

ABSTRACT
Purified retroviral Gag proteins can assemble in vitro to form immature virus-like particles (VLPs). By cryoelectron tomography, Rous sarcoma virus VLPs show an organized hexameric lattice consisting chiefly of the capsid (CA) domain, with periodic stalk-like densities below the lattice. We hypothesize that the structure represented by these densities is formed by amino acid residues immediately downstream of the folded CA, namely, the short spacer peptide SP, along with a dozen flanking residues. These 24 residues comprise the SP assembly (SPA) domain, and we propose that neighboring SPA units in a Gag hexamer coalesce to form a six-helix bundle. Using in vitro assembly, alanine scanning mutagenesis, and biophysical analyses, we have further characterized the structure and function of SPA. Most of the amino acid residues in SPA could not be mutated individually without abrogating assembly, with the exception of a few residues near the N and C termini, as well as three hydrophilic residues within SPA. We interpret these results to mean that the amino acids that do not tolerate mutations contribute to higher-order structures in VLPs. Hydrogen-deuterium exchange analyses of unassembled Gag compared that of assembled VLPs showed strong protection at the SPA region, consistent with a higher-order structure. Circular dichroism revealed that a 29mer SPA peptide shifts from a random coil to a helix in a concentration-dependent manner. Analytical ultracentrifugation showed concentration-dependent self-association of the peptide into a hexamer. Taken together, these results provide strong evidence for the formation of a critical six-helix bundle in Gag assembly.
IMPORTANCE The structure of a retrovirus like HIV is created by several thousand molecules of the viral Gag protein, which assemble to form the known hexagonal protein lattice in the virus particle. How the Gag proteins pack together in the lattice is incompletely understood. A short segment of Gag known to be critical for proper assembly has been hypothesized to form a six-helix bundle, which may be the nucleating event that leads to lattice formation. The experiments reported here, using the avian Rous sarcoma virus as a model system, further define the nature of this segment of Gag, show that it is in a higher-order structure in the virus particle, and provide the first direct evidence that it forms a six-helix bundle in retrovirus assembly. Such knowledge may provide underpinnings for the development of antiretroviral drugs that interfere with virus assembly.
INTRODUCTION
The orthoretroviral Gag protein self-associates to form the immature retroviral core during assembly. Gag is a polyprotein consisting of the major domains MA (matrix), CA (capsid), and NC (nucleocapsid), as well as several minor domains that differ among retroviruses. During the late stages of the virus life cycle, retroviral particles bud from the infected cell and maturation takes place. The transition from the immature Gag core to the mature CA capsid is brought about by the retroviral protease (PR)-mediated cleavage of Gag during or soon after budding. Proteolysis of Gag releases its constituent protein domains. The liberated CA protein then reassembles to form the shell of the mature capsid (1, 2).
Both immature Gag assembly and mature CA assembly can be recapitulated in vitro using purified proteins. Mature in vitro assembly has been established using purified CA protein from lentiviruses (3, 4) and alpharetroviruses (5–8). The products of mature assembly can take on a variety of morphologies, including cones, tubes, or smaller polyhedral structures under specific pH and salt conditions. Although the tubes are not outwardly similar to mature capsids, they conserve the CA lattice observed in retroviral protein cores (9, 10). Immature virus-like particles (VLPs) have been successfully assembled using purified Gag-derived proteins from members of the Lentiviridae (1, 11–15), Alpharetroviridae (16–20), Betaretroviridae (21–23), and Gammaretroviridae (24, 25) genera. Immature assembly usually yields spherical particles, with the exception of Mason-Pfizer monkey virus (MPMV), which is able to form tubes under certain conditions (26, 27). VLPs formed by in vitro assembly are structurally comparable to immature virions collected from infected cells (1, 2, 10, 28–30), lacking only the viral membrane.
The immature Gag viral core and the mature CA capsid have a similar protein lattice composed of hexameric subunits. The immature Gag lattice curves into an incomplete sphere in the immature viral core (30–32), while the mature CA lattice closes into a fullerene structure by incorporating 12 pentameric CA subunits into the mature core shell (7, 9, 10, 33). In both the immature and mature lattices, the two independently folding domains of CA, the N-terminal domain (CANTD) and the C-terminal domain (CACTD), are the main constituents of the hexameric lattice subunits, and these same domains also make up the pentameric lattice subunits in the mature capsid (7, 27, 32, 34–37). Although the general lattice geometry is similar in immature and mature retroviral cores, the details of the CA lattice show several differences. First, the spacing between hexamers is 8 nm in the immature lattice and 9 nm in the mature lattice (1, 38). Second, in VLPs and hexamers reconstructed using a variety of techniques, including cryoelectron tomography (cryo-ET) and crystallography, immature CANTD hexamers differ from their mature counterparts both in intrahexameric protein-protein contacts and interhexameric lattice contacts (1, 7, 27, 30, 34–43). For example, the extensive CANTD-CACTD contacts observed in the mature hexamer are not present in the immature hexamer. Also, in the mature hexamer the intrahexamer contacts are mediated by CANTD-CANTD contacts, while interhexamer contacts are formed by CACTD-CACTD interactions. In contrast, the immature hexamer uses primarily CACTD-CACTD for interhexamer interactions and both CACTD-CACTD and CANTD-CANTD interactions for intrahexamer interfaces (27, 30, 34, 36, 37, 40, 44, 45). These differences in protein-protein contacts result in part from intermolecular interactions that are absent from the cleaved CA protein. In the case of Rous sarcoma virus (RSV), mutational studies in Gag, including disulfide cross-linking followed by structural modeling, showed that one such critical interaction takes place between p10, the domain immediately upstream of CA, and CANTD; this interaction serves to stabilize the immature hexamer (17, 20, 46, 47).
The spacer peptide is a minor cleavage product of Gag found in alpharetroviruses and lentiviruses. In RSV Gag, the spacer peptide, known as the SP domain, is located immediately downstream of CA, while in HIV-1 the homologous peptide is called SP1. Kräusslich et al. were the first to propose that HIV-1 SP1 plays a part in forming a transient assembly domain leading to a critical intermolecular interaction during immature assembly (48). This hypothesis was further extended by Accola et al., who proposed that the disruption of a helix spanning the HIV-1 CA-SP1 junction is required for proper mature assembly (49). Later studies using cryo-ET reconstructions of immature RSV and HIV-1 VLP lattices revealed a pillar-like density located at the center of each immature hexamer below the densities assigned to CACTD (32, 35). This pillar-like density was originally hypothesized by Wright et al. to be formed by a six-helix bundle (6HB) that contributes to the stability of an immature Gag hexamer (35). For both RSV and HIV-1, secondary structure predictions assign amino acids including and immediately flanking the spacer peptide domain as a single amphipathic helix (32, 49–52). The prediction that sequences downstream of the known structured region of CACTD form an alpha helix is not limited to alpharetroviruses and lentiviruses. In gammaretroviruses, a highly charged region at the very end of CA also has been inferred to form an α-helix (53), although there is no cleaved spacer peptide. Amino acids in this region can be deleted without affecting viral assembly, as long as the deletions allow conservation of the original helical phase.
The nuclear magnetic resonance (NMR) structure of a peptide corresponding to HIV-1 SP1 and eight adjoining amino acids revealed that this region has the ability to form an amphipathic helix under certain conditions, although the biological relevance of this structure has been questioned, as it was solved in the presence of 30% trifluoroethanol (TFE), a helix-stabilizing solvent (50). A more recent study using a different HIV-1 peptide encompassing the last eight amino acids of CA and the N-terminal two-thirds of SP1 showed a clear shift in the circular dichroism (CD) spectrum from unstructured peptide to alpha helix when the peptide concentration was increased to a level comparable to the high local protein concentration found in an assembling virion. The authors interpreted that finding to mean that SP1 acts as a conformational switch that is relatively unstructured until viral assembly increases the local concentration of Gag, at which point SP1 adopts a more helical conformation to form essential interacting surfaces that promote assembly (52).
Previous work using RSV allowed our laboratory to define a similar critical region for immature assembly, starting just downstream of the structured CACTD and extending into SP. Using mutational analyses combined with baculovirus-mediated Gag expression in insect cells, the SP assembly (SPA) domain was found to include the last eight amino acids of CA, all of SP, and the first four residues of NC (51). Although RSV SPA and HIV-1 SP1 share little sequence homology, their proposed structural role as a molecular switch in stabilizing the immature retroviral hexamer is likely to be similar.
In this paper, we mapped the predicted RSV SPA amphipathic helix by mutating single amino acids to alanine or serine and identified residues that may be involved in the putative helical bundle. We also performed biophysical analyses on in vitro-assembled VLPs, unassembled Gag protein, and a peptide encompassing the SPA domain. The data imply that a stable ultrastructural element in the SP region of assembled VLPs self-associates in a concentration-dependent manner into a hexamer. Based on these data, we have modeled the SPA helix onto an existing 6HB structure and have shown that the residues critical to immature assembly, as identified by our mutational studies, appear able to interact with each other. These findings lend strong support for the role of 6HB downstream of the structured CACTD in the formation of the immature viral core.
MATERIALS AND METHODS
DNA constructs.
