

ABSTRACT
To identify novel stimulators of the innate immune system, we constructed a panel of eight HEK293 cell lines double positive for human Toll-like receptors (TLRs) and an NF-κB-inducible reporter gene. Screening of a large variety of compounds and cellular extracts detected a TLR3-activating compound in a microsomal yeast extract. Fractionation of this extract identified an RNA molecule of 4.6 kb, named nucleic acid band 2 (NAB2), that was sufficient to confer the activation of TLR3. Digests with single- and double-strand-specific RNases showed the double-strand nature of this RNA, and its sequence was found to be identical to that of the genome of the double-stranded RNA (dsRNA) L-BC virus of Saccharomyces cerevisiae. A large-scale process of production and purification of this RNA was established on the basis of chemical cell lysis and dsRNA-specific chromatography. NAB2 complexed with the cationic lipid Lipofectin but neither NAB2 nor Lipofectin alone induced the secretion of interleukin-12(p70) [IL-12(p70)], alpha interferon, gamma interferon-induced protein 10, macrophage inflammatory protein 1β, or IL-6 in human monocyte-derived dendritic cells. While NAB2 activated TLR3, Lipofectin-stabilized NAB2 also signaled via the cytoplasmic sensor for RNA recognition MDA-5. A significant increase of RMA-MUC1 tumor rejection and survival was observed in C57BL/6 mice after prophylactic vaccination with MUC1-encoding modified vaccinia virus Ankara (MVA) and NAB2-Lipofectin. This combination of immunotherapies strongly increased at the injection sites the percentage of infiltrating natural killer (NK) cells and plasmacytoid dendritic cells (pDCs), cell types which can modulate innate and adaptive immune responses.
IMPORTANCE Virus-based cancer vaccines offer a good alternative to the treatment of cancer but could be improved. Starting from a screening approach, we have identified and characterized an unexplored biological molecule with immunomodulatory characteristics which augments the efficacy of an MVA-based immunotherapeutic agent. The immune modulator consists of the purified dsRNA genome isolated from a commercially used yeast strain, NAB2, mixed with a cationic lipid, Lipofectin. NAB2-Lipofectin stimulates the immune system via TLR3 and MDA-5. When it was injected at the MVA vaccination site, the immune modulator increased survival in a preclinical tumor model. We could demonstrate that NAB2-Lipofectin augments the MVA-induced infiltration of natural killer and plasmacytoid dendritic cells. We suggest indirect mechanisms of activation of these cell types by the influence of NAB2-Lipofectin on innate and adaptive immunity. Detailed analysis of cell migration at the vaccine injection site and the appropriate choice of an immune modulator should be considered to achieve the rational improvement of virus vector-based vaccination by immune modulators.
INTRODUCTION
We set out to identify novel stimulators of the innate immune system which would augment the efficacy of active immunotherapies against cancer or chronic viral infections. Ideally, such stimulators or adjuvants should modulate in a rational and understandable manner the course and intensity of an immune response generated against disease-specific antigens and thus improve disease outcomes (1).
The concept of active immunotherapy is based on using one's own immune system to specifically target tumor-specific antigens (reviewed in reference 2). Tumor antigens can be delivered as autologous tumor cell preparations, in the form of tumor antigen-derived peptides, or via gene transfer vectors like the attenuated poxvirus modified vaccinia virus Ankara (MVA) (3). MVATG9931, named TG4010 in clinical trials, is an MVA-based therapeutic vaccine delivering an expression cassette for the tumor antigen mucin 1 (MUC1) (4) and human interleukin-2 (IL-2). Phase II clinical data suggest that the combination of chemotherapy with TG4010 enhances the therapeutic effects against advanced non-small cell lung cancer (NSCLC) (5). The efficacy of therapeutic MVA-based vaccines was also demonstrated in chronic human hepatitis C virus (HCV)-infected patients (6) and in patients with human papillomavirus (HPV)-associated cervical intraepithelial neoplasia (7). In these studies, the MVA vector expressing the HCV- or HPV-specific proteins was shown to generate adaptive immune responses against the respective expressed virus proteins. As such, a clinical proof of concept for MVA-based therapeutic vaccines has been provided, yet the therapeutic efficacy of such vaccines is calling for improvement of their potency.
In order to initiate or promote antigen-specific reactions, active immunotherapy approaches include those that use adjuvants which stimulate various arms of the immune cascade. It has been demonstrated that pathogen-associated molecular patterns (PAMPs) which are recognized by receptors of the innate immune system act as adjuvants for tumor vaccines (8). The efficacy of PAMP-derived adjuvants depends on their ability to activate antigen-presenting cells (APCs) through their pattern recognition receptors (PRRs) in order to shape the adaptive immune response.
PRRs were tailored by nature to sense PAMPs specific for invading microorganisms or endogenous danger-associated molecular patterns (DAMPs) (9). Major classes of PRRs are the Toll-like receptors (TLRs), the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and the nucleotide-oligomerization domain (NOD)-like receptors (NLRs).
While RLRs and NLRs are cytoplasmic sensors, TLRs are membrane-associated proteins. To date, at least 11 different TLRs have been identified in humans (10). In immune cells, TLRs 1, 2, 4, 5, and 6 are exposed on the cell surface, and they mainly recognize bacterial components, such as lipopeptides and peptidoglycans (TLRs 1, 2, and 6), lipopolysaccharides (LPSs; TLR4), or flagellin (TLR5). TLRs 3, 7, 8, and 9 are predominantly located in intracellular vacuoles or endosomes, and they respond to nucleic acid structures, such as double-stranded RNA (dsRNA; TLR3), single-stranded RNA (ssRNA; TLRs 7 and 8), and unmethylated CpG motifs in bacterial DNA or synthetic oligonucleotides (TLR9) (11). TLR-mediated signaling pathways involve recruitment of Toll/interleukin-1 receptor (TIR) domain-containing adaptors, such as myeloid differentiation primary response protein 88 (MyD88) or TIR domain-containing adaptor-inducing beta interferon (IFN-β) (TRIF), thereby leading to the activation of transcription factors, such as nuclear factor-κB (NF-κB) or IFN regulatory factors (IRFs), which regulate the expression of genes that encode proinflammatory cytokines and type I IFNs, respectively.
Clinical studies have shown that the recombinant therapeutic MAGE-A3 vaccine, directed against a commonly expressed tumor antigen, can be reinforced by coinjecting the adjuvant AS02B, consisting of the TLR4 ligand monophosphoryl lipid A (MPL) formulated with the saponin QS21 (12): the presence of adjuvant AS02B was a prerequisite for the development of MAGE-A3-specific immunity in NSCLC cancer patients. An ongoing phase III clinical trial in NSCLC patients, the MAGRIT trial, is based on the coinjection of MAGE-A3 with the adjuvant AS15, comprising MPL, QS21, and a TLR9 ligand (ClinicalTrials.gov identifier NCT00480025 [13]).
Due to their viral nature, MVA-based vaccines import their intrinsic PAMPs: MVA was described to stimulate the TLR2/6 receptor, the AIM-2-containing (14, 15) and NLRP3-containing (16) inflammasomes, and the cytoplasmic RLR melanoma differentiation antigen 5 (MDA-5) (16). The detection of viral DNA via cyclic GMP (cGMP)-AMP synthase (cGAS), which generates the second messenger cGAMP(2′-5′), which in turn activates STING, was recently suggested (17). Despite the presence of internal immune modulators, it was observed that an external adjuvant, the TLR1/2 ligand Pam3Cys, can further augment the immunotherapeutic efficacy of an MVA-based vaccine in a mouse model of MUC1-positive tumors (18).
Our goal was to identify and characterize new adjuvants which augment the efficacy of an MVA vaccine against MUC1-positive tumors. Using our human embryonic kidney 293 (HEK293) cell-based TLR screening platform, we have identified, out of a crude microsomal Saccharomyces cerevisiae yeast extract, an RNA molecule of 4.6 kb, called nucleic acid band 2 (NAB2), which stimulated TLR3-positive HEK-TLR-luc cells. When it was associated with the cationic lipid Lipofectin, NAB2 stimulated all HEK-TLR-luc cell lines, which we ascribed to the stimulation of the ubiquitous receptor MDA-5. We could show that NAB2 is double stranded and that its sequence is identical to the one of the genomic dsRNA L-BC virus of Saccharomyces cerevisiae. A production and purification process based on dsRNA-specific chromatography was established. NAB2-Lipofectin induced the maturation of human monocyte-derived dendritic cells (moDCs) and the secretion of IL-12(p70) and IFN-α, gamma interferon-induced protein 10 (IP-10), macrophage inflammatory protein 1β (MIP-1β), and IL-6. Finally, injection of NAB2-Lipofectin 24 h after prophylactic vaccination with MVATG9931 significantly increased RMA-MUC1 tumor rejection and survival in a mouse model. To delineate the mode of action, we characterized tumors at the onset of regression; vaccination with MVATG9931 increased the percentage of CD45+, CD8+, and cleaved caspase-3-positive cells in the tumor, while the number of CD4+ cells decreased. The injection of NAB2-Lipofectin did not alter these profiles. In contrast, compared to the injection of MVATG9931 alone, injection of NAB2-Lipofectin 24 h after injection of MVATG9931 increased the percentages of NK cells and activated plasmacytoid dendritic cells (pDCs) at the vaccination site.
MATERIALS AND METHODS
HEK293 cell lines stably transfected with hTLR and NF-κB-inducible luciferase expression cassette.
HEK293 cells expressing TLRs of human origin (hTLRs) were obtained from InvivoGen (293/hTLR2/CD14, 293/hTLR3, 293/hTLR4/MD2CD14, 293/hTLR5, 293/hTLR6, 293/hTLR7, 293/hTLR8, and 293/hTLR9). Cells were cultured in Dulbecco's minimal Eagle's medium (DMEM) supplemented with 10% inactivated fetal calf serum (FCS), 40 μg/ml gentamicin sulfate (Schering-Plough), 2 mM l-glutamine (Sigma), 1 mM sodium pyruvate (Sigma), and 0.1 mM nonessential amino acids (minimal essential medium). All cell lines were grown in the presence of 10 μg/ml blasticidin S (InvivoGen); hygromycin B (Roche) was added at a concentration of 100 μg/ml to the cell lines 293/hTLR2-CD14 and 293/hTLR4-MD2-CD14.
