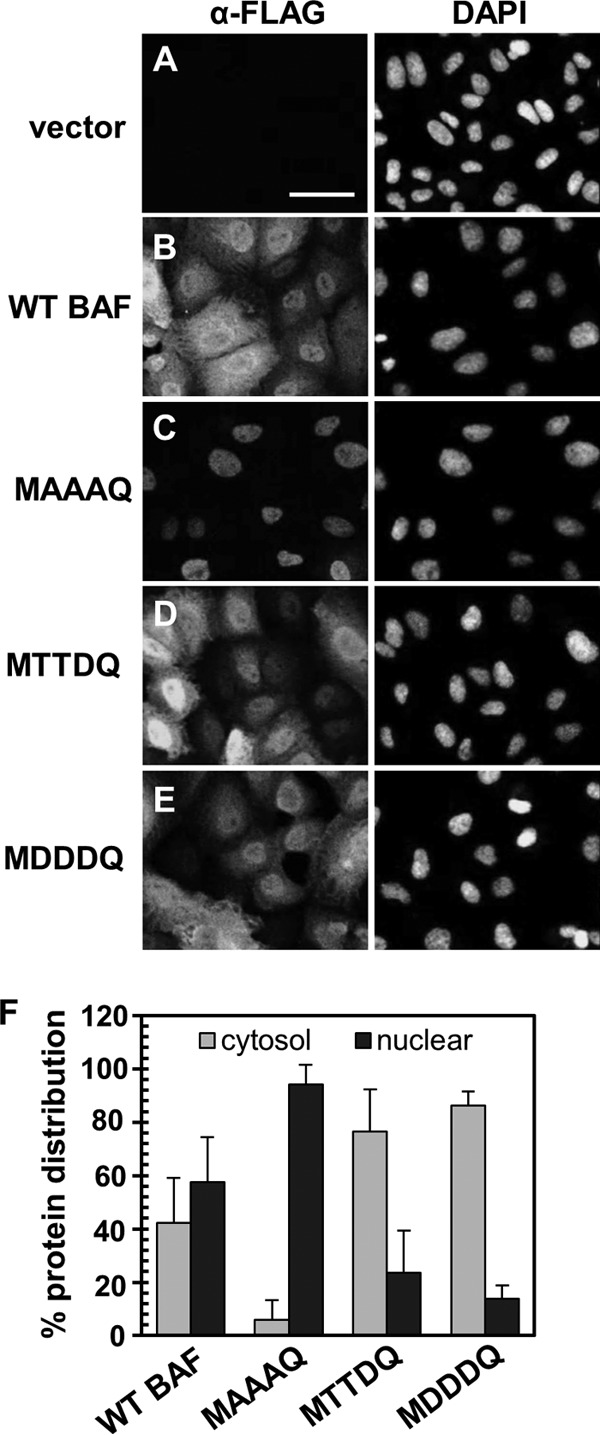
Subcellular distribution of BAF phosphorylation mutants. (A to E) Immunofluorescence analyses of WT BAF and BAF mutants with an anti-FLAG antibody (Alexa Fluor 488 conjugated). Nuclei were visualized by DAPI staining. Cells expressing empty vector (A), WT BAF (B), MAAAQ (C), MTTDQ (D), and MDDDQ (E) were analyzed. Scale bar, 50 μm. (F) Subcellular fractionation analyses of cells indicated in panels A to E. Fractionated cell lysates were analyzed by immunoblotting with an anti-FLAG antibody and quantified with ImageLab software (Bio-Rad). The percent protein distribution was derived from the amount of protein in either the cytosol (gray bars) or nuclear (black bars) fraction relative to the total protein in both fractions (n = 3). Error bars represent standard deviations.
BAF hyperphosphorylation inhibits its dimerization.
Structural studies of BAF have demonstrated its ability to homodimerize and, in the presence of DNA molecules, to cross-bridge DNA to form a higher-order nucleoprotein complex (2, 3, 42). In addition, functional studies of a BAF mutant with an inability to form dimers showed that BAF dimerization contributes to its DNA binding and is also necessary for its antipoxviral activity (29). However, the effect of phosphorylation on BAF's ability to homodimerize is currently unknown. To address this question, lysates from CV1 cells expressing empty vector, WT, or BAF mutants were subjected to anti-FLAG immunoprecipitation. These immunoprecipitations were performed in the presence of nuclease to ensure that DNA was not available to augment BAF-BAF interaction. Input lysates and immunoprecipitated fractions were then subjected to immunoblotting with an anti-BAF antibody to measure dimerization by assessing the coprecipitation of endogenous BAF. As expected, WT BAF was able to coprecipitate endogenous BAF (Fig. 3). Similarly, MAAAQ and MTTDQ can also coprecipitate endogenous BAF. Interestingly, only the fastest-migrating (unphosphorylated) form of endogenous BAF was detected in each of these immunoprecipitates, indicating that phosphorylation impairs dimerization of BAF. Consistent with this model, no endogenous BAF was immunoprecipitated with MDDDQ. Together, these data indicate for the first time that BAF hyperphosphorylation inhibits its dimerization.
FIG 3.

Dimerization assay of BAF by immunoprecipitation (IP). Cell lysates were subjected to immunoprecipitation using an anti-FLAG antibody followed by Western blot analyses using anti-BAF antibody. Input lysates (left) were also analyzed to illustrate the presence and migration pattern of endogenous BAF in each lysate. The bracket indicates the epitope-tagged BAF proteins, while the arrowhead indicates endogenous BAF.
BAF hyperphosphorylation reduces its DNA binding activity against transfected plasmid DNA and chromatin DNA.
Building on previous evidence that BAF phosphorylation at Ser4 resulted in reduced affinity for DNA in vitro (28), we next sought to examine whether the MTTDQ protein also exhibited reduced interaction with DNA in cells. In parallel, we also wanted to test our hypothesis that the “hyperphosphorylated” mutant MDDDQ would result in a complete loss of its DNA binding activity. To address these goals, we examined the DNA binding activity of BAF mutants via chromatin immunoprecipitation (ChIP) analysis. CV1 cells expressing empty-vector control, WT, or BAF mutants were transfected with 150 ng pG8-Luc plasmid for 24 h and subsequently fixed with paraformaldehyde. This pG8-Luc plasmid contains the promoter of the vaccinia virus G8R gene driving the expression of a luciferase gene (23, 25). Our recent study utilizing this plasmid demonstrated that pG8-Luc coprecipitates with BAF in ChIP analysis (25), validating this plasmid as a source of foreign DNA that can be bound by BAF. After harvest and lysis of fixed cells, lysates were subjected to immunoprecipitation of FLAG-tagged BAF followed by reverse cross-linking of protein-DNA complexes and qPCR analysis. qPCR was performed on purified DNA by using primers specific for either the G8 promoter region or the GAPDH locus, which we used as a representative region of nuclear chromatin (as described in reference 25). Fold enrichment was calculated relative to an empty-vector control. From these analyses, we found that WT BAF can interact with plasmid DNA with a fold enrichment of ∼140 (Fig. 4A). Similarly, MAAAQ also interacts with plasmid DNA with a slightly higher fold enrichment of ∼200 (Fig. 4A). In contrast, both MTTDQ and MDDDQ exhibited little or no binding of plasmid DNA, with only 3.5- and 1.0-fold enrichment, respectively, which is 40- and 140-fold less than that for the WT BAF (Fig. 4A). With respect to binding to chromatin DNA, we found a similar trend of DNA binding. Specifically, MTTDQ and MDDDQ also did not interact with chromatin DNA, with ∼20- to 25-fold less binding than for WT BAF (Fig. 4B). In contrast, MAAAQ precipitated 3-fold more chromatin DNA than the WT BAF (Fig. 4B).
FIG 4.