All single-amino-acid mutations were made using site-directed mutagenesis and the previously described ΔMBD.ΔBlpI.pET3xc construct (47), which was derived from the ΔMBD.pET3xc plasmid (17). The mutations D472A-I475A were made by replacing the BlpI/SacII fragment of ΔMBD.ΔBlpI.pET3xc with the products of site-directed mutagenesis. A476S-A478S mutations were made by replacing the BlpI/KpnI fragment using two-step PCR products containing the desired mutation. M479A-N492A mutations were made by replacing the SacII/KpnI fragment with products of site-directed mutagenesis. All PCR products were amplified using ΔMBD.ΔBlpI.pET3xc as the template. All final constructs used for protein expression were confirmed by sequencing.
Protein purification, in vitro assembly, and electron microscopy (EM).
Single-amino-acid mutants of ΔMBDΔPR were expressed and purified as previously described (47). Concentrations of the purified proteins were determined by spectrophotometric measurements using either a Pharmacia Biotech Ultrospec 2000 or a Thermo Scientific NanoDrop 2000. When concentrations were equal to or above 5 mg/ml, the protein was assembled by dilution to 1 mg/ml in 50 mM morpholineethanesulfonic acid (MES), pH 6.5, after additions of 10% (wt/wt) of the single-stranded DNA (ssDNA) GT50 (a 50mer oligonucleotide composed of 25 GT repeats). Assembly reactions were complete after incubating at room temperature for 15 min. When the protein concentration was below 5 mg/ml, assembly was performed by first diluting the protein to 1 mg/ml in 50 mM MES, 100 mM NaCl, pH 6.5. GT50 was added (10%, wt/wt), and 50 μl of the protein-oligonucleotide mix was dialyzed overnight at 4°C against 50 mM MES, 100 mM NaCl, pH 6.5. Ten microliters of each assembly reaction was spotted onto a carbon-Formvar-coated copper grid for 1 min and then stained using a 2% uranyl acetate solution for 1 min. The samples were examined using a Morgagni 268 transmission electron microscope.
Hydrogen-deuterium exchange.
In order to isolate assembled ΔMBDΔPR (hereafter referred to as ΔMBD) particles, assembly solutions were pelleted at 5,000 × g for 5 min and resuspended to a concentration of ∼1 mg/ml in assembly buffer (37.5 mM MES, 100 mM NaCl, pH 6.5). Monomeric proteins were also diluted to 1 mg/ml (in 37.5 mM MES, 500 mM NaCl, pH 7.5) prior to the exchange experiments. All exchange experiments were performed in a randomized, automated manner with a LEAP HDX robot (LEAP Technologies, Carrboro, NC). To initiate hydrogen/deuterium (H/D) exchange, 2 μl of assembled or monomeric ΔMBD samples were diluted into 20 μl of deuterated buffer (37.5 mM MES, pDmeasured 6.12) containing 100 mM NaCl for assembled samples and 500 mM for monomeric samples (to prevent assembly during the exchange period) at 20°C. Following a set period of exchange, the exchange reaction was quenched via the addition of formic acid (1% final concentration) at 1°C prior to digestion with pepsin (1 mg/ml) for 2 min. Digested samples were then separated on a Synergi Fusion-RP column (50 by 2 mm, 1.5-μm volume) with a 15-min gradient (at increments of 50 μl/min; 0 to 70% acetonitrile [ACN], 0.1% formic acid) and electrosprayed into a 7T LTQ-FT mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Exchanged peptides were identified via exact mass measurements for the H/D exchange experiments based on peptide identifications determined by tandem mass spectrometry (MS/MS) sequencing of nonexchanged peptides.
The level of deuterium incorporation at each time point of exchange was determined by calculating the centroid from the isotopic distribution for each peptide with the HD Desktop software package (54). Exchange plots were then plotted for the measured values of deuteron incorporation versus time to allow for comparisons to be made between peptides from the monomeric and assembled ΔMBD samples.
Peptide synthesis and circular dichroism.
The 28- or 29-amino-acid SPApep, which encompasses residues P469 to D496 of RSV Gag with or without an exogenous cysteine, was purchased from the Tufts University Core Facility. The peptide was synthesized using FastMoc chemistry on an ABI 431 peptide synthesizer and further purified by high-performance liquid chromatography (HPLC). For TFE-dependent structural studies, the purified, lyophilized peptide was resuspended in distilled H2O to a concentration of 10 mg/ml. The peptide concentration was calculated by measuring absorbance at 205 nm and using an extinction coefficient of 31 for 1 mg/ml (55). SPApep was diluted into 10 mM sodium phosphate buffer, pH 6.5, 0 to 30% TFE just prior to recording the CD spectrum. Data were collected in the region of 190 to 250 nm with a 1-mm-path-length Hellma quartz cell and an AVIV 202 CD spectrometer (25°C, 1-nm wavelength step, 10-s averaging time). For concentration-dependent studies in aqueous buffer, SPApep and SPAmut, the 28-amino-acid peptide bearing the I475A mutation, were diluted into phosphate-buffered saline (PBS) and measured using a Jasco J-810 CD spectrometer (25°C or 37°C, 1-nm wavelength step, 8-s averaging time) and a 1-mm-path-length Hellma quartz cell. For CD measurements at high peptide concentrations (10 mg/ml), a quartz cell with a shorter path length (0.1 mm) was used.
The percent helicity was calculated using an Excel version of PEPFIT, generously provided by Michael Amon (56, 57). For each CD spectrum, the best fit was obtained by selecting a counter setting of 4 for the initial round of analysis and eliminating reference curves that do not contribute to the spectrum. A second round of fitting was performed using a counter setting of 1. For each peptide concentration, the percent α-helix value was taken from the best fit.
Model of six-helix bundle.
The model was built using Swiss PDB Viewer. The raw amino acid sequence of the 28-residue SPApep (without the exogenous cysteine) was imported into Swiss PDB Viewer. The program automatically assigned residues L470 to R495 as α-helical and showed SPApep as a single helix. The SPApep helix was spatially aligned with each individual chain of CChex (Protein Data Bank [PDB] code 3R47) using the Iterative Magic Fit tool, taking all atoms into consideration for the initial alignment. The six resulting SPApep helices were then subjected to energy minimization three times to minimize side chain clashes.
Analytical ultracentrifugation.
Lyophilized SPApep was dissolved in PBS to a final concentration of 11 mg/ml. Dilutions of 1, 5, and 10 mg/ml were prepared from the stock solution with a total volume of 400 μl per concentration. Centrifugation was performed with epon centerpieces (Spin Analytical) and sapphire windows. The transmittance of light through the sapphire windows in the far-UV spectrum was confirmed prior to performing the experiment. In each centrifuge cell, 380 μl of each SPApep dilution and 400 μl of reference (PBS) were combined. Centrifugation was performed at 40,000 rpm in an An60-Ti rotor in a Beckman-Coulter XL-A analytical ultracentrifuge, monitoring sedimentation of the 1, 5, and 10 mg/ml samples at 240, 245, and 248 nm, respectively. Scans were collected every 4 min. The data were analyzed using Peter Schuck's SEDFIT (www.analyticalultracentrifugation.com) using the c(s) and c(s,ff0) models. The density and viscosity of PBS were estimated with SEDNTERP (http://bitcwiki.sr.unh.edu/index.php/Main_Page) to be 1.0053 g/ml and 0.010189 P, respectively. The partial specific volume of SPApep was estimated from the peptide sequence to be 0.741 ml/g using SEDNTERP.
Secondary structure prediction.
Secondary structure predictions were performed using Psipred (http://bioinf.cs.ucl.ac.uk/psipred/). The Gag sequence spanning the N terminus of CA to the first cysteine of the first zinc knuckle of NC was selected and analyzed for at least one species from each retroviral genus.
RESULTS
Single-amino-acid mutations in the RSV SPA domain.
To guide structural studies of the RSV SPA domain, a string of amino acids, including SP and adjoining sequences, was submitted to PsiPred (http://bioinf.cs.ucl.ac.uk/psipred/) for secondary structure prediction. Similar to previously reported results (51), 21 residues, including the C-terminal five residues of CA, all of SP, and the N-terminal four residues of NC, were predicted to form an α-helix (Fig. 1A). We have termed this predicted structure the SPA helix. The region surrounding SP and its role in immature assembly have been studied previously using five- or two-amino-acid insertion mutations coupled with baculovirus-mediated expression of Gag in insect cells (51), that study showed the entire domain to be indispensable for assembly and defined the boundaries of the SPA domain.
FIG 1.

RSV SP predicted helix and point mutations. (A) Schematic diagram of the RSV SP predicted helical sequence. Each residue within the predicted helix was mutated to alanine or serine. All mutations were made within ΔMBDΔPR, a truncated version of RSV Gag that contains a deletion of the membrane-binding domain (MBD) of MA and of the PR domain. (B to D) Examples of observed in vitro assembly using negative-stain EM. ΔMBDΔPR, wild-type protein (control); A478S, assembly-negative mutant; M488A, assembly-positive mutant. Scale bars are 500 nm.
