

ABSTRACT
Herpes simplex virus 1 (HSV-1)-infected cell protein 0 (ICP0) is a multifunctional protein that plays a key role in overcoming numerous facets of host innate immunity. A key function of ICP0 that requires an intact RING finger domain is that of an ubiquitin E3 ligase: ICP0 interacts with at least three ubiquitin-conjugating enzymes of which one, UbcH5a, is required for degradation of PML and SP100. A preceding report showed that ICP0 is highly unstable at very early times after infection but becomes stable at later times. We report here that (i) the degradation of ICP0 is not infected cell specific, (ii) the degradation does not require the interaction of ICP0 with either UbcH5a, UbcH6, or UbcH9, (iii) ICP0 is degraded both early and late in cells infected with a mutant lacking the UL13 protein kinase, (iv) ICP0 encoded by wild-type virus or the ΔUL13 mutant is stable in cells transfected with a plasmid encoding UL13 before infection, (v) ICP0 carrying mutations in the RING finger domain is stable both early and late in infection, and, finally, (vi) in cells infected with both wild type and RING finger mutant only the wild-type ICP0 is rapidly degraded at early times. The results suggest that the stability of ICP0 is mediated by the UL13 protein kinase and that the target of proteolysis is a site at or near the RING domain of ICP0.
IMPORTANCE ICP0, a major regulatory protein of HSV-1, turns over rapidly early in infection but becomes stable at late times. We report that stabilization requires the presence of UL13 protein kinase and that an ICP0 with mutations in RING finger is stable. In mixed infections mutant ICP0 is stable, whereas the wild-type ICP0 is degraded. Our findings suggest that the lifestyle of HSV-1 requires an ICP0 that turns over rapidly if late proteins are absent.
INTRODUCTION
The infected cell protein 0 (ICP0) is a multifunctional α protein that plays a key role in the biology of herpes simplex virus 1 (HSV-1) (1). At low multiplicities of infection, ICP0 is instrumental in overcoming the innate immune responses to infection. Its key functions include the degradation of promyelocytic leukemia protein (PML) and SP100 (2–5), derepression of post α genes by displacement of the HDAC1 or 2/CoREST/REST repressor complex (6–11), recruitment of cyclin D3 and CLOCK histone acetyltransferase (12–14), and inhibition of activation of interferon-dependent genes (1, 15–18). An earlier publication from our laboratory reported that HSV-1 ICP0 has a short half-life of approximately 1 h during the first several hours after infection but then becomes stable at late times after infection (19). The key findings were that (i) the cleavage was independent of the virus strain and (ii) the initial cleavage generated several discrete polypeptides by a proteasome-independent process. In a second step, the products of the initial cleavage were degraded by a proteasome-dependent process. The persistence of the products of the initial cleavage raised the possibility that they may perform specific functions. These findings raised several questions. The first and operationally definable question concerns the mechanism that renders ICP0 resistant to degradation at late times after infection. The second and less operable question is why ICP0 is so unstable at early times given the importance of the protein in enabling viral replication?
The focus of this report is on the requirements for the cleavage and stabilization of ICP0. We report that (i) the degradation of ICP0 is cell line independent, (ii) the degradation of ICP0 does not involve the ubiquitin-conjugating enzymes with which it interacts to perform its function as a ubiquitin ligase, (iii) ICP0 carrying amino acid substitutions in the RING domain which inactivate the E3 ligase activity is not degraded in singly infected cells or in cells infected with both mutant and wild-type (WT) virus, and (iv) ICP0 requires the UL13 protein kinase to become resistant to degradation.
MATERIALS AND METHODS
Cells and viruses.
HEp-2, Vero, HeLa, and HEK293T cell lines were obtained from the American Type Culture Collection (Manassas, VA), and the human embryonic lung (HEL) was a gift from Thomas E. Shenk (Princeton University). All cell lines were grown in Dulbecco modified Eagle medium supplemented with 5% fetal calf serum (HEp-2 cells), 5% newborn calf serum (Vero and HeLa cells), or 10% fetal bovine serum (HEK 293T and HEL cells). U2OS cells were grown in McCoy 5A medium (Gibco-BRL) supplemented with 10% fetal calf serum. HSV-1 strain F [HSV-1(F)] is a prototype HSV-1 strain used in this laboratory (20). The RING-finger (RF) mutant (C116A/C156A) virus was kindly provided by Saul Silverstein (Columbia University) (21). HSV-1 mutant viruses R7356 (ΔUL13 virus) (22), R7041 (ΔUS3 virus) (23), and R8515 encoding a ICP0-EGFP chimeric protein (19) were as described elsewhere.
Construction of recombinant virus carrying RF-EGFP chimeric protein.
The enhanced green fluorescent protein (EGFP) fragment was obtained from SalI-digested plasmid pRB8536 and cloned into the SalI site of plasmid pRB8537 in which ICP0 carries the C116A/C156A substitutions (RF mutations) (19). The plasmid expressing the in-frame fusion of RF-GFP protein was designated pRB538. The PmeI and PstI doubly digested fragment from pRB538 was cloned between the ScaI and PstI sites of pKO5 and generated pKO538. The pKO538 was transformed into BAC-ΔICP0 to generate BAC538. The BAC538 DNA was then transfected into U2OS cells to generate recombinant virus R538. The strategy of virus construction has been reported elsewhere (7).
