

ABSTRACT
In cells infected with herpesviruses, two capsid-associated, or inner tegument, proteins, UL37 and UL36, control cytosolic trafficking of capsids by as yet poorly understood mechanisms. Here, we report the crystal structure of the N-terminal half of UL37 from pseudorabies virus, an alphaherpesvirus closely related to herpes simplex viruses and varicella-zoster virus. The structure—the first for any alphaherpesvirus inner tegument protein—reveals an elongated molecule of a complex architecture rich in helical bundles. To explore the function of the UL37 N terminus, we used the three-dimensional framework provided by the structure in combination with evolutionary trace analysis to pinpoint several surface-exposed regions of potential functional importance and test their importance using mutagenesis. This approach identified a novel functional region important for cell-cell spread. These results suggest a novel role for UL37 in intracellular virus trafficking that promotes spread of viral infection, a finding that expands the repertoire of UL37 functions. Supporting this, the N terminus of UL37 shares structural similarity with cellular multisubunit tethering complexes (MTCs), which control vesicular trafficking in eukaryotic cells by tethering transport vesicles to their destination membranes. Our results suggest that UL37 could be the first viral MTC mimic and provide a structural rationale for the importance of UL37 for viral trafficking. We propose that herpesviruses may have co-opted the MTC functionality of UL37 to bring capsids to cytoplasmic budding destinations and further on to cell junctions for spread to nearby cells.
IMPORTANCE To move within an infected cell, viruses encode genes for proteins that interact with host trafficking machinery. In cells infected with herpesviruses, two capsid-associated proteins control the cytosolic movement of capsids by as yet poorly understood mechanisms. Here, we report the crystal structure for the N-terminal half of one of these proteins, UL37. Structure-based mutagenesis revealed a novel function for UL37 in virus trafficking to cell junctions for cell-cell spread. The unexpected structural similarity to components of cellular multisubunit tethering complexes, which control vesicular traffic, suggests that UL37 could be the first viral MTC mimic and provides a structural basis for the importance of UL37 for virus trafficking.
INTRODUCTION
The ability to undergo directional movement within a host cytoplasm is essential for replication of many viruses. During viral entry, genomes must be delivered to the replication compartment, while later, newly assembled viral particles need to escape the cell to initiate new rounds of infection. To move within the infected cell, viruses typically encode genes for proteins that interact with the host trafficking machinery.
Herpesviruses are large enveloped viruses that cause lifelong, latent infections from which viruses can reactivate, causing diseases ranging from mucocutaneous eruptions to encephalitis, cancers, and disseminated disease. All herpesviruses share a morphology consisting of four components: a double-stranded DNA genome, an icosahedral capsid, a lipid envelope, and the tegument, a protein layer between the capsid and the envelope. Herpesvirus replication occurs in the nucleus, requiring incoming particles to traverse the cytoplasm following entry into a cell and progeny virions to traffic back to the cell surface during the egress phase of infection (1). Following genome replication and encapsidation, nascent capsids first bud into the inner nuclear membrane. The resulting perinuclear viral particles fuse their envelopes with the outer nuclear membrane, releasing unenveloped capsids into the cytosol. Cytosolic capsids next travel to tubular vesicles thought to originate from intracellular membranes (trans-Golgi network [TGN] or early endosomes) (1, 2), where the final round of budding—secondary envelopment—takes place. Cytosolic capsids bud into these vesicles and acquire lipid envelopes and outer tegument proteins. The resulting mature virions are subsequently released into the extracellular milieu by exocytosis or directed to cell junctions for spread into uninfected cells.
The movement of capsids within the cytosol is controlled by a subset of tegument proteins that are capsid associated during both the entry and egress phases of infection. These tegument proteins include the herpesvirus protein UL37, which is a highly conserved component of the capsid-proximal, or inner, tegument (3–5). UL37 interacts with capsids indirectly by binding another conserved tegument protein, UL36 (6–13), which in turn binds capsids through a direct interaction with the capsid proteins UL25 (14–16), UL19, and UL17 (17). During transport to the nuclear pore, UL37 remains attached to the incoming capsids (18–21), and although UL37 is not required for capsid trafficking to the nucleus (13), in its absence the incoming transport is delayed (22).
The UL37 protein is also necessary for herpesvirus egress. In UL37-null viruses, unenveloped capsids accumulate in the cytosol of infected cells (4, 6, 13, 23–25) and the release of infectious virions is either completely blocked, as seen in herpes simplex virus 1 (HSV-1) (23, 24), or is strongly impaired, as observed in pseudorabies virus (PRV), a herpesvirus closely related to HSV and varicella-zoster virus (4, 23). The directed movement of HSV-1 and PRV UL37-null capsids is substantially impeded, which supports the idea that efficient capsid transport to the organelle of secondary envelopment requires assembly of UL37 onto capsids (6, 26). In contrast, the role of UL37 in cytoplasmic envelopment is less clear. UL37 is not required for the formation of the so-called light, or L, particles, which lack capsids and contain only enveloped tegument (4, 13), and, therefore, is not required for the assembly of the outer tegument or for membrane deformation during cytoplasmic budding. However, UL37 may be necessary for the correct addition of the outer tegument proteins to the capsid, which is essential for secondary envelopment (4).
The C-terminal half of HSV-1 UL37 interacts with UL36 (27, 28) and dystonin/BPAG1 (29). While the N-terminal half of UL37 has been proposed to have a nuclear export signal (30) and a small self-association domain (27), the role of the N-terminal half of UL37 in replication remains largely unknown. To gain insight into the mechanism by which UL37 enables capsid trafficking and cytoplasmic capsid envelopment, we determined the crystal structure of the N-terminal half of UL37 (UL37N), residues 1 to 479, from PRV. UL37N is an elongated molecule composed of multiple helical bundles. The structure revealed an unanticipated similarity to the structures of several components of the complexes associated with tethering containing helical rods (CATCHR) family of eukaryotic multisubunit tethering complexes (MTCs), which direct vesicular trafficking in cells by tethering vesicles to their target organelles. Furthermore, mutagenesis of a surface cluster identified in the UL37N structure using evolutionary trace analysis (ETA) reduced the spread of PRV without impeding the rate of viral production. This finding suggests an additional, novel role for UL37 in intracellular trafficking that promotes the subsequent spread of virus between cells. Collectively, these observations offer new insights into the function of UL37 and provide a structural basis for its essential role in intracellular virus trafficking. We propose that herpesviruses may utilize the tethering function of UL37 to bring capsids to their cytoplasmic budding sites perhaps using UL37 to specify their budding destination and to promote trafficking of mature virions inside transport vesicles to cell junctions for spread to nearby cells.
MATERIALS AND METHODS
Cloning.
Plasmid pGS3610 encodes the PRV Becker UL37 gene fused to an N-terminal His6-SUMO tandem tag. This was made by cutting the pETDuet-SUMO vector (a derivative of pETDuet-1 and a gift from Thomas Schwartz) and the pGS1740 subclone of UL37 with BamHI and HindIII. The pJP4 plasmid, which contains a His6-SUMO-PreScission tag in frame with the BamHI restriction site of the multiple-cloning site in a pET24b vector, was made through PCR of the His6-SUMO-PreScission tag from pETDuet-SUMO using the primers 5′-GGGAATTCCATATGGGCAGCAGCCATCACCATCA and 3′-CTAGGGATCCGGGCCCCTGGAACAGAACTT (restriction sites are underlined). The PCR product was subcloned into pET24b using NdeI and BamHI restriction sites. The PRV UL37 gene for Escherichia coli expression was synthesized by GeneArt. The N-terminal half (residues 1 to 496) of codon-optimized PRV UL37 (referred to as UL37N) was amplified by PCR from the full-length PRV codon-optimized UL37 gene using the primers 5′-CTAGGGATCCATGGAAGCACTGGTTCGTGC and 3′-CTAGAAGCTTCTAGGCTGCGCTGGTCGGTG (restriction sites are underlined). The PCR product was subcloned into pJP4 using the BamHI and HindIII restriction sites to yield plasmid pJP23.
Virus construction.
All recombinant PRV (strain Becker) isolates were derived from a variant of the pBecker3 infectious clone, pGS4284, that encodes the mCherry red fluorescent protein fused in frame to the UL25 capsid protein (31). Viruses were produced by electroporation of infectious clones into the pig kidney epithelial cell line PK15, as previously described (20). PK15 cells were maintained in Dulbecco modified Eagle medium (DMEM; Invitrogen) supplemented with 10% bovine growth supplement (BGS; HyClone), which was reduced to 2% during transfection and infection. The harvested virus was passaged once to produce a high-titer stock by infecting a 10-cm dish of PK15 cells with 1 μl virus. Transfection of pGS4284 resulted in PRV-GS4284, which upon passage propagated to titers of >5 × 108 PFU/ml.
