

ABSTRACT
The mechanisms of increased memory CD4+ T cell cycling in HIV disease are incompletely understood but have been linked to antigen stimulation, homeostatic signals, or exposure to microbial products and the inflammatory cytokines that they induce. We examined the phenotype and Vβ family distribution in cycling memory CD4+ T cells among 52 healthy and 59 HIV-positive (HIV+) donors. Cycling memory CD4+ T cells were proportionally more frequent in subjects with HIV infection than in controls, more often expressed CD38 and PD-1, and less frequently expressed OX40 and intracellular CD40L. OX40 expression on memory CD4+ T cells was induced in vitro by anti-CD3, interleukin-2 (IL-2), IL-7, or IL-15 but not by Toll-like receptor ligands. In HIV+ donors, memory CD4+ T cell cycling was directly related to plasma lipopolysaccharide (LPS) levels, to plasma HIV RNA levels, and to memory CD8+ T cell cycling and was inversely related to peripheral blood CD4+ T cell counts but not to the levels of IL-2, IL-7, or IL-15, while in HIV-negative donors, memory CD4+ T cell cycling was related to IL-7 levels and negatively related to the plasma levels of LPS. In both controls and HIV+ donors, cycling memory CD4+ T cells had a broad distribution of Vβ families comparable to that of noncycling cells. Increased memory CD4+ T cell cycling in HIV disease is reflective of generalized immune activation and not driven primarily by cognate peptide stimulation or exposure to common gamma-chain cytokines. This cycling may be a consequence of exposure to microbial products, to plasma viremia, or, otherwise, to proinflammatory cytokines.
IMPORTANCE This work provides evidence that the increased memory CD4+ T cell cycling in HIV infection is not a result of cognate peptide recognition but, rather, is more likely related to the inflammatory environment of HIV infection.
INTRODUCTION
The fundamental mechanisms whereby human immunodeficiency virus (HIV) infection results in progressive immune deficiency are incompletely understood. The progressive depletion of circulating CD4+ T cells is the hallmark of immune deficiency in chronic HIV infection (1). Although infection of CD4+ T cells by cytopathic virus undoubtedly contributes to cellular losses, other mechanisms, such as activation-induced cellular turnover, have been implicated as well (2). Acute HIV infection rapidly depletes the majority of gut effector CD4+ CCR5+ T cells by direct infection (3), yet despite a similar effect on gut CD4+ T cells, simian immunodeficiency virus (SIV) infection of sooty mangabeys is uncommonly associated with systemic CD4+ T cell depletion, despite high-level SIV replication (4). In HIV infection, the CD4+ T cell decline has been linked to persistent immune activation (5) and less so to the magnitude of HIV replication (6). The degree to which immune activation in HIV infection is a consequence of CD4+ T cell cytopenia as opposed to a driver of it is not clear. Nonetheless, pathways of CD4+ T cell cytopenia have been linked to activation mechanisms (7) as well as to impaired homeostatic proliferation (8), cell cycle arrest (9), and cell sequestration (10). Here, we investigated potential mechanisms that may be driving memory CD4+ T cell cycling in HIV infection and found that cycling CD4+ T cells in HIV infection more frequently express antigens of activation (CD38) and exhaustion (PD-1) but less frequently express markers of recent T cell receptor (TCR) or common gamma-chain cytokine engagement (CD40L and OX40). In HIV infection, the cycling frequencies of memory CD4+ T cells are inversely correlated with circulating CD4+ T cell numbers and are directly correlated with the levels of bacterial lipopolysaccharide (LPS) and HIV RNA in plasma but not with the plasma levels of the homeostatic cytokine interleukin-7 (IL-7), which are correlated with CD4+ T cell cycling in healthy controls.
MATERIALS AND METHODS
Study subjects.
This study was approved by the institutional review board at University Hospitals/Case Medical Center. All subjects provided written informed consent in accordance with the Declaration of Helsinki. Plasma samples were prepared from whole blood collected into tubes containing EDTA and were stored at −80°C until they were thawed once for study. The clinical characteristics of the subjects are shown in Table 1. CD4 T cell counts were similar in the antiretroviral therapy (ART)-treated and untreated patients (P = 0.27), but the plasma levels of HIV RNA were higher in the untreated subjects than in the treated subjects (P < 0.04).
TABLE 1.
Clinical characteristics
| Donor group | Total no. of subjects | No. of females/males | Agea (yr) | CD4+ T cell counta (no. of cells/mm3) | Plasma HIV RNA levela (no. of copies/ml) | Duration of ARTa (mo) |
|---|---|---|---|---|---|---|
| HIV− | 52 | 33/19 | 36 (29–44) | |||
| HIV+/ART treated | 29 | 11/18 | 45 (37–48) | 337 (179–551) | 3,660 (<48–103,043) | 163 (76–244.5) |
| HIV+/ART naive | 31 | 15/16 | 46 (45–48) | 438 (301–584) | 33,270 (10,365–97,184) |
Data are medians (interquartile ranges).
Cell preparation.
Peripheral blood mononuclear cells (PBMCs) were isolated over a Ficoll-Hypaque cushion. In some assays, CD45RA-depleted PBMCs were prepared using anti-CD45RA microbeads (AutoMACS; Miltenyi Biotec, Bergisch Gladbach, Germany). CD45RA-depleted cells were examined for purity by flow cytometry (>95% purity) and for viability (>98% viability) by trypan blue staining. PBMCs or CD45RA-depleted PBMCs were subsequently cultured in 96-well plates (0.4 × 106 cells/200 μl) in fresh complete medium with or without anti-CD3 (50 ng/ml; BD, San Jose, CA), human cytomegalovirus (CMV) lysate (1:40 dilution; BioWhittaker, Walkersville, MD), recombinant human IL-2, IL-7, or IL-15 (50 ng/ml; R&D Systems, Minneapolis, MN), or a mixture of microbial elements consisting of bacterial LPS (20 ng/ml, Escherichia coli; Sigma), bacterial peptidoglycan (PGN; 1 μg/ml; Imgenex, San Diego, CA), bacterial CpG oligodeoxynucleotide (ODN2395; 3 μg/ml; Coley Pharmaceutical Group Inc., Wellesley, MA), or recombinant human IL-6 (50 ng/ml; R&D Systems), IL-1β (50 ng/ml, R&D Systems), or alpha interferon (IFN-α; 500 units/ml; PBL Piscataway, NJ) for 20 h. Cellular phenotypes were then evaluated by flow cytometry.