Our current interest is the structural contribution of the predicted SPA helix to the immature core. More specifically, we wanted to know if the SPA helix is able to form a 6HB. To further probe the structural function of the SPA helix, we performed alanine scanning mutagenesis on its 21 constituent residues (existing alanines were mutated to serines). Each single-amino-acid mutation was made in a well-characterized, truncated version of Gag known as ΔMBDΔPR (Fig. 1A) that has been previously shown to assemble robustly in vitro and to form VLPs nearly identical to wild-type immature virions (2, 16, 17, 47, 58). The mutant proteins were expressed in Escherichia coli, purified, and placed in assembly conditions in the presence of a 50-nucleotide (nt) DNA oligonucleotide. Negative-stain transmission electron microscopy (TEM) was used to evaluate the ability of the proteins to assemble. The mutants were scored as either positive or negative for assembly based on the presence or absence of VLPs, and each protein purification and assembly was performed at least twice.
Table 1 summarizes the results of this mutagenesis. Assembly-positive mutants were defined as mutations that resulted in at least five VLPs per EM grid. Assembly of the wild-type protein ΔMBDΔPR was used as a control and invariably resulted in many VLPs per field (Fig. 1B). The assembly-negative phenotype usually appeared as large regions of protein aggregation with no sign of VLPs (representative example is shown in Fig. 1C). Some assembly-positive mutants could assemble at near wild-type efficiency, but more often they produced fewer particles than ΔMBDΔPR assembly (Fig. 1D). None of the mutations resulted in intermediate morphology, for example, tubular assembly.
TABLE 1.
Mutations in the SP predicted helix and their effects on in vitro assemblya
| Mutation | Assembly |
|---|---|
| D472A | + |
| Q473A | + |
| G474A | + |
| I475A | − |
| A476S | − |
| A477S | + |
| A478S | − |
| M479A | − |
| S480A | + |
| S481A | + |
| A482S | − |
| I483A | − |
| Q484A | − |
| P485A | − |
| L486A | − |
| I487A | − |
| M488A | + |
| A489S | − |
| V490A | − |
| V491A | + |
| N492A | + |
ΔMBDΔPR containing point mutations was assembled in vitro. Mutations that did not affect assembly are marked as plusses, while mutants that abrogated assembly are marked as minuses.
Residues around and including RSV SP form an amphipathic helix when plotted into a helical wheel (32). We generated a helical wheel using the predicted SPA helix sequence and found that this longer sequence also forms an amphipathic helix with a large hydrophobic face and a smaller hydrophilic face (Fig. 2A). With the exception of D472A (the first residue on the helical wheel), all of the mutations that did not disrupt assembly (indicated by asterisks) are located on the hydrophilic face of the helix. Furthermore, it appears that every residue on the hydrophilic face except Q484 can be mutated without compromising assembly. This finding is in agreement with the idea that the SPA helix contributes to immature assembly by forming an amphipathic helix that mediates Gag-Gag interaction through hydrophobic contacts.
FIG 2.
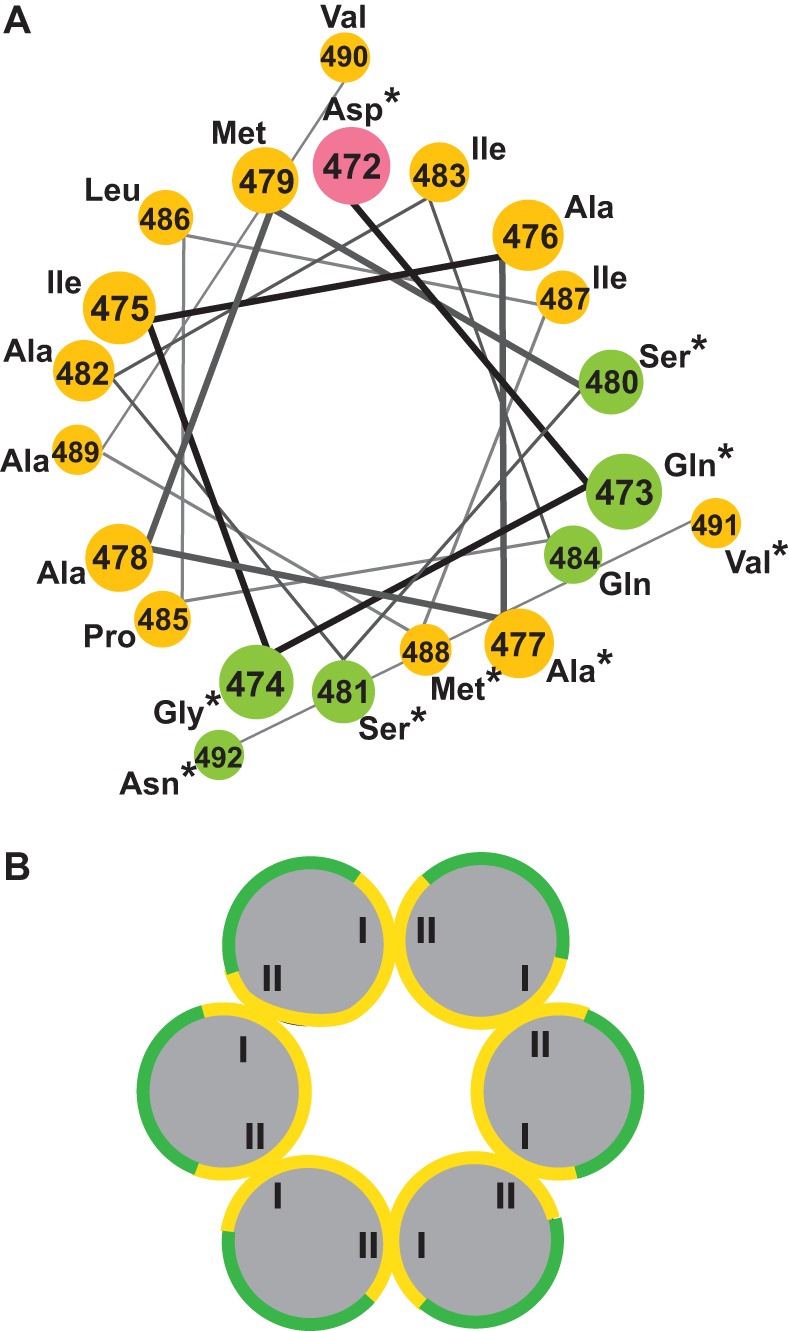
Model of the RSV SPA amphipathic helix and putative six-helix bundle. (A) A helical wheel representation of the predicted SP helix (enumerated according to each residue's position within full-length RSV Gag). Polar, uncharged residues are green, negatively charged residues are pink, and nonpolar/hydrophobic residues are yellow. Residues indicated by asterisks can be mutated without abrogating in vitro assembly. (B) The proposed organization of the putative six-helix bundle. The Roman numerals I and II designate opposite but putatively interacting sides of the large hydrophobic face.
Hypothetical model of putative RSV SPA six-helix bundle.
When RSV VLPs are studied using electron microscopy, the immature Gag hexamer appears to consist of six Gag molecules interacting laterally (32). This arrangement is a conserved property of Gag assembly and has been confirmed for other retroviruses that have been studied at this level, notably MPMV and HIV-1 (27, 30–32, 35, 42, 59, 60). As the results from our mutations indicate that the SPA helix can form an amphipathic helix in immature assembly, we hypothesized that six of these helices organize into a parallel six-helix bundle, as originally suggested for HIV-1 by Wright et al. (35), and that opposing sides of the large hydrophobic face are involved in contacts that tie the bundle together (Fig. 2B; opposing sides are designated I and II). In this model, the hydrophilic face of each helix is oriented toward the outside of the helical bundle; therefore, it is not involved in interhelix contacts, explaining its lack of sensitivity to mutation.
Hydrogen-deuterium exchange on ΔMBD protein and VLPs.
The data from our point mutations suggest that the SPA helix is capable of forming homotypic interactions with neighboring SPA helices in the context of an assembled RSV VLP. To further probe the structural contribution of the SPA helix in assembly, we used hydrogen-deuterium exchange mass spectrometry (HDX MS) to examine this segment of Gag, comparing free ΔMBDΔPR protein to assembled VLPs. HDX MS allows the monitoring of protein structure and dynamics by measuring the rates of exchange of protein backbone amide hydrogens with hydrogens from the solvent. By diluting protein into a deuterated buffer, these exchange events result in a 1-Da mass shift (H to D) for each exposed amide, which is readily detectable with mass spectrometry. The rate of this exchange is dependent on secondary, tertiary, and quaternary structure. H atoms in regions of a protein involved in more stable folds and interactions will exchange more slowly than H atoms in regions that form less stable structural elements. While this technique cannot provide explicit structural detail, comparisons of exchange patterns provide valuable insight into the dynamics, stability, and intermolecular interfaces of protein assemblies, especially in those cases where the atomic-level structure of a protein is unknown.
