

ABSTRACT
The replication of coronaviruses occurs in association with multiple virus-induced membrane structures that evolve during the course of infection; however, the dynamics of this process remain poorly understood. Previous studies of coronavirus replication complex organization and protein interactions have utilized protein overexpression studies and immunofluorescence of fixed cells. Additionally, live-imaging studies of coronavirus replicase proteins have used fluorescent reporter molecules fused to replicase proteins, but expressed from nonnative locations, mostly late-transcribed subgenomic mRNAs, in the presence or absence of the native protein. Thus, the timing and targeting of native replicase proteins expressed in real time from native locations in the genome remain unknown. In this study, we tested whether reporter molecules could be expressed from the replicase polyprotein of murine hepatitis virus as fusions with nonstructural protein 2 or 3 and whether such reporters could define the targeting and activity of replicase proteins during infection. We demonstrate that the fusion of green fluorescent protein and firefly luciferase with either nonstructural protein 2 or 3 is tolerated and that these reporter-replicase fusions can be used to quantitate replication complex formation and virus replication. The results show that the replicase gene has flexibility to accommodate a foreign gene addition and can be used directly to study replicase complex formation and evolution during infection as well as to provide highly sensitive and specific markers for protein translation and genome replication.
IMPORTANCE Coronaviruses are a family of enveloped, positive-sense RNA viruses that are important agents of disease, including severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus. Replication is associated with multiple virus-induced membrane structures that evolve during infection; however, the dynamics of this process remain poorly understood. In this study, we tested whether reporter molecules expressed from native locations within the replicase polyprotein of murine hepatitis virus as fusions with nonstructural proteins could define the expression and targeting of replicase proteins during infection in live cells. We demonstrate that the replicase gene tolerates the introduction of green fluorescent protein or firefly luciferase as fusions with replicase proteins. These viruses allow early quantitation of virus replication as well as real-time measurement of replication complexes.
INTRODUCTION
Coronaviruses (CoVs) are a family of RNA viruses that are important agents of human and animal diseases (1), including severe acute respiratory syndrome coronavirus (SARS-CoV) and the recently emerged Middle East respiratory syndrome coronavirus (MERS-CoV) (2–5). The genome of the CoV murine hepatitis virus (MHV) is one of the largest known replicating RNA molecules, at 31.3 kb (1). The 5′-most replicase gene is composed of two open reading frames (ORFs), ORF1a and ORF1b, and comprises approximately two-thirds of the genome. Translation begins upon entry into a host cell, first of replicase ORF1a and then of ORF1ab following a −1 ribosomal frameshift. The replicase polyproteins are proteolytically processed by papain-like protease 1 (PLP1) and PLP2 in nsp3 and by the nsp5 protease (3CLpro) to generate 16 nonstructural proteins (nsp1 to nsp16); functions include RNA-dependent RNA polymerase, helicase, primase, cap methylation, and a novel proofreading exonuclease (1). The replicase proteins nsp3, -4, and -6 have been demonstrated to be involved in membrane modifications leading to the formation of double-membrane vesicles (DMVs) (6). Each MHV nsp studied has been shown to localize to virus-induced DMVs and other modified host membranes, collectively referred to as replication complexes (RCs) (7–13). While much has been learned about virus-induced host cell modifications, little is known of the process of RC formation and how RCs change over time. It is known that nucleocapsid is associated with new sites of RNA synthesis but also sites of virus assembly in the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) and in the Golgi compartment distinct from sites of replication (14). The mechanisms by which RCs form, RNA synthesis occurs, and nucleocapsids transit to sites of virion assembly, however, remain unknown.
To date, studies of CoV replication complex formation have involved immunofluorescence imaging of fixed cells using antibodies against native proteins (10, 12, 15, 16). For assessment of kinetics of replication, fluorescent and luminescent reporters have been expressed with either replicase proteins from expression plasmids, reporter proteins replacing nonessential accessory ORFs, or replicase protein-reporter fusions expressed in place of accessory ORFs (17–20). Reporters have also been utilized within CoV replicon genomes (21). Studies with such constructs have provided insights into the function and interaction of replicase proteins during viral replication, and the constructs have also served as reporters for studies of CoV inhibitors (21–24). While these strategies have been useful for reports on overall virus replication, they were not designed to test the expression or localization of specific proteins, nor were they designed to report replicase gene expression. A replicase reporter virus has been constructed for equine arteritis virus, an arterivirus with a genome size less than half that of MHV, with the insertion of enhanced green fluorescent protein (EGFP) between nsp1 and nsp2 (25). The capacity of the CoV replicase gene to accept foreign genes, however, is not known, nor has foreign gene insertion within the replicase gene of any replicating CoV been tested without a compensatory deletion of viral genetic material. In this study, we describe the quantitative measurement of MHV replicase gene expression and the formation of replication complexes using engineered reporter viruses expressing green fluorescent protein (GFP) and firefly luciferase (FFL) as in-frame fusions with viral replicase proteins nsp2 and nsp3. nsp2 is a 65-kDa protein that has been shown to localize to CoV replication complexes but is dispensable for virus replication in culture (26). nsp3 is a 210-kDa protein that contains two essential proteases and other functional protein domains and is required for both virus-induced membrane modifications and virus replication (6). We demonstrate that GFP and FFL reporter fusions with nsp2 and nsp3 permit efficient virus replication, target replication complexes, provide the earliest indicators of MHV replication, and allow direct measurements of replication complex formation. These results also demonstrate the capacity of the MHV genome to tolerate expansion and identify sites for possible virus-mediated expression of foreign proteins within the replicase polyprotein.
MATERIALS AND METHODS
MHV and previously described mutant viruses, cells, and antibodies.
Recombinant murine hepatitis virus strain A59 (GenBank accession number AY910861) was used as the wild type (WT) for all experiments (27, 28). MHV-Δnsp2, with an in-frame deletion of nsp2, and MHV-ΔCS2, with a deletion of the nsp2-3 cleavage site P1 Glu, were described previously (26). Delayed brain tumor (DBT) cells (29) and baby hamster kidney cells expressing the MHV receptor (BHK-MHVR) (30, 31) were grown in Dulbecco's modified Eagle medium (DMEM) that contained 10% fetal bovine serum (FBS), 1% 1 M HEPES, 100 units/ml of penicillin, 100 μg/ml streptomycin, and 0.23 μl/ml amphotericin B for all experiments. Medium for BHK-MHVR cells was supplemented with G418 (0.8 mg/ml) for selection of cells expressing the receptor. The polyclonal antibodies used in biochemical and imaging experiments were described previously. All polyclonal antibodies were raised in rabbits unless otherwise indicated. For MHV, these included antibodies specific for nsp1 (VU221) (16), nsp2 (VU154) (15), nsp3 (VU164) (26), and nsp8 (VU123) (11). Mouse monoclonal anti-firefly luciferase (FFL), clone LUC-1, was purchased from Sigma-Aldrich. Mouse monoclonal antibody specific for full-length GFP was purchased from SantaCruz Biotechnology. Mouse monoclonal antibody against the viral membrane protein (M) was generously provided by J. Fleming (University of Wisconsin, Madison).
Construction of mutant MHV cDNA plasmids.
Insertions of reporter genes in place of the nsp2 coding sequences of MHV were engineered by using PCR with primers shown in Table 1. For ABCDEF primer sets, primers A and B generated an A/B PCR product, primers C and D generated a C/D PCR product, and primers E and F generated an E/F PCR product. AB, CD, and EF PCR amplicons were ligated into an ABCDEF product by using the class IIs restriction enzyme method, and ligation products were cloned into the appropriate fragment A vector by using unique sites: 5′-SacII and 3′-NdeI. Successful insertions of reporter gene sequences were confirmed by restriction digestion and sequencing. The infectious cDNA fragment A construct (pCR-XL-Topo-A), which consists of genome nucleotides (nt) 1 to 4882, was used as the template DNA (33). Reporter genes were cloned from pEGFP-C1 (GFP; Clontech) and pGEM-Luc (firefly luciferase; Promega).