Flow cytometry.
Cells were stained with fluorochrome-labeled monoclonal antibodies at room temperature for 10 min. After surface staining, cells were washed and intracellular staining was performed after permeabilization using Cytoperm/Cytofix reagents (BD) following the manufacturer's protocol. The following antibody conjugates were used: anti-CD4–Pacific Blue (BD), anti-CD8–allophycocyanin (APC)–Cy7 (Biolegend, San Diego, CA), anti-CD45RO–phycoerythrin (PE)–Cy7 (Biolegend), anti-OX40–PE–Cy5 (BD), anti-CD38–APC (BD), anti-CD40L–PE (BD), anti-PD-1–PE (BD), anti-CD27–APC (BD), and anti-Ki67–fluorescein isothiocyanate (FITC; BD). After staining, samples were examined by flow cytometry (MACSQuant; Miltenyi Biotec). Appropriate isotype control antibodies were used where noted.
LPS measurement.
Plasma samples were diluted to 10% with endotoxin-free water and then were heated to 85°C for 12 min to inactivate inhibitory plasma proteins. We then quantified plasma LPS levels with a commercially available Limulus amebocyte lysate assay (Cambrex, Walkersville, MD) according to the manufacturer's protocol. We ran samples in triplicate, and background attributable to the turbidity of the diluted plasma was subtracted.
Plasma levels of IL-2, IL-7, and IL-15.
Commercial enzyme-linked immunosorbent assays were used to measure the plasma concentrations of IL-2 (high sensitivity; eBioscience, San Diego, CA), IL-7 (high sensitivity; R&D Systems), and IL-15 (eBioscience) following the manufacturers' protocols.
Vβ distribution analysis.
Vβ distribution analyses were performed using an IOTest Beta Mark TCR V kit (Coulter, Brea, CA). Frozen PBMCs were thawed in RPMI supplemented with 10% human serum, rested for 1 h at 37°C, and stained with anti-CD4–Pacific Blue (clone RPA-T4; Biolegend), anti-CD3–peridinin chlorophyll protein (clone SK7; BD), APC-H7-CD45RA (clone HI100; BD), rat anti-human CCR7-PE-Cy7 (clone 3D12; BD), and the FITC- and PE-conjugated anti-Vβ antibodies. These antibodies are specific for Vβ families 1, 2, 3, 4, 5.1, 5.2, 5.3, 7.1, 7.2, 8, 9, 11, 12, 13.1, 13.2, 13.6, 14, 16, 17, 18, 20, 21.3, 22, and 23, and together they detect approximately 70% of the circulating T cell repertoire. Cells were fixed, permeabilized (BD), and stained with anti-Ki67–Alexa 647 (clone B56l BD). The use of eight tubes, each containing a mixture of 3 different antibodies conjugated to PE, FITC, or PE and FITC, permitted the simultaneous analysis of 3 Vβ families per tube.
Statistical methods.
The distribution of the percentage of cycling memory CD4+ T cells or the expression levels of various markers were compared between the HIV-negative (HIV−) and HIV-positive (HIV+) groups using the Wilcoxon rank-sum test. Spearman's correlation test was used to explore associations between the percentage of cycling memory CD4+ T cells and the plasma levels of LPS, IL-7, IL-2, and IL-15; peripheral CD4+ T cell counts; or HIV RNA levels. A two-sided significance level of 0.05 was used for each test. To compare the Vβ distributions between Ki67-positive (Ki67+) and Ki-67-negative (Ki67−) cells in a given subject, we performed a Spearman correlation of the expression levels of the Vβ families in our panel in the two subsets. To test this more formally, we also performed a principal component analysis on the expression levels of the Vβ families in our panel and used the first principal component as a measure of the breadth of the Vβ response. Using repeated-measures analysis of variance on the principal component scores (which accounted for 23% of the variability in the Vβ response), we then compared the breadth of the Vβ response between the two subsets within HIV− and HIV+ subjects and tested if the difference in the breadth of the Vβ response between the two subsets differed by HIV status. Analyses were performed using SPSS, v20 (IBM Corp., Armonk, NJ), and Prism software. All tests were two-sided, and P values of ≤0.05 were considered statistically significant.
RESULTS
Increased memory CD4+ T cell cycling in HIV− infection.
Memory CD4+ T cell cycling was measured by determination of the intracellular expression of Ki67, a marker of ongoing or recent cell cycle entry, among CD45RO+ CD4+ T cells. We found that the percentage of cycling memory CD4+ T cells was increased in HIV+ donors compared to the frequencies in HIV− controls (P < 0.0001; Fig. 1); the mean ± standard deviation (SD) percentage of cycling memory CD4+ T cells was 10.9% ± 5.8% and 3.9% ± 2.0% in HIV+ patients and HIV− controls, respectively, confirming earlier reports (5, 7). When patients were divided according to the receipt of antiretroviral therapies, both the treated and untreated patient groups had higher percentages of cycling memory CD4+ T cells than the controls (11.1% ± 5.7% and 10.8% ± 6.1% in ART-naive and ART-treated patient groups, respectively; P < 0.0001 for each compared to the values for the healthy controls). In patients, cycling was increased in memory CD4+ T cells among both CD27high and CD27low cells. Cycling of memory CD8+ T cells was also increased in HIV+ patients compared to the cycling among the controls' memory CD8+ T cells (10.9% ± 5.9% versus 4.5% ± 2.5%, P < 0.0001). There was no difference in the proportions of cycling memory CD8+ T cells in treated and nontreated patients (P = 0.99).