Plasmids.
Constructions of plasmids expressing UL13 and US3 protein kinases, respectively, were performed according to standard procedures. The DNA sequence encoding UL13 was PCR amplified with the primers 5′-ATGAATTCTCACGACAGCGCGTGCCG-3′ and 5′-TAGCGGCCGCATGGATGAGTCCCGCAG-3′, digested by using EcoRI+NotI, and then cloned into pcDNA3.1 (+) vector to generate the UL13 expression plasmid pcDNA3.1-UL13. The DNA sequence encoding US3 was PCR amplified with the primers 5′-ATGAATTCTATATTACAGGCCCGTGTCC-3′ and 5′-TAGCGGCCGCCAACTAGATACCACCG-3′, digested by using EcoRI+NotI, and then cloned into pcDNA3.1 (+) vector to generate the US3 expression plasmid pcDNA3.1-US3.
shRNA lentiviral transduction.
MISSION short hairpin RNA (shRNA) lentiviral plasmids (pLKO.1-puro; Sigma-Aldrich, Poole, United Kingdom) were used to knockdown UbcH5a, UbcH6, and UbcH9 expression in HEp-2 cells according to the manufacturer's protocol. Stable gene knockdown was established by cellular resistance to puromycin (1,000 ng/ml). Clones were isolated and several clonal cell lines were established. The nontarget shRNA control vector (Sigma-Aldrich) containing an insert sequence that target an unrelated gene served as a negative control.
Transfection.
Cells grown in 25-cm2 flasks were transfected at 60 to 70% confluence with 1 μg of DNA per flask with Lipofectamine transfection reagent, as specified by the supplier (Invitrogen).
Immunoblots.
Cells grown in 25-cm2 flasks were transfected or infected as indicated. Harvested cells were rinsed twice with ice-cold phosphate-buffered saline (PBS), resuspended in 150 μl of triple-detergent buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 100 μg of phenylmethylsulfonyl fluoride/ml) supplemented with protease inhibitor cocktail (Sigma), and briefly sonicated. The total protein concentrations were measured by using Bradford reagent (Bio-Rad). Protein lysates were then mixed with a 1/3 volume of 4× sodium dodecyl sulfate loading buffer, boiled for 5 min, electrophoretically separated in 10% denaturing polyacrylamide gels, and transferred to preequilibrated nitrocellulose membranes. The membranes were blocked in PBST (PBS supplemented with 0.1% [vol/vol] Tween 20) and 5% nonfat dry milk and then reacted overnight at 4°C with appropriate primary antibodies diluted in PBST–1% nonfat milk. After several rinses with PBST, the membranes were reacted with appropriate secondary antibody conjugated with horseradish peroxidase. Protein bands were visualized by using ECL Western blotting detection reagents (Amersham Biosciences) according to the manufacturer's instructions.
Antibodies.
The rabbit polyclonal antibody to ICP0 exon 2 was from Goodwin Institute for cancer research (Plantation, FL). The mouse monoclonal antibodies to UbcH5a, UbcH6, and UbcH9 were from Abnova. The monoclonal antibody to β-actin was from Sigma. Rabbit monoclonal antibodies to UL13 and US3 were purchased from Santa Cruz.
RESULTS
The degradation of ICP0 early after infection is not cell line dependent.
In this series of experiments, replicate cultures of HEK293T, HEp-2, Vero, HEL, or HeLa cells were exposed to 10 PFU of HSV-1(F) per cell. The inoculum was replaced after 1 h. At 3 h after infection, the cells were incubated in medium containing 100 μg of cycloheximide per ml. Replicate culture harvested at 1-h intervals until 7 h after infection were solubilized, subjected to electrophoresis in denaturing gels, and probed with antibody to ICP0. The results shown in Fig. 1 indicate that a substantial decrease in the amounts of ICP0 was noted as early as 1 h after exposure to cycloheximide. In the present study, the cellular protein migrated at ∼160 kDa and served as a loading control.
FIG 1.

ICP0 protein was degraded in a cell-type-independent manner during the early stages of HSV-1(F) infection. (A) Schematic diagram of the experimental design. (B) Replicate cultures of HEK293T, HEp-2, Vero, HEL, or HeLa cells were infected at 10 PFU/cell with HSV-1(F). At 3 h after infection, the cells were exposed to medium containing cycloheximide (CHX; 100 μg/ml). The cultures were harvested at 3, 4, 5, 6, or 7 h after infection as indicated and processed as described in Materials and Methods. Cell lysates were subjected to electrophoresis on denaturing polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted with ICP0 antibody.
Depletion of UbcH5a, UbcH6, or UbsH9 individually has no discernible effect on rapid turnover of newly made ICP0 early in infection.