To make PRV encoding the pentuple mutations D79A/D81A/E82A/D382A/D383A in the calcium-binding region (Ca) of UL37, codon changes were introduced through two rounds of en passant mutagenesis of pGS4284 (32). The first set of primers, 5′-CTCGCCGAGAACCTGGCCGGCCTGGCGCTGTGGCGCCTGCGCCACGCCTGGGCCGCGGGCACGGCCCCGCTGAGGATGACGACGATAAGTAGGG and 5′-GTCGCCGTTGACGACCCCCAGGAGCTCCAGCAGCGGGGCCGTGCCCGCGGCCCAGGCGTGGCGCAGGCGCCACAACCAATTAACCAATTCTGATTAG, was used to generate the D382A/D383A mutations (mutated bases are in bold, the sequence from template plasmid pEPkan-S is underlined) and produced pGS5456. The second set of primers, 5′-GTCGGCTGCACGGCGGTCGTCGGCGGCGTCGTGCACCGCCTCCTCGCCGCCTACGGGCCCGGGCTGAGGATGACGACGATAAGTAGGG and 5′-CGCGACGTCCGTGTAGGCGCGCACGTAGTCCAGCCCGGGCCCGTAGGCGGCGAGGAGGCGGTGCACCAACCAATTAACCAATTCTGATTAG, was used to generate the D79A/D81A/E82A mutations, which were introduced into pGS5456 to produce the final mutant, pGS5476. PRV-GS5476 typically propagated to a titer of >5 × 108 PFU/ml.
The region 2 (R2) and region 3 (R3) mutant viruses were produced in the same manner as the Ca mutant just described. For R2, mutations were introduced into pGS4284 in three sequential rounds using primers 5′-CTCGACCACACGCAGGTGGACGCCACGGGCGTGTGGGAGGCGGTGGCGGCCAGCGCCTCGCCGAGGATGACGACGATAAGTAGGG and 5′-CGCGGTCACGAGCGCCTCCACGACCTGCAGCGGCGAGGCGCTGGCCGCCACCGCCTCCCACACCAACCAATTAACCAATTCTGATTAG (encoding Q324A), 5′-GACCTCCTCGAGCGCGCCGTGCTGGACCGCGCGCCCCGCCTGACGGCCGCGCAGGCTGCCGTCGGCTGCACGAGGATGACGACGATAAGTAGGG and 5′-GAGGCGGTGCACGACGCCGCCGACGACCGCCGTGCAGCCGACGGCAGCCTGCGCGGCCGTCAGGCGGGGCGCCAACCAATTAACCAATTCTGATTAG (encoding D362A/R365A), and 5′-GGGGACGTGACGGCGGCGCTGGGGCTCCCCGAGAAGGGCGTGGAGGCCGTGGTGCGCGCTTGCATGGCGCCGCGCAGGATGACGACGATAAGTAGGG and 5′-GCGCGCCGCGCCCACGTGCTCCGTGGGCGGGCGCGGCGCCATGCAAGCGCGCACCACGGCCTCCACGCCCTTCTCCAACCAATTAACCAATTCTGATTAG (encoding H421A/H425A). The first PCR product was recombined into pGS4284, resulting in pGS5483. The second PCR product was then recombined into pGS5483, resulting in pGS5558. The final recombination was made into pGS5558, resulting in pGS5604. PRV-GS5604 typically propagated to a titer of >5 × 108 PFU/ml. The R3 mutations were introduced in two rounds using primers 5′-CTGCCGCTGGCGTTGGCGGTGCGCCAGATGCAGAACGAGGGCCTGGCGCAGCTGACGCGCGCGCTCAGGATGACGACGATAAGTAGGG and 5′-GAAGAACTCGTCGGCGATCGTGAGGGCAAAGAGCGCGCGCGTCAGCTGCGCCAGGCCCTCGTTCTGCAACCAATTAACCAATTCTGATTAG (encoding D239A/E240A) and primers 5′-AACCCGACGCTGCGCGAGCAGTTCGCCGAGGCGGCGCGGGCCGTGGCCGCGGCGGCGCTGGTGCCCAGGATGACGACGATAAGTAGGG and 5′-CGTGCGCGGCGTGGCGTTGACCTCGCCCACGGGCACCAGCGCCGCCGCGGCCACGGCCCGCGCCGCCAACCAATTAACCAATTCTGATTAG (encoding K203A/P204Q). The first PCR product was recombined into pGS4284, resulting in pGS5242. The second PCR product was then recombined into pGS5242, resulting in pGS5350. PRV-GS5350 typically propagated to a titer of >5 × 108 PFU/ml.
The region 1 (R1) mutant virus (V249R/R254A/R285A/D287A/H311A) was generated using a modified two-step recombination. The region of the UL37 gene encoding amino acids 249 to 311 was first replaced with the kanamycin resistance cassette of pEPkan-S using primers 5′-CCGAGGCGGCGCGGGCCGTGGACGAGGCGGCGCTGGTGCCCGTGGGCGAGACGCAGGTGGACGCCACGGGAGGATGACGACGATAAGTAGGG and 5′-GAGGCGCTGGCCTGCACCGCCTCCCACACGCCCGTGGCGTCCACCTGCGTCTCGCCCACGGGCACCAGCGCAACCAATTAACCAATTCTGATTAG. The PCR product was recombined into pGS4284, resulting in the intermediate construct pGS5313. The deletion in pGS5313 was then repaired using a 489-bp synthetic DNA encoding the missing UL37 sequence with the five codon changes and 150 bp of flanking homologous sequence to each side (pGS5267; Integrated DNA Technologies). The synthetic DNA was released from a pIDTSmart vector using flanking HindIII sites and recombined into pGS5313. Recombination was carried out by growing E. coli strain GS1783 harboring pGS5313 in 30 ml of Luria Broth (LB) supplemented with 20 μg/ml chloramphenicol to an optical density at 600 nm (OD600) of 0.6 at 32°C in a baffled flask. At this point, 20 ml of LB supplemented with 20 μg/ml chloramphenicol and 2% l-arabinose was added, and the culture was incubated with shaking at 32°C for 70 min. The culture was then transferred to a 42°C shaking water bath for 15 min, and the contents were then transferred to a 50-ml conical tube and chilled on ice. The chilled bacteria were washed three times, and the final pellet was suspended in 300 ml double-distilled H2O, of which 48 μl was used in an electroporation with 2 μl of the pGS5267 synthetic fragment. After recovery, the reaction mixture was plated on LB agar plates supplemented with 20 μg/ml chloramphenicol and 2% l-arabinose. The resulting isolate was saved as pGS5321. PRV-GS5321 typically propagated to a titer of >5 × 108 PFU/ml. The sequences of all genetic modifications in the infectious clones were confirmed.
Viral propagation kinetics, viral titers, and plaque size analysis.
Quantitation of viral propagation kinetics was assessed by single-step growth in PK15 cells infected at a multiplicity of infection (MOI) of 10 for each viral stain. Viral titers from cells or medium supernatants harvested at 2, 5, 8, 12, or 24 h postinfection (hpi) were determined in duplicate by plaque assay, as previously described (33).
Measurements of plaque diameters were obtained by infection of PK15 cells in 6-well trays with serial 10-fold dilutions for each virus. At 4 days postinfection, images were captured with a ×4 objective on a Nikon TE2000 inverted fluorescence microscopy (Nikon Instruments) fitted with a CoolSnap HQ2 camera (Photometrics). Two orthogonal diameter measurements of each fluorescent plaque were obtained using the Metamorph software package (Molecular Devices) and averaged. The reported plaque diameters represented an average of more than 50 plaques per virus. Measurements of the plaque diameters of mutant viruses were always conducted side by side with measurement of the plaque diameter of PRV-GS4284 (the virus encoding wild-type [WT] UL37), and the diameters of the mutant viruses were normalized to that diameter. Single-step growth and plaque diameters were plotted using the Prism software package (GraphPad Software).
Virion protein incorporation.