We compared the HDX profiles of the RSV ΔMBD protein in unassembled and assembled (VLP) forms. The exchange plots illustrate notable variations in assembly across the two states. Several peptides arising from helices 1 to 3 in the CANTD and helices 8 and 9 in the CACTD showed reduced exchange in the assembled state (data not shown), consistent with current models of the intermolecular interfaces present in the capsid lattice. This reduced rate of exchange is very similar to the level of protection observed by HDX of HIV-1 VLPs (61). Most important, comparisons of peptides from the SP domain showed very high levels of protection following assembly (Fig. 3). This level of protection is suggestive of a very stable ultrastructural element and is consistent with the proposed helical bundle motif (32). Although we were not able to find a peptide that spanned the entire length of the SPA helix, we were able to examine two relevant peptides: a longer fragment spanning residues 477 to 488 (AAMSSAIQPLIM) and a shorter one including residues 482 to 488 (AIQPLIM). Both showed very similar exchange and protection patterns (Fig. 3). This result indicates that the structural stability afforded by the assembled lattice entails the complete SP domain and not just the N- or C-terminal portion. Furthermore, the low level of protection of the same region observed in the unassembled protein suggests that this region is unstructured before assembly, consistent with previously published structures of CA or CA fragments that showed an unstructured coil downstream of CACTD helix 11 (6, 62, 63).
FIG 3.
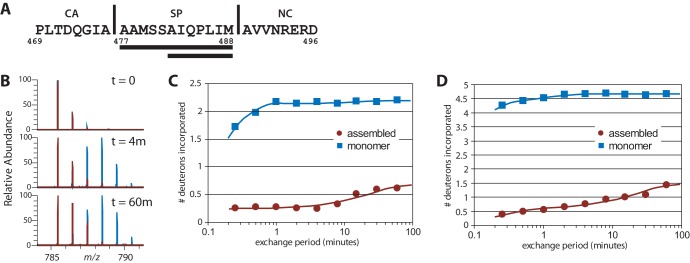
Hydrogen-deuterium exchange analysis of unassembled and assembled ΔMBDΔPR. (A) SP predicted helix sequence. The two peptic fragments corresponding to regions in RSV SP are underlined. Vertical lines correspond to natural protease cleavage sites in Gag. (B) Mass-to-charge spectrum for the peptide of residues 477 to 488 in unassembled ΔMBDΔPR protein (blue) and assembled VLP (red). (C) Plot of deuterium incorporation in the peptide of residues 482 to 488. (D) Plot of deuterium incorporation in the peptide of residues 477 to 488 at different time points.
Circular dichroism spectroscopy of SPApep and SPAmut.
To investigate the mechanism underlying the structural transformation of the SPA helix during assembly, we performed CD spectroscopy on a 29-amino-acid peptide termed SPApep (PLTDQGIAAAMSSAQIPLIMAVVNRERDC). It includes the last eight residues of CA, all of SP, the first eight residues of NC, and an extra C-terminal ectopic cysteine residue for potential future labeling experiments. Preliminary experiments (Fig. 4A) in the presence and absence of 1 mM tris(2-carboxyethyl)phosphine (TCEP; a reducing agent) revealed no difference in the CD spectra. Therefore, we inferred that the additional cysteine residue does not interfere with peptide structure (data not shown).
FIG 4.

Circular dichroism spectroscopy of SPApep. (A) Amino acid sequence of SPApep. Residues are numbered based on their position in full-length RSV Gag. The exogenous C-terminal cysteine is shown in red. (B) Spectra of SPApep in PBS at various concentrations, measured at 25°C. A comparison of the spectra of SPApep and SPAmut at the highest concentrations, measured at 37°C, is shown in the inset. (C) Quantification of the percent helicity of each spectrum shown in panel B. (D) Spectra of SPApep at 0.20 mM concentration with increasing concentrations of TFE. (E) Quantification of the percent helicity of each spectrum shown in panel D. (F) Spectra of SPApep at 0.033 mM with increasing concentrations of TFE. (G) Quantification of the percent helicity of each spectrum shown in panel F.
We measured the CD spectra of SPApep at various concentrations in aqueous buffer, from 0.033 mM (0.1 mg/ml) to 3.3 mM (10 mg/ml). Previously published work showed that the CD spectra of a similar peptide based on the HIV-1 SP1 region became more helical as peptide concentration was increased (52). Due to the structural similarity between HIV-1 and RSV VLPs (2, 32), we predicted that SPApep should exhibit similar behavior when its concentration is increased. Indeed, the CD spectrum of SPApep began to shift from coil to helix at 1.6 mM (5 mg/ml) and became significantly more helical at 3.3 mM (Fig. 4B and C). At 3.3 mM, a noticeable maximum at 197 nm and local minima at 206 nm and 222 nm appeared in the spectrum, with the calculated helicity of the peptide increasing to 23% (Fig. 4C). SPApep became more α-helical at a concentration similar to what was previously reported using the HIV-1 SP1 peptide (52). Our finding suggests that the SPA helix region becomes more structured within an assembling virus particle, where the local Gag concentration might reach values around 4 mM, as roughly calculated using an average immature viral diameter of 129 nm and 2,500 Gag proteins per virion (2). To definitively rule out any contribution of cysteine cross-linking leading to unwanted nucleation, we repeated the CD measurements using a version of SPApep that did not contain the exogenous cysteine and saw nearly identical CD spectra for the cysteine-free peptide (data not shown).
The structure of peptides can be very different in isolation compared to the structure that the same sequence may adopt as part of a full-length protein. To validate our hypothesis that the concentration-driven increase in helicity in SPApep is biologically relevant, we measured the CD spectra of a 28-amino-acid mutant peptide, SPAmut (PLTDQGAAAAMSSAIQPLIMAVVNRERD), bearing the I475A point mutation shown to abrogate assembly using ΔMBDΔPR in vitro assembly. Upon dilution of the lyophilized SPAmut into PBS to a concentration of 10 mg/ml, SPAmut formed a thick gel. Upon further dilution or increasing temperature, SPAmut became more soluble but remained highly viscous. We did not observe this phenomenon with SPApep even at the highest concentrations. Therefore, it was immediately obvious that SPAmut forms a different secondary structure than SPApep and likely forms additional intermolecular interactions in solution to form a peptide gel. We performed CD on SPAmut and SPApep at 37°C and saw a clear difference in the structures of the two peptides at the highest concentration. While SPApep adopted a more helical conformation, as expected, SPAmut took on a beta-sheet-like conformation with a negative band near 220 nm (Fig. 4A, inset).
To further probe the concentration-dependent structural transition of SPApep, we analyzed the conformation of SPApep in the presence and absence of trifluoroethanol (TFE) with various peptide concentrations (Fig. 4D to G). It had been shown that another peptide that includes HIV-1 SP1 takes on an amphipathic α-helical structure in 30% TFE, as determined by NMR (50). Although TFE is often used as a helix-stabilizing solvent in NMR and CD studies (64), it can induce nonnative α-helical structures (65). This becomes particularly relevant with peptides, where nonlocal interactions that stabilize the native protein conformation are nonexistent (66–68). We diluted SPApep into solvent containing 0% to 30% TFE to a final concentration of either 0.033 mM (Fig. 4F and G) or 0.20 mM (Fig. 4D and E) and measured the CD spectra. At 0.033 mM, SPApep showed significantly increased helicity in 20% TFE, which is lower than the concentration used to induce helix formation in the aforementioned HIV-1 SP1 study. At a peptide concentration of 0.20 mM, a clear coil-to-helix transition took place in 15% TFE. In summary, these results reveal that with SPApep, both the increase in peptide concentration and addition of TFE can independently induce helix formation, and that these two variables have additive effects.
Analytical ultracentrifugation of SPA peptide.
We employed sedimentation velocity analytical ultracentrifugation experiments to directly probe SPApep oligomerization in solution. Using a one-dimensional c(s) fit, the sedimentation coefficient distributions for SPApep at 1, 5, and 10 mg/ml showed an increasing weight-average sedimentation coefficient with increasing concentration (Fig. 5A). This behavior is expected for species in rapid equilibrium, suggesting that SPApep does self-associate in solution (69). However, the one-dimensional c(s) fit does not allow us to accurately determine the molecular weights of the sedimenting species, as it uses a weight-average diffusion coefficient to perform the fit. To accurately calculate the molecular weight of the fastest sedimenting species, the sedimentation boundaries from the 10 mg/ml sample were fit with a two-dimensional c(s,ff0) model (Fig. 5B). In this model, no assumptions about the diffusion coefficient are made and the data are fit for both sedimentation and diffusion coefficient distributions. Consistent with the one-dimensional fit shown in Fig. 5A, the two-dimensional fit showed that the predominant species in the 10 mg/ml sample has a weight-average sedimentation coefficient of 1.65 S. The weight-average molecular mass of this species was determined to be 17.9 kDa, very close to the expected mass of the six-helix bundle derived from SPApep (17.8 kDa), providing strong evidence that SPApep does readily form a six-helix bundle in solution.
FIG 5.