All hTLR cell lines were stably transfected with the NF-κB-inducible reporter plasmid pNiFty2-luc (InvivoGen) encoding the firefly luciferase gene under the control of an engineered ELAM1 promoter which combines five NF-κB sites and the proximal ELAM promoter, as well as a zeocin resistance gene. Emerging clones, named HEK-hTLR-luc, were tested for luciferase activity after stimulation with the respective TLR-specific ligands. Subclones were obtained by limiting dilutions.
Stimulation with specific ligands or test compounds and luciferase assay.
HEK-hTLR-luc clones were seeded in 96-well plates and on the next day were stimulated with the test compounds and their respective specific ligands (all ligands were purchased from InvivoGen): synthetic diacetylated lipoprotein FSL-1 (Pam2CGDPKHPKSF; hTLR2 and hTLR2/6), LPS (hTLR4), poly(I·C) (hTLR3), flagellin (hTLR5), R848 (hTLR7, hTLR8), or ODN2006 (hTLR9) (see Table 1). On the day after that, cells were lysed in 100 μl buffer containing 25 mM Tris, pH 7.8, 2 mM EDTA, 1 mM dithiothreitol (DTT), 0.5% Triton X-100, and 10% glycerol. Firefly luciferase activity was quantified in 10 μl of lysate by integral measurement of flash luminescence over 1 s following injection of 50 μl luciferase revelation buffer (20 mM Tris, pH 7.8, 1 mM MgCl2, 2.7 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 470 μM d-luciferin firefly [Euromedex], 530 μM ATP, 270 μM coenzyme A) using Berthold Tristar LB941 and Greiner white 96-well plates.
TABLE 1.
EC50s and fold induction of selected HEK-hTLR-luc clones using the indicated control ligands at the indicated cell densitiesa
| HEK293 hTLR-luc cell line | Ligand | EC50 | Fold induction | No. of cells/96-well plate |
|---|---|---|---|---|
| HEK-hTLR2-luc | FSL-1 | 2.9 nM | 157 | 1 × 104 |
| HEK-hTLR3-luc | Poly(I·C) | 1.8 ng/ml | 62 | 9 × 103 |
| HEK-hTLR4-luc | LPS | 0.9 ng/ml | 17 | 2.5 × 103 |
| HEK-hTLR5-luc | Flagellin | 66.2 ng/ml | 392 | 1.5 × 104 |
| HEK-hTLR2/6-luc | FSL-1 | 0.64 nM | 140 | 7.5 × 104 |
| HEK-hTLR7-luc | R848 | 2.9 × 10−7 M | 126 | 4 × 104 |
| HEK-hTLR8-luc | R848 | 3 × 10−5 M | 261 | 6 × 104 |
| HEK-hTLR9-luc | ODN2006 | 0.68 μM | 11 | 5 × 104 |
The TLR screening platform was based on HEK293 cells stably transfected with hTLR 2, 3, 4, 5, 2/6, 7, 8, or 9 and an NF-κB-controlled luciferase expression cassette.
The relative light units (RLU) obtained with escalating doses of specific ligands were analyzed with GraphPad Prism (version 5) software (GraphPad Software, San Diego, CA) using an equation for sigmoid dose-response to determine the median (50%) effective dose (EC50) and fold induction (ratio between maximum and minimum induction [max/min]) (see Fig. S1 in the supplemental material).
Identification of NAB2 from yeast microsomal extract.
Yeast strain S. cerevisiae JC7 was grown in YPDA medium (1% yeast extract, 2% peptone, 2.5% glucose, 100 μg/ml adenine) at 28°C to a final cell density of an optical density at 600 nm (OD600) of 2 ± 0.5. Cells were harvested by centrifugation, and the cell pellet was dissolved in phosphate-buffered saline (PBS) to an OD600 of the resulting suspension of about 100. Cells were broken by vigorous mixing with glass beads. The cell lysate was centrifuged at 3,400 × g for 10 min at 4°C to eliminate membrane debris and nuclei. The supernatant was ultracentrifuged for 90 min at 287,000 × g and 4°C. The pellet was dissolved in PBS, resulting in a suspension of the microsomal fraction named SN.
To extract nucleic acids, SN was treated twice with equal volumes of Tris-buffered phenol (Amresco), followed by two extractions with dichloromethane. Nucleic acids were recovered from the aqueous phase by ethanol precipitation; the pellet was dissolved in TE buffer (10 mM Tris, pH 7.5, 1 mM EDTA). The resulting solution of microsomal nucleic acids was named NA.
NA was fractionated by gel electrophoresis in a 1% native agarose gel in the presence of ethidium bromide. Individual bands (NAB1, NAB2, etc.) were cut out under mild UV. The nucleic acids present in excised agarose blocks were extracted after 2 freeze-thaw cycles by centrifugation across a cotton barrier. The nucleic acids in the flowthrough were ethanol precipitated.
RNase profiling of NAB2.
To assess the presence of double- or single-stranded regions, NAB2 (4 μg) was submitted to hydrolysis by single-strand-specific RNase T2 and by double-strand-specific RNase V1 and RNase III. NAB2 was first denatured in water for 1 min at 95°C, followed by 1 min at 4°C. Then, the RNA was allowed to refold for 15 min at 37°C in buffer containing 50 mM sodium cacodylate, pH 7.5, 300 mM KCl, and 5 mM MgCl2. Hydrolysis was performed after the addition of 2 μl of RNase III (3 μM), RNase T2 (0.025 U/μl), or RNase V1 (0.004 U/μl) in a final volume of 140 μl. Control and RNase III digests were incubated for 15 min at 37°C, the RNase T2 digest was incubated for 10 min at 20°C, and the RNase V1 digest was incubated for 5 min at 20°C. All reaction mixtures were mixed with 5 μl of 7 M urea containing dyes, and the mixtures were then migrated on a 6% polyacrylamide–8 M urea–TBE (Tris-borate-EDTA) gel. The RNA was revealed by gel staining with Stains-All (Sigma-Aldrich).
Electron microscopy.
Electron microscopy analysis of NAB2 was performed by depositing 4 μl of the NAB2 preparation at a concentration of 35 μg/ml on a carbon-coated electron microscopy grid that had been freshly glow discharged in amylamine. After 40 s of adsorption, the sample was stained with a solution of uranyl acetate (2% [wt/vol] uranyl acetate in double-distilled water) for 40 s, and the excess stain was washed away with distilled water. After drying, the grid was placed in a platinum evaporator and rotary shadowed at an angle of 15°. The platinum-shadowed nucleic acid molecules were observed in a transmission electron microscope (model CM120; FEI) equipped with an LaB6 filament and operating at 100 kV (1 K = 1024 × 1024 pixels). Images were recorded on a Peltier cooled 1K slow-scan charge-coupled-device camera (model 794; Gatan, Pleasanton, CA) at a magnification of ×19,000. Length and curvature measurements were performed using Digital Micrograph software.
Scaled-up purification of NAB2.
S. cerevisiae yeast strain JC7 grown in YPDA medium was harvested by centrifugation and lysed as described previously (19), with the following modifications. Cell pellets were washed with 50 mM EDTA, pH 8.3, and then stirred in lysis buffer (50 mM Tris-HCl, 50 mM EDTA, 3% SDS, 1% β-mercaptoethanol; the pH was adjusted to 8.0) for 2 h at 20°C. After centrifugation for 20 min at 5,000 × g, ethanol was added to the supernatant to a final concentration of 15%. Double-stranded RNA-specific CF11 phase (Whatman) was added, and the suspension was incubated at 20°C for 90 min under gentle mixing. CF11 was recovered by centrifugation at 5,000 × g and resuspended in STE buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA) containing 15% ethanol. CF11 was then packed in an XK column (GE Healthcare) which was plugged into an Äkta purifier (GE Healthcare). The CF11 was washed overnight, and the dsRNA molecule NAB2 was eluted from the CF11 using STE buffer. Peak fractions were pooled and submitted to a second CF11 purification cycle. The pool of fractions containing NAB2 was then loaded on a CIM DEAE disk monolith column (BIA Separations) (20) equilibrated in buffer A (25 mM Tris-HCl, pH 7.5). NAB2 was then eluted by a linear gradient of buffer A to buffer B (25 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 1.5 M NaCl). Fractions containing dsRNA were pooled and dialyzed (Pierce Slide-A-Lyser dialysis cassette; molecular weight cutoff, 10,000) against TE buffer overnight at 4°C. NAB2 was quantified by determination of the absorbance at 260 nm using a dsRNA extinction coefficient of 45 ml · μg−1 · cm−1. The endotoxin level was determined using a portable test system (PTS; Charles River) according to the provider's recommendations. The dsRNA was stored at −20°C until use.
Complexation of NAB2 with Lipofectin.
NAB2 was complexed with the transfection reagent Lipofectin (Invitrogen). Lipofectin is a 1:1 (wt/wt) liposome formulation of the cationic lipid N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA) and dioleoylphosphatidyl-ethanolamine (DOPE) in filtered water. Typically, 0.3 μg NAB2 and 1.5 μl Lipofectin were mixed in 30 μl TE buffer, pH 7.5, incubated for 20 min, and either added to cell cultures or injected into mice.
Determination of sequence identity of NAB2 by reverse transcription-PCR (RT-PCR).
For first-strand cDNA synthesis, 100 ng of NAB2 purified from Saccharomyces cerevisiae strain JC7 was reverse transcribed using an Advantage RT-for-PCR kit and random hexamer primers (Clontech).