PK15 cells were infected with either PRV-GS4284 (WT) or PRV-GS604 (R2) at an MOI of 3. Infections were carried out in 15-cm dishes of confluent cells. Infected cells and extracellular media were harvested once all cells displayed a cytopathic effect, which was typically at 18 hpi. Cellular debris was removed by centrifugation at 5,000 × g, and virions were concentrated from the supernatant by pelleting through a 10% Nycodenz cushion at 13,000 rpm in an SW28 rotor (Beckman). The resulting pellet was resuspended in 100 μl of TNE buffer (150 mM NaCl, 50 mM Tris [pH 7.4], 10 mM EDTA). Viral particles were dispersed by 10 1-s pulses of sonication in a cup horn ultrasonic processor (VCX-500; Sonics and Materials, Newtown, CT). The sample was loaded onto a 12 to 32% dextran gradient and centrifuged at 20,000 rpm for 1 h at 4°C. The heavy viral band was collected and spun at 25,000 rpm in a Beckman SW50.1 rotor at 4°C for 30 min. The final pellet was resuspended in final sample buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100) containing 10% β-mercaptoethanol, and the samples were boiled for 5 min prior to electrophoresis of 5 μl of each sample through an 8% sodium dodecyl sulfate (SDS)-polyacrylamide gel. Proteins were subsequently transferred onto an Immobilon polyvinylidene difluoride membrane (Millipore), and VP5 was detected using the 3C10 mouse monoclonal antibody (a gift of Lynn Enquist) at a 1:1,000 dilution. UL37 was detected using D1789, a rabbit antiserum raised against a peptide derived from the PRV UL37 sequence (REAADRVLGDYHE), at a 1:2,500 dilution. The secondary goat antimouse and antirabbit dye-labeled antibodies (LiCor) were used at 1:5,000 dilutions. Proteins were visualized and quantitated using an Odyssey Fc imager and ImageStudio software (LiCor). The ratio of UL37 to VP5 was quantified for four independent experiments and normalized to the average value obtained for the UL37-to-VP5 ratio for WT virus. Data were plotted using the Prism software package (GraphPad Software), and significance was determined using an unpaired Student's t test.
Protein expression and purification.
Both UL37 and UL37N constructs were expressed as N-terminal His6-SUMO fusions in T7 Express E. coli (New England BioLabs). Freshly transformed cells were incubated at 37°C overnight in 5 ml LB starter culture supplemented with 50 μg/ml kanamycin. The starter culture was diluted into 1 liter LB supplemented with 50 μg/ml kanamycin and grown at 37°C until the OD600 reached 0.8 to 1.0. At this point, the temperature was shifted to 16°C and the cells were induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG).
For production of UL37N, expression was induced for 16 to 20 h. Cells were harvested by centrifugation at 12,000 × g for 40 min, resuspended in 25 ml 20 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), pH 7.0, 50 mM NaCl, 0.1% Igepal CA-630 (Sigma), 5% glycerol, 10 mM imidazole, 0.1 mM tris(2-carboxyethyl)phosphine (TCEP), and 1 EDTA-free cOmplete protease inhibitor cocktail tablet (Roche), and lysed by use of a French press. The insoluble fraction was removed by centrifugation of the whole-cell lysate at 14,000 × g for 30 min at 4°C. Soluble lysate was loaded onto a 5-ml Ni-Sepharose 6B FF column (GE Healthcare). The column was subsequently washed with 10 column volumes (CVs) of 20 mM PIPES, pH 7.0, 50 mM NaCl, 0.1 mM TCEP (buffer A) containing increasing amounts of imidazole at 10 mM or 25 mM. Protein was eluted in buffer A containing 100 mM imidazole. The eluate was immediately concentrated, and the imidazole was removed by buffer exchange into buffer A using an Ultra-15 50-kDa-cutoff concentrator (Millipore). The protein concentration was determined from the absorbance at 280 nm using a calculated extinction coefficient. Glutathione S-transferase (GST)-tagged PreScission protease was added to the protein solution at a 1:50 protease-to-protein ratio, and the protein was cleaved overnight at 4°C to remove the His6-SUMO tag. The protease-protein solution was sequentially applied to glutathione-Sepharose 4B (GE Healthcare) and Ni-Sepharose 6B to remove the GST-tagged PreScission protease and the His6-SUMO tag, respectively. Cleaved protein was present in the unbound and wash fractions. UL37N was further purified by size exclusion chromatography using a Superdex 200 column (GE Healthcare) and concentrated to 3.5 to 4.0 mg/ml using an Ultra-15 30-kDa-cutoff concentrator (Millipore). Protein purity was assessed by SDS-polyacrylamide gel electrophoresis (PAGE) and Coomassie G-250 staining. The final yield was 18 mg of pure protein per 1 liter of E. coli culture. All UL37N protein samples used for crystallization and biochemical studies were stored in 20 mM PIPES, pH 7.0, 50 mM NaCl, and 0.5 mM TCEP.
A BL21 E. coli strain expressing GST-tagged PreScission protease was a gift from Peter Cherepanov (London Research Institute, London, United Kingdom). Protein expression was induced with 0.5 mM IPTG at 30°C for 4 h before the cells were harvested and lysed. The PreScission protease was purified over glutathione-Sepharose in a buffer containing 20 mM Tris, pH 8.0, 200 mM NaCl, and 1 mM TCEP. The column was washed 3 times with 10 CVs of the binding buffer, and protein was eluted from the column in binding buffer containing 5 mM reduced glutathione. The eluted protein was concentrated in a 30-kDa-cutoff concentrator (Millipore) and further purified over a Superdex 200 size exclusion column equilibrated with the binding buffer. The protein was concentrated to 1 mg/ml, flash frozen, and stored at −80°C.
Thermofluor assay.
The optimal buffer composition and the optimal NaCl concentration for the stability of the UL37N protein (PIPES, pH 7.0, and 50 mM NaCl) were determined using the Thermofluor method (34). Protein was diluted to 0.15 mg/ml in the storage buffer, and a fluorescent dye, SYPRO orange (Invitrogen), was added at a 1:1,000 dilution. Ten microliters of the protein-dye solution was pipetted into each well of a 96-well PCR microplate. Next, 10 μl of buffer (from a custom-made screen containing buffers at pH 4.5 to 10.5 and NaCl concentrations ranging from 0 to 500 mM) was added to wells containing the protein-dye solution. The plate was sealed and centrifuged for 1 min at 500 × g and 25°C. Samples were analyzed on a Roche LightCycler 480 quantitative PCR machine using an excitation wavelength of 465 nm and detection of emission at 610 nm. The emission signal was analyzed from 25°C to 95°C at a continuous acquisition rate of 3 measurements per °C. Data were analyzed using the ThermoQ software program (http://jshare.johnshopkins.edu/aherna19/thermoq/). Conditions that stabilized UL37N further increased its solubility.
Mass spectrometry.
For mass spectrometry analysis, the UL37 protein was analyzed using sinapinic acid (Agilent Technologies) as the matrix. Mass spectrometry measurements were performed on a Voyager DE-Pro matrix-assisted laser desorption ionization–time of flight mass spectrometer (Applied Biosystems).
Crystallization and structure determination.
Crystals of UL37N were grown by vapor diffusion at room temperature in hanging drops using 1 μl protein and 1 μl well solution containing 24 to 26% polyethylene glycol 1000, 0.3 M Ca(CH3COO)2, and 0.1 M imidazole, pH 8.0. Large plates formed in 3 to 8 days and were harvested 2 to 4 weeks later. For data collection, crystals were incubated in a solution identical to the well solution plus 10% glycerol for 30 s to 2 min prior to flash freezing in liquid N2. Heavy atom derivative crystals were obtained by soaking native crystals in well solution containing 5 mM thimerosal (Na salt of ethylmercurithiosalicylic acid or C9H9HgNaO2S) for 12 to 16 h. Derivative crystals were harvested and frozen using the protocol developed for the native crystals.
X-ray diffraction data were collected at 100 K at the X25 beam line at the National Synchrotron Light Source. The data were processed using HKL2000 (35) and indexed in space group P21 (Table 1). The native data set was processed up to a 2.0-Å resolution, and the single-wavelength anomalous dispersion (SAD) Hg data set was processed to a 2.3-Å resolution (Table 1). All 12 heavy atom sites were found using the phenix.autosol program, and the experimental density allowed the tracing of ∼70% of the residues in the phenix.autobuild program. Additional residues were manually built using the Coot program (36). There are two UL37N molecules in the asymmetric unit.
TABLE 1.
Data collection and refinement statistics
| Parameter | Value fora: |
|
|---|---|---|
| Native crystal | Native crystal soaked in thimerosal | |
| Data collection | ||
| Space group | P21 | P21 |
| Unit cell dimensions | ||
| a, b, c (Å) | 51.67, 156.59, 67.38 | 51.53, 156.30, 66.34 |
| α, β, γ (°) | 90, 91.33, 90 | 90, 91.78, 90 |
| Resolution (Å) | 43.12–2.00 (2.07–2.00) | 48.91–2.05 (2.12–2.05) |
| Rsym or Rmerge | 0.086 (0.516) | 0.097 (0.280) |
| I/σI | 20.32 (2.74) | 13.87 (2.18) |
| Completeness (%) | 89.4 (49.5) | 85.1 (35.31) |
| Redundancy | 6.3 (4.4) | 3.9 (2.1) |
| Refinement statistics | ||
| Resolution range (Å) | 43.12–2.00 | |
| No. of reflections (free) | 64,342 (2,347) | |
| Rwork/Rfree | 17.30/22.01 | |
| No. of atoms | 7,983 | |
| Protein | 7,342 | |
| Ligand/ion | 45 | |
| Water | 596 | |
| B-factorsb | 35.05 | |
| Protein | 35.03 | |
| Ligand/ion | 42.8 | |
| Water | 37.5 | |
| RMSD | ||
| Bond length (Å) | 0.007 | |
| Bond angle (°) | 0.96 | |
Values in parentheses are for the highest-resolution shell.