FIG 1.

The percentage of cycling memory CD4+ T cells is increased in HIV+ donors compared to the percentages in HIV− controls. Blood samples were tested for intracellular Ki67 staining by flow cytometry. (A) Gating strategy and representative experiments. SSC, side scatter; FSC, forward scatter. (B) Median (horizontal line) and observed percentages of cycling memory CD4+ T cells in 52 HIV− donors and 59 HIV+ subjects (open circles, 30 ART-treated subjects; solid circles, 29 ART-naive subjects) and CD8+ T cells in 28 HIV− donors and 34 HIV+ subjects (18 ART-treated subjects, 16 ART-naive subjects).
Expression of CD38 and PD-1 is increased in cycling memory CD4+ T cells in HIV+ donors, while OX40 and CD40L expression is diminished.
To gain further insight into the drivers of cycling memory CD4+ T cells in HIV infection, we sought to characterize the phenotype of these cells. Surface expression of OX40 and intracellular CD40L expression have been reported to be reasonably specific markers of recent TCR engagement in CD4+ T cells (11–15). On the other hand, CD38 is a recognized nonspecific marker of T cell activation in HIV disease (16). PD-1 is a marker linked to T cell exhaustion that is associated with impaired T cell function (17) and suboptimal reconstitution of CD4+ T cells after highly active ART (18). We therefore examined the expression of these four markers in cycling memory CD4+ T cells in HIV− controls and HIV+ patients. We found that expression of both OX40 and CD40L on cycling memory CD4+ T cells was reduced in HIV+ donors compared to their expression among controls' cycling CD4+ T cells (Fig. 2A and B); on the other hand, expression of both CD38 and PD-1 on cycling memory CD4+ T cells was increased in the HIV+ patients compared to their expression on the controls' cells (Fig. 2C and D). Thus, cycling memory CD4+ T cells in HIV infection show evidence of increased activation and exhaustion, but the lower level of CD40L and OX40 expression suggests that fewer of these cells are likely to have been activated through recent TCR engagement.
FIG 2.

Cycling memory CD4+ T cells of HIV+ donors less frequently express OX40 and intracellular CD40L but more often express CD38 and PD-1. Blood samples were tested for intracellular Ki67 expression as well as for the surface markers OX40, CD38, and PD-1 and intracellular CD40L by flow cytometry. For each marker, the top figure displays the results of the representative experiment and the bottom figure displays the median (horizontal line) and observed expression levels in HIV− and HIV+ donors. (A) Surface OX40 expression among gated cycling (Ki67+) memory CD4+ T cells from 27 HIV− donors and 24 HIV+ donors; (B) intracellular CD40L expression among gated cycling (Ki67+) memory CD4+ T cells in 23 HIV− donors and 24 HIV+ donors; (C) surface CD38 expression among gated cycling (Ki67+) memory CD4+ T cells in 15 HIV− donors and 19 HIV+ donors; (D) surface PD-1 expression among gated cycling (Ki67+) memory CD4+ T cells in 31 HIV− donors and 38 HIV+ donors. Open circles, ART-treated HIV+ donors; solid circles, ART-naive HIV+ donors.
OX40 expression is induced on memory CD4+ T cells in vitro by TCR stimulation or exposure to IL-2, IL-7, or IL-15 but not by exposure to LPS or the inflammatory cytokines IL-1β, IL-6, and IFN-α.
Although OX40 expression has been linked to recent TCR engagement (14, 15), we next explored the in vitro effects of different activation pathways on the activation phenotype of cycling memory CD4+ T cells using PBMCs obtained from healthy HIV− control donors. PBMCs were stimulated with anti-CD3 or CMV lysates, with selected common gamma-chain cytokines known to drive T cell proliferation, with microbial elements that we had earlier shown could drive memory CD4+ T cells into the cell cycle, and with proinflammatory cytokines that are known to be increased in the circulation or lymphoid tissues of persons with HIV infection. As expected, compared to cultivation in medium alone, anti-CD3 and CMV lysate stimulation increased OX40 expression on memory CD4+ T cells, but so also did the common gamma-chain cytokines IL-2, IL-7, and IL-15 (Fig. 3B). In contrast, bacterial LPS, IFN-α, IL-6, or IL-1β did not increase OX40 expression. As expected, compared to cultivation in medium alone, CD38 expression on cycling memory CD4+ T cells was induced by stimulation through the TCR, by IL-7, and by exposure to microbial elements (Fig. 3C). Thus, OX40 and CD40L are upregulated after T cell activation through TCR stimulation or by exposure to gamma-chain cytokines, whereas CD38 expression can mark T cell activation through a wider range of stimuli in vitro.
FIG 3.

OX40 expression on memory CD4+ T cells is induced by T cell receptor engagement and by IL-2, IL-7, and IL-15 but not by a TLR ligand (LPS) or by proinflammatory cytokines (IL-6, IL-1β, or IFN-α), and induction is not impaired in HIV infection. PBMCs from HIV− control donors were cultured in medium alone (med) or medium supplemented with anti-CD3, CMV lysate, IL-2, IL-7, IL-15, LPS, IL-6, IFN-α, or IL-1β for 20 h. (A) Surface OX40 expression on memory CD4+ T cells was examined by flow cytometry. The results of a representative experiment are shown. (B) Means and SDs of expression levels in 10 different experiments. (C) Surface CD38 expression on memory CD4 T cells after PBMCs from healthy controls (n = 10) were cultivated with medium, anti-CD3, IL-7, or a mixture of microbial elements: LPS, CpG DNA, and bacterial peptidoglycan (see Materials and Methods). P values reflect comparisons between medium alone and each stimulus. (D) CD45RA-depleted PBMCs from 7 HIV− controls and 7 ART-naive HIV+ patients were cultured in medium supplemented with anti-CD3, IL-2, IL-7, or IL-15 for 20 h. The mean ± SD induction of OX40 on CD45RO+ memory CD4+ T cells compared with OX40 expression on cells incubated in medium alone was examined by flow cytometry.