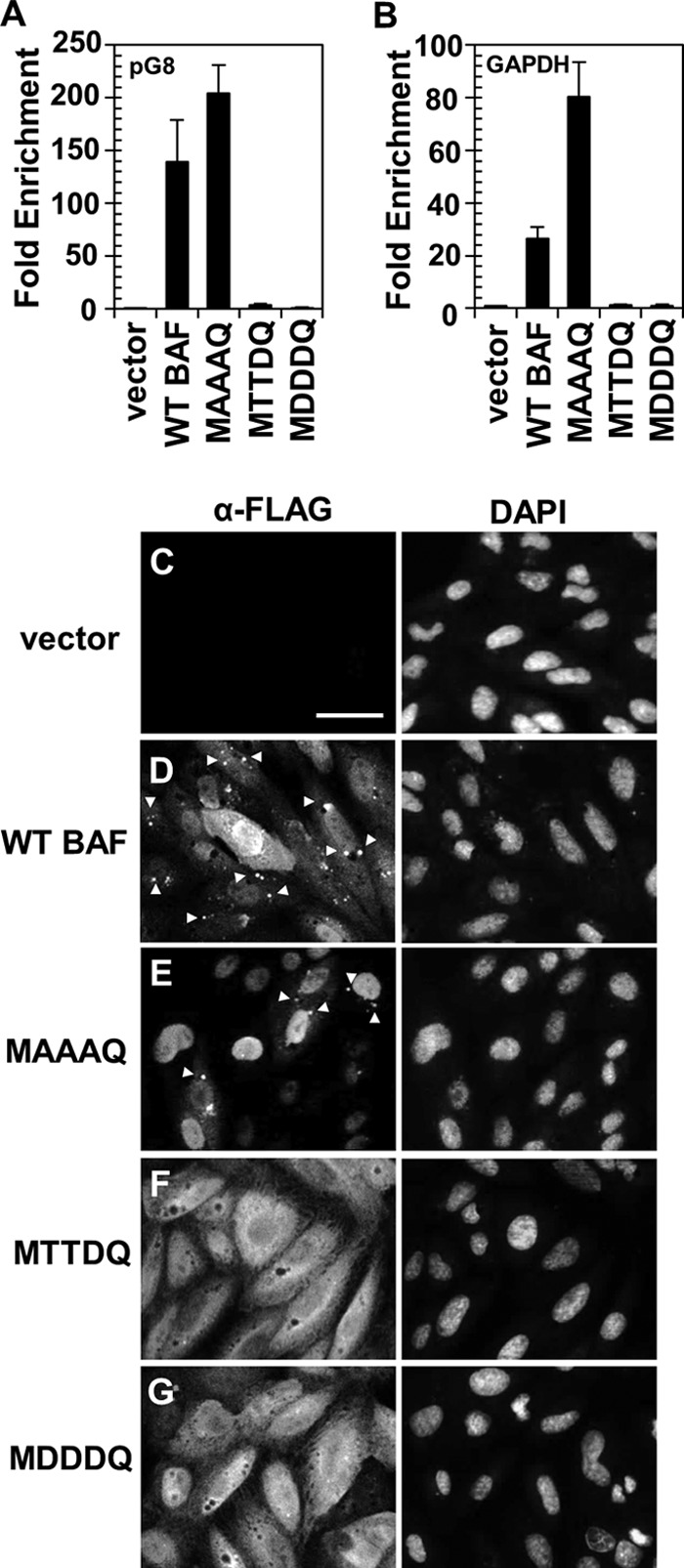
DNA binding activity of WT BAF and its mutants to plasmid DNA. (A and B) ChIP analyses of WT BAF and BAF mutants. Cells were transfected with 150 ng of pG8-Luc plasmid DNA for 24 h, followed by fixation, immunoprecipitation with anti-FLAG antibody, and reverse cross-linking of protein-DNA complexes. Purified DNA was analyzed by qPCR using primers specific for the G8 promoter region of pG8-Luc plasmid (A) or the GAPDH locus of chromatin DNA (B). Fold enrichment was obtained relative to an empty-vector control and normalized to input DNA (n = 3). Error bars represent standard deviations. (C to G) Immunofluorescence analyses of cells indicated in panels A and B that were transfected with 1 μg pUC19 plasmid DNA prior to cell costaining with an anti-FLAG antibody (Alexa Fluor 488 conjugated) and DAPI. Cells expressing empty vector (C), WT BAF (D), MAAAQ (E), MTTDQ (F), and MDDDQ (G) were analyzed. Scale bar, 50 μm. Puncta formations are indicated by arrowheads.
To complement these ChIP data, we also performed immunofluorescence analysis on these aforementioned cells that had been transfected with plasmid DNA. This was done to assay the ability of each BAF mutant to relocalize to cytoplasmic puncta containing foreign DNA, as previously established (29). Like endogenous BAF's relocalization to plasmid DNA (29), cells expressing WT BAF also respond to the presence of foreign plasmid DNA by forming numerous puncta throughout the cytoplasm (Fig. 4D, arrowheads). Interestingly, although MAAAQ is clearly capable of binding foreign DNA (Fig. 4A), the nuclear localization of this protein appears to severely reduce its cytoplasmic binding ability because the number of cytoplasmic puncta observed is less than 20% of what was observed with WT BAF (Fig. 4E). Finally, despite the clear cytoplasmic presence of MTTDQ and MDDDQ, the number of cytoplasmic puncta observed in either MTTDQ- or MDDDQ-expressing cells is only 3% of that seen in WT BAF-expressing cells (Fig. 4F and G). Altogether, we have shown that BAF phosphorylation disrupts its DNA binding activity in live cells, as evident from our studies of both phosphomimetics MTTDQ and MDDDQ. In addition, Ser4 phosphorylation is largely sufficient to inhibit DNA binding in these assays, since similar effects were observed for both MTTDQ- and MDDDQ-expressing cells. Finally, we have also shown that unphosphorylated BAF can interact with either chromatin DNA or plasmid DNA, although its binding likely occurs predominantly in the nucleus rather than the cytoplasm.
Both unphosphorylated and hyperphosphorylated BAF exhibit a reduced ability to relocalize to vaccinia virus DNA upon infection.
Our current working model is that BAF's antipoxviral activity depends on its ability to relocalize to viral replication factories in the cytoplasm and to bind and condense viral DNA therein (27, 29). However, in the presence of the vaccinia virus B1 kinase, BAF is phosphoinactivated, and no relocalization of BAF is observed (27, 29). Building on these data, we wanted to examine how relocalization of BAF during vaccinia virus infection is impacted by its phosphorylation status. Based on our earlier observations of the lack of DNA binding when BAF is phosphorylated, we hypothesized that both phosphomimetic mutants MTTDQ and MDDDQ also lack the capability to relocalize to viral DNA replication factories upon infection with vaccinia virus. To address this hypothesis, we performed immunofluorescence analysis on cells that were infected with the Cts2 mutant vaccinia virus. The temperature-sensitive Cts2 mutant carries a point mutation in the B1 locus, resulting in the production of an unstable and nonfunctional B1 protein at nonpermissive temperatures (21, 22). For these studies, CV1 cells expressing empty-vector control, WT, or BAF mutants were infected with Cts2 virus at an MOI of 5 at 32°C for 16 h, followed by a shift to 40°C for 3 h. This temperature shift protocol allows first for viral factory formation at the permissive temperature, followed by arrest of viral DNA synthesis at the nonpermissive temperature and potential BAF relocalization to the cytoplasmic viral factories (22, 27). The colocalization of BAF mutants and viral DNA replication factories was then assessed by costaining cells with anti-FLAG (green) and anti-I3 (red) antibodies (Fig. 5). I3 is a vaccinia virus single-stranded DNA binding protein that is often found throughout the cytosol but is highly enriched at active viral DNA replication sites, thus allowing us to identify those sites in this assay (43–45). For example, our immunofluorescence analyses showed that cells that were expressing the empty-vector control contained some I3 throughout the cell, but most was localized to viral factories (Fig. 5A, arrowheads). These results are indicative of normal infection and serve as a positive control. Next, in 1× FLAG-WT BAF cells, while we observed a reduction in the number of I3 foci due to the increased amount of WT BAF, the colocalization of FLAG-tagged BAF to the I3-positive factories was often observed (Fig. 5B, arrows). Meanwhile, in MAAAQ-expressing cells, we observed I3 staining similar to that in the control cells, with no obvious localization of MAAAQ to the I3 sites in the cytoplasm (Fig. 5C, arrowheads). While this result is somewhat incongruent with the data shown in Fig. 4E, the fact that MAAAQ could be detected relocalizing to plasmid DNA in that assay may be due to the larger amount of DNA introduced by transfection than by infection. These data in Fig. 5C suggest that because MAAAQ is localized predominantly in the nucleus, interaction between the nucleus-retained MAAAQ proteins and the cytoplasmic viral DNA likely becomes spatially restricted. Finally, in the phosphomimetic MTTDQ- and MDDDQ-expressing cells, we also observed I3 staining at multiple viral factories (Fig. 5D and E, arrowheads), with little relocalization of these BAF mutant proteins to the I3 sites (Fig. 5D and E, arrows). This is consistent with our previous observation that, despite their cytoplasmic localization, the weakened DNA binding activities of these two mutants likely limits their response to vaccinia virus.
FIG 5.