TABLE 1.
Primers used for generation of reporter insertions
| Primer | Sequencea | Amplification target | Sense | Description |
|---|---|---|---|---|
| MHV A | 5′-AACGGCACTTCCTGCGTGTCCATG | MHV nsp1 | + | Common MHV left nsp1 primer |
| MHV B | 5′-CGTCTCCCTTAACACCGCGATAGCCCTTAAGAAGAG | MHV nsp1 | − | Common MHV right nsp1 primer |
| MHV-GFP C | 5′-CGTCTCCTAAGATGGTGAGCAAGGGCGAGGAGCTGT | GFP | + | |
| MHV-GFP D | 5′-CGTCTCGTGCCCTTGTACAGCTCGTCCATGCCGAGAGT | GFP | − | MHV-Δ2-GFP3 |
| MHV-GFP D | 5′-CGTCTCGTAACCTTGTACAGCTCGTCCATGCCGAGAGT | GFP | − | MHV-GFP2 |
| MHV-FFL C | 5′-CGTCTCCTAAGATGGAAGACGCCAAAAACATAAAGAAAG | FFL | + | |
| MHV-FFL D | 5′-CGTCTCGTGCCCAATTTGGACTTTCCGCCCTTCTT | FFL | − | MHV-Δ2-FFL3 |
| MHV-FFL D | 5′-CGTCTCGTAACCAATTTGGACTTTCCGCCCTTCTT | FFL | − | MHV-FFL2 |
| MHV E | 5′-CGTCTCGTATCGCGGTGTTAAGAAAGTCGAGTTTAAC | MHV nsp3 | + | Common MHV left nsp3 primer |
| MHV F | 5′-ACTTGCACATATGAGACACAACGTCCCCA | MHV nsp3 | − | Common MHV right nsp3 primer |
Underlining indicates residues added for cloning or mutagenesis purposes.
Generation of MHV mutant viruses.
Viruses containing PCR-generated insertions within the viral coding sequence were produced by using infectious cDNA assembly strategies for MHV as previously described, with modifications (27, 28, 33, 34). Plasmids containing the seven cDNA cassettes of the MHV genome were digested by using MluI, BsmBI, and SfiI for fragment A; BglI and BsmBI for fragments B and C; BsmBI and NciI for fragments D and E; BsmBI for fragment F; and BsmBI and SfiI for fragment G. Digested, gel-purified fragments were ligated together in a total reaction volume of ∼100 μl overnight at 16°C. Following chloroform extraction and isopropanol precipitation of ligated DNA, capped, polyadenylated, full-length RNA transcripts of MHV infectious cDNA were generated in vitro by using the mMessage mMachine T7 transcription kit (Ambion) according to the manufacturer's protocol, with modifications. Twenty-microliter reaction mixtures were supplemented with 3 μl of 30 mM GTP, and transcription was performed at 40.5°C for 25 min, 37.5°C for 50 min, and 40.5°C for 25 min. In parallel, capped, polyadenylated RNA transcripts encoding the corresponding nucleocapsid (N) proteins were generated in vitro by using N cDNA generated by PCR (33, 34). N transcripts and mutant viral transcripts were then mixed and electroporated into BHK-MHVR cells. Cells were grown to subconfluence, trypsinized, and then washed twice with phosphate-buffered saline (PBS) and resuspended in PBS at a concentration of 107 cells/ml. Six hundred microliters of cells was then added to RNA transcripts in a 4-mm-gap electroporation cuvette (BTX), and three electrical pulses of 850 V at 25 μF were delivered with a Bio-Rad Gene Pulser Xcell electroporator. Transfected cells were then seeded onto a layer of 106 uninfected DBT cells in a 75-cm2 flask and incubated at 37°C for 30 to 90 h. Virus viability was determined by syncytium formation. RNA was recovered from infected cell monolayers by using TRIzol (Invitrogen) according to the manufacturer's instructions, and retention of the introduced mutations was verified by reverse transcription-PCR (RT-PCR) and sequencing.
Microscopy.
DBT cells grown to 60% confluence on 12-mm glass coverslips were infected with reporter viruses. At 10 h postinfection (p.i.), medium was aspirated from cells, and cells were fixed and permeabilized in methanol at −20°C overnight. Cells were rehydrated in PBS for 20 min and blocked in PBS containing 5% bovine serum albumin (BSA). Blocking solution was aspirated, and cells were washed with immunofluorescence (IF) assay wash solution (PBS containing 1% BSA and 0.05% Nonidet P-40) at room temperature. Cells were incubated with primary antibodies where indicated (anti-nsp8 at a 1:250 dilution, anti-M at 1:1,000, or anti-FFL [Sigma-Aldrich] at 1:1,000) for 45 min. Cells were washed in IF wash solution 3 times for 5 min per wash. Cells were incubated in secondary antibodies (goat anti-rabbit Alexa Fluor 488 [1:1,000] or Alexa Fluor 546 [1:1,500]; Invitrogen Molecular Probes) for 30 min. Cells were washed 3 times for 5 min per wash, followed by a final wash in PBS, and then rinsed in distilled water. Coverslips were mounted with Aquapolymount (Polysciences) and visualized by confocal immunofluorescence microscopy on a Zeiss LSM 510 laser scanning confocal microscope at 488 and 543 nm with a 40× oil immersion lens. Images were processed and assembled by using Adobe Photoshop CS2 (9.0.2). For live-cell fluorescence microscopy, DBT cells were seeded onto 35-mm glass-bottom culture dishes (MatTek). Forty-eight hours later, cells were infected with MHV-GFP2 at a multiplicity of infection (MOI) of 1 PFU/cell. At 5 h p.i., plates were transferred to the live-cell incubator surrounding the objective stage of a Nikon Eclipse TE-2000S wide-field fluorescence microscope. Cells were imaged by using a 40× oil immersion lens through differential interference contrast (DIC) and fluorescein isothiocyanate (FITC) filters, with images being captured at 30-s intervals over the course of 4 h. Resulting images were merged and assembled by using Nikon Elements, ImageJ, and Adobe Photoshop CS2.
Protein immunoprecipitations.
For protein labeling and immunoprecipitation experiments, cells were infected with MHV at an MOI of 10 and incubated at 37°C. At 4 h p.i., medium was aspirated and replaced with medium lacking methionine and cysteine and supplemented with actinomycin D (Sigma) at a final concentration of 20 μg/ml. At 5 h p.i., cells were labeled with [35S]Met-Cys at a concentration of 0.08 mCi/ml. Radiolabeled cells were lysed in 1 ml no-SDS lysis buffer (1% NP-40, 0.5% sodium deoxycholate [DOC], 150 mM sodium chloride [NaCl], and 50 mM Tris [pH 8.0]) at 7 to 10 h p.i. Cellular debris and nuclei were pelleted by centrifugation at 14,000 × g for 3 min at room temperature (RT), and the supernatant was transferred into a fresh tube. One hundred microliters of cell lysate was subsequently used per 400 μl of immunoprecipitation reaction buffer. The lysate was combined with protein A-Sepharose beads and a 1:200 dilution of antibody in no-SDS lysis buffer supplemented with 1% SDS. After incubation at 4°C for 18 h, beads were pelleted and washed with low-salt lysis buffer (0.1% SDS lysis buffer with 150 mM NaCl), followed by high-salt lysis buffer (no-SDS lysis buffer with 1 M NaCl) and a final low-salt wash (0.5% SDS lysis buffer with 150 mM NaCl). After rinsing, proteins were eluted by the addition of 2× LDS (lithium dodecyl sulfate)–1× DTT (dithiothreitol) buffer (NuPage; Invitrogen) to the pelleted beads, which were heated at 70°C for 10 min prior to electrophoresis of the supernatant on 4 to 12% Bis-Tris or 3 to 8% Tris-acetate gels (NuPage; Invitrogen). A full-range rainbow ladder (GE Healthcare) and a 14C ladder (PerkinElmer LAS) were used as molecular weight standards.
Viral replication assays.