For the PCR analysis, two sets of primers pairs were designed for the specific identification of Saccharomyces cerevisiae virus (ScV) strain L-A (GenBank accession number J04692) or L-BC (GenBank accession number U01060). For the detection of ScV L-A, primers OTG19482 (GGGTCGGTGAGCCTGGGCCTGATGCTCTG) and OTG19483 (CGCGCATTTATCCGGTGCATAGCG) were used, while primers OTG19484 (GCTATTCATCATGGCGAGGGCCGAAGGG) and OTG19485 (CGCGAACTCCCGTGGCTGCGGCGTTT) were used for the detection of ScV L-BC. All PCRs were performed in a final volume of 50 μl. Each reaction mixture contained 5 μl of cDNA, 5 μl of 10 μM each primer, 5 μl of 10× PCR buffer (Qiagen), 1.5 μl of a 10 mM deoxynucleoside triphosphate mix, 1 μl Taq polymerase (Qiagen), and 27.5 μl of distilled water. Following denaturation at 95°C for 5 min, DNA was amplified using 35 cycles at 95°C for 45 s, 55°C for 45 s, and 72°C for 75 s. A final extension step was performed at 72°C for 7 min.
To sequence NAB2, reverse and forward PCR primers were synthesized on the basis of the published sequence of ScV L-BC. Four primers pairs (OTG19708 [GAATTTTTCGGTGAACCGGAATTAT] and OTG19720 [CTCGATTCATAACAGGCCGTCCTTG], OTG19710 [TCCGGGCGGTATACTCACGAGGATG] and OTG19719 [GCATATTGCTGCCCGACAAACTTGG], OTG19712 [CAACAAGACGCATACACGCGCTTGG] and OTG19485 [CGCGAACTCCCGTGGCTGCGGCGTTT], OTG19713 [TTCACGTCCAGAAGGCCGGAAATAC] and OTG19715 [TGCGTAGTATCGTATGTACTCTTTG]) were identified as allowing the amplification of overlapping PCR fragments. JC7 NAB2 dsRNA sequences were determined by sequencing their four corresponding amplified PCR fragments.
Stimulation of human IGROV cells knocked down for RIG-I or MDA-5.
The human ovarian cell line IGROV was stably transfected with lentiviral vectors carrying short hairpin RNA (shRNA) against RIG-I or against MDA-5. Clones with permanent integration of the viral vector were seeded, and knockdown of RIG-I and MDA-5 was confirmed to be less than 5% by RT-PCR (G. Hartmann, unpublished data). Cells were kept in DMEM containing 10% FCS, 1.5 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Cells were stimulated by transfection with deoxyribosyladenine- deoxyribosylthymine (dAdT), 5′-triphosphate single-stranded RNA (3P-RNA) (from in vitro transcription [IVT]), or poly(A). Nucleic acids (0.2 μg) were mixed with 0.5 μl Lipofectamine LF2000 in 50 μl Opti-MEM, incubated for 20 min, and added to the well. For transfection of poly(I·C), 0.2 μg nucleic acids and 0.5 μl LT2 transfection reagent (Mirus) were mixed in 50 μl Opti-MEM, incubated for 20 min, and added to the well. For NAB2, 0.5 μg was mixed with 1.5 μl of Lipofectin in 20 μl of TE buffer, and the mixture was incubated for 20 min and added to the well.
The amount of IP-10 in cell supernatant taken 24 h after stimulation was determined by enzyme-linked immunosorbent assay (BD Biosciences).
Isolation and stimulation of human primary immune cells.
Human peripheral blood mononuclear cells (PBMCs) were isolated from whole human blood of healthy donors (Etablissement Français du Sang, Strasbourg, France) by Ficoll-Hypaque density gradient centrifugation. Human monocytes, pDCs, and NK cells were obtained by negative depletion from PBMCs according to the manufacturer's instructions (Miltenyi Biotec, Germany). Cells were cultured in RPMI (Gibco) supplemented with 10% inactivated fetal calf serum, 40 μg/ml gentamicin (Sigma), and 2 mM l-glutamine (Sigma). Monocytes were differentiated in dendritic cells (moDCs) by 3 days of incubation in granulocyte-macrophage colony-stimulating factor (20 ng/ml) and IL-4 (10 ng/ml) (Peprotech).
For stimulation assays, cells were plated in 24- or 96-well plates and incubated overnight with the test substances. On the next day, the amount of secreted cytokines was determined by FlowCytomix assays (eBioscience) performed according to the manufacturer's protocol and by using a FACSCanto A flow cytometer (Becton, Dickinson [BD]). The concentration of cytokines was determined from the standard curve obtained using known amounts of recombinant cytokines and the software FlowCytomix Pro (version 2.4; eBioscience).
Human moDC maturation markers CD83 and CD86 were detected by flow cytometry using a multicolor kit for CD86 phycoerythrin (PE)/CD209 peridinin chlorophyll protein (PerCP)-Cy5.5/CD83 allophycocyanin (APC) (BD) according to the manufacturer's protocol and by using a FACSCanto A flow cytometer and the analysis software Diva (Becton, Dickinson).
The functionality of NK cells which encounter target cells was measured by a CD107a degranulation assay: 1 × 106 NK cells were incubated overnight with the test substances. On the next day, 1 × 106 major histocompatibility complex (MHC) class I-deficient K562 cells were added and the mixture was incubated at 37°C in 5% CO2 in the presence of APC-conjugated anti-CD107a antibody (BD), monensin (1:1,500; BD), and brefeldin A (1:1,000) to monitor NK degranulation. Living cells were identified using LIVE/DEAD stain (fixable near-infrared dead cell stain kit; Invitrogen), and cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-human CD56 (clone AF12-7H3; Miltenyi Biotec) and PE-conjugated anti-human CD69 (clone FN50; Miltenyi Biotec).
Murine RMA-MUC1 tumor cells.
RMA-MUC1 tumor cells are derived from C57BL/6 RMA lymphoma cells (21) transfected with an expression plasmid for the human MUC1 gene (RMA-MUC1) (22). Cells were cultured in DMEM supplemented with 10% inactivated FCS, 40 μg/ml gentamicin sulfate, 2 mM l-glutamine, 1 mM sodium pyruvate, and 0.1 mM nonessential amino acids in the presence of 550 μg/ml hygromycin B. Immediately before subcutaneous injection, the cells were washed twice in PBS and suspended in PBS at 107 cells/ml.
Recombinant MVA-MUC1 vector.
MVATG9931 is a highly attenuated MVA-based recombinant vaccinia virus expressing human MUC1 sequences and human IL-2. This vector was generated by homologous recombination between the two expression cassettes and the empty vector MVATGN33.1 in primary chicken embryo fibroblasts (CEFs) as described earlier (23). The synthetic MUC1 gene containing five tandem repeats corresponding to cDNA sequences obtained from the human breast carcinoma cell line T47D was inserted downstream of the natural pH5R vaccinia virus promoter (24). The human IL-2 gene was inserted downstream of the p7.5K promoter (25). Both expression cassettes were introduced in deletion II of the viral genome.
MVATG9931 and the empty MVA vector MVATGN33.1 were produced in primary CEFs. The purified virus was resuspended in storage S08 buffer containing 10 mM Tris-HCl, pH 8, 5% (wt/vol) sucrose, 10 mM sodium glutamate, and 50 mM NaCl and stored at −80°C. All virus stocks were plaque titrated on CEF cells and were typically present at between 5 × 108 and 109 PFU per ml. Viruses were diluted in S08 buffer to the final concentrations required for the in vivo studies.
RMA-MUCI tumor model.
C57BL/6 mice were obtained from Charles River (L'Arbresle, Les Oncins, France). Animals were used when they were between 7 and 10 weeks age. Mice (12 animals per group) were vaccinated by three subcutaneous injections (one injection per week for 3 weeks) of 1 × 103 PFU MVATG9931. NAB2-Lipofectin (0.3 μg and 1.5 μl, respectively) was injected at the same site either simultaneously or with a delay of 6 h or 24 h. Twenty-one days after the first injection, mice received 1 × 106 RMA-MUC1 tumor cells by subcutaneous injection. All injections were carried out on the same flank. During the following 40 to 60 days, tumor rejection and animal survival were monitored.
Tumor growth was monitored with calipers twice per week and estimated according to the formula (4/3) × π[(length/2) × (width/2) × (thickness/2)] and expressed in mm3. Tumor rejection and mouse survival were recorded. Mice were sacrificed for ethical reasons when the tumor volume was greater than 2,000 mm3.
This study was conducted in compliance with CEE directive 86/609 of 24 November 1986 on the harmonization of laws, regulations, or administrative provisions relating to the protection of animals used for experimental or other scientific purposes. It was also conducted in compliance with French décret no. 87.848 of 19 October 1987, modified by décret no. 2001-464 of 29 May 2001 and décret no. 2001-486 of 6 June 2001. From 1 February 2013, CEE directive 2010/63/UE of 22 September 2010 and French décret no. 2013-118 of 1 February 2013 were applied.
Infiltration studies. (i) Skin.
The flanks of C57BL/6 mice were shaved and subcutaneously injected with S08 buffer or 5 × 105 PFU MVATG9931 on days 1 and 8; on days 2 and 9, 0.3 μg NAB2 mixed with 1.5 μg Lipofectin was injected. Mice were sacrificed at day 10, and approximately 1 cm2 of skin was excised around the injection site. Skin samples were cut down to small pieces of 1 mm3, transferred into PBS-containing C-type tubes (Miltenyi Biotec), and mechanically dissociated (GentleMACS; Miltenyi Biotec) by twice applying program B. Cells were passed through filters (pore size, 70 μm), washed twice in PBS, and prepared for immunofluorescence staining and flow cytometric analysis.
(ii) Tumor.
Vaccinated C57BL/6 mice were sacrificed between days 11 and 14 after tumor cell implantation, at the onset of palpable tumor regression. Tumors were isolated, and 9 to 10 tumors per group were pooled, cut into small pieces, and transferred into GentleMACS C-type tubes (Miltenyi Biotec) containing 8 ml PBS. Tumors were dissociated by applying the ImpTumor1 program. Cells were passed through filters (pore size, 70 μm), layered over Lympholyte-M cell separation medium (Cedarlane Laboratories), and centrifuged. Lymphocytes collected from the interphase and the cell pellet were washed and prepared for immunofluorescence staining and flow cytometric analysis.