Relocalization of BAF and its mutants to viral replication factories in B1-deficient Cts2 vaccinia virus-infected cells. (A to E) Immunofluorescence analyses of cells stably expressing empty vector (A), WT BAF (B), MAAAQ (C), MTTDQ (D), and MDDDQ (E). Cells were infected with Cts2 vaccinia virus at an MOI of 5 at 32°C for 16 h, followed by a shift to 40°C for 3 h. Cells were then costained with anti-FLAG antibody (Alexa Fluor 488 conjugated; green), anti-I3 antibody (Alexa Fluor 594 conjugated; red), and DAPI (blue). Scale bar, 50 μm. In the rightmost panels (α-FLAG + α-I3), I3-positive FLAG-negative puncta are indicated by arrowheads, and evidence of colocalization (I3-positive, FLAG-positive) sites are indicated by arrows.
BAF hyperphosphorylation mutants cannot suppress vaccinia virus DNA replication and yield.
Based on the observation that BAF phosphorylation led to the loss of DNA binding, we posited that BAF phosphomimetic mutant proteins would not be functional in responding to vaccinia virus infection. Specifically, we hypothesized that neither the MTTDQ nor the MDDDQ will disrupt viral DNA replication and/or productive infection of the Cts2 virus. To accurately analyze the effect of BAF mutant proteins on vaccinia virus, we expressed each BAF mutant in cells that lack endogenous BAF. This was accomplished by first depleting endogenous BAF using shRNA, followed by expression of shRNA-resistant WT BAF or BAF mutant genes. Upon antibiotic selection of these stable cell lines, their protein expression and growth characteristics were assessed. Averaging from three independent transduction events, we were able to express the BAF protein relative to shControl in WT BAF by as much as ∼5.5-fold and either MTTDQ or MDDDQ by ∼3-fold (Fig. 6A). Furthermore, the MTTDQ-shBAF and MDDDQ-shBAF cells displayed a growth rate similar to that of the vector-shBAF control.
FIG 6.
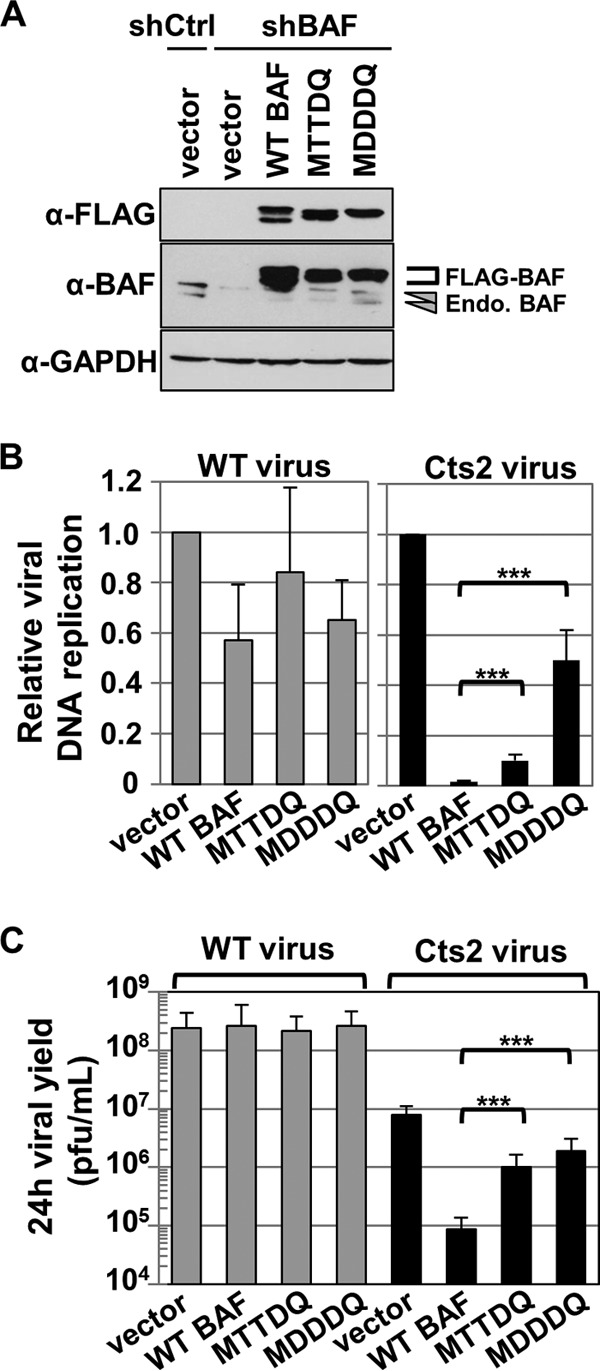
Phosphomimetic mutants MTTDQ and MDDDQ minimally suppress Cts2 viral DNA replication and viral production. (A) Western blot analyses of cells stably expressing vector-shControl and vector-shBAF, WT BAF-shBAF, or BAF mutant-shBAF. Protein expression was detected with anti-FLAG, anti-BAF, or anti-GAPDH antibody. Endogenous BAF proteins are indicated by arrowheads, and FLAG-tagged proteins are indicated by a bracket. (B and C) Cells from panel A, with the exception of vector-shControl, were infected with WT (gray bars) or Cts2 (black bars) virus at an MOI of 5 at 40°C for 24 h. Following harvest, cells were analyzed for viral DNA replication (B) or viral production (C). (B) qPCR were performed by using vaccinia DNA-specific primers, and viral DNA replication was measured relative to the vector-shBAF control (n = 3). (C) Viral titer was measured by plaque titration assay on BSC40 cells at 32°C (n = 3). Error bars represent standard deviations (***, P < 0.005 from unpaired t test analysis).
Next, we tested the ability of each BAF protein to repress vaccinia virus DNA replication and viral production. Control cells (vector-shBAF), as well as WT BAF-shBAF, MTTDQ-shBAF, and MDDDQ-shBAF cells were infected with WT or Cts2 vaccinia virus at an MOI of 5 at 40°C for 24 h. Cells were then harvested and utilized in parallel DNA replication or viral yield analyses. First, viral DNA replication was measured by real-time qPCR. In our positive control, WT vaccinia virus-infected cells showed comparable viral DNA replication among all cells tested (Fig. 6B, gray bars), which indicates that, regardless of the presence of mutant BAF, these cells are susceptible to vaccinia virus infection and can support viral DNA replication. In contrast, in Cts2-infected cells, we observed a 57-fold reduction in viral DNA replication in WT BAF-shBAF relative to control cells (Fig. 6B, black bars). This confirms that the WT BAF protein is functional in suppressing viral DNA replication during Cts2 infection. Interestingly, in Cts2 infection of cells expressing BAF phosphomimetic mutants, we found that the repressive activity of each of these mutants was significantly weaker than that of the WT BAF. Specifically, only a partial reduction of viral DNA replication (10-fold) was observed in MTTDQ-shBAF cells, and even less (2-fold) was found in MDDDQ-shBAF cells compared to the vector-shBAF control (Fig. 6B, black bars).
In parallel analyses, we also assayed viral production from the same samples described above using a plaque titration assay. During WT vaccinia virus infection, viral production was comparable among all cell lines tested, which is consistent with the DNA replication results shown above (Fig. 6C, gray bars). In Cts2-infected cells, we observed a dramatic reduction in viral yield (∼90-fold) in WT BAF-shBAF cells relative to that in control vector-shBAF cells (Fig. 6C, black bars) which is also consistent with the DNA replication results. In contrast to the functional WT BAF, we found only a partial reduction (8-fold) in viral yield in Cts2-infected MTTDQ and even less reduction (4-fold) in MDDDQ cells compared to control cells. These results confirm our hypothesis that hyperphosphorylated BAF is nonfunctional against vaccinia virus in a manner correlating with impaired DNA binding activity.
Unphosphorylatable BAF also cannot suppress vaccinia virus DNA replication and yield.
The BAF nonphosphorylatable mutant MAAAQ displays some interesting properties which contrast with those of the other mutants we produced. It binds DNA at least as efficiently as WT BAF, is predominantly nuclear, relocalizes to cytoplasmic puncta much less efficiently than WT BAF upon introduction of foreign DNA, and did not relocalize to cytoplasmic viral replication factories upon vaccinia virus infection. Based on these observations, we hypothesized that despite possessing DNA binding activity and due to its localization, MAAAQ would be nonfunctional against vaccinia virus. To address this question, we also expressed MAAAQ in BAF-depleted cells (Fig. 7A) using the same method described in the preceding section. Upon infection with WT vaccinia virus, we found comparable viral DNA replication and virus production in MAAAQ-shBAF and control vector-shBAF cells (Fig. 7B and C, gray bars). Interestingly, with respect to Cts2 vaccinia virus infection, we found only a very modest inhibition of viral DNA replication in MAAAQ-shBAF cells (<3-fold) relative to control cells, which was ∼20-fold less inhibition than in WT BAF-shBAF cells (Fig. 7B, black bars). This indicates that MAAAQ does not repress viral DNA replication as efficiently as WT BAF does. In addition, we found only partial reduction in the viral yield of Cts2-infected MAAAQ-shBAF cells (7.5-fold) relative to control cells, as opposed to the ∼90-fold reduction in WT BAF-shBAF cells (Fig. 7C, black bars). Overall, these analyses confirm our hypothesis that the predominantly nuclear unphosphorylated BAF, despite its ability to bind DNA, is only minimally effective at inhibiting vaccinia viral DNA replication and new virus production compared to WT BAF.