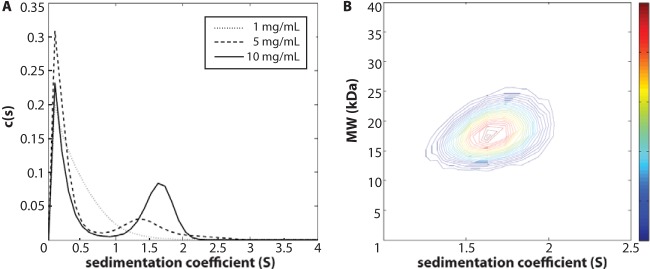
Analytical ultracentrifugation. (A) c(s) distributions of SPApep at 1, 5, and 10 mg/ml loading concentrations. An increasing weight-average sedimentation coefficient with increasing concentration is consistent with SPApep existing in rapid equilibrium. (B) Two-dimensional c(s,M) fit for the 10 mg/ml loading concentration SV data. The peak s-value was found at 1.65 S, consistent with the one-dimensional fit.
Homology model of SPA peptide into six-helix bundle.
Currently, three parallel, homotypic 6HBs have been described at atomic resolution. The first 6HB was discovered and solved as part of a 3.2-Å X-ray structure of the cucumber mosaic virus (CMV) capsid (PDB code 1F15) (70). The second 6HB forms the central pore of a hexameric tyrosine-coordinated heme protein (HTHP) from Silicibacter pomeroyi and is part of a 2.5-Å X-ray structure (PDB code 2OYY) (71). The most recent 6HB comes from the assembly of a synthetic peptide named CChex, which was designed with the goal of engineering protein pores (PDB code 3R3K) (72). The constituent helices in the CMV 6HB each have 17 residues, those in the HTHP 6HB have 21 residues, and those in CChex have 32 residues. We selected CChex as the reference structure for our homology model, as it is the longest characterized 6HB and can accommodate the entirety of the putative SPA helix within its defined structure. We then aligned the cysteine-free SPApep onto the structure of CChex using the Iterative Magic Fit tool in Swiss PDB Viewer (http://www.expasy.org/spdbv/) to generate a model of what the SPApep 6HB might look like. Briefly, the Iterative Magic Fit tool superimposed all of the atoms of the 28-amino-acid (aa)-predicted SPApep helix onto the 32-aa CChex reference helix, resulting in a slightly offset structural alignment between the two helices where Gly1 of CChex is aligned to Leu2 of SPApep.
Figure 6 shows our model of the putative SPApep 6HB next to a ribbon representation of the atomic-level model of CChex. As expected, the two structures are highly similar, with a seam of interacting residues along the length of each set of adjacent helices (shown in stick format). The putative interacting residues in the modeled SPA 6HB are mostly hydrophobic, with the exception of a predicted salt bridge at the base of the 6HB between the R493 and E494 side chains (Fig. 6A). The Swiss PDB Viewer model of RSV SPApep is also organized similarly to the predicted model of the RSV 6HB shown in Fig. 2B. Residues that do not tolerate mutations are either found inside the helical bundle or mediate contacts between helices. Mutable residues do not appear to contact neighboring helices, while the I475A mutation appears to eliminate a hydrophobic contact with the neighboring helix (Fig. 6A). Each individual helix interacts with two adjacent helices using two opposing sides of the helix, creating the same heterotypic I-II interaction illustrated in Fig. 2B around the entire 6HB. In summary, despite using a de novo approach to model the putative RSV 6HB, the result corresponds neatly to the model of a parallel 6HB hypothesized based on our point mutations.
FIG 6.

Model of the putative SPA 6HB. (A) The SPApep 6HB (green ribbon) was modeled using Swiss PDB Viewer. The model was built based on structural alignments with the published crystal structure of a synthetic 6HB, CChex (pink ribbon). Putative interacting residues are shown in stick format and are colored according to amino acid type (yellow, hydrophobic; red, negatively charged; blue, positively charged). A seam of hydrophobic residues lines the interface between two adjacent helices. There is also a predicted salt bridge between residues R493 and E494. (B) Top-down view of the putative SPA 6HB (green ribbon) highlighting potential interacting residues. The top-down view of CChex is shown for comparison.
DISCUSSION
The data presented in this study strongly support the hypothesis that a predicted amphipathic helix overlaps the RSV SPA domain. We identified residues within the SPA helix that are critical for immature assembly and that may be involved in a putative six-helix bundle that promotes immature virus formation. The existence of this helical bundle is supported by results of biophysical analyses, demonstrating that a stable, self-associating, hexameric element is able to take on helical conformation in a concentration-dependent manner in the SPA region of assembled VLPs. Our results strongly support a model of the SPA domain forming a six-helix bundle in immature assembly but do not prove that the model is correct.
Currently, the best-studied immature retroviral assembly systems are those using some variation of the Gag protein from HIV-1, RSV, MPMV, and murine leukemia virus (MLV). Purified Gag-derived proteins from these four retroviruses can assemble in vitro into morphologically similar spherical particles (12, 14–17, 20, 21, 23, 24, 28, 29, 52, 58, 73), although MPMV Gag can be coaxed into tubes bearing an immature lattice (27). In vitro assembly has proven to be a very useful tool for the study of retroviral core structures, as amino acids critical for core formation can be easily identified through site-directed mutagenesis followed by in vitro assembly as the readout. In vitro assembly can also generate VLPs free of cellular debris or contamination from unassembled protein or misfolded aggregates, and these VLPs can be used for cryoelectron microscopy (cryo-EM), HDX MS, or other analyses that require high sample purity.
The morphological similarity between immature VLPs likely is due to the structural homology between CANTD and CACTD in all retroviral CA proteins described so far (74). However, specific contacts that promote immature assembly differ between retroviral species and can be found in the regions immediately adjacent to the CA domain. These have come to be known as conformational switches or molecular switches for immature assembly (28, 47, 52), since their presence seems to constrain the CA domain into its immature conformation. For some retroviruses, including HIV-1, the upstream switch requires nothing more than residues upstream of the CA domain that prevent the formation of a critical salt bridge between P1 and a conserved Asp (D52 in RSV, D51 in HIV-1) of the processed CA protein (75). Disruption to this beta-hairpin through an N-terminal extension results in immature assembly (11). The downstream switch seems to be amino acid specific, possibly due to its role in forming a higher-order structure, as mutations generally lead to disruption of immature assembly (23, 28, 48, 49, 51–53). Although HIV-1 does not have a well-characterized upstream switch, a Gag-derived protein encompassing only CA through NC (CA-NC) assembles into tubes and cones in vitro (3, 10, 18). RSV CA-NC also assembles into tubes in vitro (18). Tubes are generally believed to be reflective of mature assembly, as free CA assembles into tubes (3–5, 11, 76). Similarly, when the downstream switch is deleted or disrupted, HIV-1 and RSV Gag-derived proteins may assemble into tubes and cones both in vitro and in mammalian or insect cell-based expression systems (28, 48, 49, 51, 52, 77). Therefore, having a single upstream or downstream switch is not sufficient to elicit immature assembly.
Perhaps the best-described molecular switches governing mature and immature assembly are found in RSV. The upstream switch consists of 25 amino acids immediately upstream of CA in p10. When p10 is deleted, the mutant Gag assembles in vitro into tubes (17, 20). The switch region was narrowed down to the last 25 amino acids in p10 using a series of truncations (20). In the crystal structure of an N-terminally extended CANTD dimer, six amino acids in p10 make side chain contacts with CANTD (46, 47). These six residues are critical for immature VLP assembly. As shown by disulfide cross-linking analysis, this switch segment of p10 acts as a bridge between neighboring CANTD domains, essentially locking the CANTD domains into a hexamer (47). To date, RSV is the only retrovirus in which a well-defined amino acid sequence upstream of CA has been identified as necessary for immature assembly. Since in HIV-1 CA is directly adjacent to MA, HIV-1 is not believed to require an upstream switch other than the cleavage of CA from MA, allowing the formation of the N-terminal beta-hairpin. Both MPMV and MLV have a 12-kDa cleavage product upstream of CA called p12. MPMV p12 does not appear to be crucial for the formation of immature particles in cells (78); similar to HIV-1, disruption of the N-terminal beta-hairpin suffices for the formation of spherical particles (22), although MPMV p12 has been shown to play a scaffolding role in Gag assembly at low concentrations (78–80). On the other hand, MLV p12 deletion mutants result in tubular rather than spherical assembly, suggesting that MLV p12 interacts with CA in a manner similar to that in RSV (81, 82).
The downstream molecular switch in RSV is the SPA domain. The cleaved spacer peptide between CA and NC is common to both RSV and HIV-1 but is not found in MPMV or MLV. Both RSV and HIV-1 spacer peptides are predicted to be part of a 12th helix in CA downstream of the last (11th) helix of CACTD (Fig. 7), and both are exquisitely sensitive to mutation (28, 48, 49, 51, 52, 77, 83–85). Cryo-ET studies of in vitro-assembled HIV-1 and RSV VLPs showed a stalk-like density underneath the organized CA-CTD lattice which can be fitted with the structure of a parallel six-helix bundle (32, 35). Although MLV Gag does not have a cleaved peptide between CA and NC, the C terminus of MLV CA is predicted to form a highly charged helix, and mutational studies have shown that this charged helix is required for proper immature assembly and infectivity (53). MPMV is also predicted to have a 12th helix spanning CA-NC (Fig. 7), which overlaps a previously characterized immature assembly domain similar to RSV SPA located at the N terminus of the MPMV NC domain (23). Although the recent high-resolution cryo-ET structure of MPMV VLPs did not show a prominent stalk-like density under the CACTD layer, the authors point out a ring of densities adjacent to helix 11 in CACTD (27) which might reflect an alternative higher-order structure involving the predicted helix. In fact, when we aligned the amino acid sequences of representative viruses from every orthoretroviral genus, we found a predicted helix downstream of the structured CACTD across all genera except the Epsilonretroviridae (Fig. 7). In alpha- and lentiviruses, the predicted helix overlaps the C terminus of CA and part or all of the spacer peptide. In beta-, gamma-, and deltaretroviruses, the predicted helix covers the cleavage site between CA and NC. The conservation of this predicted structural element suggests a crucial role in immature assembly, for example, through homologous interactions with other Gag molecules to constrain CA, thereby exposing surfaces favorable for the immature hexamer.