For viral replication analysis, DBT cells were infected in triplicate with WT MHV or mutant viruses at the MOIs indicated for each experiment. Following a 30-min adsorption with rocking at RT, medium was aspirated, and cells were washed 3 times with PBS and then incubated with prewarmed medium at 37°C. Aliquots of medium were collected from 0 to 24 h p.i., and virus titers were determined by plaque assays on DBT cells in duplicate, as described previously (35).
Competition assay.
Confluent monolayers of DBT cells in T25 flasks were infected with WT MHV or MHV-GFP2 alone or at a 1:1, 1:10, or 10:1 ratio of WT MHV to MHV-GFP2 at a total MOI of 0.1 PFU/cell. Each flask was passaged 10 times, as described above for serial virus passages, and 1 ml supernatant was collected and stored at −80°C. DBT cells were seeded onto glass coverslips and infected at an MOI of 1 PFU/cell with passage 1 (P1), P3, and P10 of each combination of viruses. At 8 h p.i., slides were fixed with cold methanol (MeOH). Coverslips were immunostained with primary antibodies specific for nsp8 and a secondary antibody, goat anti-rabbit Alexa Fluor 546, as described above. Coverslips were mounted with Aquapolymount (Polysciences) and visualized by immunofluorescence microscopy on a Nikon Eclipse TE-2000S wide-field fluorescence microscope. Cells were imaged by using a 40× oil immersion lens through DIC, FITC, Cy3, and 4′,6-diamidino-2-phenylindole (DAPI) filters. The resulting images were merged and assembled by using Nikon Elements, ImageJ, and Adobe Photoshop CS2.
Luciferase activity versus viral replication assay.
DBT cells in 6-well plates were infected with MHV-Δ2-FFL3 or MHV-FFL2 at an MOI of 1 PFU/cell and incubated at 37°C for 30 min. At 0.5 h p.i., medium was aspirated to remove the inoculum, and cells were washed three times with PBS and supplemented with prewarmed medium. At time points through 24 h p.i., the supernatant was harvested or cells were lysed in reporter lysis buffer (Promega) and subjected to one freeze-thaw cycle. Following freeze-thaw, lysates were vortexed and centrifuged briefly at top speed, and supernatants were transferred into new tubes. To measure luciferase activity, 20 μl of lysate was added to each well of an opaque 96-well plate. Plates were loaded onto a Chameleon luminometer, and an automatic injector was used to add 100 μl of reconstituted luciferase assay reagent (Promega) to each well as the samples were being read, for 5 s per well without delay. Viral replication was determined from harvested supernatants by a plaque assay, as described above.
RESULTS
Generation and recovery of MHV replicase reporter viruses.
Since nsp2 is dispensable for replication (26), we first tested the tolerance for reporter expression in place of deleted nsp2. This approach was also used because the engineered genome length would not exceed that of WT MHV. We engineered constructs in which enhanced green fluorescent protein (EGFP) (0.71 kb/26.8 kDa) or firefly luciferase (FFL) (1.65 kb/60.7 kDa) (Fig. 1) replaced the nsp2 coding sequence. All constructs were initially designed with PLP1-specific cleavage sites at the N and C termini of the reporter, based on the hypothesis that processing of the reporter from nsp1 and nsp3 would be required for viability. Following electroporation of in vitro-transcribed genome RNA into BHK cells layered onto permissive DBT cells, cytopathic effect (CPE) of syncytia was detected by 24 h postelectroporation for both recombinant viruses. Medium supernatants from electroporated cultures were used to infect fresh DBT cells, and RNA was harvested and amplified by RT-PCR for sequencing of the entire genome. Viral genome sequencing demonstrated the retention of reporter genes without additional mutations in the genome. Passage of the P0 virus in culture demonstrated the stability of the inserted GFP and FFL sequences for >5 passages (data not shown). This confirmed the capacity of MHV to accept proteins of differing sizes and structures in place of deleted nsp2. These viruses are referred to as MHV-Δ2-GFP3 and MHV-Δ2-FFL3. We next determined the capacity of the MHV replicase to accept additional genetic material into its genome without deletion of the viral sequence. GFP and FFL were engineered between nsp1 and nsp2 while retaining all viral proteins. Since we sought to test whether reporter fusions with nsp2 could be recovered, we retained the cleavage site between nsp1 and the reporter but deleted the cleavage site between the reporter and the nsp2 amino terminus (Fig. 1B). Both recombinant viruses were recovered, their sequences were confirmed, and they were passaged for >5 passages, with retention of the introduced sequences (data not shown). These viruses are referred to as MHV-GFP2 and MHV-FFL2.
FIG 1.

Generation and recovery of MHV replicase reporter viruses. (A) Schematic of MHV genome organization with ORF1a and ORF1b connected by a ribosomal frameshift (RFS) and downstream structural/accessory ORFs. S, spike; E, envelope; M, membrane; N, nucleocapsid. The ORF1ab polyprotein of MHV is shown with nonstructural protein domains 1 to 16 (nsp). Papain-like proteinase domains PL1 and PL2 are indicated by gray and white boxes, respectively, in nsp3. The nsp5 protease (3CLpro and Mpro) is indicated by the black box. Cleavages mediated by each domain are indicated by correspondingly colored arrowheads (CS1, cleavage site 1). Deletion is indicated by △. (B) Design of reporter fusion viruses. The top schematic shows WT MHV-A59 nsp1-nsp3 with cleavage site residues P5-P2′, with cleavage sites marked by carets. The next schematics show previously described MHV-ΔCS2 (lacking P1 Gly at the nsp2-3 cleavage site) and MHV-Δnsp2 with an in-frame deletion of nsp2 and a functional engineered nsp1-nsp3 cleavage site. GFP and FFL reporter gene insertions are indicated in gray. All confirmed functional cleavages are indicated by gray arrowheads. MHV-Δ2-GFP/FFL3, MHV with a deletion of nsp2 and fusion of GFP or FFL to nsp3; MHV-GFP/FFL2, MHV with the addition of GFP or FFL as a fusion to nsp2.
Polyprotein expression and processing in MHV replicase reporter viruses.
MHV-Δ2-GFP3 and MHV-Δ2-FFL3 mutant viruses were engineered with predicted functional cleavage sites at the nsp1 and nsp3 junctions. To define the processing of nsp1-reporter-nsp3 from the replicase polyprotein, DBT cells were infected with WT MHV, MHV-Δ2-GFP3, or MHV-Δ2-FFL3 and radiolabeled, and proteins were immunoprecipitated from lysates of infected cells with antibodies specific for nsp1, -2, -3, and -8 (Fig. 2A). Immunoprecipitation of lysates from WT MHV-infected cells detected nsp1, -2, -3, -2-3, and -8. Immunoprecipitation of proteins from cell lysates of MHV-Δ2-FFL3- and MHV-Δ2-GFP3-infected cells detected both nsp1 and nsp8 but not nsp2, as expected. This indicated that processing was occurring at the junction between nsp1 and FFL/GFP and that expression and processing of downstream proteins from the replicase polyprotein were intact. Immunoprecipitation with anti-nsp3 in WT-infected cells detected the 210-kDa nsp3 as well as the known nsp2-nsp3 intermediate precursor (275 kDa). In contrast, a 210-kDa nsp3 protein was barely detectable following infection with MHV-Δ2-FFL3 and not detected from cells infected with MHV-Δ2-GFP3. Instead, anti-nsp3 antibodies detected a protein of ∼271 kDa in MHV-Δ2-FFL3-infected cells (the predicted mobility of nsp3 plus FFL) and a protein of ∼237 kDa in MHV-Δ2-GFP3-infected cells (the predicted mobility of nsp3 plus GFP). These results indicate that reporter molecules are efficiently processed at the nsp1-reporter junctions but minimally or not processed at the engineered cleavage sites between the reporter and nsp3. This suggested that while P1′ to P5′ residues are required for cleavage, the context of P5-P1 is also important. This result serendipitously demonstrated that viable recombinant MHV could be engineered with a reporter fusion with the 210-kDa nsp3 protein.
FIG 2.