FIG 7.
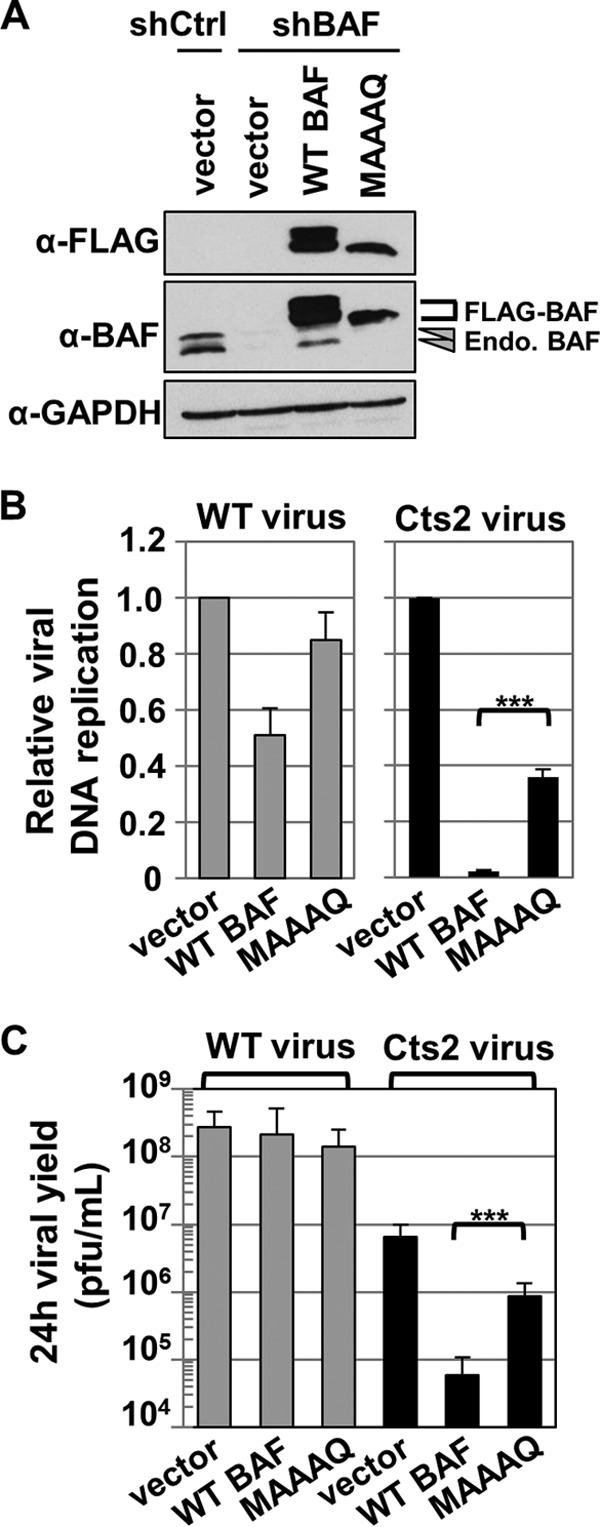
Nonphosphorylatable mutant MAAAQ does not suppress Cts2 vaccinia viral DNA replication and viral production. (A) Western blot analyses of cells stably expressing vector-shControl, vector-shBAF, WT BAF-shBAF, and MAAAQ-shBAF. Protein expression was detected with anti-FLAG, anti-BAF, or anti-GAPDH antibody. Endogenous BAF proteins are indicated by arrowheads, and FLAG-tagged proteins are indicated by bracket. (B and C) Cells from panel A, with the exception of vector-shControl, were infected with WT or Cts2 vaccinia virus at an MOI of 5 at 40°C for 24 h. Following harvest, cells were analyzed for viral DNA replication (B) or viral production (C). (B) qPCR was performed by using vaccinia virus DNA-specific primers, and viral DNA replication in either WT or Cts2 infection was measured relative to the vector-shBAF control (n = 3). (C) Viral titer was measured by plaque titration assay on BSC40 cells at 32°C (n = 3). Error bars represent standard deviations (***, P < 0.005 from unpaired t test analysis).
Phosphatase inhibitor treatment leads to BAF relocalization from the nucleus to the cytoplasm.
Our data above demonstrate that phosphorylation of BAF is necessary for its cytoplasmic localization; however, constitutively phosphorylated BAF is unable to act as a host defense against poxviral DNA replication. This leads us to posit that WT BAF is dephosphorylated in the cytoplasm to “activate” its antiviral activity. In support of this model, it has recently been observed that at least two phosphatases, PP2A and PP4, can act on BAF (32, 34). Those studies showed that siRNA-mediated depletion of the catalytic subunit of either PP2A or PP4 led to the accumulation of hyperphosphorylated BAF in cell culture and C. elegans (32, 34). Therefore, we tested whether BAF phosphatases were capable of modulating BAF phosphorylation, localization, or antipoxviral activity in our cell system. Our first aim was to confirm the involvement of phosphatases in affecting BAF's phosphorylation via treatments with phosphatase inhibitors. This method is advantageous since the effects of inhibitors are rapid and the substrates are largely known. Our initial study was to determine the minimal time in which BAF becomes completely phosphorylated. CV1 cells were treated for a period of 15 min up to 3 h with a 50 nM concentration of the phosphatase inhibitor calyculin A. Calyculin A is a potent inhibitor of PP1 and PP2A and is a useful tool to demonstrate the involvement of these phosphatases in cell culture systems (46). In this analysis, we observed an increase in BAF phosphorylation after 1 h of drug treatment and complete hyperphosphorylation (as judged from the disappearance of the fastest-migrating form of BAF) upon 2 h of treatment (Fig. 8A). Next, to determine the dose-dependent response of calyculin A and its effects on BAF's subcellular distribution, CV1 cells were treated with 12.5, 25, 50, or 100 nM calyculin A at 37°C for 2 h, followed by subcellular fractionation with 0.5% saponin (cytoplasmic fraction) and subsequently with 0.5% Triton X-100 (nuclear fraction). The degree of BAF phosphorylation was analyzed by immunoblot analysis using an anti-BAF antibody. In DMSO control-treated cells, different BAF species were present in both the cytosol and the nuclear fractions (Fig. 8B). Upon calyculin A treatment, we observed an increase in the amount of phosphorylated BAF at 25 nM and complete BAF hyperphosphorylation at 50 nM, followed by a clear shift in protein distribution from the nuclear to the cytoplasmic fraction (Fig. 8B). Specifically, ∼65% of BAF accumulated in the cytosol at 25 nM and ∼88% of BAF was present in the cytosol at 50 nM, as opposed to the ∼35% of BAF found in the cytosol with the DMSO control (Fig. 8C). As a complement to these analyses, we also treated CV1 cells with okadaic acid, another natural inhibitor of PP1 and PP2A (47), and found increasing BAF phosphorylation in a dose-dependent manner, along with a clear shift in BAF's distribution to the cytosol as it becomes phosphorylated (data not shown). Overall these results suggest that BAF is the target of calyculin A and an okadaic acid-sensitive phosphatase(s) and demonstrate for the first time the impact of BAF dephosphorylation in regulating its subcellular distribution.
FIG 8.