FIG 7.
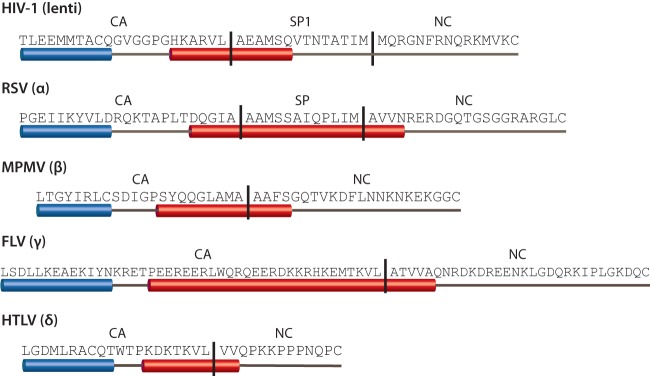
Conserved 12th helix spans the cleavage site between CA and the downstream domain. Sequences show Gag from the last helix of CACTD to the first cysteine in the first zinc finger of NC. The sequences are aligned with one another at the end of the 11th helix of CA (blue cylinder). The putative 12th helix of CA is shown as a red cylinder and is found in all retroviral genera except epsilonretroviruses. Protease cleavage sites (both predicted and known) are indicated by vertical lines.
One proposed structure for the RSV SPA helix is a 6HB. Although parallel, homotypic 6HBs are relatively rare, one naturally occurring 6HB exists in the CMV capsid (70), and another is at the core of HTHP (71). The only example of a free-standing 6HB is from a study of synthetic helical bundles illustrating the fundamental concepts behind helical pore formation (72). Similar to both existing structures, we have modeled the RSV putative SPA helix bundle with hydrophobic interactions on the inside and along helix-helix contacts and with hydrophilic residues on the outside (Fig. 2B and 6). Hydrophobic interactions are known to contribute critical contacts in well-defined α-helical tertiary structures, such as leucine zippers and coiled coils. Our mutation and modeling data suggest that if the SPA domain is a 6HB in immature virions, the interactions that hold the bundle together form a hydrophobic seam along the length of two neighboring helices, similar to previously described 6HBs.
Interestingly, charged residues downstream of the predicted SPA helix also may play a role in assembly. Keller et al. mapped the SPA domain to include the first four residues of NC and excluded the four charged residues that followed (51). However, there are indications that at least a couple of these four residues play a structural role in assembly. The two Arg residues were previously shown to be critical for assembly of a truncated, purified version of ΔMBDΔPR, since they did not tolerate mutation to Ala (16). A similar requirement for at least two basic residues downstream of an SP-like domain was identified in MPMV (23), while in HIV-1, two specific Arg residues in the so-called I domain (localized to the N terminus of the NC domain) are required for proper VLP formation (86). We included these four residues in SPApep to enhance peptide solubility, but it is conceivable that they play a role in stabilizing the higher-order structure of the peptide hexamer. In fact, our Swiss PDB Viewer model of the putative RSV 6HB places R493 and E494 at the seam between two SPApep helices, suggesting that these two residues form an interhelix salt bridge.
What might a 6HB be doing under the organized CA lattice? One hypothesis is that the 6HB is a transient structure that helps stabilize the upstream CA domain into its immature conformation. However, no helix has ever been observed in the region downstream of CACTD, and our data as well as data presented by Datta et al. suggest that helical structure in that region only forms in a concentration-dependent or chemically induced manner (52). Interestingly, the metastable nature of this region may be the key to the function of SPA in RSV assembly and maturation. We speculate that during the early steps of assembly, Gag converges to the plasma membrane, increasing the local concentration and allowing SPA to shift from unstructured to helical. The CA domain is highly flexible due to the floppy CANTD-CACTD linker and may sample many possible conformations during initial dimerization and multimerization. Formation of the 6HB may force the CACTD to expose surfaces favorable for immature assembly, and these downstream spatial constraints could propagate upstream into CANTD. The CANTD hexamer then is locked in place by its interactions with the upstream p10 residues, securely fastening both ends of the Gag hexamer. The 6HB itself likely also has considerable flexibility, which may explain why even at peptide levels approaching virion Gag levels, the time-averaged percent helicity of the SPApep does not exceed 23% (Fig. 4C). This may reflect significant “breathing” of the helix to allow protease to release CA during maturation. The relatively high level of order at the CA-SP junction also may explain why, in both RSV and HIV-1, cleavage of SP and SP1 from CA is the last step in maturation (87, 88).
Can one 6HB functionally replace another? Although the answer is ambiguous, it is heavily weighted toward no. The first experiments that attempted to address this question used an assembly-competent chimeric bovine immunodeficiency virus Gag in which domains downstream of the p3 spacer were replaced with their cognate HIV-1 Gag domains. When p3 was replaced with SP1, virus production was abolished. However, p3 can be replaced by short, five-residue-long sequences derived from the spacer domains from feline immunodeficiency virus, equine infectious anemia virus, and an N-terminal NC fragment from Visna virus without severely compromising virus production and reverse transcriptase activity (85). A similar set of experiments using RSV Gag showed that sequences downstream of the structured CACTD cannot be replaced by homologous sequences from HIV-1 without compromising VLP assembly (51), but when five amino acids at the beginning of the SPA helix are exchanged with seven residues downstream of the structured HIV-1 CA having the highest predicted helical propensity, the Gag chimera assembles in vitro (data not shown). In MPMV, when the SP-like region at the N terminus of NC is replaced by HIV-1 SP1, no virus assembly is observed (23). The amino acid specificity of the spacer region across retroviral genera suggests that there are contacts besides the putative homotypic interhelix interactions that exist between SP and other Gag domains. In fact, using the maturation inhibitor PF-46396, Waki et al. mapped a novel binding pocket formed by the major homology region (MHR) of HIV-1 CA and SP1 and predicted that these two distant regions are in close proximity in an assembled virus (89). This inference is supported by data from Bowzard et al. that showed the rescue of a lethal MHR mutation in RSV CA by a second-site suppressor mutation in SP (90). Given the apparent conservation of an SP-like domain and the absolute conservation of the MHR across all retroviral genera, perhaps this putative interaction surface between SP and the MHR should be further investigated for its role in stabilizing the immature retrovirus.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM107013 to V.M.V. and by NSF grant 1051715 to P.E.P.