Our earlier work indicated that intracellular CD40L expression by memory CD4+ T cells is impaired in HIV infection after TCR stimulation in vitro (19). To ascertain whether the reduced expression of OX40 on cycling memory CD4+ T cells in HIV+ donors (Fig. 2) might be related to impaired OX40 induction in this setting, we stimulated CD45RA-depleted PBMCs with antibodies to CD3 or with IL-2, IL-7, or IL-15 for 20 h and then measured OX40 surface expression on CD45RO+ CD4+ T cells. OX40 induction by TCR stimulation was comparable in memory CD4+ T cells of HIV− controls and HIV-infected patients (Fig. 3D). Interestingly, induction of OX40 by IL-2, IL-7, or IL-15 tended to be greater in memory CD4+ T cells from HIV+ donors than in those from HIV− controls, but these differences in seven experiments were not statistically significant. These results suggest that the reduced OX40 expression in cycling memory CD4+ T cells in HIV+ donors is not likely due to an impaired OX40 response after in vivo stimulation through the TCR or through exposure to homeostatic cytokines.
LPS levels are related to memory CD4+ T cell cycling in HIV infection, while IL-7 levels are related to memory CD4+ T cell cycling in healthy donors.
We have earlier found that a number of different Toll-like receptor (TLR) ligands can drive memory CD4+ T cells to enter the cell cycle (20). To explore further the possible mechanisms of increased memory CD4+ T cell cycling in HIV infection, we examined the relationships between the percentage of cycling memory CD4+ T cells and the plasma levels of bacterial LPS and the plasma levels of the homeostatic common gamma-chain cytokines IL-2, IL-7, and IL-15. In HIV+ donors, the percentage of cycling memory CD4+ T cells was positively related to the plasma levels of LPS (r = 0.37, P = 0.03; Fig. 4A) but not to the plasma levels of IL-2, IL-7, or IL-15 (P > 0.05; Fig. 4B to D), suggesting that memory CD4+ T cell cycling may be driven (at least in part) by the elevated systemic levels of microbial TLR agonists such as LPS but less so by the common gamma-chain cytokines IL-2, IL-7, and IL-15. These relationships differed in healthy controls, such that the percentage of cycling memory CD4+ T cells was negatively related to the plasma levels of LPS (r = −0.40, P = 0.01; Fig. 4A) and positively related to the plasma levels of IL-7 (r = 0.30, P = 0.05). Interestingly, the percentage of cycling memory CD4+ T cells was positively related to the percentage of cycling memory CD8+ T cells in patients but not in controls (Fig. 4E), suggesting that in patients with HIV disease, but not in healthy subjects, the important drivers of CD4+ and CD8+ T cell cycling may be linked. In this regard and as shown in Fig. 4F and G, the percentage of cycling memory CD4+ T cells was inversely related to circulating CD4+ T cell counts (r = −0.43, P = 0.001) and positively related to the plasma levels of HIV RNA (r = 0.37, P = 0.004). CD4+ T cell data were available only for the HIV-infected patients, and only they were viremic, so the data in Fig. 4F and G are from HIV-infected patients only.
FIG 4.

Among HIV+ donors, the percentage of cycling memory CD4+ T cells is positively related to the plasma levels of LPS and HIV RNA and negatively related to peripheral CD4+ T cell counts but not to the plasma levels of IL-2, IL-7, or IL-15. Among HIV− control donors, the percentage of cycling memory CD4+ T cells is negatively related to the plasma levels of LPS and positively related to the plasma levels of IL-7 but not to the plasma levels of IL-2 or IL-15. (A) The relationship between the percentage of cycling memory CD4+ T cells and the plasma levels of LPS was positive in HIV+ donors (r = 0.37, P = 0.03) but negative in HIV− controls (r = −0.40, P = 0.01). (B) The percentage of cycling memory CD4+ T cells is not related to the plasma levels of IL-7 in HIV+ donors but is positively related to the plasma levels of IL-7 in HIV− controls. (C, D) The percentage of cycling memory CD4+ T cells is not related to the plasma levels of IL-2 (C) or to the plasma levels of IL-15 (D) in HIV− controls or in HIV+ donors. (E) The percentage of cycling memory CD4+ (mCD4+) T cells is directly related to the percentage of cycling memory CD8+ (mCD8+) T cells in HIV+ donors (r = 0.56, P = 0.002) but not in the controls. (F, G) Data for the HIV+ subjects only. The percentage of cycling memory CD4+ T cells is negatively related to peripheral CD4+ T cell counts (r = −0.43, P = 0.001) (F) and positively related to the plasma levels of HIV RNA in HIV+ donors (r = 0.37, P = 0.004) (G). Open circles, ART-treated (ART+) HIV+ donors; solid circles, ART-naive (ART−) HIV+ donors. VL, viral load.
Broad Vβ repertoire on Ki67-positive memory CD4+ T cells.
If cycling is driven by peptide-induced expansion of cells in the setting of chronic infection, one would expect a difference in T cell receptor distributions between cycling and resting cells. On the other hand, as our data suggest that cycling cells in HIV infection tend to have the characteristics of cells expanded by bystander mechanisms, they should have T cell receptor distributions similar to those of resting cells and the Vβ repertoires would be broad. With this in mind, we investigated the Vβ distribution on Ki67-positive and -negative memory CD4+ T cells using a panel of Vβ antibodies. To examine Vβ distributions, we used a commercial panel of antibodies that contains directly conjugated antibodies specific for 24 different Vβ families. These antibodies detect approximately 70% of the normal circulating T cell repertoire.