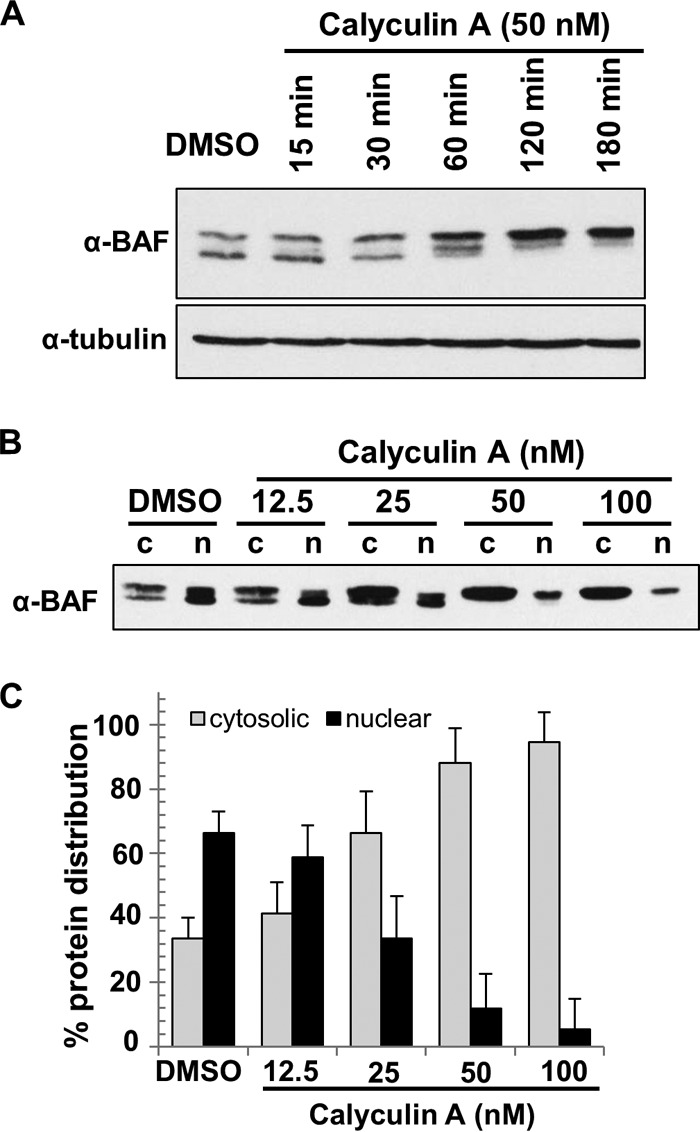
Calyculin A treatment promotes BAF relocalization to the cytosol. (A) Time-dependent analyses of BAF phosphorylation upon calyculin A treatment of CV1 cells. Cells were treated with 1% DMSO or 50 nM calyculin A at 37°C for the indicated times. Western blot analysis was performed using anti-BAF and anti-tubulin (loading control) antibodies. (B) Dose-dependent response and subcellular fractionation of CV1 cells treated with calyculin A. Cells were treated with the indicated concentration of calyculin A at 37°C for 2 h, followed by subcellular fractionation into cytosolic (c) and nuclear (n) fractions. Western blot analysis was performed by using anti-BAF antibody. (C) The percent protein distribution was calculated based on Western blot results from panel B. Error bars represent standard deviations. All data were obtained from three independent experiments.
PP2A is involved in dephosphorylating BAF and can counteract B1 activity on BAF in vaccinia virus-infected cells.
Although the use of calyculin A and okadaic acid clearly upregulated BAF phosphorylation, we noticed that extended treatment with these inhibitors was cytotoxic. These effects have been previously reported in cell cultures and have been linked to global effects of phosphatase inhibition on cytoskeleton actin and myosin phosphorylation (48–50). Therefore, we pursued the possibility of targeting an individual phosphatase(s) in an effort to reduce the possibility of pleiotropic effects in our system. To this end, we knocked down several phosphates using siRNA oligonucleotides directed against PP1, PP2A, PP4, and PP6. Briefly, cells were transfected with a 50 nM concentration of each siRNA for 72 h, and protein depletions were measured by immunoblotting using antibodies specific to each phosphatase. In some cases, we also measured the mRNA transcript levels by qPCR following 48 h posttransfection. First, we analyzed the depletion of PP1 isoforms α, β, and γ. Despite efficient depletion of each isoform, we found no changes in BAF phosphorylation profiles in these cells compared to control cells (data not shown). The same was also true in cells that were depleted of all 3 PP1 isoforms, which suggests that PP1 likely does not play a role in BAF dephosphorylation in CV1 cells. Next, we depleted the catalytic subunit of PP2A, PP4, or PP6. In cells that were depleted of PP2A, we found an increased amount of phosphorylated BAF (Fig. 9A). However, neither the PP4 nor the PP6 depletion enhanced BAF phosphorylation levels despite efficient depletion of each of these phosphatases (data not shown). Furthermore, codepletion of PP2A, PP4, and PP6 also did not result in additional enhancement of BAF phosphorylation compared to that in cells depleted of PP2A alone (data not shown). Thus, these studies show that PP2A is involved in dephosphorylating BAF in CV1 cells, as has been demonstrated in HeLa cells (32), while no evidence could be found that PP1, PP4, or PP6 targets BAF in this model system.
FIG 9.

PP2A depletion is not sufficient to rescue Cts2 vaccinia viral DNA replication. (A) CV1 cells were treated with 50 nM control or PP2A siRNA at 37°C for 72 h. Whole-cell lysates were used for Western blot analyses using anti-BAF or anti-PP2A antibodies. Bands shown were all taken from the same blot with the same exposure times; however, some irrelevant lanes have been removed for clarity. (B) Western blot analysis of BAF phosphorylation in various cell treatments. CV1 cells were transfected with 50 nM control, PP2A, or PP4 siRNA at 37°C for 72 h. Cells were then infected with WT or Cts2 vaccinia virus at an MOI of 5 at 40°C for 24 h. Western blot analyses were performed using the indicated antibodies. (C) Viral DNA replication of CV1 cells transfected with 50 nM control or PP2A siRNA at 37°C for 72 h followed by infection with WT (gray bars) or Cts2 (black bars) virus at an MOI of 5 at 40°C for 24 h. qPCR was performed on purified DNA by using vaccinia virus DNA-specific primers. Viral DNA replication was calculated relative to control siRNA in each infection set (n = 3). Error bars represent standard deviations. (D) Model of BAF phosphoregulation by cellular and viral enzymes. Filled arrows indicate movement of BAF proteins within the cell, while open arrows indicate BAF's (de)phosphorylation by cellular and viral enzymes.
Since BAF phosphorylation by the B1 kinase occurs during vaccinia virus infection, we raised the question of whether BAF dephosphorylation by cellular PP2A may counteract B1 activity. Thus, we hypothesize that in the absence of PP2A, BAF phosphorylation in vaccinia virus-infected cells would increase. To test this, CV1 cells were transfected with 50 nM control, PP2A, or PP4 siRNA for 72 h at 37°C, followed by infection with WT or Cts2 virus at an MOI of 5 at 40°C for 24 h. Total cell lysates were then analyzed by immunoblot analysis using anti-BAF and anti-phospho-BAF (specific for phosphorylated Ser4) antibodies. First, as a control, in the absence of PP2A depletion, we observed an increase in the Ser4 phosphorylation in cells that were infected with WT vaccinia virus (Fig. 9B, lane 2) compared to uninfected control cells (Fig. 9B, lane 1). This is consistent with the activity of B1 in phosphorylating BAF upon infection. In contrast, there was only a slight change in BAF phosphorylation in Cts2 vaccinia virus-infected cells (Fig. 9B, lane 6), which is consistent with the lack of active B1 expressed from this virus. Next, upon infections of cells that were depleted of PP2A, we observed increased hyperphosphorylation of BAF in WT vaccinia virus-infected cells (Fig. 9B, lane 3). This suggests that PP2A can play a role in counteracting B1 kinase activity on BAF. Meanwhile, we also observed an increase in phosphorylated BAF in PP2A-depleted cells that were infected with Cts2 vaccinia virus (Fig. 9B, lane 7). In addition, we also tested BAF phosphorylation in PP4-depleted cells infected with either WT or Cts2 vaccinia virus and found no changes in BAF phosphorylation patterns (Fig. 9B, compare lanes 2 and 4 or lanes 6 and 8).
Treatment of cells with siPP2A is not sufficient to rescue Cts2 viral DNA replication/yield.
Because PP2A depletion in infected cells led to an increase in BAF phosphorylation, we hypothesized that this increase in phosphorylation may enhance vaccinia viral DNA replication and viral production, especially in Cts2 vaccinia virus-infected cells. To test this, CV1 cells were transfected with 50 nM control or PP2A siRNA for 72 h at 37°C, followed by infection with WT or Cts2 virus at an MOI of 5 at 40°C for 24 h. Following harvest of cells, qPCR was performed on viral DNA isolated from infected cells. In WT vaccinia virus-infected cells, we observed similar viral DNA replication in control and PP2A-depleted cells, as we expected (Fig. 9C, gray bars). In Cts2-infected cells, rather than a rescue of DNA replication, we observed a slight reduction (2-fold) in viral DNA replication in PP2A-depleted cells relative to the control cells (Fig. 9C, black bars). Overall, these results show that BAF-induced phosphorylation via PP2A depletion is not sufficient to enhance Cts2 viral DNA replication. These results either may be explained by our inability to completely deplete PP2A from cells, thus leaving some enzyme to activate BAF, or suggest the existence of another, unknown BAF phosphatase which can activate BAF.