B-factor, isotropic displacement parameter.
Before refinement of the heavy atom model, 10% of the data was set aside for cross-validation. The model was refined against the SAD Hg data set to 2.3-Å resolution using the phenix.refine program. Next, test set flags were transferred to the native data set; additionally, 10% of the native data between 2.3 and 2.0 Å was set aside for cross-validation. After several cycles of refinement in the phenix.refine program (37) and rebuilding in Coot (36), Rwork was 17.3% and Rfree was 22.0%. The final model contained all amino acids from residues 1 to 479, including 3 of the 4 linker residues left after protease cleavage of the N-terminal tag. The final model is missing residues 480 to 496 in both chains. The MolProbity server (38) was used to assess the stereochemical quality of all models. According to MolProbity, 99.0% of the residues lie in the most favored regions of the Ramachandran plot and 1% lie in the additionally allowed regions of the Ramachandran plot. Final statistics are listed in Table 1.
Structure analysis.
The sequence alignment was generated and analyzed using the ClustalW (39) and ESPRIPT (40) programs. Interfaces were analyzed using the PISA program (41). Structural homology searches were performed using the Dali server (42), and the top hits were superposed onto the UL37N protein using the Dalilite pairwise comparison tool. The Evolutionary Trace server was used for evolutionary trace analysis (http://www-cryst.bioc.cam.ac.uk/∼jiye/evoltrace/evoltrace.html). All structure figures were made in the PyMOL program (http://www.pymol.org).
Protein structure accession number.
Atomic coordinates and structure factors for the UL37N structure have been deposited in the RCSB Protein Data Bank under accession number 4K70.
RESULTS
Characterization of UL37N.
Initially, we expressed full-length PRV UL37 with an N-terminal His6-SUMO tag in E. coli (Fig. 1A). During expression, this protein underwent spontaneous proteolysis, which generated a fragment containing the His6-SUMO tag and the N terminus of UL37 (Fig. 1B). Using mass spectrometry, the proteolytic site was localized around residue 498, which is approximately in the middle of the UL37 sequence. The difficulty in separating full-length UL37 from the truncated UL37 resulted in a very low yield of the purified full-length UL37, ∼200 μg/liter cell culture. Unlike full-length UL37, which was prone to aggregation, the N-terminal product of proteolytic cleavage was readily soluble and was pursued further. We expressed a fragment containing residues 1 to 496 of UL37 (UL37N) plus an N-terminal His6-SUMO tag in E. coli (Fig. 1A), purified it to homogeneity, and cleaved off the His6-SUMO tag (Fig. 1B), obtaining a yield of ∼18 to 20 mg per liter of cell culture. The optimal buffer composition and NaCl concentration for protein stability, PIPES, pH 7.0, and 50 mM NaCl, were determined using the Thermofluor method (34). These conditions further increased the solubility of UL37N. All UL37N protein samples used for crystallization and biochemical studies were stored in 20 mM PIPES, pH 7.0, 50 mM NaCl, and 0.5 mM TCEP.
FIG 1.
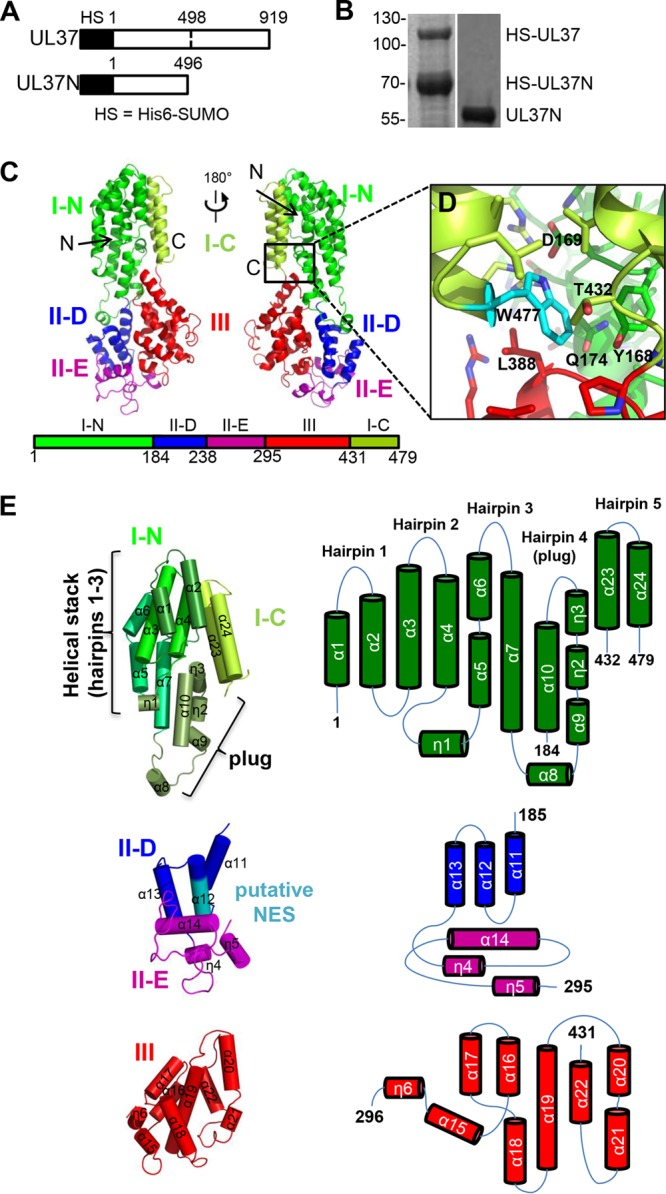
UL37N structure. (A) Linear diagram of UL37 constructs expressed in this work. HS, the His6-SUMO tag. (B) Coomassie-stained SDS-polyacrylamide gel showing purified His6-SUMO-tagged UL37 contaminated with the His6-SUMO-tagged UL37N proteolytic cleavage product and purified monodisperse UL37N. (C) Crystal structure of a UL37N monomer shown in two orientations related by a 180-degree rotation around the vertical axis. (D) A close-up view of residue W477 and its surroundings. Domains are colored as in panel C. (E) UL37N domains are shown individually. The color scheme is the same as that in panels C and D, except that the layers of domain 1 are highlighted by different shades of green and the putative nuclear export signal (NES) region in domain II is shown in teal. Orientations were chosen to show all secondary structure elements. Topology diagrams displaying individual domain organization of helices are also shown.
Architecture of UL37N.
The crystal structure of UL37N was determined using single anomalous dispersion and refined against a 2.0-Å native data set (Table 1). There are two monomers in the asymmetric unit, and the final model included residues 1 to 479 plus N-terminal linker residues PGS in both monomers (Table 1). The two monomers adopted very similar conformations, with the root mean square deviation (RMSD) being 0.4 for 482 common C-α residues (42).
UL37N is an elongated molecule with dimensions of 99 by 42 by 26 Å composed of 24 α helices and 6 310 helices arranged into a series of helical bundles (Fig. 1C). The structure can be divided into three domains: domain I, residues 1 to 184 and 432 to 479; domain II, residues 185 to 295; and domain III, residues 296 to 431 (Fig. 1C and E).
Domain I is formed by two noncontiguous segments of the polypeptide chain, residues 1 to 184 and residues 432 to 479 (Fig. 1E). Residue 479 is the last resolved residue; no electron density was observed for residues 480 to 496, and they are likely disordered. Domain I consists of five helical hairpins with the up-down topology which are formed by 12 α helices (α1 to α10, α23, and α24) and 3 310 helices (η1 to η3). Linker residues GS, which precede the start methionine, form the N terminus of helix α1. Hairpins 1 through 3 form a helical stack (Fig. 1E). Hairpin 1 consists of two short antiparallel helices, while hairpin 2 consists of two longer kinked helices, and hairpin 3 has two up helices followed by a loop and a single down helix. Helix η1 connects hairpins 2 and 3. The last two helices, α23 and α24, form hairpin 5 (Fig. 1E). Only the top part of hairpin 5 interacts with hairpins 1 to 3, an arrangement that results in a large U-shaped groove within domain I. Hairpin 4, formed by helix α10 running antiparallel to helices α9 and η2, forms a plug in the U-shaped groove in domain I. Helix η3 forms the tip of the plug. At the opposite end of the plug, a solitary helix, α8, at the tip of a long extension interacts with domain II. The Dali structural homology search (42) revealed that domain I bears a structural resemblance to the helical bundle domains of several subunits of multisubunit tethering complexes.