The Vβ distributions were analyzed on cycling versus noncycling memory CD4+ T cells in 6 healthy donors (Fig. 5). The distributions of Vβ families were similar between the cycling and noncycling populations (Fig. 5b; P = 0.061 on the basis of principal component analysis), indicating that homeostatic or bystander activation rather than antigenic peptide-induced expansion underlies the vast majority of memory CD4+ T cell cycling in healthy donors.
FIG 5.


Vβ distribution on Ki67-positive and -negative memory CD4+ T cells. (a) Healthy donor PBMCs were stained with anti-CD3, anti-CD4, anti-CD45RO, and three anti-Vβ antibodies: Vβ5.3-PE, Vβ7.1 PE-FITC, and Vβ3-FITC (A), Vβ9-PE, Vβ17-PE-FITC, and Vβ16-FITC (B), Vβ18-PE, Vβ5.1-PE-FITC, and Vβ20-FITC (C), Vβ13.1-PE, Vβ13.6-PE-FITC, and Vβ8-FITC (D), Vβ5.2-PE, Vβ2-PE-FITC, and Vβ12-FITC (E), Vβ23-PE, Vβ1-PE-FITC, and Vβ21.3-PE (F), Vβ11-PE, Vβ22-PE-FITC, and Vβ14-FITC (G), and Vβ13.2-PE, Vβ4-PE-FITC, and Vβ7.2-FITC (H). (Top) Distributions among Ki67-negative CD4+ CD45RO+ cells; (bottom) distributions among Ki67-positive CD4+ CD45RO+ cells. (b) Polyclonal distribution of Vβ expression on Ki67-negative and Ki67-positive CD4+ T cells in 6 healthy individuals.
To ensure that we could detect Vβ skewing in the setting of antigen-induced expansion in HIV infection, PBMCs from two ART-treated HIV-infected patients were labeled with the fluorescent tracking dye carboxyfluorescein succinimidyl ester (CFSE) and were stimulated with HIV-1 gag overlapping peptides and with lysates prepared from cytomegalovirus-infected cells, as both CMV and HIV are plausible drivers of cellular expansion in chronic HIV infection (21–24). On day 6, the Vβ distributions of the expanded (CFSE low) and unexpanded (CFSE high) cells were compared. As shown in Fig. 6, both Gag and CMV stimulation induced the selective expansion of Vβ families in each patient.
FIG 6.

Oligoclonal expansion of Vβ families upon antigen-induced stimulation. (Top) PBMCs from two HIV-infected, ART-treated patients (patients 8001 and 4345) were labeled with CFSE and stimulated with CMV lysate or overlapping Gag peptides supplemented with 10 U/ml IL-2. On day 6, cells were stained with anti-CD3, anti-CD4, and anti-Vβ antibodies. NS, nonstimulated. (Bottom) Vβ frequencies among CFSElow and CFSEhigh cells.
These data suggest that even a short exposure to antigens derived from these 2 prevalent viruses can drive selective cycling and expansion of particular Vβ families. We next evaluated the Vβ distributions in Ki67-positive and -negative memory CD4+ T cells in viremic HIV-infected ART-naive patients (Fig. 7). As it was in the healthy controls, the distribution of Vβ families was similar in resting and cycling memory CD4+ T cells in viremic HIV-infected patients (P = 0.60 on the basis of principal component analysis).
FIG 7.


Vβ distributions are similar in resting and cycling memory CD4+ T cells in HIV infection. PBMCs from 10 HIV-infected patients who were not receiving antiretroviral therapies were stained with anti-CD3, anti-CD4, anti-CD45RO, and anti-Vβ antibodies and examined by flow cytometry, as described in the legend to Fig. 6. Shown is the distribution of the Vβ staining on Ki67+ (red bars) and Ki67− (blue bars) populations.
To compare any minimal degrees of skewing between these two patient populations, we estimated the Spearman correlation between the proportions of cycling and noncycling memory CD4+ T cells in each Vβ family in each subject, reasoning that even subtle skewing would decrease the magnitude of the resulting correlation coefficients. As shown in Fig. 8, the correlations between Vβ distributions among cycling and noncycling cells were nearly superimposable in the 6 healthy controls and in the 10 HIV-infected viremic patients (r = 0.8978 and 0.8974, respectively), indicating that in both populations most cycling does not appear to be skewed by antigenic stimulation. Similarly, our principal component analysis also showed that there was no evidence that the difference in the breadth of the Vβ response between cycling and noncycling cells differed between HIV− and HIV+ donors (Fig. 8, P = 0.20).
FIG 8.
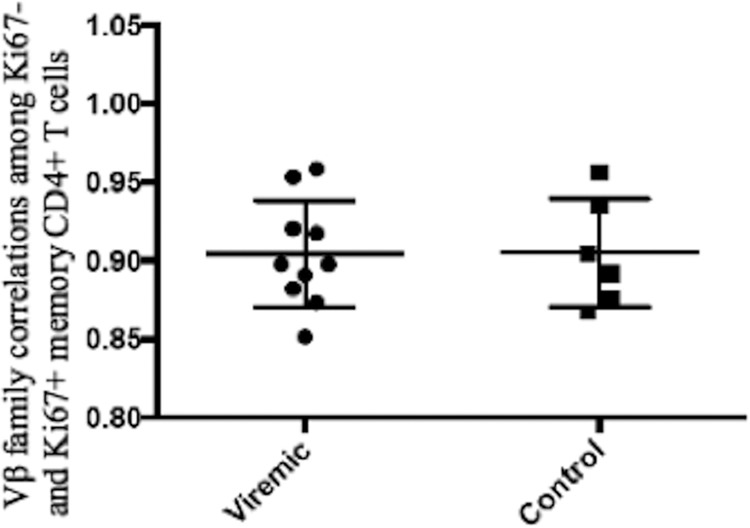
The distributions of T cell receptor Vβ families between cycling (Ki67+) and noncycling (Ki67−) memory CD4+ T cells are similar in subjects with untreated HIV infection and in healthy controls. To examine for potential skewing of the repertoires among cycling cells, the correlations between the proportions of each Vβ family among noncycling (Ki67−) and cycling (Ki67+) memory CD4+ T cells were calculated for each subject studied (Fig. 5b and 7) using Spearman's correlation. For each subject, the correlations were highly significant, and the mean correlations in the HIV+ viremic patients (r = 0.8974) and healthy controls (r = 0.8978) were nearly superimposable.