Living cells were identified using LIVE/DEAD staining. FITC-conjugated rat anti-mouse murine PDCA-1 (mPDCA-1; clone JF05-1C2.4.1) was purchased from Miltenyi Biotec (Germany), FITC-conjugated rat anti-mouse 7/4 (clone ab53453) was purchased from Abcam (United Kingdom), and APC-conjugated anti-mouse Langerin (clone 929F3.01) was purchased from Dendritics (France). APC-conjugated rat anti-mouse CD4 (clone GK1.5) and rat anti-mouse F4/80-PE were obtained from eBioscience (France). PE-CF594-conjugated hamster anti-mouse CD3e (clone 145-2C11), FITC- or V450-conjugated rat anti-mouse CD8a (clone 53-6.7), V500-conjugated hamster anti-mouse CD3 (clone 500A2), PE-Cy7-conjugated hamster anti-mouse CD11c (clone HL3), V500-conjugated rat anti-mouse CD11b (clone M1/70), V450- or PerCP-Cy5.5-conjugated rat anti-mouse CD19 (clone ID3), PerCP- or PE-Cy7-conjugated anti-mouse CD45 (clone 30-F11), PE-conjugated rat anti-mouse CD45R (clone RA3-6B2), V450-conjugated rat anti-mouse CD86 (clone GL1), V450-conjugated anti-mouse Ly6C (clone AL-21), PE-conjugated rat anti-mouse Gr1 (clone RB6-8C5), APC-conjugated anti-mouse Ly6G (clone 1A8), and V450- or AF700-conjugated rat anti-mouse NKp46 (clone 29A1.4) were purchased from BD Biosciences (Le Pont de Claix, France). BV711-conjugated rat anti-mouse CD11b (clone M1/70) was obtained from BioLegend (San Diego, CA). PE-conjugated rat anti-mouse cleaved caspase 3 (clone 5A1E) was obtained from Cell Signaling (Boston, MA).
Depending on the experiments, cells were acquired on a FACSCanto A or FACSAria III flow cytometer (Becton, Dickinson) or a Navios cytometer (Beckman Coulter). Accordingly, analyses were performed with Diva or Kaluza (Beckman Coulter) software.
Statistical analyses.
Mouse survival was analyzed by a log-rank test using Statistica software (StatSoft). Hazard ratio calculations were carried out to identify significant differences between groups. Mann-Whitney tests were performed for individual comparisons of two independent groups. Wilcoxon tests were performed for individual comparisons of paired groups. Statistical analysis was performed with GraphPad Prism (version 5) software. P values of <0.05 were considered significant.
RESULTS
Generation of a TLR screening platform.
To generate a TLR ligand screening platform, HEK293 cell lines expressing human TLR (hTLR) 2, 3, 4, 5, 2/6, 7, 8, or 9 were stably transfected with a firefly luciferase expression cassette under the control of an NF-κB-inducible ELAM1 promoter. Emerging clones were probed at various cell densities by stimulation with the respective specific control ligand at escalating doses. Clones with the lowest EC50, the highest fold induction, and the best growth characteristics (defined as a doubling time of less than 48 h and robust adherent phenotype) were chosen. The characteristics of the selected HEK-hTLR-luc clones using the indicated control ligands are summarized in Table 1.
The specificity of each cell line was determined by testing its stimulation by every other TLR ligand in a dose-response experiment (see Fig. S1 in the supplemental material). Specificity was observed for all ligand/HEK-hTLR-luc combinations except poly(I·C), which stimulated not only HEK-hTLR3-luc but also HEK-hTLR5-luc and HEK-hTLR7-luc. We ascribed this to the endogenous TLR3 expression described for a variety of human and murine tumor cell lines (26, 27).
The 4.6-kb RNA molecule NAB2 stimulates TLR3.
The TLR screening platform was used to analyze more than 3,000 compounds, among which there was a microsomal yeast extract. This extract was generated by crushing yeast cells by vigorous mixing with glass beads. The supernatant obtained after clearance from the cell debris and nuclei was ultracentrifuged, and the resulting pellet was dissolved (schematically depicted in Fig. 1A). This extract, referred to as SN, stimulated luciferase activity 60-fold in the HEK-hTLR3-luc cell line. Such a level of activation was comparable to that observed with the poly(I·C) TLR3 control ligand (Table 1).
FIG 1.
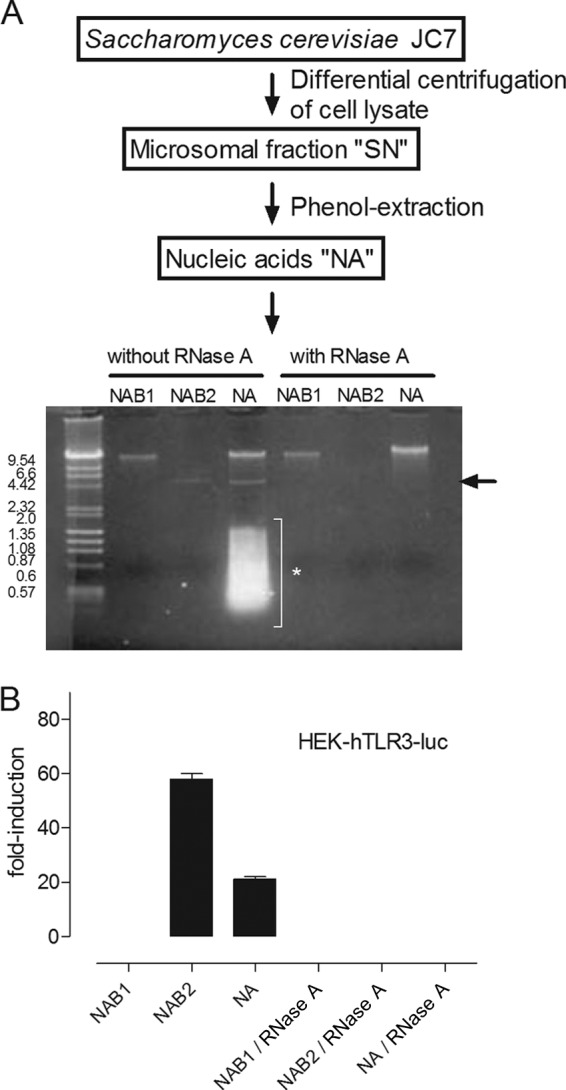
(A) (Top) Schematic representation of fractionation of yeast strain S. cerevisiae JC7. Cell lysis followed by differential centrifugation generated SN, a suspension of yeast microsomes. Phenol extraction of SN removed lipids and proteins and resulted in a nucleic acid solution (NA). (Bottom) Next, NA was fractionated by electrophoresis in a native 1% agarose gel. The first and second bands were isolated from the gel and digested in the presence of high-dose RNase A (50 ng/μl). NAB1 was RNase A resistant, while NAB2 and the lower-molecular-mass molecules appearing as smears were digested. Arrow, NAB2; asterisk, lower-molecular-mass molecules. (B) NA, NAB1, and NAB2 were treated with RNase A or not and tested on the HEK-hTLR3-luc cell lines. NAB2, like NA, increased the fold induction of luciferase activity. NAB1 had no effect. Treatment with RNase A abolished TLR3 activation by NA and NAB2. The means ± SEMs of two independent experiments are shown.
Ligands for TLR3 are synthetic or viral dsRNA molecules. Assuming that the component in SN stimulating luciferase activity in HEK-hTLR3-luc cells was of a nucleic acid nature, we isolated nucleic acids from SN by phenol-chloroform extraction (schematically depicted in Fig. 1A). The resulting nucleic acid preparation (referred to as NA) was separated in a native agarose gel. We could distinguish a band of more than 20 kb (NA band 1 [NAB1]), another one of about 4.6 kb (NA band 2 [NAB2]), and low-molecular-mass nucleic acids heterogeneous in size (between 1.8 and 0.2 kb). All bands except NAB1 disappeared after a high dose of RNase A (Fig. 1A). When they were tested in the HEK-hTLR-3-luc cell line, NA and NAB2, but not NAB1, induced luciferase activity. This activation was not detected when NA or NAB2 was treated with RNase A before incubation with the HEK-hTLR3-luc cell line (Fig. 1B).
Hence, we have identified an approximately 4.6-kb RNA molecule, NAB2, out of a complex microsomal yeast extract that stimulated the HEK-hTLR3-luc cell line.
NAB2 stimulates TLR3, and NAB2-Lipofectin stimulates MDA-5.
In order to stabilize NAB2 and to favor its cellular uptake, we mixed NAB2 with the cationic lipid Lipofectin. NAB2-Lipofectin and NAB2 or Lipofectin alone were tested in dose-response experiments on all HEK-TLR-luc cell lines. The fold inductions of luciferase activity are shown in Fig. S3 in the supplemental material. NAB2 associated with Lipofectin activated all HEK-hTLR-luc cell lines. In cell lines which could be stimulated with NAB2 alone, e.g., the HEK-hTLR3-luc cell line, levels of luciferase activity equal to or even higher than those achieved with NAB2 alone were observed with 81-fold less NAB2 when it was associated with Lipofectin. Lipofectin on its own had no effect on any of the cell lines at any concentration tested (see Fig. S3 in the supplemental material).
We anticipated that NAB2-Lipofectin-dependent transfection of HEK-hTLR-luc cells could stimulate cytoplasmic RLRs, such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation antigen 5 (MDA-5). We could detect transcripts for both genes by RT-PCR in all HEK-hTLR-luc cell lines (see Fig. S4 in the supplemental material). RIG-I and MDA-5 are helicases which recognize distinct viral and synthetic RNA (28). Both recognize cytoplasmic dsRNA but differ in the length of the molecule that they recognize: dsRNA longer than 2,000 bp is preferentially recognized by MDA-5, while shorter dsRNA and 5′-triphosphate single-stranded RNA (3P-RNA) are ligands for RIG-I (29, 30).