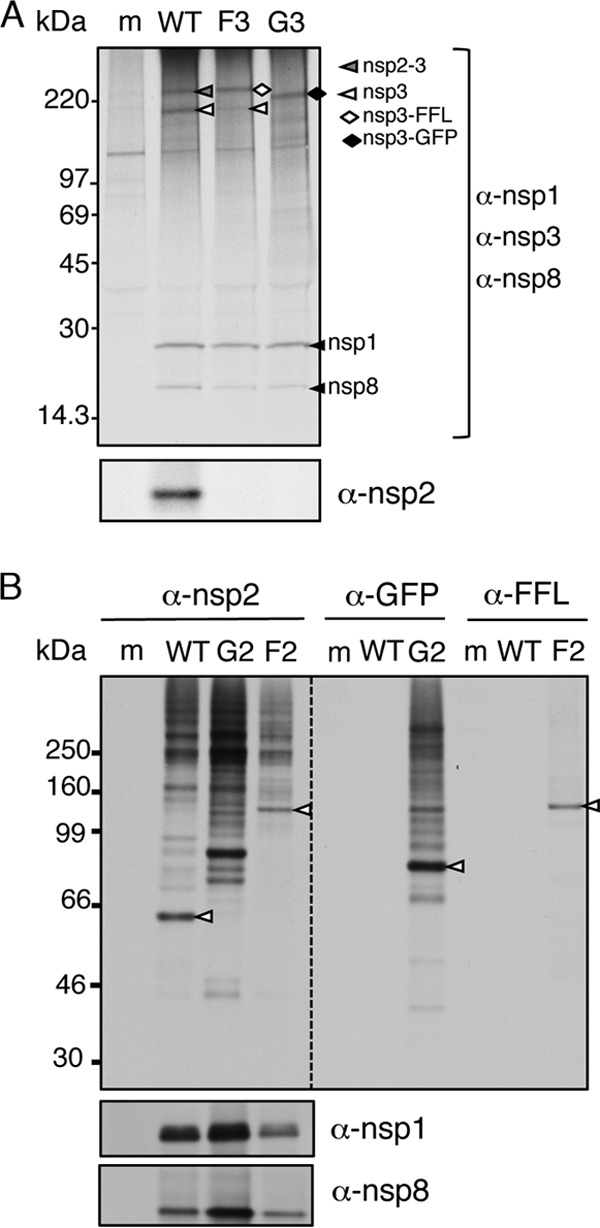
Polyprotein expression and processing in MHV replicase reporter viruses. (A) DBT cells were infected with WT MHV, MHV-Δ2-GFP3 (G3), and MHV-Δ2-FFL3 (F3). Radiolabeled proteins were immunoprecipitated by combined antibodies for nsp1, nsp3, and nsp8 and detected by fluorography. Molecular mass markers are indicated on the left. nsp3, nsp2-3, FFL-nsp3, and GFP-nsp3 are indicated. m, mock. (B) DBT cells were infected with WT MHV, MHV-GFP2 (G2), and MHV-FFL2 (F2), as indicated by the top labels, and radiolabeled proteins were harvested by immunoprecipitation and detected by fluorography. Antibodies used are indicated to the right or at the top. The dashed line represents the same gel with different exposure times: overnight on the left and 1 week on the right. Molecular mass markers are indicated on the left. nsp2, GFP-nsp2, and FFL-nsp2 are indicated by white arrowheads.
This outcome served as the rationale for the design of the “second-generation” reporters, with a retained cleavage site between nsp1 and the reporter and a deletion of the cleavage site between the reporter and nsp2. For MHV-GFP2 and MHV-FFL2, immunoprecipitation of infected cell lysates with anti-nsp1 and anti-nsp8 detected the respective proteins. Antibodies against nsp2 detected the 65-kDa nsp2 in WT-infected lysates but did not detect nsp2 in either reporter virus. Instead, anti-nsp2 immunoprecipitated proteins of 92 kDa or 126 kDa, consistent with the predicted sizes of the GFP-nsp2 and FFL-nsp2 fusion proteins, respectively (Fig. 2B). To confirm this, we immunoprecipitated lysates with anti-GFP and anti-FFL antibodies. Anti-FFL detected a protein from MHV-FFL2-infected cells with a mobility identical to that detected by anti-nsp2. The anti-GFP immunoprecipitation of MHV-GFP2-infected lysates detected a protein with a mobility differing slightly from that of anti-nsp2 but consistent with a fusion of nsp2-GFP. The reason for the difference in mobility is not clear but suggests different migrating forms of nsp2-GFP with different available epitopes recognized by anti-nsp2 and anti-GFP. The complete lack of nsp2 and the detection of new proteins consistent with nsp2-GFP and nsp2-FFL strongly supported the expression of stable fusion proteins that are not cleaved during infection. Overall, these results indicated that the recombinant viruses expressed the reporters as fusions with nsp3 or with nsp2.
Replication of MHV-Δ2-GFP3, MHV-Δ2-FFL3, MHV-GFP2, and MHV-FFL2.
Previous work in our laboratory has shown that MHV-Δnsp2 replicates with kinetics similar to those of WT MHV but with a lower peak titer (26), while MHV mutants engineered to abolish cleavage site 2 between nsp2 and nsp3 (MHV-ΔCS2) demonstrated a prolonged eclipse phase but ultimately achieved WT-like peak titers (36). Finally, a loss of cleavage between nsp1 and nsp2 has been demonstrated to result in a decreased viral yield but WT-like timing of exponential replication (37). To test the effect of reporter fusions with nsp2 or nsp3, we infected murine DBT cells with MHV-Δ2-GFP3, MHV-Δ2-FFL3, MHV-GFP2, or MHV-FFL2 in direct comparison with WT MHV, MHV-Δnsp2, and MHV-ΔCS2, with measurement of supernatant virus titers by plaque assays at multiple time points postinfection (Fig. 3). MHV-Δnsp2 replicated with a 1-log10 impairment in viral titers but normal timing of exponential replication. MHV-ΔCS2 (nsp2-3 fusion) also replicated as previously reported, with delayed exponential replication but WT-like virus yield. MHV-Δ2-GFP3 and MHV-Δ2-FFL3 replication resulted in a cumulative phenotype: a 4-h delay in exponential replication similar to that of MHV-ΔCS2 and a decreased viral yield similar to that of MHV-Δnsp2. In contrast, both MHV-GFP2 and MHV-FFL2 demonstrated replication in culture with exponential replication and viral yields similar to those of WT MHV. This suggests that the replication phenotypes of MHV-Δ2-GFP3 and MHV-Δ2-FFL3 result from the deletion of nsp2 and the fusion of nsp2 with nsp3 and not from the insertion of foreign genes themselves. These results support our previous studies proposing that cleavage at the nsp3 N terminus is required for efficient onset of exponential replication (36). Similarly, the WT-like replication of MHV-GFP2 and MHV-FFL2 further shows that it is cleavage at the C terminus of nsp1 that is required for a WT-like yield. Finally, the results demonstrate that expansion of the replicase gene and polyprotein can still allow for WT-like replication.
FIG 3.