DISCUSSION
Untreated HIV infection is associated with sustained increased turnover of both CD4+ and CD8+ memory T cells (25) as well as less dramatic cycling of naive T cells (26). In the setting of immunologic failure, even with control of HIV replication, cycling memory CD4+ T cells remain elevated (7). Moreover, Hazenberg et al. (25) and our group (27) have found that immune activation and T cell cycling fall rapidly with application of ART long before circulating CD4+ T cell numbers recover, suggesting that factors beyond homeostatic proliferation likely contribute to CD4+ T cell turnover in chronic HIV infection. This study was undertaken to examine the phenotype of the cycling memory CD4+ T cells in HIV infection to ascertain if these cells had the characteristics of cells driven into the cell cycle as a homeostatic response, through bystander activation, or through engagement of the T cell receptor by a cognate peptide. As demonstrated earlier by us and other investigators (7, 25), we found that cycling memory CD4+ T cells were increased in HIV infection. We also found that compared to the phenotype of cycling memory CD4+ T cells in healthy controls, cycling memory CD4+ T cells in HIV infection had higher expression of the activation antigen CD38 and the exhaustion marker PD-1 (Fig. 2). This might reflect the more durable expression of these antigens on recently cycling cells or, alternatively, that the pathways by which cells are activated to enter the cell cycle may be somewhat different in patients with HIV infection than in healthy subjects. We propose that the latter is plausible, since in HIV infection, cycling CD4+ memory T cells had lower expression of both OX40 and CD40L, two T cell markers that are increased after TCR engagement (15, 28). We also show here that in vitro exposure to the common gamma-chain cytokines IL-2, IL-7, and IL-15 also increases OX40 expression, while exposure to LPS or to an inflammatory cytokine (IL-6, IL-1β, or IFN-α) does not. In earlier work, we showed that the induction of CD40L expression after TCR engagement is impaired in HIV infection (19) but that this is not the case for induction of OX40. The relatively lower level of expression of both OX40 and CD40L on cycling CD4+ T cells in HIV infection suggests that the relative contributions to memory CD4+ T cell cycling of TCR engagement and responses to homeostatic cytokines may be lower in subjects with HIV infection than in healthy subjects, although it is possible that the more activated (CD38+), more exhausted (PD-1-positive) cycling cells in HIV infection may be less capable of upregulating OX40 expression or that OX40 expression is lost more rapidly in HIV infection. In both healthy subjects and in subjects with chronic HIV infection, cycling CD4+ T cells and noncycling CD4+ T cells have a nearly superimposable distribution of Vβ T cell receptor families, suggesting that in both settings, most memory CD4+ T cell cycling is not a consequence of antigen-induced expansion. This may be different in the setting of an acute infection (such as with HIV), but in chronic HIV infection, where sustained cycling and turnover of memory T cells are thought to play roles in disease pathogenesis, we found that the magnitude of HIV replication was linked to the frequency of CD4+ T cell cycling but could not find any skewing of the T cell receptor Vβ repertoire to support the concept that this was mediated through cognate peptide recognition.
The relative contributions of homeostatic signals and bystander activation to memory CD4+ T cell cycling in HIV infection are not clear. In the present study, we found that memory CD4+ T cell cycling was inversely related to circulating CD4+ T cell counts in patients with HIV infection, yet in contrast to our findings in healthy persons, we could not find a relationship between the plasma levels of IL-7 and the percentages of cycling memory CD4+ T cells in HIV-infected patients. This may be attributable in part to the relatively early stage of disease in our patients, as the plasma levels of IL-7 typically tend to rise most dramatically as circulating CD4+ T cells descend to less than 100 cells/μl (29, 30), yet the diminished expression of CD127, the IL-7 receptor alpha chain seen in HIV infection (31), and an impaired responsiveness to IL-7 (8, 32) might be expected to enhance the inverse relationship between the plasma levels of IL-7 and cell cycling in HIV disease. For these reasons, we suspect that additional factors may be important contributors to the increased cycling of memory CD4+ T cells in HIV infection. In this regard, the levels of IL-2 or IL-15 were not related to the percentage of cycling memory CD4+ T cells in either HIV− or HIV+ donors.
Here we show that in HIV infection, memory CD4+ T cell cycling was associated with the plasma levels of HIV and also with the plasma levels of bacterial LPS, a consequence of microbial translocation from the damaged gut (33). In earlier work, we showed that a variety of microbial TLR agonists, including bacterial LPS and single-stranded RNAs that, like HIV, can bind and signal through TLR7 and TLR8, could indirectly activate memory CD4+ T cells to enter the cell cycle (20). In more recent work, we found that the inflammatory cytokines IL-6 and IL-1β, which are inducible by these microbial products, can also drive the cycling of CD4+ T cells in vitro and can block IL-7 responsiveness (34). The significance of the negative correlation between memory CD4+ T cell cycling and plasma LPS levels in healthy controls is less clear. This modest relationship could be spurious; alternatively, low levels of LPS-induced inflammatory cytokines may block homeostatic responses to IL-7 (34), which may be relatively more important in driving the physiologic memory CD4+ T cell cycling in healthy subjects than it is in patients with HIV infection.