To assess the involvement of RLR detection pathways, we tested NAB2-Lipofectin in the TLR7-negative human ovarian cell line IGROV knocked down for either RIG-1 or MDA-5 expression due to the permanent integration of respective shRNA lentiviral vectors (Hartmann, unpublished). Activation of IGROV cells was measured by quantifying secreted IP-10, a surrogate marker for a type I IFN response (31). The lack of IP-10 induction by the RIG-I ligand 3P-RNA in RIG-I-knockdown IGROV cells confirmed the functional knockdown of RIG-I (Fig. 2). The similar levels of induction of IP-10 with and without RIG-I knockdown by poly(I·C) confirmed that RIG-I-knockdown IGROV cells showed no generally impaired IP-10 response. IP-10 induction by NAB2-Lipofectin was slightly diminished but not abrogated in RIG-I-knockdown cells. As expected, IP-10 induction by poly(I·C) was almost abrogated in MDA-5-knockdown IGROV cells, confirming that the knockdown was functional. The induction of IP-10 secretion observed for NAB2-Lipofectin was abolished in MDA-5-knockdown IGROV cells.
FIG 2.

Stimulation of IGROV cells knocked down for either RIG-1 or MDA-5. Cells were incubated with NAB2-Lipofectin and transfected with the MDA-5 ligand poly(I·C), the RIG-I ligand 3P-RNA (in vitro-transcribed 5′-triphosphate RNA), and the negative-control RNA poly(A) or poly(dA·dT) ligand to assess TLR-independent stimulation by DNA. Secreted IP-10, a surrogate marker for type I IFN induction, was measured in IGROV cells in which RIG-I (A) and MDA-5 (B) were knocked down, and the results were compared with those for the wild type (wt). The activity of NAB2-Lipofectin to induce IP-10 was abolished in MDA-5-knockdown IGROV cells. The means ± SEMs of two independent experiments are shown. w/o, no ligand treatment.
These data indicate that NAB2 alone stimulated TLR3, while NAB2-Lipofectin also stimulated MDA-5.
Configuration of NAB2.
The activation of TLR3 and MDA-5 by NAB2 and NAB2-Lipofectin, respectively, strongly suggested that NAB2 is mainly double stranded, as are the control ligands for these receptors. To address the existence of single-stranded subdomains, we treated NAB2 with double-strand- or single-strand-specific RNases. The digestions were carried out under nearly physiological concentrations with divalent cations (5 mM MgCl2) (32). Double-strand-specific RNase III and RNase V1 completely digested NAB2, while NAB2 treated with the single-strand-specific RNase T2 migrated similarly to the untreated control (Fig. 3A). This experience confirmed that NAB2 is double stranded and argues against the existence of exposed single-stranded loop domains.
FIG 3.

NAB2 configuration. (A) NAB2 was heat denatured in water and renatured in high-concentration MgCl2 buffer. Then, NAB2 was digested with the dsRNA-specific RNase III and RNase V1 and single-strand RNA-specific RNase T2. Urea and dyes were added, and the samples were run on a 6% polyacrylamide-urea slab gel under denaturing conditions. Lane 1, NAB2; lane 2, NAB2 digested with RNase III; lane 3, NAB2 digested with RNase T2; lane 4, NAB2 digested with RNase V1; lanes L, RNA ladder (numbers on the left right are in bases). NAB2 is sensitive to the two dsRNA-specific RNases. (B) Electron microscopy analysis of NAB2. Platinum-shadowed nucleic acid molecules were observed in a transmission electron microscope. Analyses revealed a linear molecule without bulges or forks.
Electron microscopy analyses revealed a linear molecule without any bulge that could result from an unpaired region or a fork that could result from a more complex hybridization pattern (Fig. 3B). Measurements of the molecule resulted in an average length of 1,414 ± 72 nm (n = 6), which corresponds to 4,503 ± 229 nucleotides, assuming a rise of 0.314 nm per nucleotide. This value is consistent with the electrophoretic mobility of NAB2.
These results indicate that NAB2 is a double-stranded and linear RNA molecule.
Sequence identity of NAB2.
Double-stranded RNA of 4.6 kb isolated from a cytoplasmic, microsomal yeast extract may be identical to yeast dsRNA viruses belonging to the Totiviridae family (33). Members of this family are S. cerevisiae viruses (ScVs) L-A and L-BC. ScVs L-A and L-BC are dsRNA viruses forming icosaedric particles encapsidating their dsRNA genomes of approximately 4.6 kb. These viruses do not lyse yeast cells, nor does their presence slow yeast growth. The viruses are transmitted from cell to cell at the moment of cell budding. The L-A virus is associated with satellite RNAs called M encoding a toxin and toxin resistance genes which confer a killer phenotype to the yeast hosting this virus. M is described to be double stranded, of approximately 1.8 kb, and present in separate cytoplasmic particles (33).
We probed NAB2 by RT-PCR with primers generated against the sequences of ScVs L-A and L-BC, which have only 42% sequence homology. A single PCR product was obtained with the primer set corresponding to ScV L-BC. The PCR amplicons were further analyzed by sequencing. An amplified fragment of 809 bp derived from NAB2 shared 99.4% identity with ScV L-BC, thus indicating that NAB2 is genomic RNA from the yeast virus L-BC (for the alignment of the NAB2 and ScV L-BC sequences, see Sequence Alignment S1 in the supplemental material).
Modified purification protocol for NAB2.
Since NAB2 is a double-stranded RNA molecule and is most likely found in viral capsids present in the cytoplasm of S. cerevisiae JC7 yeast cells, the purification protocol was modified in order to increase the yield and purity for further immunological studies in vitro and in vivo.
Briefly, yeast cells and capsids were chemically lysed in the presence of 3% SDS and 1% β-mercaptoethanol. Released dsRNA was purified from cleared cell lysate by affinity chromatography using dsRNA-specific CF11 phase (34), followed by one ion-exchange chromatography step. Fractions with peaks at OD260 were run on a 1% agarose gel. Figure 4 shows the enrichment of a single nucleic acid band in the course of the purification process. Purified dsRNA ran at 4.6 kb, and RT-PCR analysis of the obtained RNA confirmed that it was NAB2. Typical yields of NAB2 were about 5 μg of NAB2 per liter of yeast culture grown to an OD600 of 1. Endotoxin levels ranged from 0.01 to 0.03 endotoxin units/μg NAB2.
FIG 4.
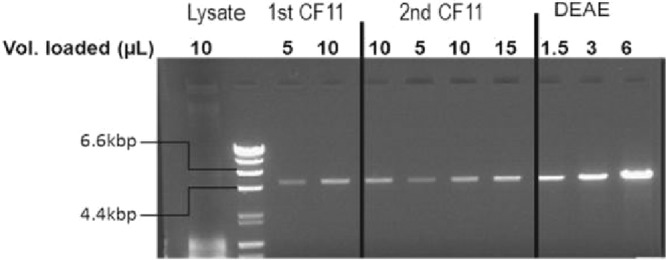
Enrichment of NAB2 following the improved purification method for NAB2. Agarose electrophoresis analysis of the different pools was performed after each step of the purification. A total of 1.5 to 15 μl of each pool was loaded on a 1% agarose gel. Lysate (204 ml), supernatant after lysis and centrifugation; 1st CF11 (190 ml), pool after the CF11 capture step; 2nd CF11 (125 ml), pool after the polishing CF11 step, DEAE (7 ml), pool after the DEAE step. The double-stranded DNA markers in the second lane are 23, 9.5, 6.6, 4.4, 2.3, 2, 1.3, 1.1, 0.9, and 0.6 kbp. The 6.6- and 4.4-kbp bands are indicated.
Thus, with this modified protocol we were able to purify NAB2 in sufficient amounts and to a sufficient quality to assess its immunostimulatory characteristics in vitro and in vivo.
Assessment in human primary immune cells.
To assess their immunomodulatory capacities, we tested NAB2 and NAB2-Lipofectin in human monocyte-derived dendritic cells (moDCs). Human moDCs are positive for the cytoplasmic RIG-I-like receptors RIG-I and MDA-5, as well as TLR3, TLR4, and TLR8 (11, 35). Therefore, the TLR control ligands LPS, R848, and poly(I·C) were included in the analysis. While these ligands activate proinflammatory cytokine responses via interaction with their respective TLRs, poly(I·C) can, in addition, induce a type I interferon response by signaling via the cytoplasmic RLRs (35).
After overnight stimulation of the moDCs from three donors, the upregulation of the maturation markers CD83 and CD86 was measured and the cytokine concentration in the cell culture supernatant was quantified (Fig. 5). NAB2-Lipofectin, but not Lipofectin or NAB2 alone, increased the levels of CD83 and CD86 on the cell surface. A comparable pattern was observed with LPS, poly(I·C), and R848 (Fig. 5A).
FIG 5.

Activation of human moDCs. (A) Human monocyte-derived dendritic cells (7.5 × 105) from three healthy donors were seeded once (donor C) or three times (donors A and B) and incubated overnight with 0.3 μg NAB2, 1.5 μg Lipofectin, or 0.3 μg NAB2 plus 1.5 μg Lipofectin. The TLR4 ligand LPS (5 ng), the TLR7/8 ligand R848 (10−4 M), and the TLR3 ligand poly(I·C) (0.3 μg) were added as controls. The dendritic cell marker CD209 and the maturation markers CD83 and CD86 were quantified by flow cytometry. (B) The secreted cytokines IFN-α, IP-10, IL12(p70), IL-6, and MIP-1β were quantified by FlowCytomix analysis. The means ± SDs for donors A and B are shown. medFI, median fluorescence intensity.
IFN-α was detected in the supernatant of moDCs treated with poly(I·C) or NAB2-Lipofectin, but it was not observed when cells were treated with NAB2 or Lipofectin alone. IP-10 and the proinflammatory cytokines IL-6, MIP-1β, and IL-12(p70) were secreted after stimulation with NAB2-Lipofectin, LPS, R848, or poly(I·C) but not after incubation with NAB2 or Lipofectin (Fig. 5B).