Conserved residue W477 plays a key role in the stability of not only domain I but also the entire UL37N because a shorter construct, UL37N from residues 1 to 476 [UL37N(1 to 476)], which lacks residue W477, has a lower thermal stability and progressively loses secondary structure during storage (Fig. 2). W477 helps anchor the hairpin 4 plug in domain I through van der Waals interactions with several hydrophobic residues and a hydrogen bond with the carboxyl of D169 (Fig. 1D), as well as van der Waals interactions with several hydrophobic residues in domain III.
FIG 2.

UL37N(1 to 496) is more stable over time than UL37N(1 to 476), as measured using circular dichroism on protein aged 1 day, 2 weeks, or 1 month. Helical content was estimated by the K2D2 program http://k2d2.ogic.ca/ using the spectrum over the range from 190 to 240 nm.
Domain II, residues 185 to 295, consists of helices α11 to α14 and two 310 helices (η4 and η5) (Fig. 1E). Helices α11 to α13 form a helical bundle, in which the last turn of helix α12 adopts a π-helix conformation. The putative nuclear export signal (NES), residues 263 to 273 in HSV-2 (30), maps to buried helix α12 (Fig. 1E) and is unlikely to be functional. Two long loops at the bottom of domain II are well structured (Fig. 1E) and adopt similar conformations in the two UL37N molecules present within the crystal asymmetric unit. Helix α14 appears to buttress both loops. Domain II does not have any structural homologs according to the Dali server (42).
Domain III, residues 296 to 431, is composed of helices α15 to α22 and one 310 helix (η6) (Fig. 1E). This domain is also a helical bundle, with the α19 central helix surrounded by the other six helices. This central helix maintains the structural integrity of domain III and is highly conserved.
UL37N is a dimer in crystals but a monomer in solution.
Two UL37N monomers in the asymmetric unit form an X-shaped dimer (Fig. 3A) that buries 1,734.8 Å2 of surface area. Four calcium ions are coordinated at the dimer interface as two symmetry-related sets of two calcium ions. Each set is coordinated by carboxyl oxygens from the side chains of Asp79, Asp81, and Glu82 of one monomer, carboxyl oxygens from the side chains of Asp382 and Asp383 plus the carbonyl oxygen of Trp379 of the other monomer, and two water molecules (Fig. 3B). As a result, one calcium ion is hexahedrally coordinated, while the second is pentahedrally coordinated.
FIG 3.
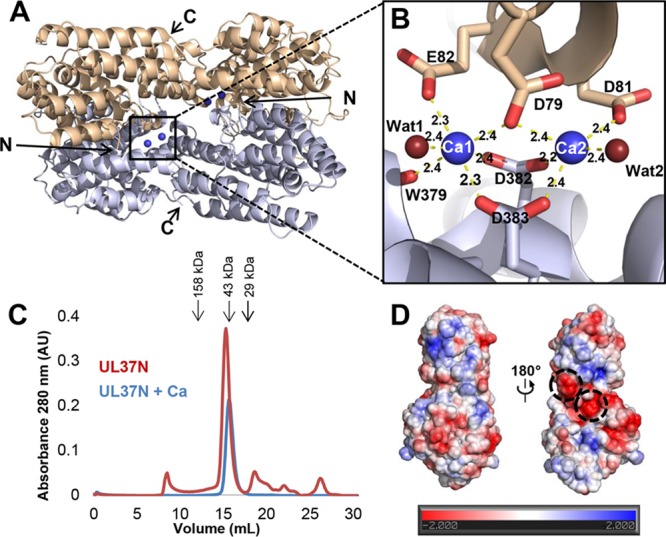
UL37N is a calcium-dependent dimer in crystals but not in solution. (A) Two UL37N monomers in the asymmetric unit are shown in gray and beige. Calcium ions at the dimer interface are shown as blue spheres. (B) A close-up view of the calcium-binding site at the dimer interface. The color scheme is the same as that in panel A. Residues coordinating calcium ions are shown as sticks, while waters (Wat1 and Wat2) involved in coordinating calcium are shown as maroon spheres. Distances (in angstroms) between calcium and coordinating atoms are shown as dashed lines and labeled. (C) Overlay of size exclusion chromatograms of UL37N with or without 0.2 M CaCl2. The elution volumes of several standards are indicated by arrows, and their molecular masses are labeled. AU, absorbance units. (D) Electrostatic surface potential map of UL37N generated using the Charmm program (http://www.charmm-gui.org/?doc=input/pbeqsolver). Negatively charged patches involved in binding calcium in crystals are circled.
Despite forming a dimer in crystals, UL37N is a monomer in solution. Crystal formation required the presence of at least 0.1 M Ca(CH3COO)2, and the best crystals were obtained in the presence of 0.3 M Ca(CH3COO)2. In solution, UL37N remained monomeric even in the presence of 0.2 M CaCl2, judging by its elution volume on size exclusion chromatography (Fig. 3C). We conclude that the dimerization of UL37N observed in crystals is likely induced by crystallization conditions (a high protein concentration and the presence of calcium). The coordination of four calcium ions at the dimer interface helps explain the importance of calcium ions in mediating crystal contacts. In the absence of calcium, the buried interface would have been smaller, 1,504.0 Å2 instead of 1,734.8 Å2. Although UL37N does not dimerize in solution, the dimerization interface features multiple grooves and several negatively charged patches (Fig. 3D). In full-length UL37, this area may perhaps participate in intramolecular contacts with the C-terminal half of UL37, which is also conserved among alphaherpesviruses and is predicted to be largely α helical.
ETA reveals several conserved surface clusters within UL37N.
To analyze sequence conservation within UL37N, we generated a sequence alignment of 15 UL37 homologs from alphaherpesviruses—a subfamily of herpesviruses that includes HSV and PRV—and identified 35 strictly conserved residues (Fig. 4). Most are located within the hydrophobic core and are probably necessary for maintaining the structural integrity of the protein, but 11 of these conserved residues are surface exposed (Fig. 3 and 5A) and are a logical choice for mutational analysis because surface-exposed conserved residues often participate in protein-protein interactions. None of these, however, clustered in a way that would help pinpoint regions of potential functional importance (Fig. 5A).
FIG 4.

Multiple-sequence alignment of UL37 homologs from 15 alphaherpesviruses. Only the alignment of residues corresponding to residues 1 to 479 of PRV UL37 is shown. Helices are indicated by rectangular boxes labeled and colored by domain with colors similar to those used in Fig. 1C. Identical residues are boxed in red, and those exposed on the surface of UL37N are marked by purple asterisks. Residues involved in binding calcium are marked by green circles. All residues specified for mutagenesis are highlighted by triangles (blue, R1; pink, R2; orange, R3) using the same color scheme used in Fig. 5C. PRV, suid herpesvirus strain Kaplan; BHV1, bovine herpesvirus 1; EHV1, equine herpesvirus 1; Felid_HV1, felid herpesvirus 1; Cerco_HV9, cercopithecine herpesvirus 9; VZVD, varicella-zoster virus serotype D; Gallid_HV2, gallid herpesvirus 2; Gallid_HV3, gallid herpesvirus 3; Meleagrid_HV1, meleagrid herpesvirus 1; Anatid_HV1, anatid herpesvirus 1; Macacine_HV1, macacine herpesvirus 1; Papiine_HV2, papiine herpesvirus 2; HSV2_HG52, herpes simplex virus 2 strain HG52; HSV1_17, herpes simplex virus1 strain 17; Saimiriine_HV1, saimiriine herpesvirus 1. PRV_UL37 (in italics) represents the secondary structure derived from the crystal structure of PRV UL37.
FIG 5.

Residue conservation on the surface of UL37N. (A) The UL37N structure is shown in surface representation. Two orientations related by a 180-degree rotation around the vertical axis are shown. Identical surface residues are in purple. (B) ETA class conserved and class specific. (C) Mutated residues in R1, R2, R3, and the calcium-binding site (Ca).
To locate potentially important functional sites on the surface of UL37N, ETA (43) was performed on the same sequence alignment (Fig. 6). ETA uses a sequence alignment of homologous proteins to generate a phylogenetic tree, which is then broken up into partitions, with more closely related sequences being grouped into classes. Within each partition, consensus sequences are generated for each set of sequences within a class. Each position within the sequence alignment is designated conserved, class specific, or neutral. Conserved residues have the same residue in all consensus sequences, whereas class-specific residues have a common residue for each closely related subgroup, but that residue is different among more divergent subgroups. Positions lacking consensus among the members of at least one subgroup are considered neutral. Clustering of conserved and class-specific residues on the protein surface may indicate regions of potential functional importance (43). This method has been used to detect functional sites in a number of proteins (44, 45).
FIG 6.

Phylogenetic tree from ETA using UL37 homologs from 15 alphaherpesviruses. ETA was carried out at partition 5 to avoid the high background levels associated with later partitions.