Footnotes
Published ahead of print 5 March 2014
REFERENCES
- 1.Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. 2004. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672–675. 10.1038/nsmb785 [DOI] [PubMed] [Google Scholar]
- 2.Briggs JA, Johnson MC, Simon MN, Fuller SD, Vogt VM. 2006. Cryo-electron microscopy reveals conserved and divergent features of gag packing in immature particles of Rous sarcoma virus and human immunodeficiency virus. J. Mol. Biol. 355:157–168. 10.1016/j.jmb.2005.10.025 [DOI] [PubMed] [Google Scholar]
- 3.Gross I, Hohenberg H, Krausslich HG. 1997. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur. J. Biochem. 249:592–600. 10.1111/j.1432-1033.1997.t01-1-00592.x [DOI] [PubMed] [Google Scholar]
- 4.Ehrlich LS, Agresta BE, Carter CA. 1992. Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 66:4874–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purdy JG, Flanagan JM, Ropson IJ, Rennoll-Bankert KE, Craven RC. 2008. Critical role of conserved hydrophobic residues within the major homology region in mature retroviral capsid assembly. J. Virol. 82:5951–5961. 10.1128/JVI.00214-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kingston RL, Fitzon-Ostendorp T, Eisenmesser EZ, Schatz GW, Vogt VM, Post CB, Rossmann MG. 2000. Structure and self-association of the Rous sarcoma virus capsid protein. Structure 8:617–628. 10.1016/S0969-2126(00)00148-9 [DOI] [PubMed] [Google Scholar]
- 7.Cardone G, Purdy JG, Cheng N, Craven RC, Steven AC. 2009. Visualization of a missing link in retrovirus capsid assembly. Nature 457:694–698. 10.1038/nature07724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey GD, Hyun JK, Mitra AK, Kingston RL. 2009. Proton-linked dimerization of a retroviral capsid protein initiates capsid assembly. Structure 17:737–748. 10.1016/j.str.2009.03.010 [DOI] [PubMed] [Google Scholar]
- 9.Li S, Hill CP, Sundquist WI, Finch JT. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407:409–413. 10.1038/35030177 [DOI] [PubMed] [Google Scholar]
- 10.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83. 10.1126/science.283.5398.80 [DOI] [PubMed] [Google Scholar]
- 11.Gross I, Hohenberg H, Huckhagel C, Krausslich HG. 1998. N-Terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J. Virol. 72:4798–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell S, Rein A. 1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 73:2270–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Affranchino JL, Gonzalez SA. 2010. In vitro assembly of the feline immunodeficiency virus Gag polyprotein. Virus Res. 150:153–157. 10.1016/j.virusres.2010.03.012 [DOI] [PubMed] [Google Scholar]
- 14.Crist RM, Datta SA, Stephen AG, Soheilian F, Mirro J, Fisher RJ, Nagashima K, Rein A. 2009. Assembly properties of human immunodeficiency virus type 1 Gag-leucine zipper chimeras: implications for retrovirus assembly. J. Virol. 83:2216–2225. 10.1128/JVI.02031-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alfadhli A, Dhenub TC, Still A, Barklis E. 2005. Analysis of human immunodeficiency virus type 1 Gag dimerization-induced assembly. J. Virol. 79:14498–14506. 10.1128/JVI.79.23.14498-14506.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu F, Joshi SM, Ma YM, Kingston RL, Simon MN, Vogt VM. 2001. Characterization of Rous sarcoma virus Gag particles assembled in vitro. J. Virol. 75:2753–2764. 10.1128/JVI.75.6.2753-2764.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell S, Vogt VM. 1997. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: identification of the p10 domain as a morphological determinant in the formation of spherical particles. J. Virol. 71:4425–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell S, Vogt VM. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 69:6487–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishna NK, Campbell S, Vogt VM, Wills JW. 1998. Genetic determinants of Rous sarcoma virus particle size. J. Virol. 72:564–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi SM, Vogt VM. 2000. Role of the Rous sarcoma virus p10 domain in shape determination of gag virus-like particles assembled in vitro and within Escherichia coli. J. Virol. 74:10260–10268. 10.1128/JVI.74.21.10260-10268.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klikova M, Rhee SS, Hunter E, Ruml T. 1995. Efficient in vivo and in vitro assembly of retroviral capsids from Gag precursor proteins expressed in bacteria. J. Virol. 69:1093–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rumlova-Klikova M, Hunter E, Nermut MV, Pichova I, Ruml T. 2000. Analysis of Mason-Pfizer monkey virus Gag domains required for capsid assembly in bacteria: role of the N-terminal proline residue of CA in directing particle shape. J. Virol. 74:8452–8459. 10.1128/JVI.74.18.8452-8459.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohmova K, Hadravova R, Stokrova J, Tuma R, Ruml T, Pichova I, Rumlova M. 2010. Effect of dimerizing domains and basic residues on in vitro and in vivo assembly of Mason-Pfizer monkey virus and human immunodeficiency virus. J. Virol. 84:1977–1988. 10.1128/JVI.02022-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hadravova R, de Marco A, Ulbrich P, Stokrova J, Dolezal M, Pichova I, Ruml T, Briggs JA, Rumlova M. 2012. In vitro assembly of virus-like particles of a gammaretrovirus, the murine leukemia virus XMRV. J. Virol. 86:1297–1306. 10.1128/JVI.05564-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datta SA, Zuo X, Clark PK, Campbell SJ, Wang YX, Rein A. 2011. Solution properties of murine leukemia virus gag protein: differences from HIV-1 gag. J. Virol. 85:12733–12741. 10.1128/JVI.05889-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nermut MV, Bron P, Thomas D, Rumlova M, Ruml T, Hunter E. 2002. Molecular organization of Mason-Pfizer monkey virus capsids assembled from Gag polyprotein in Escherichia coli. J. Virol. 76:4321–4330. 10.1128/JVI.76.9.4321-4330.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bharat TA, Davey NE, Ulbrich P, Riches JD, de Marco A, Rumlova M, Sachse C, Ruml T, Briggs JA. 2012. Structure of the immature retroviral capsid at 8 A resolution by cryo-electron microscopy. Nature 487:385–389. 10.1038/nature11169 [DOI] [PubMed] [Google Scholar]
- 28.Gross I, Hohenberg H, Wilk T, Wiegers K, Grattinger M, Muller B, Fuller S, Krausslich HG. 2000. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 19:103–113. 10.1093/emboj/19.1.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilk T, Gross I, Gowen BE, Rutten T, de Haas F, Welker R, Krausslich HG, Boulanger P, Fuller SD. 2001. Organization of immature human immunodeficiency virus type 1. J. Virol. 75:759–771. 10.1128/JVI.75.2.759-771.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. 2009. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. U. S. A. 106:11090–11095. 10.1073/pnas.0903535106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Marco A, Muller B, Glass B, Riches JD, Krausslich HG, Briggs JA. 2010. Structural analysis of HIV-1 maturation using cryo-electron tomography. PLoS Pathog. 6:e1001215. 10.1371/journal.ppat.1001215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Marco A, Davey NE, Ulbrich P, Phillips JM, Lux V, Riches JD, Fuzik T, Ruml T, Krausslich HG, Vogt VM, Briggs JA. 2010. Conserved and variable features of Gag structure and arrangement in immature retrovirus particles. J. Virol. 84:11729–11736. 10.1128/JVI.01423-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78:2545–2552. 10.1128/JVI.78.5.2545-2552.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganser-Pornillos BK, Cheng A, Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79. 10.1016/j.cell.2007.08.018 [DOI] [PubMed] [Google Scholar]
- 35.Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 26:2218–2226. 10.1038/sj.emboj.7601664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pornillos O, Ganser-Pornillos BK, Yeager M. 2011. Atomic-level modelling of the HIV capsid. Nature 469:424–427. 10.1038/nature09640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey GD, Hyun JK, Mitra AK, Kingston RL. 2012. A structural model for the generation of continuous curvature on the surface of a retroviral capsid. J. Mol. Biol. 417:212–223. 10.1016/j.jmb.2012.01.014 [DOI] [PubMed] [Google Scholar]
- 38.Briggs JA, Wilk T, Welker R, Krausslich HG, Fuller SD. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22:1707–1715. 10.1093/emboj/cdg143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortuza GB, Dodding MP, Goldstone DC, Haire LF, Stoye JP, Taylor IA. 2008. Structure of B-MLV capsid amino-terminal domain reveals key features of viral tropism, gag assembly and core formation. J. Mol. Biol. 376:1493–1508. 10.1016/j.jmb.2007.12.043 [DOI] [PubMed] [Google Scholar]
- 40.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292. 10.1016/j.cell.2009.04.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. 2009. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790. 10.1016/j.cell.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng X, Zhao G, Yufenyuy E, Ke D, Ning J, Delucia M, Ahn J, Gronenborn AM, Aiken C, Zhang P. 2012. Protease cleavage leads to formation of mature trimer interface in HIV-1 capsid. PLoS Pathog. 8:e1002886. 10.1371/journal.ppat.1002886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. 10.1038/nature12162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, Prevelige PE., Jr 2003. Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J. Mol. Biol. 325:759–772. 10.1016/S0022-2836(02)01245-7 [DOI] [PubMed] [Google Scholar]
- 45.Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M, Prevelige PE., Jr 2004. Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 11:676–677. 10.1038/nsmb790 [DOI] [PubMed] [Google Scholar]
- 46.Nandhagopal N, Simpson AA, Johnson MC, Francisco AB, Schatz GW, Rossmann MG, Vogt VM. 2004. Dimeric Rous sarcoma virus capsid protein structure relevant to immature Gag assembly. J. Mol. Biol. 335:275–282. 10.1016/j.jmb.2003.10.034 [DOI] [PubMed] [Google Scholar]