Next, we analyzed the effect of NAB2, Lipofectin, and NAB2-Lipofectin in isolated human pDCs and NK cells. Direct stimulation of NK cells with these test compounds (Fig. 6) did not augment the CD107a degranulation induced by MHC class I-deficient K562 cells. In contrast, an increase of the percentage of CD107ahi cells was observed when NK cells were incubated with the supernatant of moDCs treated with NAB2-Lipofectin (Fig. 6). Plasmacytoid dendritic cells are TLR3 negative and TLR7 and TLR9 positive. They do not signal via cytoplasmic RLR signaling pathways (28). In agreement, isolated human pDCs were not stimulated in the presence of NAB2 or NAB2-Lipofectin (see Fig. S5 in the supplemental material).
FIG 6.

CD107a degranulation assay with human NK cells. Human NK cells (1 × 106) isolated from two healthy donors (donors D and E) were stimulated directly with 0.3 μg NAB2, 1.5 μg Lipofectin, 0.3 μg NAB2 plus 1.5 μg Lipofectin, R848 (10−4 M), or poly(I·C) (0.3 μg) or indirectly with 50 μl of supernatant from moDCs (donors A and B) treated with the same substances (Fig. 5). The percentages of CD107ahigh NK cells in the presence of K562 cells is shown. The percentage of CD107ahigh NK cells in the absence of K562 for all conditions was below 2% (data not shown).
We conclude that NAB2 stabilized with Lipofectin is capable of inducing the maturation of human moDCs and the secretion of IFN-α, IP-10, and proinflammatory cytokines. Human pDCs and NK cells are not a direct target of NAB2-Lipofectin-mediated immune modulation. CD107a degranulation in NK cells, however, can indirectly be activated by the supernatant of moDCs treated with NAB2-Lipofectin.
Assessment in murine RMA-MUC1 tumor model.
Next, we tested the capacity of NAB2-Lipofectin to enhance MVA-mediated vaccination against an MUC1-positive tumor.
To do this, mice were subcutaneously injected with 1 × 103 PFU of MVATG9931. At this dose, tumor rejection and survival rates were about 30 to 10%, allowing monitoring of further improvement; an empty MVA vector had no effect (see Fig. S6 in the supplemental material). NAB2 or NAB2-Lipofectin was injected subcutaneously 24 h after each injection of the MVA vector. Figure 7A shows that tumor rejection increased significantly when NAB2-Lipofectin was combined with MVATG9931 compared to the level of tumor rejection achieved with MVATG9931 alone. Combining MVATG9931 with NAB2 did not result in statistically significant effects (Fig. 7A), nor did combining MVATG9931 with Lipofectin (data not shown). NAB2-Lipofectin on its own had no effect (data not shown). The increase of efficacy obtained by combining MVATG9931 with NAB2-Lipofectin, determined by reporting of mouse survival, was seen in four independent experiments (Fig. 7B).
FIG 7.
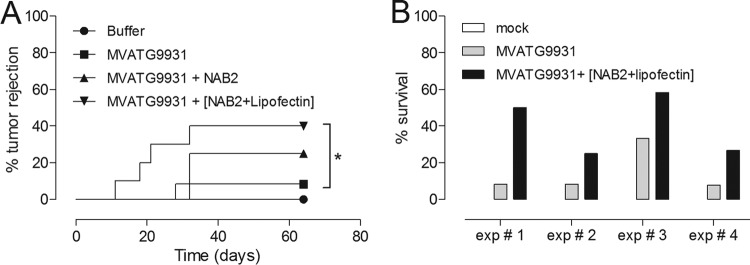
(A) Tumor rejection in C57BL/6 mice vaccinated with MVATG9931 and NAB2-Lipofectin (12 mice per group). (B) NAB2-Lipofectin increases the efficacy of MVATG9931 in four independent experiments. The effect was statistically significant, according to hazard ratio analysis.
Thus, these results demonstrate that the external immune modulator NAB2-Lipofectin can increase the beneficial effect of an MVA-based tumor vaccine.
Mode of action in vivo.
We had observed that NAB2-Lipofectin had to be injected several hours after the viral vector in order to augment tumor regression (see Fig. S7 in the supplemental material); in contrast, coinjection of the MVA vector and NAB2-Lipofectin reduced the rate of survival compared to that achieved with the vector alone. While the latter observation could be due to the induction of an antiviral state (28), we assumed that the action of NAB2-Lipofectin injected at a later time was dependent on the infiltration of receptive cell populations.
Therefore, we set out to analyze the cellular infiltration at the MVA injection site. The skin around the injection site was cut out according to the schedule depicted in Fig. 8A. The samples were mechanically dissociated, and isolated cells were prepared for immunofluorescence analyses. pDCs were identified as a Ly6C+ mPDCA-1+ CD45R+ CD11b− CD11clow subpopulation within living CD45+ CD3 CD19− NKp46− cells. In the same subpopulation, macrophages were identified as CD11c− CD11b+ Ly6G− Ly6C+ F4/80+ cells, and neutrophils were as identified as Ly6G+ Ly6C+ 7/4+ cells. NK cells were identified as the CD11c− CD11b− CD3− NKp46+ cell population. The percentage of these various cell types within the total cell population was calculated, and the results are expressed as the fold induction on the basis of the values obtained with the buffer-injected control group.
FIG 8.
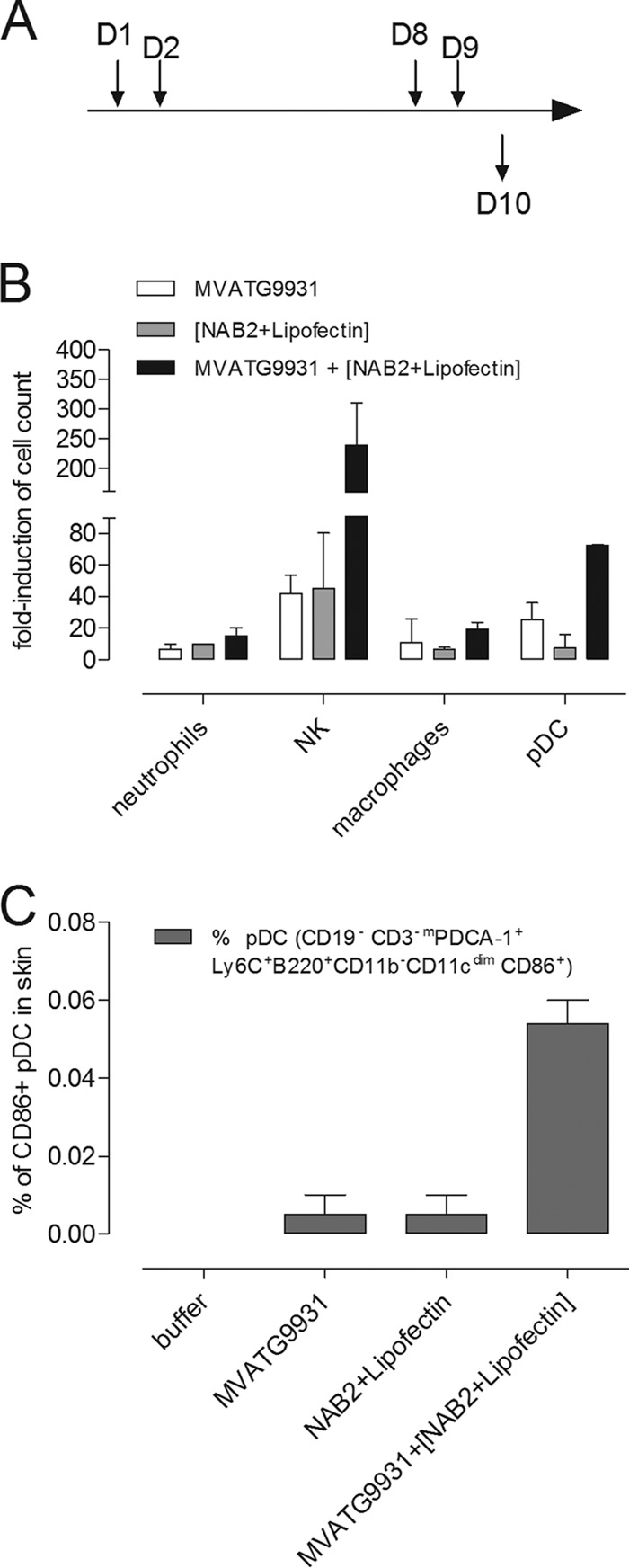
Skin infiltration assay. (A) Mice were subcutaneously injected on day 1 (D1) and day 8 (D8) with 5 × 105 PFU of MVATG9931. NAB2-Lipofectin (0.3 μg and 1 μg, respectively) was injected on day 2 (D2) and day 9 (D9). Mice were sacrificed on day 10 (D10). Skin around the injection site was cut out and mechanically dissociated. Suspensions of cells from 10 to 14 injection sites were immunofluorescently stained and analyzed by flow cytometry. (B) The percentages of pDCs macrophages, neutrophils, and NK within the total population were calculated, and the fold induction is expressed on the basis of the values obtained with the negative-control group (buffer injection). (C) The percentage of CD86+ pDCs in the total cell population was determined. The means ± SEMs of two independent experiments are shown.
Twenty-four hours after the injection of MVATG9931, we observed the infiltration of pDCs, NK cells, neutrophils, macrophages, and a subpopulation of dermic dendritic cells (E. Schaedler, C. Remy-Ziller, J. Hortelano, M.-C. Claudepierre, A. Carpentier, X. Preville, and K. Rittner, unpublished data). The same picture emerged 48 h after the injection of MVATG9931 (Fig. 8B): neutrophils, NK cells, macrophages, and pDCs were 5- to 40-fold more abundant at the injection site than they were in the buffer-injected control. When MVA injection was followed by NAB2-Lipofectin treatment, the infiltration of pDCs and NK cells was augmented during the next 24 h (Fig. 8B): compared to the percentage of NK cells in the buffer-injected control, the percentage of NK cells increased more than 200-fold, 6 times greater than the percentage observed with MVATG9931 or NAB2-Lipofectin. About 70 times more pDCs infiltrated at the injection site, representing more than twice as many pDCs as the amount that infiltrated after MVA injection. More interestingly, among the infiltrating pDCs, the percentage of activated CD86-positive cells (36) was augmented (Fig. 8C). The infiltration level of macrophages and neutrophils remained unchanged after injection of NAB2-Lipofectin (Fig. 8B).