DISCUSSION
As a DNA binding protein with important functions in mitosis, the antiviral response, and likely other cellular processes, it is necessary that BAF is tightly regulated. In recent years, it has become increasingly evident that dynamic phosphorylation is likely a key contributor to BAF's regulation. Indeed, phosphorylation has been clearly demonstrated to affect multiple molecular properties of BAF in vitro, including its DNA binding activity and its interaction with LEM proteins (28, 35). A link between BAF phosphorylation by the kinase VRK1 and its presence in the cytoplasm has also been discovered, suggesting that modification of BAF can control its localization as well. Despite this evidence of the importance of the phosphoregulation of BAF, much remains to be learned about how phosphorylation modulates BAF properties in live cells and in response to infection. In this study, we assessed the effects of BAF phosphorylation on multiple BAF properties, including DNA binding, BAF dimerization, and localization. As a model of constitutive phosphorylation or dephosphorylation, we generated and analyzed cells stably expressing BAF phosphorylation mutants: phosphomimetic MTTDQ and MDDDQ and unphosphorylatable MAAAQ mutants. These mutants served as powerful tools in studying the direct effects of BAF phosphorylation. Ultimately, our goal was to better understand how phosphorylation coordinately regulates several BAF properties likely required for its antipoxviral activity, while simultaneously gaining insights into how BAF is regulated in uninfected cells.
In most cell types, BAF localizes to the nucleus, the cytoplasm, and the nuclear envelope, although its relative level at each of these locations can be cell type and age dependent (40). From our examination of the BAF phosphomimetic mutants, we found that phosphorylated BAF shifts markedly from the nucleus to the saponin-soluble cytosolic fraction in comparison to WT BAF. We observed this localization change not only in CV1 cells as we described here but also in other cell types, including mouse fibroblast L929 cells and HeLa cells (data not shown), suggesting that the regulation of BAF localization through phosphorylation is conserved across species and cell lines. Furthermore, we consistently found that the unphosphorylatable mutant MAAAQ is highly concentrated in the nucleus in CV1 cells as shown here, as well as in L929 and HeLa cells (data not shown). These findings confirm and significantly extend prior evidence that BAF phosphorylation leads to changes in its localization. For example, transient expression of BAF-S4E in HeLa cells (35) and of GFP-BAF/VRK1 in U2OS cells (28) first suggested that BAF phosphorylation alters the protein's localization in the cell. Meanwhile, studies of BAF-MAAAQ in Drosophila oocytes (33) and VRK1 depletion in C. elegans embryo and breast cancer cell lines (30, 31) also indicated that dephosphorylated BAF becomes less mobile in the nucleus. Thus, our data are consistent with these reports and form the basis for our working model (Fig. 9D) that BAF phosphorylation mobilizes the protein, allowing it to relocalize within the nucleus or to transit to the cytoplasm. At this time, it remains to be determined whether BAF's movement between nucleus and cytoplasm occurs via active transport or passive diffusion through the nuclear pore complex. However, BAF's small size and lack of a canonical nuclear localization sequence suggest that diffusion may be a likely possibility.
Mobilization of BAF likely requires disruption of its protein-DNA and/or protein-protein interactions (2). Indeed, BAF's ability to interact with the LEM protein emerin is impaired by phosphorylation (35). However, while BAF has been known to form homodimers for some time, to date no report has yet examined the impact of BAF phosphorylation on its homodimerization. Here we demonstrate that hyperphosphorylated BAF (from our studies of MDDDQ) exhibits a sharply reduced ability to interact with endogenous BAF. Interestingly, our study also indicates that Ser4-phosphorylated BAF (MTTDQ) was still capable of immunoprecipitating endogenous BAF, leading us to speculate that phosphorylation of both Thr residues (or all residues, including Ser4) may be required to impact BAF-BAF interaction. Together, these data demonstrate for the first time that like BAF's interaction with a LEM domain partner, its homodimeric interaction is also phosphoregulated. As current structural studies of BAF show no direct contact between its sites of phosphorylation and its dimerization interface (3, 51), additional studies will be needed to determine how phosphorylation might modulate BAF-BAF contacts.
Based on both direct in vitro studies (28) and indirect evidence in live cells (30–33), phosphorylation is a potent inhibitor of BAF interaction with DNA. Here, we have examined BAF-DNA interaction of our mutant proteins by utilizing a ChIP protocol. This approach allows us to monitor how phosphorylation regulates BAF binding to cellular chromatin DNA as well as to foreign plasmid DNA in a more direct and quantitative manner than was previously possible. In these ChIP analyses, we found that BAF phosphorylation abolishes its DNA binding activity independent of the DNA sequence or sources (chromatin DNA versus plasmid DNA). Consistent with these results, our immunofluorescence analyses also showed significantly reduced relocalization of phosphorylated BAF to transfected plasmid DNA puncta in the cytoplasm. It is worth noting that the similar DNA binding inhibition observed for MTTDQ and MDDDQ suggests that Ser4 phosphorylation is the major contributor to the regulation of BAF's DNA binding activity. Together with previous reports demonstrating Ser4 phosphorylation by both VRK1 and B1 kinase (28, 31), the data shown here continue to support the model that VRK1/B1-mediated BAF phosphorylation releases BAF from the DNA, as first proposed by Nichols et al. (28). Finally, our ChIP experiments revealed a modest but detectable increase in the chromatin DNA bound to MAAAQ compared to the WT BAF. Enhanced chromatin binding by unphosphorylated BAF is also likely to contribute to the reduced mobility of BAF observed by other researchers and probably influences BAF's subcellular localization by limiting its movement within and out of the nucleus.
If it is correct that BAF phosphorylation releases it from the DNA and mobilizes it out of the nucleus, it would result in an increased proportion of phosphorylated BAF in the cytosol. In this event, we posited that Cts2 vaccinia viral DNA replication would be enhanced, since the cytoplasmic pool of phosphorylated BAF would be incapable of binding viral DNA. Indeed, we found that the cytoplasmic hyperphosphorylated BAF pool, as shown in MDDDQ, was unable to suppress Cts2 viral DNA replication (only a 2-fold reduction) as well as viral production (4-fold reduction) as opposed to the WT BAF activity of 50-fold reduction in viral DNA replication and 90-fold reduction in viral production. Somewhat surprisingly, despite a clear loss in MTTDQ DNA binding activity shown by ChIP analysis, we found that MTTDQ could still partially suppressed Cts2 viral DNA replication (10-fold reduction) as well as viral production (8-fold reduction). Thus, in the absence of phosphorylated Thr residues, BAF remains capable of antipoxviral defense, albeit much more weakly than WT BAF. Importantly, with respect to the unphosphorylatable MAAAQ mutant, we found that it was incapable of suppressing viral DNA replication (∼2.5-fold reduction) compared to the WT BAF activity (50-fold reduction) and could only partially suppress production of new virus. Therefore, although unphosphorylated BAF has DNA binding activity, its localization likely restricts its activity to the nucleus, allowing Cts2 viral DNA replication to continue unhindered in the cytoplasm. This demonstrates that the cellular localization of BAF also is a critical determinant of its antiviral activity against vaccinia virus, which forms the basis for our model that BAF must be only transiently phosphorylated to reach the cytoplasm and interfere with the viral life cycle (Fig. 9D).