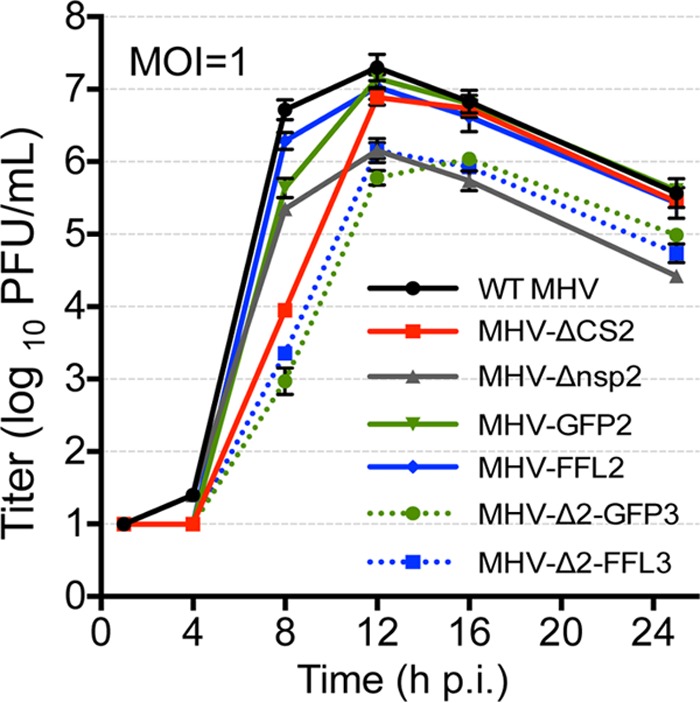
Replication of MHV reporter fusion viruses. DBT cells were infected with recombinant WT MHV, MHV-Δnsp2, MHV-ΔCS2, MHV-Δ2-GFP3, MHV-Δ2-FFL3, MHV-GFP2, and MHV-FFL2 at an MOI of 1 PFU/cell. The supernatant was sampled at the indicated times p.i., and titers were determined by plaque assays. Titers reported are the averages of three replicates ± standard deviations.
Subcellular localization of nsp2 and nsp3 fusion reporters.
We next tested the expression and localization of nsp2 and nsp3 reporter fusions in virus-infected cells. DBT cells on glass coverslips were infected with reporter-expressing MHV at an MOI of 1 PFU/cell, fixed at 10 h p.i., stained for replicase proteins, and examined by confocal microscopy (Fig. 4). DBT cells infected with WT MHV and stained with antibodies for nsp2, nsp3, nsp8, or membrane (M) protein demonstrated the established punctate cytoplasmic pattern of localization (Fig. 4A). Cells infected with MHV-Δ2-GFP3 or MHV-Δ2-FFL3 also exhibited a punctate, perinuclear cytoplasmic localization of reporter molecules by native fluorescence (GFP) or by IF with anti-FFL. Both GFP and FFL colocalized with nsp8 (Fig. 4B), a replicase protein known to localize to replication complexes, but were distinct from M, which localizes to sites of viral assembly in the ERGIC/Golgi compartment (Fig. 4A and data not shown for FFL). The punctate nature of the reporter localization suggests a mechanism for specific targeting to replication complexes. In addition, these results demonstrate that GFP fluorescence is intact when the reporter is fused to the N terminus of nsp3. DBT cells infected with MHV-GFP2 (Fig. 4C) also exhibited a punctate, perinuclear cytoplasmic localization and colocalized with nsp2 and nsp8, again demonstrating specific targeting to replication complexes and indicating that native fluorescence is intact in the nsp2-reporter fusion protein.
FIG 4.
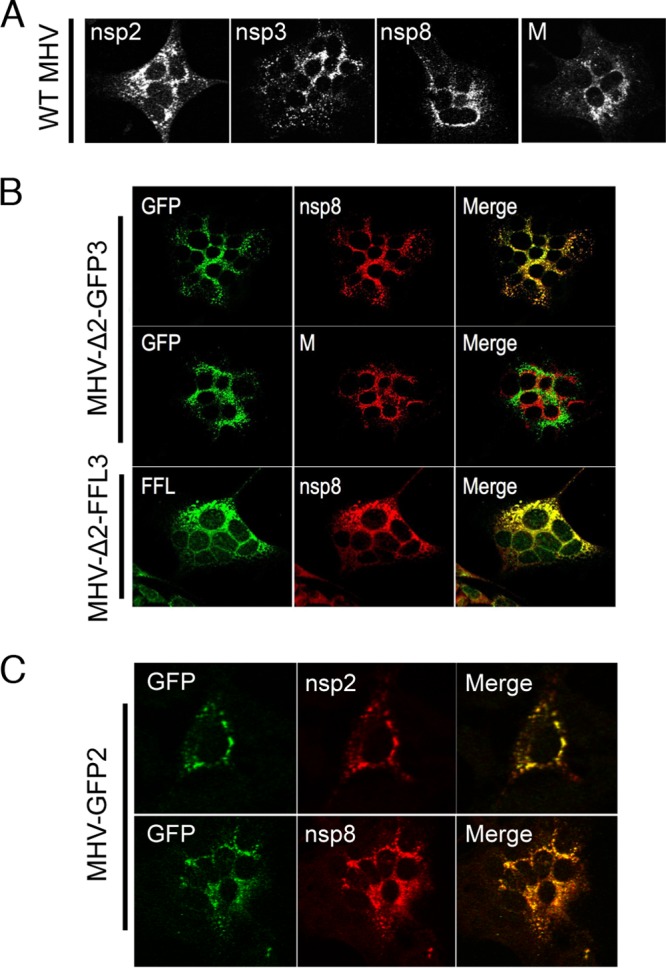
MHV nsp2 and nsp3 fusion reporters localize to replication complexes. DBT cells were infected with WT MHV (A), MHV-Δ2-GFP3 or MHV-Δ2-FFL3 (B), or MHV-GFP2 (C). At 10 h p.i., cells were fixed, stained with the indicated antibodies (anti-FFL, anti-nsp2, anti-nsp3, anti-nsp8, and anti-M), and imaged by confocal microscopy. GFP was detected by native fluorescence.
Competitive fitness of WT MHV versus MHV-GFP2.
Since MHV-FFL2 and MHV-GFP2 demonstrated WT-like kinetics during replication in culture, we tested whether there was a fitness cost associated with the introduction of a foreign protein into the replicase polyprotein. Cells were infected with WT MHV or MHV-GFP2 alone or at a WT MHV/MHV-GFP2 ratio of 1:1, 1:10, or 10:1 at a combined MOI of 0.1 PFU/cell, followed by 10 passages of supernatant into fresh flasks of DBT cells. We then compared GFP expression with the expression of the replicase protein nsp8 to determine the relative competitiveness of MHV-GFP2. DBT cells seeded onto glass coverslips were infected with P1, P3, and P10 passages of each combination of viruses for 8 h. Following immunostaining for nsp8, coverslips were imaged and scored for nsp8 (indirect immunofluorescence [red]) and GFP (native green fluorescence). At all passages, WT MHV-infected cells exhibited only an nsp8 (red) signal, while MHV-GFP2-infected cells demonstrated colocalized nsp8 (red) and native GFP signals in all cells (Fig. 5). In competition experiments, at P1, MHV-GFP2-infected cells were detected but not at levels equivalent to the input ratios. By P3 to P10, even when a 10-fold advantage was given to MHV-GFP2, the recombinant virus could not compete with the WT. Thus, while GFP as a fusion with nsp2 had no effect on replication as measured by plaque assays, the insertion of the gene was associated with a fitness cost compared to the WT virus.
FIG 5.
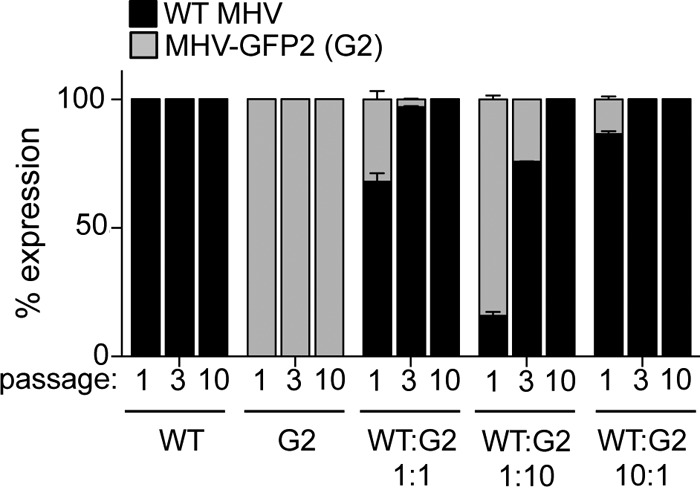
Competitive fitness of WT MHV and MHV-GFP2. DBT cells were infected with WT MHV or MHV-GFP2 alone or at a 1:1, 1:10, or 10:1 ratio of WT MHV to MHV-GFP2. DBT cells on glass coverslips were infected at an MOI of 1 PFU/cell with P1, P3, and P10 passages of each combination. At 8 h p.i., cells were fixed and imaged for nsp8 (red) and GFP (green). Cells containing green (MHV-GFP2) or red (WT MHV or MHV-GFP2) replication complexes were scored. Thirty images from two independent experiments were obtained and scored for cells infected only with WT MHV or only with MHV-GFP2, while for the samples infected with WT MHV and MHV-GFP2 at 1:1, 1:10, and 10:1 ratios, 50 images from two independent experiments were scored.