ETA on UL37N revealed several surface clusters which contained both conserved and class-specific residues (Fig. 5A and B). Several of these were not considered further because they either contained salt bridges, which are likely essential for protein stability alone, or contained several residues with only partially exposed side chains. Three clusters were chosen for further analysis. To probe their functional roles, we mutated 4 to 5 residues within each cluster to either eliminate a bulky side chain or, in two cases, to replace a small side chain with a bulky one. Three mutants with the following mutations were generated: V249R/R254A/R285A/D287A/H311A (R1 mutant), Q324A/D362A/R365A/H421A/H425A (R2 mutant), and K203A/P204Q/D239A/E240A/D295A (R3 mutant) (Fig. 5C). Mutated residues define three regions of potential functional importance, referred to as regions 1 through 3 (R1 to R3, respectively). R1 and R3 are located in domain II of UL37N, and R2 is located in domain III of UL37N. Additionally, residues involved in calcium binding were mutated to confirm that potential calcium-induced dimerization of UL37 is not essential for function, generating the mutant D79A/D81A/E82A/D382A/D383A (Ca).
UL37 region 2 is required for efficient viral spread.
All mutations were introduced into the PRV strain Becker background, and each virus was propagated to wild-type titers. To further investigate these mutants, viral replication and spread were measured in single-step growth and plaque formation assays, respectively. The Ca, R1, and R3 mutants did not display any reduction in plaque size or viral replication. In contrast, the R2 mutant plaques were restricted to about half the diameter of virus encoding wild-type UL37 (Fig. 7A). A defect in plaque formation can be the result of a defect in cell-cell spread or propagation kinetics. To address this question, the rates of cell-associated virus production and virus release into the supernatant were measured. The amount of virus released into the supernatant by the UL37 R2 mutant virus was similar to the amount released by the WT virus, suggesting that the R2 mutations cause a defect in cell-cell spread (Fig. 7B). There was no reduction in the structural incorporation of the UL37 R2 mutant protein relative to that of wild-type UL37 on the basis of immune detection in purified extracellular virions (Fig. 7C). In fact, the R2 mutant protein was slightly increased in virions relative to the amount of wild-type UL37 (161%; n = 4), but this was not statistically significant. We hypothesize that the R2 cluster serves as a potential binding site for as of yet unidentified cellular or viral proteins important for UL37 function in virus trafficking, which is essential in cell-cell spread.
FIG 7.

Propagation and spread of PRV encoding mutant forms of UL37. (A) Relative plaque diameters of mutant viruses Ca (PRV-GS5476), R1 (PRV-GS5321), R2 (PRV-GS5604), and R3 (PRV-GS5350). Plaque diameters were compiled from three individual experiments per virus and are plotted as a percentage of the average wild-type plaque diameter determined in parallel. Mean diameters are indicated by a horizontal bar, with error bars representing standard deviations. (B) Single-step growth curves comparing propagation of PRV-GS4284 (UL37 WT) and PRV-GS5604 (UL37 R2 mutant). Virus was harvested from cell medium (supernatants [Sups]; dashed lines) and cells (solid lines), and titers were measured by plaque assay. (C) Western blot analysis of UL37 protein incorporation into WT and R2 mutant extracellular virions. Virions were probed with antibodies raised against UL37 or VP5, with the latter serving as a loading control. Molecular mass standards are indicated on the left. A representative blot from four independent experiments is shown (see the text).
UL37 shares structural similarity with subunits of the CATCHR family of tethering complexes.
The UL37 proteins have no notable sequence homology to any viral or host proteins. Surprisingly, a Dali search (42) revealed that domain I resembles several subunits of eukaryotic multisubunit tethering complexes (MTCs) (46, 47). In intracellular trafficking pathways, MTCs tether vesicles to the target organelles to which they localize both to bring the vesicles closer to their target membranes and to help ensure the delivery of the vesicle to the correct target organelle (47). Several subunits of four MTCs, the Dsl1 complex, the exocyst complex, the Golgi-associated retrograde protein (GARP) complex, and the conserved oligomeric Golgi protein (COG) complex, share strong structural similarities, despite low sequence identity (46, 48–50), a finding which points to their common evolutionary origin and mechanistic similarities. Their structures consist of one to five helical bundle domains of similar folds (48–50). UL37N shares the highest structural similarity with several subunits of the Dsl1 complex and the exocyst, with Dali Z-scores of 4.2 to 5.4 (Fig. 8 and Table 2), while similarity to other MTC subunits is less pronounced. Although these similarity scores for the top hits are modest, they are comparable to the scores for some of the more distantly related MTC subunits. The structural similarity to MTC subunits is particularly remarkable because the sequence identity is under 10% (42).
FIG 8.
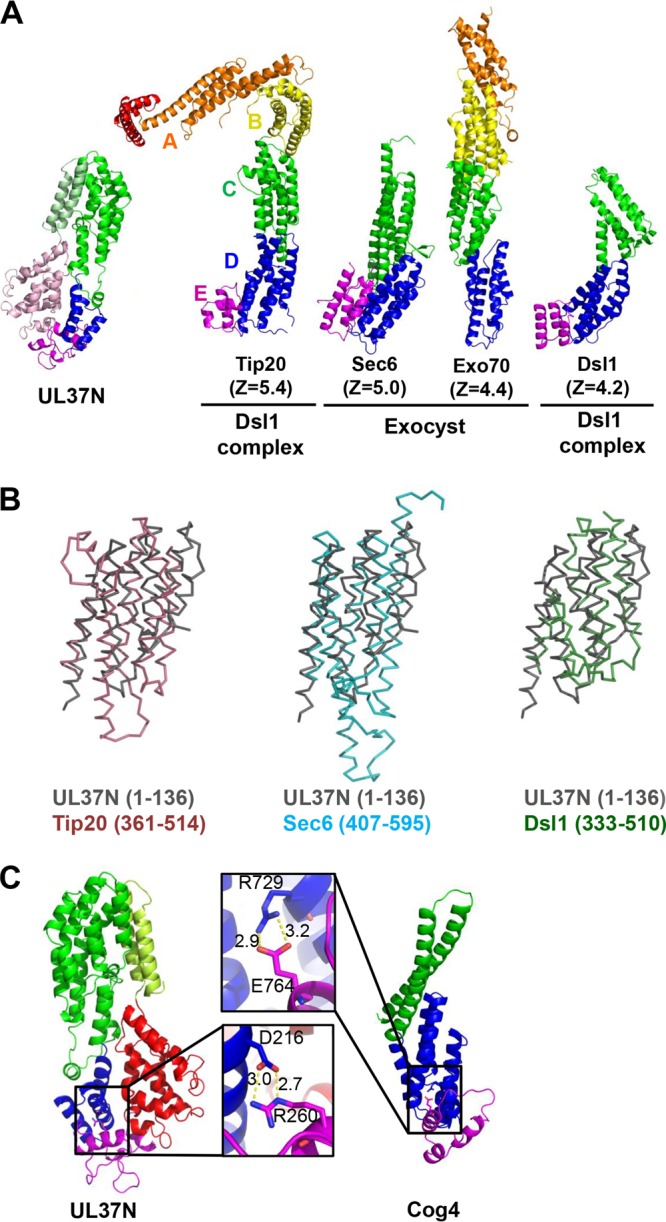
UL37N shares structural similarities with several subunits of cellular MTCs. (A) UL37N is shown side by side with the Tip20 (PDB accession no. 3FHN) and Dsl1 (PDB accession no. 3K8P) subunits of the Dsl1 complex and the Sec6 (PDB accession no. 2FJI) and Exo70 (PDB accession no. 2B7M) subunits of the exocyst complex, with the Z-score for each alignment displayed. Structure alignments were carried out using the Dali server (42). Domain I of UL37N aligns with domain C of these proteins (green). The overall J shape of domains I and II of UL37N is reminiscent of these MTC components. (B) Overlays of regions of Tip20, Sec6, and Dsl1 that align onto residues 1 to 136 of UL37N. (C) The salt bridge in domain II between putative D and E subdomains of UL37N is strictly conserved among alphaherpesviruses and likely plays a structural role. This salt bridge resembles the interdomain salt bridge between residues R729 and E764 of Cog4 subunit (PDB accession no. 3HR0) of the COG complex (48).
TABLE 2.
Alignments of UL37N with components of MTCsa
| Protein | Z-score | RMSD | No. of aligned residues | % identity |
|---|---|---|---|---|
| Tip20 | 5.4 (3.8) | 10.1 (3.8) | 186 (92) | 4 (14) |
| Sec6 | 5.0 (5.1) | 3.9 (3.7) | 145 (111) | 7 (8) |
| Exo70 | 4.4 (5.8) | 15.7 (3.3) | 182 (123) | 6 (9) |
| Dsl1 | 4.2 (5.7) | 11.1 (3.8) | 159 (98) | 6 (7) |
| Cog4 | 3.6 (3.8) | 4.7 (4.6) | 135 (107) | 10 (10) |
| Sec15 | 3.2 (4.4) | 3.4 (3.5) | 72 (97) | 4 (4) |
| Exo84 | 2.5 (5.4) | 4.1 (4.0) | 75 (102) | 4 (9) |
All alignments were carried out using the Dali server (42). Either the entire UL37N or just the helical bundle from residues 1 to 136 (for which the data are given in parentheses) was used in the Dali search.