Thus, in untreated HIV infection, memory CD4+ T cell cycling is increased. Cycling memory CD4+ T cells in HIV infection have increased markers of activation but decreased expression of both CD40L and OX40. As in healthy controls, the distribution of Vβ families in cycling and noncycling memory CD4+ T cells in patients with HIV infection is virtually identical, suggesting that recent T cell receptor engagement accounts for very little memory CD4+ T cell expansion in both of these settings. Because CD40L and OX40 expression is diminished in cycling memory CD4+ T cells in HIV infection and activation markers are increased, this phenotype suggests that bystander activation by microbial ligands or other inflammatory cytokines may have a relatively larger contribution to memory CD4+ T cell cycling in patients with chronic HIV infection than it does in healthy controls.
ACKNOWLEDGMENTS
This work was supported by the Center for AIDS Research at Case Western Reserve University (AI 36219), by grants from the National Institutes of Health (AI 76174, AI 91526), and by the Fasenmyer Foundation.
Footnotes
Published ahead of print 12 February 2014
REFERENCES
- 1.Fauci AS. 1988. The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science 239:617–622. 10.1126/science.3277274 [DOI] [PubMed] [Google Scholar]
- 2.Silvestri G, Feinberg MB. 2003. Turnover of lymphocytes and conceptual paradigms in HIV infection. J. Clin. Invest. 112:821–824. 10.1172/JCI200319799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M. 2004. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200:761–770. 10.1084/jem.20041196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon SN, Klatt NR, Bosinger SE, Brenchley JM, Milush JM, Engram JC, Dunham RM, Paiardini M, Klucking S, Danesh A, Strobert EA, Apetrei C, Pandrea IV, Kelvin D, Douek DC, Staprans SI, Sodora DL, Silvestri G. 2007. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J. Immunol. 179:3026–3034 http://www.jimmunol.org/content/179/5/3026.long [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM. 2002. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J. Immunol. 169:3400–3406 http://www.jimmunol.org/content/169/6/3400.long [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez B, Sethi AK, Cheruvu VK, Mackay W, Bosch RJ, Kitahata M, Boswell SL, Mathews WC, Bangsberg DR, Martin J, Whalen CC, Sieg S, Yadavalli S, Deeks SG, Lederman MM. 2006. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA 296:1498–1506. 10.1001/jama.296.12.1498 [DOI] [PubMed] [Google Scholar]
- 7.Lederman MM, Calabrese L, Funderburg NT, Clagett B, Medvik K, Bonilla H, Gripshover B, Salata RA, Taege A, Lisgaris M, McComsey GA, Kirchner E, Baum J, Shive C, Asaad R, Kalayjian RC, Sieg SF, Rodriguez B. 2011. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J. Infect. Dis. 204:1217–1226. 10.1093/infdis/jir507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bazdar DA, Kalinowska M, Sieg SF. 2009. Interleukin-7 receptor signaling is deficient in CD4+ T cells from HIV-infected persons and is inversely associated with aging. J. Infect. Dis. 199:1019–1028. 10.1086/597210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakai K, Dimas J, Lenardo MJ. 2006. The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc. Natl. Acad. Sci. U. S. A. 103:3369–3374. 10.1073/pnas.0509417103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bucy RP, Hockett RD, Derdeyn CA, Saag MS, Squires K, Sillers M, Mitsuyasu RT, Kilby JM. 1999. Initial increase in blood CD4(+) lymphocytes after HIV antiretroviral therapy reflects redistribution from lymphoid tissues. J. Clin. Invest. 103:1391–1398. 10.1172/JCI5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boisvert J, Edmondson S, Krummel MF. 2004. Immunological synapse formation licenses CD40-CD40L accumulations at T-APC contact sites. J. Immunol. 173:3647–3652 [DOI] [PubMed] [Google Scholar]
- 12.Chattopadhyay PK, Yu J, Roederer M. 2005. A live-cell assay to detect antigen-specific CD4+ T cells with diverse cytokine profiles. Nat. Med. 11:1113–1117. 10.1038/nm1293 [DOI] [PubMed] [Google Scholar]
- 13.Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M, Scheffold A, Thiel A. 2005. Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat. Med. 11:1118–1124. 10.1038/nm1292 [DOI] [PubMed] [Google Scholar]
- 14.Zaunders JJ, Munier ML, Seddiki N, Pett S, Ip S, Bailey M, Xu Y, Brown K, Dyer WB, Kim M, de Rose R, Kent SJ, Jiang L, Breit SN, Emery S, Cunningham AL, Cooper DA, Kelleher AD. 2009. High levels of human antigen-specific CD4+ T cells in peripheral blood revealed by stimulated coexpression of CD25 and CD134 (OX40). J. Immunol. 183:2827–2836. 10.4049/jimmunol.0803548 [DOI] [PubMed] [Google Scholar]
- 15.Gramaglia I, Weinberg AD, Lemon M, Croft M. 1998. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 161:6510–6517 [PubMed] [Google Scholar]