MHV-GFP2 allows quantitation of replication complex formation in live cells.
We tested whether reporter-nsp fusions could be used to track the quantity and movement of these proteins within a single infected cell over time. DBT cells cultured in glass-bottomed dishes were infected with MHV-GFP2, and at 5 h p.i., they were imaged in a live-imaging chamber incubator at 37°C on a wide-field fluorescence microscope. Images were collected in the differential interference contrast (DIC) and green filter sets every 30 s for 2.5 h from the same field (Fig. 6; see also Fig. S1 in the supplemental material). Every 10th image was analyzed for corrected total cell fluorescence (CTCF) by selecting the cell as the region of interest (ROI) and correcting for the background within each individual image (38). Green fluorescence was evident from the first frame, and the CTCF value nearly tripled throughout infection, until it peaked at 6.75 h p.i. At the conclusion of imaging at 7.5 h p.i., the CTCF value decreased from the peak value by 30%. Throughout the collection of images, green fluorescence was localized perinuclearly, with increasing numbers and intensities of GFP foci from 5 to 7.5 h p.i. The increase in fluorescence intensity during infection indicates that more copies of GFP-nsp2 are being produced and accumulating, as each copy of nsp2 is fused to only one fluorescent molecule, in contrast to indirect fluorescence, where multiple fluorescent molecules have the capacity to bind to one viral protein. These results indicate that the reporter viruses MHV-GFP2 and MHV-Δ2-GFP3 can be used to directly measure the progression of RC formation in real time in single infected cells as well as to track the localization and movement of nsp2 and nsp3 over time.
FIG 6.
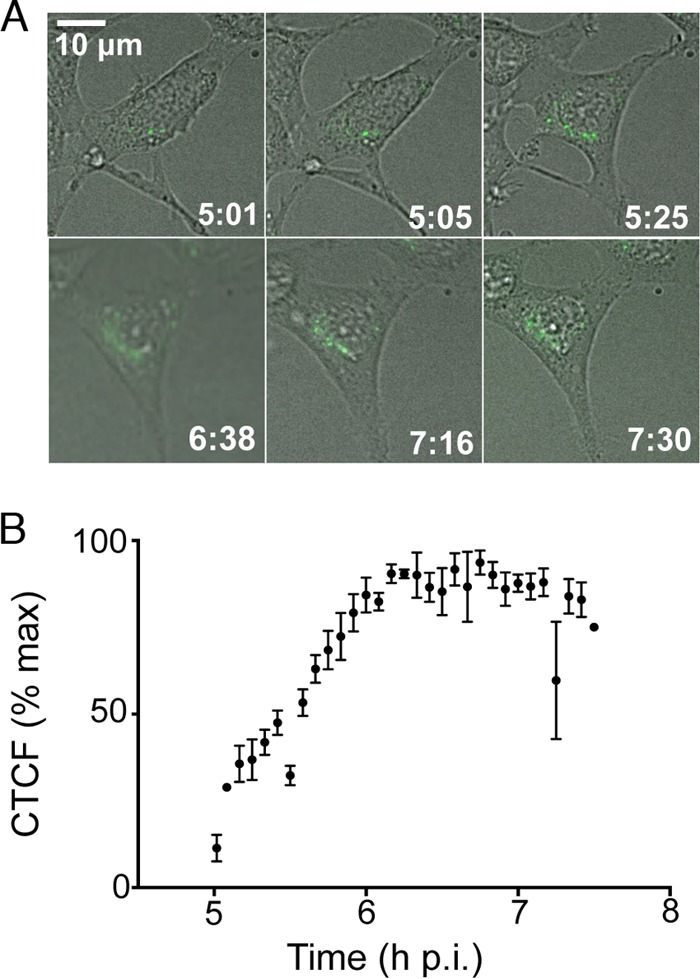
MHV-GFP2 quantitation of replication complex formation in live cells. (A) DBT cells on glass-bottom culture dishes were infected with MHV-GFP2 at an MOI of 1 PFU/cell. At 5 h p.i., dishes were transferred to a chamber incubator at 37°C, with imaging of DIC and GFP every 30 s for 2.5 h. Individual frames were used to generate this panel. The video sequence corresponding to this figure can be found in Fig. S1 in the supplemental material. (B) Corrected total cell fluorescence (CTCF) was measured for the cell by utilizing ImageJ. CTCF was calculated for every 10 frames and plotted over time. Data shown were calculated from three independent infected cells.
Use of MHV-FFL2 and MHV-Δ2-FFL3 for quantitation of early virus replication.
Translation of input genome RNA is the first step in virus replication, and ORF1a, encoding the nsp2 and nsp3 domains, is translated 100% of the time. We hypothesized that this would allow translation of luciferase to provide an earlier marker of viral replication. We inquired whether FFL expressed as fusions with nsp2 or nsp3 could be used to quantitatively measure replication and, if so, whether it was as sensitive as determination of virus titers. DBT cells were infected with MHV-FFL2 or MHV-Δ2-FFL3 at an MOI of 1 PFU/cell, supernatants were collected for plaque assays, and cells were harvested for luciferase assays at multiple times postinfection (Fig. 7). Both MHV-FFL2 and MHV-Δ2-FFL3 replicated as expected, with MHV-Δ2-FFL3 demonstrating delayed exponential replication and decreases in virus yield compared to MHV-FFL2. Luciferase activity (yield in relative light units [RLU]) for MHV-FFL2 showed a signal >3 log10 higher than the corresponding titer at 4 h p.i. while increasing in signal over time. At 8 h p.i., measurements of luciferase activity and titers were very similar. MHV-Δ2-FFL3-infected cells also demonstrated a >3-log10 increase in FFL signal compared to viral titers at 4 h p.i., but this increased signal was retained until 16 h p.i. Most importantly, for both viruses, FFL activity was detected earlier and was 100-fold higher than the virus titer during the eclipse phase. The amplification trend for MHV-FFL2 luminescence mirrors replication, as does the trend for MHV-Δ2-FFL3, suggesting that this assay can serve as a robust early surrogate for viral replication. Furthermore, the results demonstrate that replicase gene translation is occurring continuously and that the genome used as a translational template is being amplified, even in virus with a delayed exponential increase in virus titers. These results establish that the expression of the FFL reporter from the MHV replicase serves as a sensitive, quantitative marker of replicase polyprotein translation and as a discriminating indicator of viral replication. These results also suggest that it may be possible to uncouple measurements of genome translation and genome replication from measurements of infectious virus, which might act as a much earlier biological marker for studies of virus inhibition or studies or constitutive or conditional mutations.
FIG 7.