Next, we characterized cell populations in implanted RMA-MUC1 tumors and draining lymph nodes at the onset of regression. To do this, C57BL/6 mice injected with buffer, MVATG9931 alone, or MVATG9931 followed by NAB2 or NAB2-Lipofectin were sacrificed between days 11 and 14 after tumor cell implantation. Tumors were dissociated and separated via density gradient centrifugation into PBMCs, polymorphonuclear cells, and a tumor cell fraction.
When mice had been treated with MVATG9931, the percentage of tumor-derived cleaved caspase 3-positive cells was significantly increased, as was the case when mice had been treated with MVATG9931 and NAB2-Lipofectin (Fig. 9B). Within the population of living tumor-derived PBMCs, the percentage of CD45+ cells was augmented significantly after treatment with MVATG9931 alone or in combination with NAB2-Lipofectin (Fig. 9A). In all MVA-treated animals, tumors showed a significant increase of CD8+ PBMCs. The significant decrease of CD4+ PBMCs in the tumors was accompanied by an increase of this cell type in the draining lymph nodes (data not shown). The infiltration of NK cells was not statistically significantly different in the treated groups (Fig. 9A). Thus, looking at tumors at the chosen time point, we could not identify a parameter which was specific for animals treated with MVATG9931 and NAB2-Lipofectin.
FIG 9.

Tumor infiltration assay. C57BL/6 mice injected with buffer, MVATG9931 alone, or MVATG9931 followed by NAB2 or NAB2-Lipofectin were sacrificed between days 11 and 14 after tumor cell implantation. Pooled tumors (10 per group) were mechanically dissociated and separated via density gradient centrifugation into PBMCs (A) and the tumor cell fraction (pellet after density gradient) (B). (A) Living PBMCs were stained for CD4, CD8a, and NKp46 (CD335). (B) Percentages of cleaved caspase 3 (casp 3)-positive cells in the tumor cell fraction. The means ± SEMs of three experiments are shown.
In conclusion, NAB2-Lipofectin influenced the MVA-dependent cell profile at the injection site, with a higher affluence of NK cells and activated pDCs.
DISCUSSION
Today's search for immune modulators or adjuvants is inspired by emerging concepts of how innate and adaptive immunity synergize (1). The TLR screening platform presented in this work offers the opportunity to discover new immune modulators and, at the same time, to have an idea of their mode of action by knowing which TLR is targeted. By applying this platform and with subsequent tests in isolated primary immune cells, we could identify an immune modulator, NAB2, which turned out to be the genome of the yeast dsRNA virus L-BC. Yeast viruses like L-BC or L-A are part of our nutritional import, since they are present in all commercially relevant yeast strains used, e.g., for the production of wine, beer, and baked goods. We have established a production and purification process which allows NAB2 to be generated in sufficient amounts and of a sufficient quality to assess this adjuvant candidate in vitro and in preclinical studies. The potential use of NAB2 in clinical studies would require further scaling up of the process.
Using our TLR screening platform, NAB2 was identified starting from an extract of S. cerevisiae strain JC7 containing a complex mixture of cell wall components, lipids, proteins, membranes, and microsomes. This extract stimulated TLR3-, TLR2-, and TLR2/6-positive HEK-TLR-luc cell lines. The nucleic acids and, more precisely, the NAB2 extracted from this suspension stimulated only TLR3-positive HEK-TLR-luc cell lines. NAB2 is a dsRNA molecule migrating at a distinct band of 4.6 kb and in this respect is different from the standard TLR3 ligand poly(I·C), which is heterogeneous in size and appears as a smear in an agarose gel (data not shown). The analysis of the accessibility of NAB2 toward double- and single-strand-specific RNases strongly suggested that NAB2 does not contain single-stranded loop structures, motifs which could stimulate TLR7 or TLR8. In accordance with this, we did not observe activation of human TLR7/8-positive pDCs. Using the TLR platform and IGROV cells knocked down for RIG-I or MDA-5, we demonstrated that NAB2 stimulates the innate immune receptors TLR3 and MDA-5 when it is associated with Lipofectin.
Interestingly, NAB2 alone could stimulate the HEK-hTLR3-luc cell lines but not TLR3-positive human moDCs. We explain this observation by the differential localization of TLR3 in both cell types. In moDCs, TLR3 resides inside the cells in endosomes but not on the cell surface (37). Entry of a naked 4.6-kb dsRNA molecule is most likely not efficient. Compacting NAB2 with the transfection reagent Lipofectin would allow cellular uptake and allow NAB2 to encounter TLR3. In contrast, TLR3 is present on the cell surface of HEK293 cells (see Fig. S8 in the supplemental material), as has been described for human fibroblasts (37), and is thus accessible for naked NAB2.
Studies with RLR-knockdown IGROV cells suggested that NAB2-Lipofectin stimulated MDA-5. We assume that the IFN-α production in moDCs was due to the stimulation of this cytoplasmic receptor for long dsRNA. Secretion of the proinflammatory cytokine IL-12(p70), IL-6, or MIP-1β in moDCs could be ascribed to TLR3 stimulation.
Thus, the association of NAB2 with Lipofectin allows NAB2 to enter the cytoplasm of moDCs to stimulate MDA-5 or to be taken up by endocytosis to stimulate TLR3. It is conceivable that the use of alternative transfection reagents with capacities to deliver NAB2 preferentially to the cytoplasm or the endosomes would allow modification of the resulting cytokine profile. In a vaccination approach, an IFN-α-prone response could promote a memory response, while augmentation of the expression of IL12(p70) could augment the CD8+ effector response (38).
NAB2-Lipofectin prolonged the survival of mice vaccinated subcutaneously with MVATG9931 and challenged subcutaneously with RMA cells expressing the vaccine antigen MUC1. NAB2-Lipofectin had to be injected several hours after the MVA vector to augment efficacy compared to that achieved with the vector alone. To explain this prerequisite, we analyzed the infiltration of various cell types at the vaccination site. Kremer et al. have reported the massive lung infiltration of monocytes, dendritic cells, neutrophils, and NK cells in the first 6 to 72 h after intranasal vaccination with MVA (39). We could demonstrate a similar scenario 24 to 48 h after subcutaneous injection of MVATG9931: neutrophils, NK cells, as well as macrophages and pDCs assembled around the injection site. When NAB2-Lipofectin was delivered into this environment, the percentage of pDCs increased further, and in particular, the percentage of activated CD86-positive pDCs was augmented. Based on the observation that NAB2-Lipofectin could not activate human pDCs and the notion that the innate receptor profile of murine pDCs is comparable to that of human pDCs (40), we suggest that pDCs are activated by TLR7 or TLR9 ligands liberated from dying cells at the injection site (DAMPs, TLR7 ligands). The toxic effects of NAB2-Lipofectin on HEK293-luc cells had been observed at high doses in titration experiments.
Activated pDCs are described to play a pivotal role in the induction of the innate immune response, e.g., by recruitment of NK cells (41). Indeed, we have observed a strong increase of infiltrating NK cells after injection of MVATG9931 and NAB2-Lipofectin 6 times higher than that observed after injection of MVATG9931 or NAB2-Lipofectin alone. We assume that these NK cells can be activated by IFN-α from activated pDCs (41) and, in analogy to the human situation, also from cytokines released from monocytes and dendritic cells activated by NAB2-Lipofectin. NAB2-Lipofectin, when injected several hours after MVATG9931, would also encounter MVA-infected cells expressing MUC1. We know from tests with empty control vectors that the expression of MUC1 is another prerequisite for tumor control. Activation of antigen presentation by NAB2-Lipofectin and cross talk of APCs with pDCs could engender positive effects on the adaptive immune response against the vectorized tumor antigen (42). Thus, the combination of subcutaneous vaccination with a tumor-encoding MVA vector and injection of NAB2-Lipofectin in a timely deferred manner at the same site could influence the adaptive as well as the innate immune response against the implanted tumor.
Characterizing tumors at the onset of regression, we found that compared to the untreated control, vaccination with MVA induced the infiltration of CD8+ cells and augmented the percentage of dying cells. A more detailed characterization of infiltrating CD8+ cells might allow detection of differences in quality which could be linked to NAB2-Lipofectin. At the chosen time point, we could not demonstrate the infiltration of NK cells into the tumor. Nevertheless, it is conceivable that NK cells present at the vaccination site might be involved in early cytotoxic effects on tumor cells which are implanted in the same flank. The analysis of tumor cell infiltration at an earlier time point might reveal the implication of activated NK cells.
In conclusion, applying a screening approach, we have identified and characterized an immune modulator naturally occurring in yeast. The identified molecule, NAB2, represents yeast virus dsRNA and is a ligand for the innate immune receptors TLR3 and MDA-5. It needs to be associated with a cationic lipid to exert effects in human primary cells and in a mouse tumor model. In a vaccination approach with an MVA vector, we could demonstrate that despite the presence of virus-inherent endogenous adjuvants, the combination with an external adjuvant improved vaccine efficiencies. It is of interest to elucidate in greater detail the sequence of events that occur with subcutaneous vaccination, the activation profile of infiltrating cells, and their interplay when exposed to immunostimulatory molecules/formulations like NAB2-Lipofectin. The acquired knowledge will facilitate the improvement of active immunotherapy with MVA vectors by appropriate modification of the innate immune response and the resulting adaptive immune response using newly identified external adjuvants.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anne-Catherine Helfer, Isabelle Farine, and Murielle Gantzer for excellent technical assistance and Bérangère Marie Bastien, Laetitia Fend, and Nathalie Silvestre for helpful discussions and suggestions.
Footnotes
Published ahead of print 26 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03819-13.