As discussed above, phosphorylation enhances the fraction of BAF present in the cytosol, which would suggest that phosphorylated BAF would be the predominant form of BAF in that compartment. However, our Western blot analyses of total BAF in the cytoplasm showed equal amounts of modified and unmodified BAF, as assessed by the fractions of shifted and unshifted BAF. This observation suggested that a cytoplasmic BAF phosphatase(s) may exist. Additionally, while the phosphorylation of BAF is needed for its cytoplasmic localization, phosphomimetic mutants representing constitutively phosphorylated forms of BAF are unable to act as a host defense against vaccinia virus. The presence of a cytoplasmic BAF phosphatase would thus also explain how WT BAF can be dephosphorylated as needed to bind viral DNA. We therefore examined how phosphatase activity may regulate BAF's localization and antiviral activity. Indeed, when CV1 cells were treated with phosphatase inhibitor, increased accumulation of hyperphosphorylated BAF correlating with increased inhibitor concentration could be observed. While these experiments were ongoing, it was discovered that BAF dephosphorylation can be mediated by two phosphatases, PP2A and PP4 (32, 34). In this study, we validated that PP2A is a BAF phosphatase and that upon its depletion, phosphorylated BAF accumulated in our CV1 cells as has been described previously (32). PP2A is proposed to act on BAF during mitotic exit (32); however, it is also possible that PP2A can dephosphorylate BAF following its modification by the B1 kinase upon vaccinia virus infection. This appears to be true, as depletion of PP2A led to accumulation of hyperphosphorylated BAF in cells that were infected with WT vaccinia virus. This demonstrates for the first time that PP2A can counteract the B1-induced phosphorylation of BAF. Next, we examined the impact of PP2A depletion during Cts2 infection. Indeed, by depleting PP2A in cells that were infected with B1-deficient Cts2 vaccinia virus, we were able to increase BAF Ser4 phosphorylation. Despite this phosphorylation increase, however, viral DNA replication was relatively unchanged. Thus, in this case, PP2A depletion was not sufficient to enhance Cts2 viral DNA replication. We speculate that the incomplete depletion of PP2A may allow for residual PP2A activity capable of dephosphorylating BAF in the cytoplasm or that another BAF phosphatase also acts on BAF in the cytoplasm. As PP4 depletion by siRNA has been shown to increase BAF phosphorylation in HEK293 cells (34), we examined it as a candidate in our system. However, we could not obtain any evidence that PP4 can act as a BAF phosphatase in our CV1 cell model. It is possible that the different cell lines utilized in the study by Zhuang et al. (34) versus our own may explain the differences in our observations.
To summarize, these data demonstrate that dynamic phosphorylation plays a pivotal role in maintaining a balance of BAF in the nucleus versus the cytoplasm, where it can interfere with poxvirus infection. BAF localization is likely modulated in part through alteration of its interaction with DNA and protein partners, which we demonstrate are both disrupted by phosphorylation. These data shed further light on mechanisms employed by cellular kinase such as VRK1 or the vaccinia virus kinase B1 to regulate BAF's functions in mitosis and antiviral host defense. Finally, as phosphorylation clearly regulates BAF at multiple levels, it is tempting to speculate that it also impacts other cellular processes recently associated with BAF, such as transcriptional regulation (5, 8, 52, 53) and the DNA damage response (6, 9, 54).
ACKNOWLEDGMENTS
This research was supported through NIH grants to M.S.W. (K22AI080941 and R56AI099062) and the Nebraska Center for Virology (P30GM10359), which supported certain aspects of these studies, in particular, the confocal microscopy core center at UNL. A.J. was partially supported by a Ruth L. Kirschstein NRSA (T32 AI060547).
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
Published ahead of print 5 March 2014
REFERENCES
- 1.Skoko D, Li M, Huang Y, Mizuuchi M, Cai M, Bradley CM, Pease PJ, Xiao B, Marko JF, Craigie R, Mizuuchi K. 2009. Barrier-to-autointegration factor (BAF) condenses DNA by looping. Proc. Natl. Acad. Sci. U. S. A. 106:16610–16615. 10.1073/pnas.0909077106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bradley CM, Ronning DR, Ghirlando R, Craigie R, Dyda F. 2005. Structural basis for DNA bridging by barrier-to-autointegration factor. Nat. Struct. Mol. Biol. 12:935–936. 10.1038/nsmb989 [DOI] [PubMed] [Google Scholar]
- 3.Umland TC, Wei SQ, Craigie R, Davies DR. 2000. Structural basis of DNA bridging by barrier-to-autointegration factor. Biochemistry 39:9130–9138. 10.1021/bi000572w [DOI] [PubMed] [Google Scholar]
- 4.Cai M, Huang Y, Zheng R, Wei SQ, Ghirlando R, Lee MS, Craigie R, Gronenborn AM, Clore GM. 1998. Solution structure of the cellular factor BAF responsible for protecting retroviral DNA from autointegration. Nat. Struct. Biol. 5:903–909. 10.1038/2345 [DOI] [PubMed] [Google Scholar]
- 5.Montes de Oca R, Andreassen PR, Wilson KL. 2011. Barrier-to-autointegration factor influences specific histone modifications. Nucleus. 2:580–590. 10.4161/nucl.2.6.17960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montes de Oca R, Shoemaker CJ, Gucek M, Cole RN, Wilson KL. 2009. Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners PLoS One 4:e7050. 10.1371/journal.pone.0007050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montes de Oca R, Lee KK, Wilson KL. 2005. Binding of barrier to autointegration factor (BAF) to histone H3 and selected linker histones including H1.1. J. Biol. Chem. 280:42252–42262. 10.1074/jbc.M509917200 [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Xu S, Rivolta C, Li LY, Peng GH, Swain PK, Sung CH, Swaroop A, Berson EL, Dryja TP, Chen S. 2002. Barrier to autointegration factor interacts with the cone-rod homeobox and represses its transactivation function. J. Biol. Chem. 277:43288–43300. 10.1074/jbc.M207952200 [DOI] [PubMed] [Google Scholar]
- 9.Brachner A, Braun J, Ghodgaonkar M, Castor D, Zlopasa L, Ehrlich V, Jiricny J, Gotzmann J, Knasmuller S, Foisner R. 2012. The endonuclease Ankle1 requires its LEM and GIY-YIG motifs for DNA cleavage in vivo. J. Cell Sci. 125:1048–1057. 10.1242/jcs.098392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haraguchi T, Koujin T, Segura-Totten M, Lee KK, Matsuoka Y, Yoneda Y, Wilson KL, Hiraoka Y. 2001. BAF is required for emerin assembly into the reforming nuclear envelope J. Cell Sci. 114:4575–4585 [DOI] [PubMed] [Google Scholar]
- 11.Furukawa K, Sugiyama S, Osouda S, Goto H, Inagaki M, Horigome T, Omata S, McConnell M, Fisher PA, Nishida Y. 2003. Barrier-to-autointegration factor plays crucial roles in cell cycle progression and nuclear organization in Drosophila. J. Cell Sci. 116:3811–3823. 10.1242/jcs.00682 [DOI] [PubMed] [Google Scholar]
- 12.Margalit A, Liu J, Fridkin A, Wilson KL, Gruenbaum Y. 2005. A lamin-dependent pathway that regulates nuclear organization, cell cycle progression and germ cell development. Novartis Found. Symp. 264:231–240. 10.1002/0470093765.ch16 [DOI] [PubMed] [Google Scholar]
- 13.Margalit A, Neufeld E, Feinstein N, Wilson KL, Podbilewicz B, Gruenbaum Y. 2007. Barrier to autointegration factor blocks premature cell fusion and maintains adult muscle integrity in C. elegans. J. Cell Biol. 178:661–673. 10.1083/jcb.200704049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segura-Totten M, Kowalski AK, Craigie R, Wilson KL. 2002. Barrier-to-autointegration factor: major roles in chromatin decondensation and nuclear assembly. J. Cell Biol. 158:475–485. 10.1083/jcb.200202019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee MS, Craigie R. 1994. Protection of retroviral DNA from autointegration: involvement of a cellular factor. Proc. Natl. Acad. Sci. U. S. A. 91:9823–9827. 10.1073/pnas.91.21.9823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MS, Craigie R. 1998. A previously unidentified host protein protects retroviral DNA from autointegration. Proc. Natl. Acad. Sci. U. S. A. 95:1528–1533. 10.1073/pnas.95.4.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki Y, Craigie R. 2002. Regulatory mechanisms by which barrier-to-autointegration factor blocks autointegration and stimulates intermolecular integration of Moloney murine leukemia virus preintegration complexes. J. Virol. 76:12376–12380. 10.1128/JVI.76.23.12376-12380.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou W, Ngo T, Gershon PD. 2012. An overview of the vaccinia virus infectome: a survey of the proteins of the poxvirus-infected cell. J. Virol. 86:1487–1499. 10.1128/JVI.06084-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Vliet K, Mohamed MR, Zhang L, Villa NY, Werden SJ, Liu J, McFadden G. 2009. Poxvirus proteomics and virus-host protein interactions. Microbiol. Mol. Biol. Rev. 73:730–749. 10.1128/MMBR.00026-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Condit RC, Motyczka A, Spizz G. 1983. Isolation, characterization, and physical mapping of temperature-sensitive mutants of vaccinia virus. Virology 128:429–443. 10.1016/0042-6822(83)90268-4 [DOI] [PubMed] [Google Scholar]