- 16.Boasso A, Hardy AW, Anderson SA, Dolan MJ, Shearer GM. 2008. HIV-induced type I interferon and tryptophan catabolism drive T cell dysfunction despite phenotypic activation. PLoS One 3:e2961. 10.1371/journal.pone.0002961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrovas C, Price DA, Mattapallil J, Ambrozak DR, Geldmacher C, Cecchinato V, Vaccari M, Tryniszewska E, Gostick E, Roederer M, Douek DC, Morgan SH, Davis SJ, Franchini G, Koup RA. 2007. SIV-specific CD8+ T cells express high levels of PD1 and cytokines but have impaired proliferative capacity in acute and chronic SIVmac251 infection. Blood 110:928–936. 10.1182/blood-2007-01-069112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakanjako D, Ssewanyana I, Mayanja-Kizza H, Kiragga A, Colebunders R, Manabe YC, Nabatanzi R, Kamya MR, Cao H. 2011. High T-cell immune activation and immune exhaustion among individuals with suboptimal CD4 recovery after 4 years of antiretroviral therapy in an African cohort. BMC Infect. Dis. 11:43. 10.1186/1471-2334-11-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espinosa E, Ormsby CE, Reyes-Teran G, Asaad R, Sieg SF, Lederman MM. 2010. Dissociation of CD154 and cytokine expression patterns in CD38+ CD4+ memory T cells in chronic HIV-1 infection. J. Acquir. Immune Defic. Syndr. 55:439–445. 10.1097/QAI.0b013e3181ef991d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Funderburg N, Luciano AA, Jiang W, Rodriguez B, Sieg SF, Lederman MM. 2008. Toll-like receptor ligands induce human T cell activation and death, a model for HIV pathogenesis. PLoS One 3:e1915. 10.1371/journal.pone.0001915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bitmansour AD, Waldrop SL, Pitcher CJ, Khatamzas E, Kern F, Maino VC, Picker LJ. 2001. Clonotypic structure of the human CD4+ memory T cell response to cytomegalovirus. J. Immunol. 167:1151–1163 http://www.jimmunol.org/content/167/3/1151.long [DOI] [PubMed] [Google Scholar]
- 22.Younes SA, Yassine-Diab B, Dumont AR, Boulassel MR, Grossman Z, Routy JP, Sekaly RP. 2003. HIV-1 viremia prevents the establishment of interleukin 2-producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J. Exp. Med. 198:1909–1922. 10.1084/jem.20031598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pitcher CJ, Quittner C, Peterson DM, Connors M, Koup RA, Maino VC, Picker LJ. 1999. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nat. Med. 5:518–525. 10.1038/8400 [DOI] [PubMed] [Google Scholar]
- 24.Bitmansour AD, Douek DC, Maino VC, Picker LJ. 2002. Direct ex vivo analysis of human CD4(+) memory T cell activation requirements at the single clonotype level. J. Immunol. 169:1207–1218 http://www.jimmunol.org/content/169/3/1207.long [DOI] [PubMed] [Google Scholar]
- 25.Hazenberg MD, Stuart JW, Otto SA, Borleffs JC, Boucher CA, de Boer RJ, Miedema F, Hamann D. 2000. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 95:249–255 http://bloodjournal.hematologylibrary.org/content/95/1/249.long [PubMed] [Google Scholar]
- 26.Di Mascio M, Sereti I, Matthews LT, Natarajan V, Adelsberger J, Lempicki R, Yoder C, Jones E, Chow C, Metcalf JA, Sidorov IA, Dimitrov DS, Polis MA, Kovacs JA. 2006. Naive T-cell dynamics in human immunodeficiency virus type 1 infection: effects of highly active antiretroviral therapy provide insights into the mechanisms of naive T-cell depletion. J. Virol. 80:2665–2674. 10.1128/JVI.80.6.2665-2674.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Funderburg NT, Andrade A, Chan ES, Rosenkranz SL, Lu D, Clagett B, Pilch-Cooper HA, Rodriguez B, Feinberg J, Daar E, Mellors J, Kuritzkes D, Jacobson JM, Lederman MM. 2013. Dynamics of immune reconstitution and activation markers in HIV+ treatment-naïve patients treated with raltegravir, tenofovir disoproxil fumarate and emtricitabine. PLoS One 8:e83514. 10.1371/journal.pone.0083514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koguchi Y, Thauland TJ, Slifka MK, Parker DC. 2007. Preformed CD40 ligand exists in secretory lysosomes in effector and memory CD4+ T cells and is quickly expressed on the cell surface in an antigen-specific manner. Blood 110:2520–2527. 10.1182/blood-2007-03-081299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fry TJ, Connick E, Falloon J, Lederman MM, Liewehr DJ, Spritzler J, Steinberg SM, Wood LV, Yarchoan R, Zuckerman J, Landay A, Mackall CL. 2001. A potential role for interleukin-7 in T-cell homeostasis. Blood 97:2983–2990. 10.1182/blood.V97.10.2983 [DOI] [PubMed] [Google Scholar]
- 30.Napolitano LA, Grant RM, Deeks SG, Schmidt D, De Rosa SC, Herzenberg LA, Herndier BG, Andersson J, McCune JM. 2001. Increased production of IL-7 accompanies HIV-1-mediated T-cell depletion: implications for T-cell homeostasis. Nat. Med. 7:73–79. 10.1038/83381 [DOI] [PubMed] [Google Scholar]
- 31.Zhang SY, Zhang Z, Fu JL, Kang FB, Xu XS, Nie WM, Zhou CB, Zhao M, Wang FS. 2009. Progressive CD127 down-regulation correlates with increased apoptosis of CD8 T cells during chronic HIV-1 infection. Eur. J. Immunol. 39:1425–1434. 10.1002/eji.200839059 [DOI] [PubMed] [Google Scholar]
- 32.Kalinowska M, Bazdar DA, Lederman MM, Funderburg N, Sieg SF. 2013. Decreased IL-7 responsiveness is related to oxidative stress in HIV disease. PLoS One 8:e58764. 10.1371/journal.pone.0058764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365–1371. 10.1038/nm1511 [DOI] [PubMed] [Google Scholar]
- 34.Shiv CL, Mudd JC, Funderburg NT, Sieg SF, Kyi B, Bazdar DA, Mangioni D, Gori A, Jacobson JM, Brooks AD, Hardacre J, Ammori J, Estes JD, Schacker TW, Rodriguez B, Lederman MM. 28 February 2014. Inflammatory cytokines drive CD4 T cell cycling and impaired responsiveness to interleukin-7: implications for immune failure in HIV disease. J. Infect. Dis. 10.1093/infdis/jiu125 [DOI] [PMC free article] [PubMed] [Google Scholar]