- 47.Phillips JM, Murray PS, Murray D, Vogt VM. 2008. A molecular switch required for retrovirus assembly participates in the hexagonal immature lattice. EMBO J. 27:1411–1420. 10.1038/emboj.2008.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kräusslich HG, Facke M, Heuser AM, Konvalinka J, Zentgraf H. 1995. The spacer peptide between human immunodeficiency virus capsid and nucleocapsid proteins is essential for ordered assembly and viral infectivity. J. Virol. 69:3407–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Accola MA, Hoglund S, Gottlinger HG. 1998. A putative alpha-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type 1 Gag precursor is crucial for viral particle assembly. J. Virol. 72:2072–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morellet N, Druillennec S, Lenoir C, Bouaziz S, Roques BP. 2005. Helical structure determined by NMR of the HIV-1 (345–392)Gag sequence, surrounding p2: implications for particle assembly and RNA packaging. Protein Sci. 14:375–386. 10.1110/ps.041087605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keller PW, Johnson MC, Vogt VM. 2008. Mutations in the spacer peptide and adjoining sequences in Rous sarcoma virus Gag lead to tubular budding. J. Virol. 82:6788–6797. 10.1128/JVI.00213-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Datta SA, Temeselew LG, Crist RM, Soheilian F, Kamata A, Mirro J, Harvin D, Nagashima K, Cachau RE, Rein A. 2011. On the role of the SP1 domain in HIV-1 particle assembly: a molecular switch? J. Virol. 85:4111–4121. 10.1128/JVI.00006-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheslock SR, Poon DT, Fu W, Rhodes TD, Henderson LE, Nagashima K, McGrath CF, Hu WS. 2003. Charged assembly helix motif in murine leukemia virus capsid: an important region for virus assembly and particle size determination. J. Virol. 77:7058–7066. 10.1128/JVI.77.12.7058-7066.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pascal BD, Chalmers MJ, Busby SA, Griffin PR. 2009. HD desktop: an integrated platform for the analysis and visualization of H/D exchange data. J. Am. Soc. Mass Spectrom. 20:601–610. 10.1016/j.jasms.2008.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scopes RK. 1974. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 59:277–282. 10.1016/0003-2697(74)90034-7 [DOI] [PubMed] [Google Scholar]
- 56.Reed J, Reed TA. 1997. A set of constructed type spectra for the practical estimation of peptide secondary structure from circular dichroism. Anal. Biochem. 254:36–40. 10.1006/abio.1997.2355 [DOI] [PubMed] [Google Scholar]
- 57.Amon MA, Ali M, Bender V, Hall K, Aguilar MI, Aldrich-Wright J, Manolios N. 2008. Kinetic and conformational properties of a novel T-cell antigen receptor transmembrane peptide in model membranes. J. Peptide Sci. 14:714–724. 10.1002/psc.987 [DOI] [PubMed] [Google Scholar]
- 58.Ma YM, Vogt VM. 2004. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J. Virol. 78:52–60. 10.1128/JVI.78.1.52-60.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayo K, Huseby D, McDermott J, Arvidson B, Finlay L, Barklis E. 2003. Retrovirus capsid protein assembly arrangements. J. Mol. Biol. 325:225–237. 10.1016/S0022-2836(02)01176-2 [DOI] [PubMed] [Google Scholar]
- 60.Keller PW, Adamson CS, Heymann JB, Freed EO, Steven AC. 2011. HIV-1 maturation inhibitor bevirimat stabilizes the immature Gag lattice. J. Virol. 85:1420–1428. 10.1128/JVI.01926-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monroe EB, Kang S, Kyere SK, Li R, Prevelige PE., Jr 2010. Hydrogen/deuterium exchange analysis of HIV-1 capsid assembly and maturation. Structure 18:1483–1491. 10.1016/j.str.2010.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853. 10.1126/science.278.5339.849 [DOI] [PubMed] [Google Scholar]
- 63.Momany C, Kovari LC, Prongay AJ, Keller W, Gitti RK, Lee BM, Gorbalenya AE, Tong L, McClure J, Ehrlich LS, Summers MF, Carter C, Rossmann MG. 1996. Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Biol. 3:763–770. 10.1038/nsb0996-763 [DOI] [PubMed] [Google Scholar]
- 64.Buck M. 1998. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 31:297–355 [DOI] [PubMed] [Google Scholar]
- 65.Shiraki K, Nishikawa K, Goto Y. 1995. Trifluoroethanol-induced stabilization of the alpha-helical structure of beta-lactoglobulin: implication for non-hierarchical protein folding. J. Mol. Biol. 245:180–194. 10.1006/jmbi.1994.0015 [DOI] [PubMed] [Google Scholar]
- 66.Thomas PD, Dill KA. 1993. Local and nonlocal interactions in globular proteins and mechanisms of alcohol denaturation. Protein Sci. 2:2050–2065. 10.1002/pro.5560021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hamada D, Kuroda Y, Tanaka T, Goto Y. 1995. High helical propensity of the peptide fragments derived from beta-lactoglobulin, a predominantly beta-sheet protein. J. Mol. Biol. 254:737–746. 10.1006/jmbi.1995.0651 [DOI] [PubMed] [Google Scholar]
- 68.Kuroda Y, Hamada D, Tanaka T, Goto Y. 1996. High helicity of peptide fragments corresponding to beta-strand regions of beta-lactoglobulin observed by 2D-NMR spectroscopy. Fold. Des. 1:255–263. 10.1016/S1359-0278(96)00039-9 [DOI] [PubMed] [Google Scholar]
- 69.Schachman HK. 1959. Ultracentrifugation in biochemistry. Academic Press, New York, NY [Google Scholar]
- 70.Smith TJ, Chase E, Schmidt T, Perry KL. 2000. The structure of cucumber mosaic virus and comparison to cowpea chlorotic mottle virus. J. Virol. 74:7578–7586. 10.1128/JVI.74.16.7578-7586.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeoung JH, Pippig DA, Martins BM, Wagener N, Dobbek H. 2007. HTHP: a novel class of hexameric, tyrosine-coordinated heme proteins. J. Mol. Biol. 368:1122–1131. 10.1016/j.jmb.2007.02.079 [DOI] [PubMed] [Google Scholar]
- 72.Zaccai NR, Chi B, Thomson AR, Boyle AL, Bartlett GJ, Bruning M, Linden N, Sessions RB, Booth PJ, Brady RL, Woolfson DN. 2011. A de novo peptide hexamer with a mutable channel. Nat. Chem. Biol. 7:935–941. 10.1038/nchembio.692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Purdy JG, Flanagan JM, Ropson IJ, Craven RC. 2009. Retroviral capsid assembly: a role for the CA dimer in initiation. J. Mol. Biol. 389:438–451. 10.1016/j.jmb.2009.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Campos-Olivas R, Newman JL, Summers MF. 2000. Solution structure and dynamics of the Rous sarcoma virus capsid protein and comparison with capsid proteins of other retroviruses. J. Mol. Biol. 296:633–649. 10.1006/jmbi.1999.3475 [DOI] [PubMed] [Google Scholar]
- 75.von Schwedler UK, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. 1998. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 17:1555–1568. 10.1093/emboj/17.6.1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barklis E, Alfadhli A, McQuaw C, Yalamuri S, Still A, Barklis RL, Kukull B, Lopez CS. 2009. Characterization of the in vitro HIV-1 capsid assembly pathway. J. Mol. Biol. 387:376–389. 10.1016/j.jmb.2009.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morikawa Y, Hockley DJ, Nermut MV, Jones IM. 2000. Roles of matrix, p2, and N-terminal myristoylation in human immunodeficiency virus type 1 Gag assembly. J. Virol. 74:16–23. 10.1128/JVI.74.1.16-23.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sommerfelt MA, Rhee SS, Hunter E. 1992. Importance of p12 protein in Mason-Pfizer monkey virus assembly and infectivity. J. Virol. 66:7005–7011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Knejzlik Z, Smekalova Z, Ruml T, Sakalian M. 2007. Multimerization of the p12 domain is necessary for Mason-Pfizer monkey virus Gag assembly in vitro. Virology 365:260–270. 10.1016/j.virol.2007.03.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sakalian M, Dittmer SS, Gandy AD, Rapp ND, Zabransky A, Hunter E. 2002. The Mason-Pfizer monkey virus internal scaffold domain enables in vitro assembly of human immunodeficiency virus type 1 Gag. J. Virol. 76:10811–10820. 10.1128/JVI.76.21.10811-10820.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yuan B, Campbell S, Bacharach E, Rein A, Goff SP. 2000. Infectivity of Moloney murine leukemia virus defective in late assembly events is restored by late assembly domains of other retroviruses. J. Virol. 74:7250–7260. 10.1128/JVI.74.16.7250-7260.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee SK, Nagashima K, Hu WS. 2005. Cooperative effect of gag proteins p12 and capsid during early events of murine leukemia virus replication. J. Virol. 79:4159–4169. 10.1128/JVI.79.7.4159-4169.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Craven RC, Leure-duPree AE, Erdie CR, Wilson CB, Wills JW. 1993. Necessity of the spacer peptide between CA and NC in the Rous sarcoma virus gag protein. J. Virol. 67:6246–6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. 2002. Characterization of a putative alpha-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 gag. J. Virol. 76:11729–11737. 10.1128/JVI.76.22.11729-11737.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo X, Hu J, Whitney JB, Russell RS, Liang C. 2004. Important role for the CA-NC spacer region in the assembly of bovine immunodeficiency virus Gag protein. J. Virol. 78:551–560. 10.1128/JVI.78.2.551-560.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sandefur S, Smith RM, Varthakavi V, Spearman P. 2000. Mapping and characterization of the N-terminal I domain of human immunodeficiency virus type 1 Pr55(Gag). J. Virol. 74:7238–7249. 10.1128/JVI.74.16.7238-7249.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, Klein CA, Swanstrom R. 1994. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 68:8017–8027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pepinsky RB, Papayannopoulos IA, Chow EP, Krishna NK, Craven RC, Vogt VM. 1995. Differential proteolytic processing leads to multiple forms of the CA protein in avian sarcoma and leukemia viruses. J. Virol. 69:6430–6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Waki K, Durell SR, Soheilian F, Nagashima K, Butler SL, Freed EO. 2012. Structural and functional insights into the HIV-1 maturation inhibitor binding pocket. PLoS Pathog. 8:e1002997. 10.1371/journal.ppat.1002997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bowzard JB, Wills JW, Craven RC. 2001. Second-site suppressors of Rous sarcoma virus Ca mutations: evidence for interdomain interactions. J. Virol. 75:6850–6856. 10.1128/JVI.75.15.6850-6856.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]