- 21.Rempel RE, Anderson MK, Evans E, Traktman P. 1990. Temperature-sensitive vaccinia virus mutants identify a gene with an essential role in viral replication. J. Virol. 64:574–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rempel RE, Traktman P. 1992. Vaccinia virus B1 kinase: phenotypic analysis of temperature-sensitive mutants and enzymatic characterization of recombinant proteins. J. Virol. 66:4413–4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovacs GR, Vasilakis N, Moss B. 2001. Regulation of viral intermediate gene expression by the vaccinia virus B1 protein kinase. J. Virol. 75:4048–4055. 10.1128/JVI.75.9.4048-4055.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vigne S, Duraffour S, Andrei G, Snoeck R, Garin D, Crance J. 2009. Inhibition of vaccinia virus replication by two small interfering RNAs targeting B1R and G7L genes and their synergistic combination with cidofovir. Antimicrob. Agents Chemother. 53:2579–2588. 10.1128/AAC.01626-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibrahim N, Wicklund A, Jamin A, Wiebe MS. 2013. Barrier to autointegration factor (BAF) inhibits vaccinia virus intermediate transcription in the absence of the viral B1 kinase. Virology 444:363–373. 10.1016/j.virol.2013.07.002; 10.1016/j.virol.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin S, Chen W, Broyles SS. 1992. The vaccinia virus B1R gene product is a serine/threonine protein kinase. J. Virol. 66:2717–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiebe MS, Traktman P. 2007. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe 1:187–197. 10.1016/j.chom.2007.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nichols RJ, Wiebe MS, Traktman P. 2006. The vaccinia-related kinases phosphorylate the N′ terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol. Biol. Cell 17:2451–2464. 10.1091/mbc.E05-12-1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ibrahim N, Wicklund A, Wiebe MS. 2011. Molecular characterization of the host defense activity of the barrier to autointegration factor against vaccinia virus. J. Virol. 85:11588–11600. 10.1128/JVI.00641-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molitor TP, Traktman P. 2014. Depletion of the protein kinase VRK1 disrupts nuclear envelope morphology and leads to BAF retention on mitotic chromosomes. Mol. Biol. Cell 10.1091/mbc.E13-10-0603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorjanacz M, Klerkx EP, Galy V, Santarella R, Lopez-Iglesias C, Askjaer P, Mattaj IW. 2007. Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly EMBO J. 26:132–143. 10.1038/sj.emboj.7601470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asencio C, Davidson IF, Santarella-Mellwig R, Ly-Hartig TB, Mall M, Wallenfang MR, Mattaj IW, Gorjanacz M. 2012. Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis. Cell 150:122–135. 10.1016/j.cell.2012.04.043 [DOI] [PubMed] [Google Scholar]
- 33.Lancaster OM, Cullen CF, Ohkura H. 2007. NHK-1 phosphorylates BAF to allow karyosome formation in the Drosophila oocyte nucleus. J. Cell Biol. 179:817–824. 10.1083/jcb.200706067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhuang X, Semenova E, Maric D, Craigie R. 2013. Dephosphorylation of barrier-to-autointegration-factor by protein phosphatase 4 and its role in cell mitosis. J. Biol. Chem. 10.1074/jbc.M113.492777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bengtsson L, Wilson KL. 2006. Barrier-to-autointegration factor phosphorylation on Ser-4 regulates emerin binding to lamin A in vitro and emerin localization in vivo. Mol. Biol. Cell 17:1154–1163. 10.1091/mbc.E05-04-0356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki Y, Ogawa K, Koyanagi Y, Suzuki Y. 2010. Functional disruption of the moloney murine leukemia virus preintegration complex by vaccinia-related kinases. J. Biol. Chem. 285:24032–24043. 10.1074/jbc.M110.116640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wassler M, Jonasson I, Persson R, Fries E. 1987. Differential permeabilization of membranes by saponin treatment of isolated rat hepatocytes. Release of secretory proteins. Biochem. J. 247:407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dechat T, Gajewski A, Korbei B, Gerlich D, Daigle N, Haraguchi T, Furukawa K, Ellenberg J, Foisner R. 2004. LAP2alpha and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J. Cell Sci. 117:6117–6128. 10.1242/jcs.01529 [DOI] [PubMed] [Google Scholar]
- 39.Shimi T, Koujin T, Segura-Totten M, Wilson KL, Haraguchi T, Hiraoka Y. 2004. Dynamic interaction between BAF and emerin revealed by FRAP, FLIP, and FRET analyses in living HeLa cells. J. Struct. Biol. 147:31–41. 10.1016/j.jsb.2003.11.013 [DOI] [PubMed] [Google Scholar]
- 40.Haraguchi T, Koujin T, Osakada H, Kojidani T, Mori C, Masuda H, Hiraoka Y. 2007. Nuclear localization of barrier-to-autointegration factor is correlated with progression of S phase in human cells. J. Cell Sci. 120:1967–1977. 10.1242/jcs.03461 [DOI] [PubMed] [Google Scholar]
- 41.Haraguchi T, Kojidani T, Koujin T, Shimi T, Osakada H, Mori C, Yamamoto A, Hiraoka Y. 2008. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J. Cell Sci. 121:2540–2554. 10.1242/jcs.033597 [DOI] [PubMed] [Google Scholar]
- 42.Zheng R, Ghirlando R, Lee MS, Mizuuchi K, Krause M, Craigie R. 2000. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. U. S. A. 97:8997–9002. 10.1073/pnas.150240197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tseng M, Palaniyar N, Zhang W, Evans DH. 1999. DNA binding and aggregation properties of the vaccinia virus I3L gene product. J. Biol. Chem. 274:21637–21644 [DOI] [PubMed] [Google Scholar]
- 44.Rochester SC, Traktman P. 1998. Characterization of the single-stranded DNA binding protein encoded by the vaccinia virus I3 gene. J. Virol. 72:2917–2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greseth MD, Boyle KA, Bluma MS, Unger B, Wiebe MS, Soares-Martins JA, Wickramasekera NT, Wahlberg J, Traktman P. 2012. Molecular genetic and biochemical characterization of the vaccinia virus I3 protein, the replicative single-stranded DNA binding protein. J. Virol. 86:6197–6209. 10.1128/JVI.00206-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Favre B, Turowski P, Hemmings BA. 1997. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J. Biol. Chem. 272:13856–13863. 10.1074/jbc.272.21.13856 [DOI] [PubMed] [Google Scholar]
- 47.Bialojan C, Takai A. 1988. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 256:283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez A, Brautigan DL, Mumby M, Lamb NJ. 1990. Protein phosphatase type-1, not type-2A, modulates actin microfilament integrity and myosin light chain phosphorylation in living nonmuscle cells. J. Cell Biol. 111:103–112. 10.1083/jcb.111.1.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chartier L, Rankin LL, Allen RE, Kato Y, Fusetani N, Karaki H, Watabe S, Hartshorne DJ. 1991. Calyculin-A increases the level of protein phosphorylation and changes the shape of 3T3 fibroblasts. Cell Motil. Cytoskel. 18:26–40. 10.1002/cm.970180104 [DOI] [PubMed] [Google Scholar]
- 50.Kreienbuhl P, Keller H, Niggli V. 1992. Protein phosphatase inhibitors okadaic acid and calyculin A alter cell shape and F-actin distribution and inhibit stimulus-dependent increases in cytoskeletal actin of human neutrophils. Blood 80:2911–2919 [PubMed] [Google Scholar]
- 51.Cai M, Huang Y, Suh JY, Louis JM, Ghirlando R, Craigie R, Clore GM. 2007. Solution NMR structure of the barrier-to-autointegration factor-Emerin complex. J. Biol. Chem. 282:14525–14535. 10.1074/jbc.M700576200 [DOI] [PubMed] [Google Scholar]
- 52.Shaklai S, Somech R, Gal-Yam EN, Deshet-Unger N, Moshitch-Moshkovitz S, Hirschberg K, Amariglio N, Simon AJ, Rechavi G. 2008. LAP2zeta binds BAF and suppresses LAP2beta-mediated transcriptional repression. Eur. J. Cell Biol. 87:267–278. 10.1016/j.ejcb.2008.01.014 [DOI] [PubMed] [Google Scholar]
- 53.Holaska JM, Lee KK, Kowalski AK, Wilson KL. 2003. Transcriptional repressor germ cell-less (GCL) and barrier to autointegration factor (BAF) compete for binding to emerin in vitro. J. Biol. Chem. 278:6969–6975. 10.1074/jbc.M208811200 [DOI] [PubMed] [Google Scholar]
- 54.Dittrich CM, Kratz K, Sendoel A, Gruenbaum Y, Jiricny J, Hengartner MO. 2012. LEM-3—a LEM domain containing nuclease involved in the DNA damage response in C. elegans. PLoS One 7:e24555. 10.1371/journal.pone.0024555 [DOI] [PMC free article] [PubMed] [Google Scholar]