Studies reported elsewhere have shown that ICP0 interacts with the ubiquitin-conjugating E2 enzymes UbcH5a, UbcH6, and UbcH9 and that UbcH5a was required for the degradation of PML (2). The degradation of PML is an early event in viral replication and coincides roughly with the interval of time in which ICP0 turns over very rapidly. In this series of experiments, we tested the hypothesis that one of these ubiquitin-conjugating enzymes was involved in the degradation of ICP0 early in infection. To test the hypothesis, we depleted UbcH5a, UbcH6, and UbcH9 from HEp-2 cells as described in Materials and Methods. The levels of UbcH5a and UbcH6 in 5 clones derived from cultures transduced with corresponding lentiviruses expressing the appropriate shRNAs are shown in Fig. 2A and B, respectively. The corresponding levels of UbcH9 in selected clones are shown in Fig. 3A.
FIG 2.
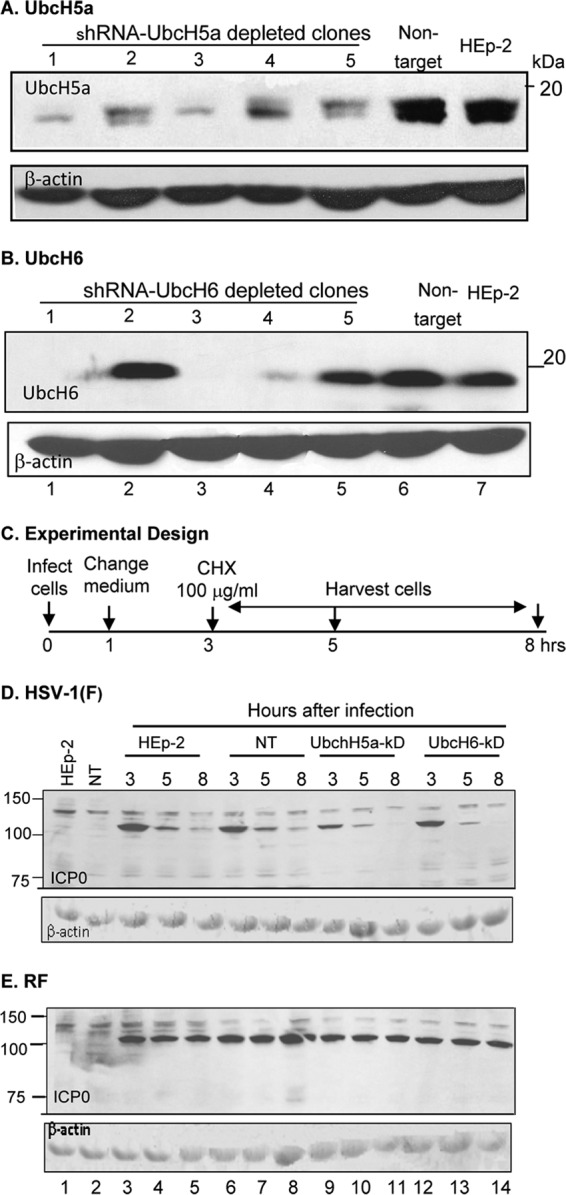
UbcH5a or UbcH6 ubiquitin-conjugating enzymes do not play a role in the degradation of ICP0 at early times after infection. (A) Immunoblots of five HEp-2 cell clones selected after transduction of lentivirus encoding anti-UbcH5a shRNA, one cell clone transduced with a lentivirus expressing a nontarget shRNA, and wild-type HEp-2 cells. The electrophoretically separated proteins were reacted with antibody to UbcH5a. (B) Same as panel A except that the shRNA was designed to knock down UbcH6 and the antibody was against UbcH6. In both panels, β-actin served as a loading control. (C) Schematic representation of the experimental design similar to that shown in Fig. 1 except that the cycloheximide-treated cells were harvested at 3, 5, or 8 h after infection. (D) Stability of wild-type ICP0 in wild-type HEp-2 cells (lanes 3 to 5), in cells transduced with nontarget shRNA (lane 6 to 8), in cells derived from clone 3 of panel A (lanes 9 to 11), or in cells from clone 3 of panel B derived by transduction with shRNA against UbcH6 (lanes 12 to 14). (E) Same as panel D except that the cells were infected with the RF mutant virus.
FIG 3.