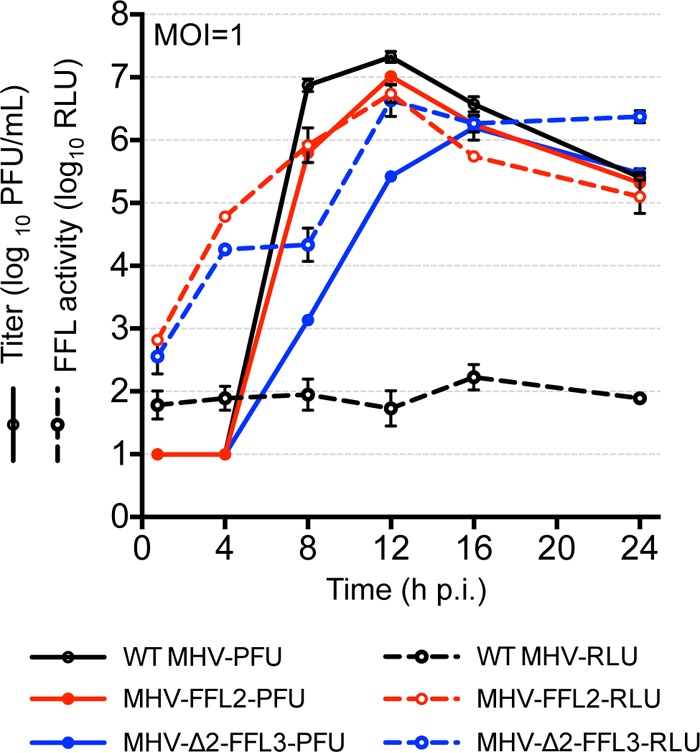
MHV-FFL2 and MHV-Δ2-FFL3 are quantitative measures of replication. DBT cells were infected with MHV-Δ2-FFL3 or MHV-FFL2 at an MOI of 1 PFU/cell in replicate wells. At the indicated times p.i., the supernatant was collected for measurement of virus titers by plaque assays, and cells were harvested for measurement of FFL activity. Titers (PFU) (solid lines) and luminescence (RLU) (dotted lines) are plotted. Data are represented as means ± standard deviations of three replicates.
DISCUSSION
In this study, we demonstrate that the CoV genome is capable of tolerating large substitutions or additions of foreign genetic material within the replicase protein as in-frame protein domains. Furthermore, we show that the fusion of reporters to replicase proteins is tolerated in efficiently replicating virus, allowing quantitative assessments of multiple stages of the virus life cycle. Specifically, these studies show that measurement of viral replication by using FFL can be a highly sensitive, early, and powerful surrogate for genome replication and that GFP fusions with nsp2 and nsp3 can allow quantitative assessments of replication complex formation and evolution during the course of infection. Finally, the results show that insertions of very different proteins are tolerated at each of these locations.
Most studies of CoV replication using the addition of reporters have involved the substitution of accessory ORFs. These ORFs have been used as general indicators of overall viral replication, mostly for testing of inhibitors or attenuation of replication (19–24, 39–41). However, these strategies are limited as direct indicators of genome replication, since they require subgenomic mRNA transcription for expression of the engineered reporters or are not contained within infectious virus. While reporters expressed from downstream ORFs are valuable as indicators of overall viral replication, they cannot distinguish genome replication from subgenomic mRNA transcription. Reporters have also been employed for analysis of coronavirus host interactions and cell biology. Specifically, the expression of nsp2-reporter or nsp4-reporter fusions as substitutions of MHV ORF4 or hemagglutinin esterase (HE) has been described and used to define the localization and movement of nsp's (42–44). While informative, these studies are limited in their interpretation by nsp expression from nonnative locations in the genome. We have shown that alternative expression of nsp2 can be detrimental to replication, even when expressed as a duplication with native nsp2 (44). Our results suggest that the context of the nsp2 location in the polyprotein is important for its interaction with adjacent replicase proteins. Although nsp2 is dispensable for replication (26), altering expression by a deletion or cleavage site alteration impacts replication and fitness.
The observation that GFP or FFL fusions with nsp2 had no detectable effect on the MHV replication cycle in culture was surprising and suggests significant flexibility in this region of the polyprotein for additional genetic information. A recent study of the evolution of the genomes of nidoviruses from the small arteriviruses (∼15 kb) to the largest coronaviruses (up to 32 kb) proposed that proto-CoVs emerged due to the incorporation of a cassette of proteins, including the proofreading exonuclease in nsp14, which allowed more stability of larger genomes (45). It was also proposed that an increased genetic robustness to mutations was also required for genome expansion and increased complexity. Our study supports that argument, with the incorporation of FFL expanding the MHV genome by ∼5.3%. The virus can be recovered at P0 and P1 with high-titer stocks for use in vitro and in vivo, and the FFL coding sequence is maintained for at least 5 passages. However, the introduction of GFP as an nsp2 fusion results in a fitness cost during direct competition with MHV. It will be interesting to see if this fitness cost is similar for FFL or other foreign genes. It will also be of interest to see whether foreign genes are retained over a long passage in the absence of competition or if the subtle fitness cost results in selection against the foreign sequences in the long term.
The implications of stable replicase nsp-reporter fusions are significant for several reasons. It was not previously possible to directly quantitate the translation of the CoV replicase/transcriptase polyproteins in infected cells. CoV ORF1a expresses a ∼495-kDa polyprotein (pp1a). Translation of ORF1b, which encodes critical replication enzymes, including nsp12–RNA-dependent RNA polymerase (RdRp) and nsp14-ExoN, requires a ribosomal frameshift between nsp10 and nsp12. Previous in vitro studies suggested that frameshift efficiencies range from 10 to 40% (46). Dual-luciferase systems have been used to examine structures and sequences in this region; however, this has never been tested by utilizing infectious virus (47, 48). Expression of different quantitative reporters from ORF1a and ORF1b could allow direct testing of the timing and stoichiometry of replicase polyprotein translation. We have recently recovered recombinant viruses expressing GFP as a fusion with nsp14 (our unpublished results), providing encouraging evidence that ORF1b reporter expression is possible. The early exponential signal from FFL-nsp2 is consistent with a rapid amplification of genome RNA prior to virus assembly and release and thus may be an early and sensitive reporter for studies of inhibitors of virus replication. Similarly, it was not previously possible to track the expression and localization of replicase proteins from their native locations in the genome. The native expression of fluorescent reporters fused to replicase proteins creates the opportunity to track replication complex formation in real time in a single cell, without potential artifacts due to cellular fixation for immunofluorescence or due to altered expression from subgenomic mRNAs or exogenous plasmids. Our results suggest that it may be possible to engineer reporter fusions with multiple replicase proteins for testing and visualization of protein-protein and protein-membrane interactions in live cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michelle Becker, E. Clinton Smith, and Dia Beachboard for advice and critical reviews of the manuscript. We thank J. Fleming (University of Wisconsin, Madison) for the kind gift of the M antibody.
Experiments were performed in part through the use of the VUMC Cell Imaging Shared Resource, supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637, and Ey008126. Support for this work was provided by National Institutes of Health grant R01 AI50083 (M.R.D.) from the National Institute of Allergy and Infectious Diseases. M.C.F. was supported by a Medical Scientist Training Program training grant through the Vanderbilt University School of Medicine (T32 GM07347). This work was also supported by the Elizabeth B. Lamb Center for Pediatric Research.
Footnotes
Published ahead of print 12 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00021-14.