Residues 1 to 136 of domain I resemble the helical bundles of MTCs the most and typically align with domain C of MTCs (46), but the structural similarity between UL37N and MTCs extends beyond domain I and includes domain II (Fig. 8A). The domain I and domain II module resembles domains C, D, and E of MTC subunits and has an overall J shape (Fig. 8A), which is found in some tethering subunits as the result of an additional domain E that follows domains C and D. Although the tip of domain II of UL37N only remotely resembles domain E of MTC subunits, the folds of domain E diverge even among MTC subunits themselves (Fig. 8A). Of note, the Cog4 subunit of the COG tethering complex has a salt bridge between domains D and E that involves a conserved arginine (48). UL37N also has a salt bridge between D216 and R260 in subdomains II-D and II-E, respectively (Fig. 8C), that is strictly conserved among alphaherpesviruses (Fig. 4).
Despite noticeable similarity, the structure of UL37N differs from the structures of MTC subunits in several aspects. First, instead of multiple helical bundles of similar topology (51), it has only one helical bundle with a topology similar to the topologies found in MTC subunits. Second, unlike in MTC subunits, where domain D or E is C terminal, the polypeptide chain in UL37N continues into domain III and a hairpin in domain I. Thus, the structural resemblance of domains I and II of UL37N to the MTCs may be the result of convergent evolution.
DISCUSSION
UL37 is one of the largest and most highly conserved herpesvirus tegument proteins, yet little is known about its role during the herpesvirus infectious cycle. Here, we report the structure of the N-terminal half of PRV UL37, UL37N, which revealed an elongated molecule rich in helical bundles that shares structural similarity to eukaryotic multisubunit tethering complexes (MTCs). This is the first structure of an inner tegument protein from an alphaherpesvirus and only the third structure available for any herpesvirus tegument protein, in addition to the structures of the transcription activator UL48 (VP16) from HSV-1 (52) and the deubiquitinase domain of the UL36 homologue of murine cytomegalovirus (53). The structure provides a three-dimensional framework for investigating the function of the UL37 N terminus and a first glimpse into the architecture of the inner tegument of alphaherpesviruses. Using evolutionary trace analysis, we identified several conserved surface regions and explored their importance using mutagenesis. We found that one of these, the R2 cluster, is dispensable for virus propagation yet important for viral cell-cell spread, which suggests that UL37 is necessary for intracellular virus trafficking to cell junctions. While UL37 is an effector of both capsid microtubule transport and secondary envelopment, these new insights into UL37N structure and function indicate that UL37 also coordinates viral spread between cells. UL37 is critical for propagation of both PRV and HSV-1, but UL37 is classified as a dispensable protein for PRV, as a small amount of residual propagation occurs in its absence; in contrast, HSV-1 fails to propagate in the absence of UL37 (4, 24). Whether R2 functionality is conserved in herpesviruses other than PRV will require formal testing.
Although the mechanism by which UL37 participates in viral trafficking to cell junctions is unknown, we speculate that the R2 cluster is a potential binding site for a viral or a cellular cofactor that promotes viral transmission between cells. The large tegument protein UL36 is the best-characterized binding partner of UL37 and tethers UL37 to capsids (6–9). The role of UL36 in viral cytosolic trafficking is well documented (6, 26, 54, 55). However, UL36 is unlikely to interact with the R2 cluster because it binds the C-terminal end of UL37 (27), and disruption of the UL36-UL37 interaction results in a severe growth defect (56) that was not observed with the R2 mutant. HSV-1 UL37 has also been shown to interact with capsid proteins UL35 and UL38 and the outer tegument protein UL46 in yeast two-hybrid screens (57), but these interactions have not yet been further validated. In any case, interactions between UL37 and capsid proteins may not be biologically relevant because UL37 recruitment to capsids is dependent upon UL36 (6, 56, 58). In addition to viral proteins, UL37 also associates with the host protein dystonin/BPAG1 (29), a protein involved in microtubule-based transport (59). Although dystonin is necessary for efficient capsid trafficking during entry and egress (29, 60), the dystonin-binding region is also located within the C terminus of UL37 (29). Thus, UL37 likely binds additional yet unidentified cellular or viral protein partners using its N terminus.
While the R2 cluster attests to the importance of UL37 for cell-cell spread and expands the repertoire of UL37 functions in intracellular virus trafficking, other conserved clusters identified on the surface of UL37N may play important functional roles, despite not displaying defects in our assays. Future studies will examine the phenotypes of the R1, R2, and R3 cluster mutants in animal infection models. In contrast, the calcium-binding site is unlikely to play any functional role because the acidic residues chelating calcium ions are not conserved among alphaherpesviruses (Fig. 4).
The cell-cell spread defect associated with the R2 cluster mutant was bolstered by the finding of a structural similarity between UL37N and components of the eukaryotic MTCs. MTCs control vesicular trafficking by tethering incoming vesicles to their target organelles and helping ensure the correct match between the vesicle and its destination (47). The functions of some MTCs may even extend beyond tethering vesicles, e.g., recruitment of dynein to kinetochores during chromosome segregation by the mammalian homolog of the Saccharomyces cerevisiae yeast Dsl1 complex (61) or interaction of the exocyst with microtubules during vesicle transport (62). The four MTC subunits with the highest structural similarity to UL37N are components of either the Dsl1 complex (Tip20 and Dsl1) or the exocyst complex (Sec6 and Exo70). Both the Dsl1 complex and the exocyst coordinate tethering of Golgi apparatus-derived vesicles to the endoplasmic reticulum or the plasma membrane, respectively. The notable structural resemblance between UL37N and MTC subunits suggests that UL37 could be the first viral MTC mimic. It will be interesting to find out whether the structure of the C terminus of UL37 reinforces the similarity to MTCs.
Considering that the herpesvirus capsids undergo cytoplasmic envelopment at tubular vesicles derived from either the TGN or early endosomes (1, 2), our data imply that herpesviruses may have co-opted the MTC functionality of UL37 to bring capsids to target membranes, perhaps even using UL37 to specify the cytoplasmic budding destination for the capsids. This idea is supported by the observation that in cells infected with UL37-null virus, cytosolic capsids do not colocalize with vesicular membranes (23, 63). Although the precise mechanism by which UL37 participates in virus trafficking remains enigmatic, its similarity to subunits of cellular MTCs will inform future work toward defining more precisely the roles of UL37 in promoting herpesvirus infection.
ACKNOWLEDGMENTS
We thank David King (University of California, Berkeley) for mass spectrometry experiments and A. Héroux (National Synchrotron Light Source) for collecting and processing X-ray diffraction data.
This work was funded by NIH grant 1DP20D001996, by the Burroughs Wellcome Fund Award for Investigators in Pathogenesis (to E.E.H.), and by NIH grant R01 AI056346 (to G.A.S.). Use of the National Synchrotron Light Source, Brookhaven National Laboratory, is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under contract no. DE-AC02-98CH10886.
J.D.P., G.A.S., and E.E.H. designed the experiments, J.D.P. expressed and purified all proteins, crystallized UL37N, and determined its structure, J.K. produced and characterized all mutant viruses, A.L.R. performed virion isolations and analyzed UL37 incorporation, E.E.H. supervised all steps of biochemical characterization, structure determination, and analysis, and all of us analyzed the data and wrote the manuscript.