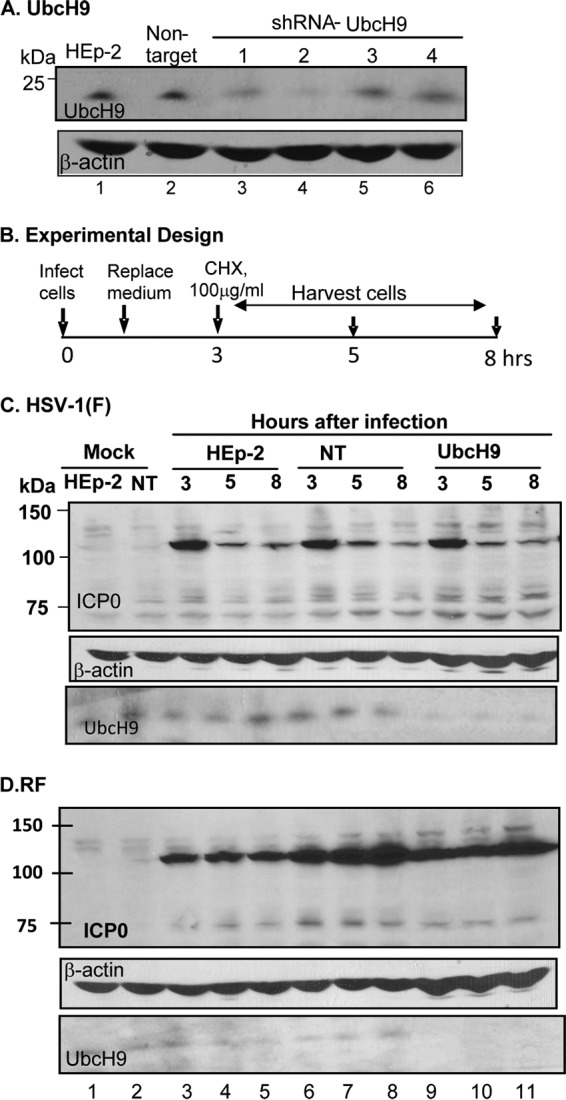
UbcH9 ubiquitin-conjugating enzyme does not play a role in the degradation of ICP0 at early times after infection. (A) Immunoblots of four HEp-2 cell clones selected after transduction of lentivirus encoding anti-UbcH9 shRNA, one clone transduced with a lentivirus expressing a nontarget shRNA, and wild-type HEp-2 cells. The electrophoretically separated proteins were reacted with antibody to UbcH9. β-Actin served as a loading control. (B) Schematic representation of the experimental design. (C and D) Stability of wild-type ICP0 (C) or RF mutant (D) in wild-type HEp-2 cells (lanes 3 to 5), nontarget cells (NT; lanes 6 to 8), or cells from clone 2 of panel A derived by transduction with shRNA against UbcH9 (lanes 9 to 11).
We tested next the stability of wild-type and ring finger mutant (RF) in several HEp-2 cell populations. These included the parent untreated HEp-2 cells, HEp-2 cells transduced with lentivirus expressing nontarget shRNA, cells derived from clone 1 of Fig. 2A that was depleted of UbcH5, cells derived from clone 1 of Fig. 2B that was depleted of UbcH6, and cells derived from clone 2 of Fig. 3A that was depleted of UbcH9. The design of the experiments is shown in Fig. 2C and Fig. 3B.
Briefly, the cells were exposed to wild-type or RF mutant virus (10 PFU/cell). The inoculum was replaced after 1 h. At 3 h after exposure to virus, the cells were exposed to 100 μg of cycloheximide per ml of medium. The replicate cultures were harvested and tested for the presence and amounts of ICP0 at the time of the addition of cycloheximide indicated in the figures. β-Actin served as a loading control. The results (Fig. 2D and E and Fig. 3C and D) were as follows. Wild-type virus ICP0 was rapidly degraded in HEp-2 untreated, nontarget transduced cells, and the HEp-2 clonal cell lines tested. In contrast, the RF ICP0 mutant was stable and was not degraded in wild-type cells or in any of the clonal cell lines tested.
We conclude that these results do not support the hypothesis that UbcH5a, UbcH6, or UbcH9 individually are involved in the rapid degradation of wild-type ICP0 at very early times after infection. The results also indicate that, in contrast to wild-type ICP0, the RF mutant virus carrying two amino acid substitutions in the RING finger domain is not degraded at early times after infection.
ICP0 remains unstable at late times in cells infected with the ΔUL13 mutant virus.
The objective of the next series of experiments was to test the hypothesis that at a late time after infection ICP0 is modified by a late HSV protein. HSV-1 encodes two protein kinases, UL13 and US3. Both are produced at late times after infection (24, 25). If either US3-PK or UL13-PK is required to stabilize ICP0 at late times after infection, ICP0 in cells infected with ΔUL13 or ΔUS3 mutant virus would be expected to remain unstable even at late times after infection.
The experimental design to test this hypothesis are shown in Fig. 4A and C. Briefly, cells were exposed to wild-type or mutant virus (10 PFU/cell). The inoculum was replaced at 1 h after infection. The cells were exposed to 100 μg of cycloheximide per ml of medium at 3 h (Fig. 4A and B) or 6 h (Fig. 4C and D) after infection. The cells were harvested 6 or 10 h after infection (Fig. 4A and B) or 10 or 12 h after infection (Fig. 4C and D). The results (Fig. 4B and D) showed that ICP0 was degraded in cells infected with wild-type or mutant viruses and exposed to cycloheximide at 3 h after infection (Fig. 4B). In cells exposed to cycloheximide 6 h after infection, the level of ICP0 remained stable in wild-type virus- and ΔUS3 virus-infected cells but was decreased in cells infected with ΔUL13 mutant virus (Fig. 4D, lanes 9 and 10).
FIG 4.