REFERENCES
- 1.Flower DR. 2012. Systematic identification of small molecule adjuvants. Expert Opin. Drug Discov. 7:807–817. 10.1517/17460441.2012.699958 [DOI] [PubMed] [Google Scholar]
- 2.Tucker ZC, Laguna BA, Moon E, Singhal S. 2012. Adjuvant immunotherapy for non-small cell lung cancer. Cancer Treat. Rev. 38:650–661. 10.1016/j.ctrv.2011.11.008 [DOI] [PubMed] [Google Scholar]
- 3.Mayr A, Stickl H, Muller HK, Danner K, Singer H. 1978. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism. Zentralbl. Bakteriol. B 167:375–390 (Authors' translation.) [PubMed] [Google Scholar]
- 4.Bansal AS, Bruce J, Devine PL, Scells B, Zimmermann PV. 1997. Serum cytokines and tumour markers in patients with non-small cell carcinoma of the lung. Dis. Markers 13:195–199 [PubMed] [Google Scholar]
- 5.Quoix E, Ramlau R, Westeel V, Papai Z, Madroszyk A, Riviere A, Koralewski P, Breton JL, Stoelben E, Braun D, Debieuvre D, Lena H, Buyse M, Chenard MP, Acres B, Lacoste G, Bastien B, Tavernaro A, Bizouarne N, Bonnefoy JY, Limacher JM. 2011. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: a controlled phase 2B trial. Lancet Oncol. 12:1125–1133. 10.1016/S1470-2045(11)70259-5 [DOI] [PubMed] [Google Scholar]
- 6.Habersetzer F, Honnet G, Bain C, Maynard-Muet M, Leroy V, Zarski JP, Feray C, Baumert TF, Bronowicki JP, Doffoel M, Trepo C, Agathon D, Toh ML, Baudin M, Bonnefoy JY, Limacher JM, Inchauspe G. 2011. A poxvirus vaccine is safe, induces T-cell responses, and decreases viral load in patients with chronic hepatitis C. Gastroenterology 141:890–899.e1–e4. 10.1053/j.gastro.2011.06.009 [DOI] [PubMed] [Google Scholar]
- 7.Nieminen PHD, Einstein MH, Garcia F, Donders G, Huh W, Wright TC, Stoler M, Ferenzy A, Rutman O, Shikman A, Leung M, Clinch B, Calleja E. 2012. Efficacy and safety of RO5217790, a therapeutic HPV vaccine, in patients with high grade cervical intraepithelial neoplasia (CIN2/3), p 206 Abstr. 28th Int. Papilloma Conf. Clin. Public Health Workshop, San Juan, Puerto Rico [Google Scholar]
- 8.Kratky W, Reis e Sousa C, Oxenius A, Sporri R. 2011. Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. Proc. Natl. Acad. Sci. U. S. A. 108:17414–17419. 10.1073/pnas.1108945108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 10.Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 11.Makela SM, Strengell M, Pietila TE, Osterlund P, Julkunen I. 2009. Multiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cells. J. Leukoc. Biol. 85:664–672. 10.1189/jlb.0808503 [DOI] [PubMed] [Google Scholar]
- 12.Atanackovic D, Altorki NK, Cao Y, Ritter E, Ferrara CA, Ritter G, Hoffman EW, Bokemeyer C, Old LJ, Gnjatic S. 2008. Booster vaccination of cancer patients with MAGE-A3 protein reveals long-term immunological memory or tolerance depending on priming. Proc. Natl. Acad. Sci. U. S. A. 105:1650–1655. 10.1073/pnas.0707140104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyagi P, Mirakhur B. 2009. MAGRIT: the largest-ever phase III lung cancer trial aims to establish a novel tumor-specific approach to therapy. Clin. Lung Cancer 10:371–374. 10.3816/CLC.2009.n.052 [DOI] [PubMed] [Google Scholar]
- 14.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. 10.1038/nature07725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. 2010. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11:395–402. 10.1038/ni.1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delaloye J, Roger T, Steiner-Tardivel QG, Le Roy D, Knaup Reymond M, Akira S, Petrilli V, Gomez CE, Perdiguero B, Tschopp J, Pantaleo G, Esteban M, Calandra T. 2009. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog. 5:e1000480. 10.1371/journal.ppat.1000480 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. 2013. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503:530–534. 10.1038/nature12640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amiset L, Fend L, Gatard-Scheikl T, Rittner K, Duong V, Rooke R, Muller S, Bonnefoy JY, Preville X, Haegel H. 2012. TLR2 ligation protects effector T cells from regulatory T-cell mediated suppression and repolarizes T helper responses following MVA-based cancer immunotherapy. Oncoimmunology 1:1271–1280. 10.4161/onci.21479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fried HM, Fink GR. 1978. Electron microscopic heteroduplex analysis of “killer” double-stranded RNA species from yeast. Proc. Natl. Acad. Sci. U. S. A. 75:4224–4228. 10.1073/pnas.75.9.4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krajacic M, Ivancic-Jelecki J, Forcic D, Vrdoljak A, Skoric D. 2007. Purification of plant viral and satellite double-stranded RNAs on DEAE monoliths. J. Chromatogr. A 1144:111–119. 10.1016/j.chroma.2006.11.081 [DOI] [PubMed] [Google Scholar]
- 21.Karre K, Ljunggren HG, Piontek G, Kiessling R. 1986. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319:675–678. 10.1038/319675a0 [DOI] [PubMed] [Google Scholar]
- 22.Graham RA, Burchell JM, Beverley P, Taylor-Papadimitriou J. 1996. Intramuscular immunisation with MUC1 cDNA can protect C57 mice challenged with MUC1-expressing syngeneic mouse tumour cells. Int. J. Cancer 65:664–670 [DOI] [PubMed] [Google Scholar]
- 23.Fournillier A, Gerossier E, Evlashev A, Schmitt D, Simon B, Chatel L, Martin P, Silvestre N, Balloul JM, Barry R, Inchauspe G. 2007. An accelerated vaccine schedule with a poly-antigenic hepatitis C virus MVA-based candidate vaccine induces potent, long lasting and in vivo cross-reactive T cell responses. Vaccine 25:7339–7353. 10.1016/j.vaccine.2007.08.020 [DOI] [PubMed] [Google Scholar]
- 24.Rosel JL, Earl PL, Weir JP, Moss B. 1986. Conserved TAAATG sequence at the transcriptional and translational initiation sites of vaccinia virus late genes deduced by structural and functional analysis of the HindIII H genome fragment. J. Virol. 60:436–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cochran MA, Puckett C, Moss B. 1985. In vitro mutagenesis of the promoter region for a vaccinia virus gene: evidence for tandem early and late regulatory signals. J. Virol. 54:30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Q, Wei H, Tian Z. 2008. Poly I:C enhances cycloheximide-induced apoptosis of tumor cells through TLR3 pathway. BMC Cancer 8:12. 10.1186/1471-2407-8-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. 2006. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 176:4894–4901 http://www.jimmunol.org/content/176/8/4894.full.pdf+html [DOI] [PubMed] [Google Scholar]
- 28.Takeuchi O, Akira S. 2008. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 20:17–22. 10.1016/j.coi.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 29.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- 30.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601–1610. 10.1084/jem.20080091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Padovan E, Spagnoli GC, Ferrantini M, Heberer M. 2002. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J. Leukoc. Biol. 71:669–676. 10.1189/jlb.1938-3673 [DOI] [PubMed] [Google Scholar]
- 32.Paillart JC, Skripkin E, Ehresmann B, Ehresmann C, Marquet R. 1996. A loop-loop “kissing” complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc. Natl. Acad. Sci. U. S. A. 93:5572–5577. 10.1073/pnas.93.11.5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wickner RB. 1996. Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol. Rev. 60:250–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castillo A, Cottet L, Castro M, Sepulveda F. 2011. Rapid isolation of mycoviral double-stranded RNA from Botrytis cinerea and Saccharomyces cerevisiae. Virol. J. 8:38. 10.1186/1743-422X-8-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabo A, Bene K, Gogolak P, Rethi B, Lanyi A, Jankovich I, Dezso B, Rajnavolgyi E. 2012. RLR-mediated production of interferon-beta by a human dendritic cell subset and its role in virus-specific immunity. J. Leukoc. Biol. 92:159–169. 10.1189/jlb.0711360 [DOI] [PubMed] [Google Scholar]
- 36.Martinez J, Huang X, Yang Y. 2010. Toll-like receptor 8-mediated activation of murine plasmacytoid dendritic cells by vaccinia viral DNA. Proc. Natl. Acad. Sci. U. S. A. 107:6442–6447. 10.1073/pnas.0913291107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, Yamamoto A, Seya T. 2003. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 171:3154–3162 [DOI] [PubMed] [Google Scholar]
- 38.Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. 2009. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood 113:5516–5525. 10.1182/blood-2008-11-188458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kremer M, Suezer Y, Volz A, Frenz T, Majzoub M, Hanschmann KM, Lehmann MH, Kalinke U, Sutter G. 2012. Critical role of perforin-dependent CD8+ T cell immunity for rapid protective vaccination in a murine model for human smallpox. PLoS Pathog. 8:e1002557. 10.1371/journal.ppat.1002557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barchet W, Wimmenauer V, Schlee M, Hartmann G. 2008. Accessing the therapeutic potential of immunostimulatory nucleic acids. Curr. Opin. Immunol. 20:389–395. 10.1016/j.coi.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 41.Guillerey C, Mouries J, Polo G, Doyen N, Law HK, Chan S, Kastner P, Leclerc C, Dadaglio G. 2012. Pivotal role of plasmacytoid dendritic cells in inflammation and NK-cell responses after TLR9 triggering in mice. Blood 120:90–99. 10.1182/blood-2012-02-410936 [DOI] [PubMed] [Google Scholar]
- 42.Nierkens S, den Brok MH, Garcia Z, Togher S, Wagenaars J, Wassink M, Boon L, Ruers TJ, Figdor CG, Schoenberger SP, Adema GJ, Janssen EM. 2011. Immune adjuvant efficacy of CpG oligonucleotide in cancer treatment is founded specifically upon TLR9 function in plasmacytoid dendritic cells. Cancer Res. 71:6428–6437. 10.1158/0008-5472.CAN-11-2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.