Footnotes
Published ahead of print 5 March 2014
REFERENCES
- 1.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234. 10.1016/j.virusres.2009.03.018 [DOI] [PubMed] [Google Scholar]
- 2.Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. 2012. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 31:4204–4220. 10.1038/emboj.2012.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmitz JB, Albright AG, Kinchington PR, Jenkins FJ. 1995. The UL37 protein of herpes simplex virus type 1 is associated with the tegument of purified virions. Virology 206:1055–1065. 10.1006/viro.1995.1028 [DOI] [PubMed] [Google Scholar]
- 4.Klupp BG, Granzow H, Mundt E, Mettenleiter TC. 2001. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 75:8927–8936. 10.1128/JVI.75.19.8927-8936.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newcomb WW, Brown JC. 2010. Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 84:9408–9414. 10.1128/JVI.00361-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandbaumhuter M, Dohner K, Schipke J, Binz A, Pohlmann A, Sodeik B, Bauerfeind R. 2013. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 15:248–269. 10.1111/cmi.12075 [DOI] [PubMed] [Google Scholar]
- 7.Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065–3071. 10.1128/JVI.76.6.3065-3071.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571. 10.1128/JVI.79.15.9566-9571.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai P, Sexton GL, Huang E, Person S. 2008. Localization of herpes simplex virus type 1 UL37 in the Golgi complex requires UL36 but not capsid structures. J. Virol. 82:11354–11361. 10.1128/JVI.00956-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stellberger T, Hauser R, Baiker A, Pothineni VR, Haas J, Uetz P. 2010. Improving the yeast two-hybrid system with permutated fusions proteins: the varicella zoster virus interactome. Proteome Sci. 8:8. 10.1186/1477-5956-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozen R, Sathish N, Li Y, Yuan Y. 2008. Virion-wide protein interactions of Kaposi's sarcoma-associated herpesvirus. J. Virol. 82:4742–4750. 10.1128/JVI.02745-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.To A, Bai Y, Shen A, Gong H, Umamoto S, Lu S, Liu F. 2011. Yeast two hybrid analyses reveal novel binary interactions between human cytomegalovirus-encoded virion proteins. PLoS One 6:e17796. 10.1371/journal.pone.0017796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105–116. 10.1128/JVI.01032-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81:11790–11797. 10.1128/JVI.01113-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610–6623. 10.1128/JVI.02655-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, Nagel CH, Bauerfeind R, Sodeik B. 2012. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86:3682–3700. 10.1128/JVI.06432-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cardone G, Newcomb WW, Cheng N, Wingfield PT, Trus BL, Brown JC, Steven AC. 2012. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J. Virol. 86:4058–4064. 10.1128/JVI.00012-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antinone SE, Shubeita GT, Coller KE, Lee JI, Haverlock-Moyns S, Gross SP, Smith GA. 2006. The herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J. Virol. 80:5494–5498. 10.1128/JVI.00026-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200–3205. 10.1128/JVI.79.5.3200-3205.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 102:5832–5837. 10.1073/pnas.0500803102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Copeland AM, Newcomb WW, Brown JC. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 83:1660–1668. 10.1128/JVI.01139-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krautwald M, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Translocation of incoming pseudorabies virus capsids to the cell nucleus is delayed in the absence of tegument protein pUL37. J. Virol. 83:3389–3396. 10.1128/JVI.02090-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leege T, Granzow H, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J. Gen. Virol. 90:1560–1568. 10.1099/vir.0.010322-0 [DOI] [PubMed] [Google Scholar]
- 24.Desai P, Sexton GL, McCaffery JM, Person S. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 75:10259–10271. 10.1128/JVI.75.21.10259-10271.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618. 10.1128/JVI.74.24.11608-11618.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209. 10.1128/JVI.80.1.201-209.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bucks MA, Murphy MA, O'Regan KJ, Courtney RJ. 2011. Identification of interaction domains within the UL37 tegument protein of herpes simplex virus type 1. Virology 416:42–53. 10.1016/j.virol.2011.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly BJ, Mijatov B, Fraefel C, Cunningham AL, Diefenbach RJ. 2012. Identification of a single amino acid residue which is critical for the interaction between HSV-1 inner tegument proteins pUL36 and pUL37. Virology 422:308–316. 10.1016/j.virol.2011.11.002 [DOI] [PubMed] [Google Scholar]
- 29.Pasdeloup D, McElwee M, Beilstein F, Labetoulle M, Rixon FJ. 2013. Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J. Virol. 87:2857–2867. 10.1128/JVI.02676-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe D, Ushijima Y, Goshima F, Takakuwa H, Tomita Y, Nishiyama Y. 2000. Identification of nuclear export signal in UL37 protein of herpes simplex virus type 2. Biochem. Biophys. Res. Commun. 276:1248–1254. 10.1006/bbrc.2000.3600 [DOI] [PubMed] [Google Scholar]
- 31.Bohannon KP, Sollars PJ, Pickard GE, Smith GA. 2012. Fusion of a fluorescent protein to the pUL25 minor capsid protein of pseudorabies virus allows live-cell capsid imaging with negligible impact on infection. J. Gen. Virol. 93:124–129. 10.1099/vir.0.036145-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- 33.Smith GA, Enquist LW. 1999. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J. Virol. 73:6405–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips K, de la Pena AH. 2011. The combined use of the Thermofluor assay and ThermoQ analytical software for the determination of protein stability and buffer optimization as an aid in protein crystallization. Curr. Protoc. Mol. Biol. Chapter 10:Unit 10.28. 10.1002/0471142727.mb1028s94 [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58:1948–1954. 10.1107/S0907444902016657 [DOI] [PubMed] [Google Scholar]
- 38.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35:W375–W383. 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 40.Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308. 10.1093/bioinformatics/15.4.305 [DOI] [PubMed] [Google Scholar]
- 41.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797. 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 42.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38:W545–W549. 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lichtarge O, Bourne HR, Cohen FE. 1996. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 257:342–358. 10.1006/jmbi.1996.0167 [DOI] [PubMed] [Google Scholar]
- 44.Sowa ME, He W, Slep KC, Kercher MA, Lichtarge O, Wensel TG. 2001. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat. Struct. Biol. 8:234–237. 10.1038/84974 [DOI] [PubMed] [Google Scholar]
- 45.Chakravarty S, Hutson AM, Estes MK, Prasad BV. 2005. Evolutionary trace residues in noroviruses: importance in receptor binding, antigenicity, virion assembly, and strain diversity. J. Virol. 79:554–568. 10.1128/JVI.79.1.554-568.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackson LP, Kummel D, Reinisch KM, Owen DJ. 2012. Structures and mechanisms of vesicle coat components and multisubunit tethering complexes. Curr. Opin. Cell Biol. 24:475–483. 10.1016/j.ceb.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brocker C, Engelbrecht-Vandre S, Ungermann C. 2010. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. 20:R943–R952. 10.1016/j.cub.2010.09.015 [DOI] [PubMed] [Google Scholar]
- 48.Richardson BC, Smith RD, Ungar D, Nakamura A, Jeffrey PD, Lupashin VV, Hughson FM. 2009. Structural basis for a human glycosylation disorder caused by mutation of the COG4 gene. Proc. Natl. Acad. Sci. U. S. A. 106:13329–13334. 10.1073/pnas.0901966106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tripathi A, Ren Y, Jeffrey PD, Hughson FM. 2009. Structural characterization of Tip20p and Dsl1p, subunits of the Dsl1p vesicle tethering complex. Nat. Struct. Mol. Biol. 16:114–123. 10.1038/nsmb.1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong G, Hutagalung AH, Fu C, Novick P, Reinisch KM. 2005. The structures of exocyst subunit Exo70p and the Exo84p C-terminal domains reveal a common motif. Nat. Struct. Mol. Biol. 12:1094–1100. 10.1038/nsmb1017 [DOI] [PubMed] [Google Scholar]
- 51.Sivaram MV, Furgason ML, Brewer DN, Munson M. 2006. The structure of the exocyst subunit Sec6p defines a conserved architecture with diverse roles. Nat. Struct. Mol. Biol. 13:555–556. 10.1038/nsmb1096 [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Gong W, Huang CC, Herr W, Cheng X. 1999. Crystal structure of the conserved core of the herpes simplex virus transcriptional regulatory protein VP16. Genes Dev. 13:1692–1703. 10.1101/gad.13.13.1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlieker C, Weihofen WA, Frijns E, Kattenhorn LM, Gaudet R, Ploegh HL. 2007. Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol. Cell 25:677–687. 10.1016/j.molcel.2007.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 82:7388–7394. 10.1128/JVI.00225-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. 10.1016/j.chom.2013.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 78:11879–11889. 10.1128/JVI.78.21.11879-11889.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. 10.1016/j.virol.2008.05.035 [DOI] [PubMed] [Google Scholar]
- 58.Ko DH, Cunningham AL, Diefenbach RJ. 2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 84:1397–1405. 10.1128/JVI.01721-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryan SD, Bhanot K, Ferrier A, De Repentigny Y, Chu A, Blais A, Kothary R. 2012. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J. Cell Biol. 196:727–742. 10.1083/jcb.201107096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McElwee M, Beilstein F, Labetoulle M, Rixon FJ, Pasdeloup D. 2013. Dystonin/BPAG1 promotes plus-end-directed transport of herpes simplex virus 1 capsids on microtubules during entry. J. Virol. 87:11008–11018. 10.1128/JVI.01633-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmitt HD. 2010. Dsl1p/Zw10: common mechanisms behind tethering vesicles and microtubules. Trends Cell Biol. 20:257–268. 10.1016/j.tcb.2010.02.001 [DOI] [PubMed] [Google Scholar]
- 62.Heider MR, Munson M. 2012. Exorcising the exocyst complex. Traffic 13:898–907. 10.1111/j.1600-0854.2012.01353.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pasdeloup D, Beilstein F, Roberts AP, McElwee M, McNab D, Rixon FJ. 2010. Inner tegument protein pUL37 of herpes simplex virus type 1 is involved in directing capsids to the trans-Golgi network for envelopment. J. Gen. Virol. 91:2145–2151. 10.1099/vir.0.022053-0 [DOI] [PMC free article] [PubMed] [Google Scholar]