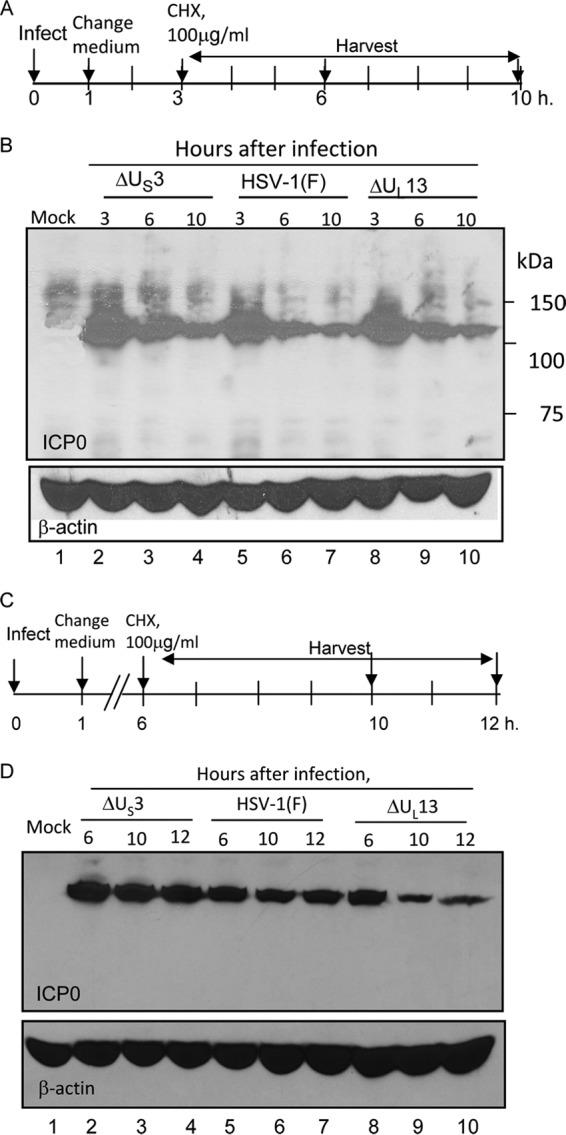
ICP0 is degraded at late times in cells infected with the ΔUL13 mutant virus. (A) The experimental design shown here is similar to those shown in Fig. 1 to 3. (B) Stability of wild-type ICP0 in HEp-2 cells infected with ΔUS3 mutant virus (lanes 2 to 4), wild-type virus (lanes 5 to 7), or ΔUL13 mutant virus (lanes 8 to 10). (C) Schematic representation of the experimental design that generated the data shown in panel D. In this experiment, the cells were exposed to the viruses indicated in panel B, cycloheximide was added at 6 h, and the cells were harvested at 6, 10, and 12 h after infection. (D) Stability of wild-type ICP0 in HEp-2 cells infected with ΔUS3 mutant virus (lanes 2 to 4), wild-type virus (lanes 5 to 7), or ΔUL13 mutant virus (lanes 8 to 10).
The ICP0 encoded by the ΔUL13 mutant virus is stabilized in cells transfected with a plasmid encoding UL13-PK before infection.
The design of this experiment is shown in Fig. 5A. In brief, HEK293T cells were mock transfected or transfected with an empty vector or plasmids encoding UL13-PK or US3-PK. At 24 h after transfection, the cells were exposed (10 PFU/cell) to wild-type virus (Fig. 5B) or ΔUL13 mutant virus (Fig. 5C). As in other experiments described here, the inoculum was replaced at 1 h after infection, and the cultures were incubated in medium containing cycloheximide (100 μg/ml) added 3 h after infection. The cultures were harvested at the time of the addition of cycloheximide and at 6 and 10 h after infection. In this experiment, the denatured electrophoretically separated proteins were probed with antibodies to ICP0, US3-PK, or UL13-PK. β-Actin served as a loading control. ICP0 encoded by wild-type virus (Fig. 5B) and ICP0 encoded by the ΔUL13 mutant virus (Fig. 5C) were rapidly degraded during the cycloheximide chase in cells transfected with empty vector (lanes 6 to 8), in cells transfected with plasmids encoding US3-PK (lanes 12 to 14), or in mock-transfected cells (lanes 3 to 5). ICP0 was stable in cells transfected with the plasmid encoding the UL13-PK (lanes 9 to 11). Both UL13-PK and US3-PK were expressed in corresponding transfected cells (Fig. 5B and C). Note that the antibody to US3-PK reacted with a host protein of the same apparent molecular weight.
FIG 5.