REFERENCES
- 1.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450. 10.1038/nrmicro2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ. 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348:1953–1966. 10.1056/NEJMoa030781 [DOI] [PubMed] [Google Scholar]
- 3.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. 10.1016/S0140-6736(03)13077-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399. 10.1126/science.1085952 [DOI] [PubMed] [Google Scholar]
- 5.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 6.Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. 2013. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4(4):pii=00524-13. 10.1128/mBio.00524-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gosert R, Kanjanahaluethai A, Egger D, Bienz K, Baker SC. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76:3697–3708. 10.1128/JVI.76.8.3697-3708.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6:e226. 10.1371/journal.pbio.0060226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ulasli M, Verheije MH, de Haan CA, Reggiori F. 2010. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 12:844–861. 10.1111/j.1462-5822.2010.01437.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snijder EJ, van der Meer Y, Zevenhoven-Dobbe J, Onderwater JJ, van der Meulen J, Koerten HK, Mommaas AM. 2006. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 80:5927–5940. 10.1128/JVI.02501-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bost AG, Carnahan RH, Lu XT, Denison MR. 2000. Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. J. Virol. 74:3379–3387. 10.1128/JVI.74.7.3379-3387.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brockway SM, Clay CT, Lu XT, Denison MR. 2003. Characterization of the expression, intracellular localization, and replication complex association of the putative mouse hepatitis virus RNA-dependent RNA polymerase. J. Virol. 77:10515–10527. 10.1128/JVI.77.19.10515-10527.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJ, Locker JK. 1999. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 73:7641–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bost AG, Prentice E, Denison MR. 2001. Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ergic at late times of infection. Virology 285:21–29. 10.1006/viro.2001.0932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sims AC, Ostermann J, Denison MR. 2000. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J. Virol. 74:5647–5654. 10.1128/JVI.74.12.5647-5654.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brockway SM, Lu XT, Peters TR, Dermody TS, Denison MR. 2004. Intracellular localization and protein interactions of the gene 1 protein p28 during mouse hepatitis virus replication. J. Virol. 78:11551–11562. 10.1128/JVI.78.21.11551-11562.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bosch BJ, de Haan CA, Rottier PJ. 2004. Coronavirus spike glycoprotein, extended at the carboxy terminus with green fluorescent protein, is assembly competent. J. Virol. 78:7369–7378. 10.1128/JVI.78.14.7369-7378.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das Sarma J, Scheen E, Seo SH, Koval M, Weiss SR. 2002. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J. Neurovirol. 8:381–391. 10.1080/13550280260422686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Haan CA, van Genne L, Stoop JN, Volders H, Rottier PJ. 2003. Coronaviruses as vectors: position dependence of foreign gene expression. J. Virol. 77:11312–11323. 10.1128/JVI.77.21.11312-11323.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer F, Stegen CF, Koetzner CA, Masters PS. 1998. Construction of a mouse hepatitis virus recombinant expressing a foreign gene. Adv. Exp. Med. Biol. 440:291–295 [DOI] [PubMed] [Google Scholar]
- 21.Hertzig T, Scandella E, Schelle B, Ziebuhr J, Siddell SG, Ludewig B, Thiel V. 2004. Rapid identification of coronavirus replicase inhibitors using a selectable replicon RNA. J. Gen. Virol. 85:1717–1725. 10.1099/vir.0.80044-0 [DOI] [PubMed] [Google Scholar]
- 22.Ge F, Luo Y, Liew PX, Hung E. 2007. Derivation of a novel SARS-coronavirus replicon cell line and its application for anti-SARS drug screening. Virology 360:150–158. 10.1016/j.virol.2006.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts RS, Yount BL, Sims AC, Baker S, Baric RS. 2006. Renilla luciferase as a reporter to assess SARS-CoV mRNA transcription regulation and efficacy of anti-SARS-CoV agents. Adv. Exp. Med. Biol. 581:597–600. 10.1007/978-0-387-33012-9_108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao G, Du L, Ma C, Li Y, Li L, Poon VK, Wang L, Yu F, Zheng BJ, Jiang S, Zhou Y. 2013. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virol. J. 10:266. 10.1186/1743-422X-10-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Born E, Posthuma CC, Knoops K, Snijder EJ. 2007. An infectious recombinant equine arteritis virus expressing green fluorescent protein from its replicase gene. J. Gen. Virol. 88:1196–1205. 10.1099/vir.0.82590-0 [DOI] [PubMed] [Google Scholar]
- 26.Graham RL, Sims AC, Brockway SM, Baric RS, Denison MR. 2005. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J. Virol. 79:13399–13411. 10.1128/JVI.79.21.13399-13411.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sperry SM, Kazi L, Graham RL, Baric RS, Weiss SR, Denison MR. 2005. Single-amino-acid substitutions in open reading frame (ORF) 1b-nsp14 and ORF 2a proteins of the coronavirus mouse hepatitis virus are attenuating in mice. J. Virol. 79:3391–3400. 10.1128/JVI.79.6.3391-3400.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denison MR, Yount B, Brockway SM, Graham RL, Sims AC, Lu X, Baric RS. 2004. Cleavage between replicase proteins p28 and p65 of mouse hepatitis virus is not required for virus replication. J. Virol. 78:5957–5965. 10.1128/JVI.78.11.5957-5965.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirano N, Fujiwara K, Matumoto M. 1976. Mouse hepatitis virus (MHV-2). Plaque assay and propagation in mouse cell line DBT cells. Jpn. J. Microbiol. 20:219–225 [PubMed] [Google Scholar]
- 30.Lin HH, Kao JH, Chen PJ, Chen DS. 1996. Mechanism of vertical transmission of hepatitis G. Lancet 347:1116. 10.1016/S0140-6736(96)90314-1 [DOI] [PubMed] [Google Scholar]
- 31.Chen W, Madden VJ, Bagnell CR, Jr, Baric RS. 1997. Host-derived intracellular immunization against mouse hepatitis virus infection. Virology 228:318–332. 10.1006/viro.1996.8402 [DOI] [PubMed] [Google Scholar]
- 32. Reference deleted.
- 33.Yount B, Denison MR, Weiss SR, Baric RS. 2002. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 76:11065–11078. 10.1128/JVI.76.21.11065-11078.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yount B, Curtis KM, Fritz EA, Hensley LE, Jahrling PB, Prentice E, Denison MR, Geisbert TW, Baric RS. 2003. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U. S. A. 100:12995–13000. 10.1073/pnas.1735582100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR. 1984. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34:597–603. 10.1212/WNL.34.5.597 [DOI] [PubMed] [Google Scholar]
- 36.Graham RL, Denison MR. 2006. Replication of murine hepatitis virus is regulated by papain-like proteinase 1 processing of nonstructural proteins 1, 2, and 3. J. Virol. 80:11610–11620. 10.1128/JVI.01428-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gadlage MJ, Denison MR. 2010. Exchange of the coronavirus replicase polyprotein cleavage sites alters protease specificity and processing. J. Virol. 84:6894–6898. 10.1128/JVI.00752-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A. 2010. Loss of human greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. U. S. A. 107:12564–12569. 10.1073/pnas.0914191107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Curtis KM, Yount B, Baric RS. 2002. Heterologous gene expression from transmissible gastroenteritis virus replicon particles. J. Virol. 76:1422–1434. 10.1128/JVI.76.3.1422-1434.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kilianski A, Mielech AM, Deng X, Baker SC. 2013. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 87:11955–11962. 10.1128/JVI.02105-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfefferle S, Schopf J, Kogl M, Friedel CC, Muller MA, Carbajo-Lozoya J, Stellberger T, von Dall'Armi E, Herzog P, Kallies S, Niemeyer D, Ditt V, Kuri T, Zust R, Pumpor K, Hilgenfeld R, Schwarz F, Zimmer R, Steffen I, Weber F, Thiel V, Herrler G, Thiel HJ, Schwegmann-Wessels C, Pohlmann S, Haas J, Drosten C, von Brunn A. 2011. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 7:e1002331. 10.1371/journal.ppat.1002331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagemeijer MC, Verheije MH, Ulasli M, Shaltiel IA, de Vries LA, Reggiori F, Rottier PJ, de Haan CA. 2010. Dynamics of coronavirus replication-transcription complexes. J. Virol. 84:2134–2149. 10.1128/JVI.01716-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagemeijer MC, Ulasli M, Vonk AM, Reggiori F, Rottier PJ, de Haan CA. 2011. Mobility and interactions of coronavirus nonstructural protein 4. J. Virol. 85:4572–4577. 10.1128/JVI.00042-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gadlage MJ, Graham RL, Denison MR. 2008. Murine coronaviruses encoding nsp2 at different genomic loci have altered replication, protein expression, and localization. J. Virol. 82:11964–11969. 10.1128/JVI.01126-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lauber C, Goeman JJ, Parquet MC, Nga PT, Snijder EJ, Morita K, Gorbalenya AE. 2013. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog. 9:e1003500. 10.1371/journal.ppat.1003500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Somogyi P, Jenner AJ, Brierley I, Inglis SC. 1993. Ribosomal pausing during translation of an RNA pseudoknot. Mol. Cell. Biol. 13:6931–6940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plant EP, Perez-Alvarado GC, Jacobs JL, Mukhopadhyay B, Hennig M, Dinman JD. 2005. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 3:e172. 10.1371/journal.pbio.0030172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plant EP, Sims AC, Baric RS, Dinman JD, Taylor DR. 2013. Altering SARS coronavirus frameshift efficiency affects genomic and subgenomic RNA production. Viruses 5:279–294. 10.3390/v5010279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.