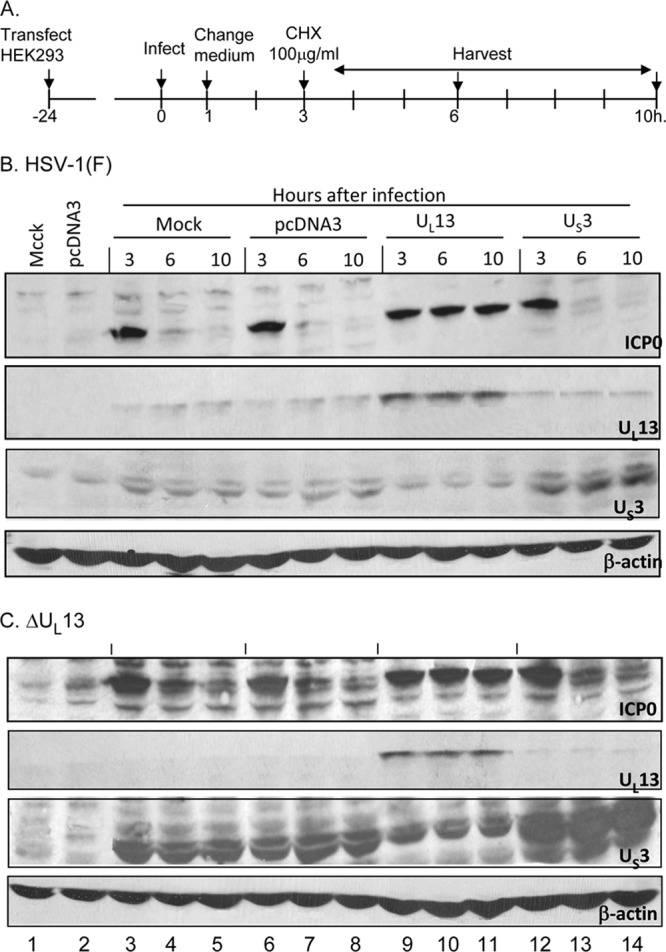
Transient transfection of UL13-PK but not of US3-PK stabilized ICP0 at early times after infection. (A) Schematic representation of experimental design. HEK293T cells were transfected with plasmids encoding UL13-PK or US3-PK 24 h before infection. The protocols for infection, the replacement of medium after 1 h, the addition of cycloheximide at 3 h after infection, and analyses of cells harvested at 3, 6, or 10 h after infection were similar to those indicated in legends to Fig. 1 to 3. (B) Immunoblots of cells infected with HSV-1(F). Lanes 1 and 2, uninfected mock-treated or pcDNA3-transfected cells, respectively; lanes 3 to 14, HSV-1(F)-infected mock treated cells (lanes 3 to 5), or cells that had been transfected with pcDNA3, or plasmids encoding UL13 or US3 protein kinases as indicated. (C) Same as panel B except that the cells were infected with the ΔUL13 mutant virus. In both panels the electrophoretically separated proteins transferred to a nitrocellulose membrane were reacted with the antibodies shown.
We conclude that the stability of ICP0 is mediated by the appearance of UL13-PK in the infected cells and that the presence of the UL13 at the time of synthesis and initial accumulation of ICP0 is sufficient to stabilize the protein.
In doubly infected cells, wild-type ICP0 is degraded independently of the ICP0 carrying mutations in the RING domain.
ICP0 forms dimers (26). The experiments described here were designed to test two hypotheses. The first hypothesis is that ICP0 possesses proteolytic activity that cleaves its partner in trans. The second hypothesis is that wild-type ICP0 induces an activity that in turn degrades both itself and the RF mutant, whereas the RF mutant is unable to do so. To this end, we used two ICP0-GFP fusion proteins. R8515 (described elsewhere) consists of wild-type ICP0 fused to GFP. The chimeric protein has the properties of wild-type ICP0 in that it is unstable at early times after infection (19). The second ICP0-GFP fusion protein designated R538 is similar to R8515 except that it carries the two amino acid substitutions that are also present in the RF mutant. The experimental design was as follows: replicate cultures of HEp-2 cells were infected with wild-type HSV-1(F), R8515, RF, R538 mutant, or virus combinations (WT+RF, WT+R538, or R8515+RF). The protocol was similar to that used in Fig. 2 in that the inocula were removed after 1 h, the cultures were exposed to cycloheximide at 3 h after infection, and the cells were harvested, solubilized, subjected to electrophoresis in denaturing gels, and probed with antibody to ICP0 or β-actin. The results presented in Fig. 6 show that ICP0 carrying the RF mutation was not degraded in either singly (lanes 8 to 13) or doubly (lanes 18 to 23) infected cells, whereas wild-type ICP0 was degraded in both singly (lanes 2 to 7) and doubly (lanes 15 to 23) infected cells.
FIG 6.
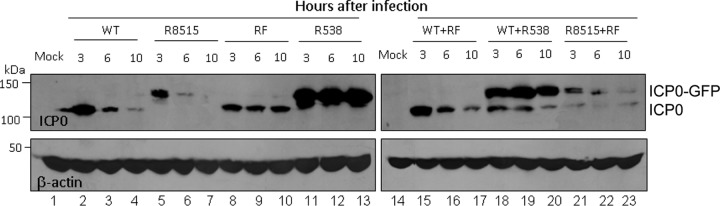
RING finger mutant ICP0 is not degraded in cells doubly infected with wild-type and RF mutant viruses. The experimental design was similar to that shown in Fig. 1 to 3 except that the cells were either singly infected with HSV-1(F) (lanes 2 to 4), R8515 (lanes 5 to 7), RF (lanes 8 to 10), or R538 (lanes 11 to 13) or doubly infected with mixtures consisting of wild-type virus and RF mutant (lanes 15 to 17), wild-type virus plus R538 (lanes 18 to 20), or R8515 and RF (lanes 21 to 23). Note that ICP0-GFP migrates more slowly than ICP0 and that ICP0 in the chimeric protein encoded by R538 carries the amino acid substitutions in the RING domain identical to those of RF virus, whereas the ICP0 component of the chimeric protein in the R8515 virus is wild type.
We conclude that the results of the experiments described above do not support either hypothesis. In essence the proteolytic activity is not selectively induced by wild-type ICP0. The data also do not support the hypothesis that the wild-type ICP0 acts in trans to cleave an inactive partner. The data suggest that the proteolytic activity discriminates between the wild-type ICP0 and ICP0 carrying the RF mutations, possibly by targeting the RING domain of ICP0.
DISCUSSION
An earlier report from this laboratory showed that in productively infected cells ICP0 accumulating within a few hours after infection has a half-life less than 1 h (19). In contrast, ICP0 present in infected cells at later times was very stable. The phenomenon was virus strain independent. This suggested that for reasons unknown ICP0 had to undergo a specific modification in order to become stabilized and that this phenomenon took place at late times after infection. Five salient features of this report are as follows.
(i) ICP0 was rapidly degraded early after infection of five cell lines. The data suggest that the degradation of ICP0 is cell line independent.
(ii) Earlier studies have shown that ICP0 interacts with UbcH5a, UbcH6, and UbcH9 ubiquitin-conjugating enzymes (3, 4). The data reported here indicate that ICP0 is degraded in cells depleted of these E2 enzymes.
(iii) The stabilization of ICP0 is linked to the presence of UL13-PK. Specifically, ICP0 encoded by ΔUL13 mutant virus is unstable both early and late in infection. Moreover, both wild-type ICP0 and ICP0 encoded by the ΔUL13 mutant virus are stable at early time after infection with the presence of UL13-PK.
ICP0 is extensively posttranslationally modified during infection. At this point, it is not clear whether the stabilization involves physical interaction of UL13 with ICP0, phosphorylation of ICP0, or a posttranslational modification of the protease responsible for the destabilization of ICP0.
(iv) In contrast to wild-type ICP0, the RF mutant carrying two amino acid substitutions in the RING finger domain is stable at early times after infection.
(v) We have examined two possible explanations for the selective degradation of wild-type ICP0 but not of RF mutant ICP0 at early times after infection. The first is that, in contrast to the RF mutant ICP0, the wild-type ICP0 induces or activates a proteolytic enzyme. The second hypothesis is that ICP0 acts in trans to degrade itself. To test this hypothesis, we used two viruses encoding ICP0-GFP fusion proteins. Whereas the ICP0 portion of the chimeric ICP0-GFP protein encoded by the R8515 is unstable and behaves like wild-type ICP0, the ICP0 moiety of the ICP0-GFP encoded by the R538 mutant carried the RING finger amino acid substitutions remain stable in both singly infected cells and in cells coinfected with the wild-type virus. The data indicate that both hypotheses are untenable.
In essence, these data indicate that ICP0 made after infection is unstable and requires a late protein, UL13-PK, for stabilization—a feature of ICP0 observed after 6 h after infection. The results also indicate that ICP0 carrying the amino acid substitutions in the RING finger domain is not subject to proteolysis, either because the RING finger domain is the site of initial cleavage of the protein or because the substitutions alter the conformation of ICP0. The hypothesis that we have not excluded is that ICP0 is itself a protease or that it interacts with an as-yet-unidentified ubiquitin-conjugating enzyme.
ICP0 plays a key role in enabling HSV-1 to overcome innate immune responses to infection. Studies on the role of the CLOCK histone acetyltransferase in productively infected cells suggest that ICP0 is the earliest protein made (12, 13). Other studies have shown that ICP0 plays a critical role in the derepression of viral DNA to enable the expression of post α genes (1, 11). Given its critical role in viral replication, why does ICP0 turn over rapidly at early times after its synthesis?
Over the course of the millennia of its evolution, ICP0 could have easily evolved into a highly stable protein, but it did not. The governing principle in virology is that the properties and functions of viral gene products have evolved to enable viruses to execute their mission with maximum efficiency. To paraphrase this principle, the instability of ICP0 at early times after infection is not the consequence of a random process, but rather it is the result of evolutionary processes that dictated the selection of an unstable ICP0. On the basis of what we currently know of the biology of HSV-1, two processes would benefit from an unstable ICP0.
The least provocative is the hypothesis that a fragment of ICP0 is required to perform an early function, that this polypeptide does not contain the RING finger domain, and that it is generated by the cleavage of ICP0 at early times.
The second hypothesis rests on the evidence that in the latent state HSV is in an equilibrium between total silence and random expression of viral genes (27, 28). Random expression of viral genes has been reported (29). We could envision, therefore, two mechanisms for reactivation of latent virus. The first is massive derepression of all viral genes observed immediately after the excision of ganglia harboring latent virus (30). The second would be the consequence of a random expression of a viral gene, such as that encoding ICP0 to initiate sequential derepression of viral genes. Reactivation of HSV follows specific stimuli—an argument against random expression of a viral gene (1, 30, 31). The accumulation of a miRNA that specifically targets ICP0 mRNA is one mechanism to prevent ICP0 synthesis (32, 33). Rendering newly synthesized ICP0 unstable would serve as additional means to prevent the reactivation of HSV-1 from becoming a totally stochastic process.
ACKNOWLEDGMENTS
This study was supported by a grant from the National Cancer Institute (CA115662).
We thank Saul Silverstein for the gift of the RF mutant.
Footnotes
Published ahead of print 26 February 2014
REFERENCES
- 1.Roizman B, Knipe DM. 2013. Herpes simplex viruses, p 1823–1897 In Knipe DM, Howley PM. (ed), Fields virology, 6th ed. Lippincott/The Williams & Wilkins Co, Philadelphia, PA [Google Scholar]
- 2.Gu H, Roizman B. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. U. S. A. 100:8963–8968. 10.1073/pnas.1533420100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagglund R, Van Sant C, Lopez P, Roizman B. 2002. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U. S. A. 99:631–636. 10.1073/pnas.022531599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boutell C, Sadis S, Everett RD. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841–850. 10.1128/JVI.76.2.841-850.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chelbi-Alix MK, de The H. 1999. Herpesvirus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941. 10.1038/sj.onc.1202366 [DOI] [PubMed] [Google Scholar]
- 6.Gu H, Roizman B. 2009. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 83:181–187. 10.1128/JVI.01940-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134–17139. 10.1073/pnas.0707266104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roizman B, Gu H, Mandel G. 2005. The first 30 minutes in the life of a virus: unREST in the nucleus. Cell Cycle 4:1019–1021. 10.4161/cc.4.8.1902 [DOI] [PubMed] [Google Scholar]
- 9.Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:7571–7576. 10.1073/pnas.0502658102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu H, Roizman B. 2009. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 83:4376–4385. 10.1128/JVI.02515-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou G, Du T, Roizman B. 2013. The role of the CoREST/REST repressor complex in herpes simplex virus 1 productive infection and in latency. Viruses 5:1208–1218. 10.3390/v5051208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalamvoki M, Roizman B. 2010. Interwoven roles of cyclin D3 and cdk4 recruited by ICP0 and ICP4 in the expression of herpes simplex virus genes. J. Virol. 84:9709–9717. 10.1128/JVI.01050-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. U. S. A. 107:17721–17726. 10.1073/pnas.1012991107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Sant C, Lopez P, Advani SJ, Roizman B. 2001. Role of cyclin D3 in the biology of herpes simplex virus 1 ICPO. J. Virol. 75:1888–1898. 10.1128/JVI.75.4.1888-1898.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chee AV, Lopez P, Pandolfi PP, Roizman B. 2003. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J. Virol. 77:7101–7105. 10.1128/JVI.77.12.7101-7105.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mossman KL, Saffran HA, Smiley JR. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 74:2052–2056. 10.1128/JVI.74.4.2052-2056.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harle P, Sainz B, Jr, Carr DJ, Halford WP. 2002. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology 293:295–304. 10.1006/viro.2001.1280 [DOI] [PubMed] [Google Scholar]
- 18.Melroe GT, Silva L, Schaffer PA, Knipe DM. 2007. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: potential role in blocking IFN-β induction. Virology 360:305–321. 10.1016/j.virol.2006.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu H, Poon AP, Roizman B. 2009. During its nuclear phase the multifunctional regulatory protein ICP0 undergoes proteolytic cleavage characteristic of polyproteins. Proc. Natl. Acad. Sci. U. S. A. 106:19132–19137. 10.1073/pnas.0910920106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364. 10.1099/0022-1317-2-3-357 [DOI] [PubMed] [Google Scholar]
- 21.Lium EK, Silverstein S. 1997. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J. Virol. 71:8602–8614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purves FC, Roizman B. 1992. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha22. Proc. Natl. Acad. Sci. U. S. A. 89:7310–7314. 10.1073/pnas.89.16.7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purves FC, Longnecker RM, Leader DP, Roizman B. 1987. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J. Virol. 61:2896–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poon AP, Benetti L, Roizman B. 2006. U(S)3 and U(S)3.5 protein kinases of herpes simplex virus 1 differ with respect to their functions in blocking apoptosis and in virion maturation and egress. J. Virol. 80:3752–3764. 10.1128/JVI.80.8.3752-3764.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith-Donald BA, Roizman B. 2008. The interaction of herpes simplex virus 1 regulatory protein ICP22 with the cdc25C phosphatase is enabled in vitro by viral protein kinases US3 and UL13. J. Virol. 82:4533–4543. 10.1128/JVI.02022-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Panagiotidis C, Silverstein S. 1992. Multimerization of ICP0, a herpes simplex virus immediate-early protein. J. Virol. 66:5598–5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du T, Zhou G, Roizman B. 2013. Modulation of reactivation of latent herpes simplex virus 1 in ganglionic organ cultures by p300/CBP and STAT3. Proc. Natl. Acad. Sci. U. S. A. 110:E2621–E2628. 10.1073/pnas.1309906110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roizman B, Whitley RJ. 2013. An inquiry into the molecular basis of HSV latency and reactivation. Annu. Rev. Microbiol. 67:355–374. 10.1146/annurev-micro-092412-155654 [DOI] [PubMed] [Google Scholar]
- 29.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. 2002. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad. Sci. U. S. A. 99:978–983. 10.1073/pnas.022301899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. U. S. A. 108:18820–18824. 10.1073/pnas.1117203108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du T, Zhou G, Roizman B. 2012. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc. Natl. Acad. Sci. U. S. A. 109:14616–14621. 10.1073/pnas.1212661109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flores O, Nakayama S, Whisnant AW, Javanbakht H, Cullen BR, Bloom DC. 2013. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J. Virol. 87:6589–6603. 10.1128/JVI.00504-13 [DOI] [PMC free article] [PubMed] [Google Scholar]