

Abstract
APOBEC3F and APOBEC3G cytidine deaminases potently inhibit human immunodeficiency virus type 1 (HIV-1) replication by enzymatically inserting G-to-A mutations in viral DNA and/or impairing viral reverse transcription independently of their deaminase activity. Through experimental and mathematical investigation, here we quantitatively demonstrate that 99.3% of the antiviral effect of APOBEC3G is dependent on its deaminase activity, whereas 30.2% of the antiviral effect of APOBEC3F is attributed to deaminase-independent ability. This is the first report quantitatively elucidating how APOBEC3F and APOBEC3G differ in their anti-HIV-1 modes.
TEXT
Human immunodeficiency virus type 1 (HIV-1), the causative agent of AIDS, hijacks lines of cellular proteins for its replication (reviewed in reference 1). On the other hand, accumulating evidence has revealed that human cells intrinsically possess anti-HIV-1 proteins (reviewed in reference 2). Human apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3) proteins are cellular cytidine deaminases, and certain APOBEC3 proteins, particularly APOBEC3F (3, 4) and APOBEC3G (5), are the intrinsic restriction factors against HIV-1. APOBEC3F and APOBEC3G are incorporated into assembling HIV-1 particles and brought into the newly infected cells. During reverse transcription (RT) in the infected cells, these APOBEC3 proteins enzymatically convert C in the viral minus-strand DNA to U, resulting in G-to-A mutation in the nascent plus-strand DNA. These mutations can become nonsynonymous or even lethal, which severely debilitates subsequent viral replication.
To antagonize these APOBEC3F- and APOBEC3G-mediated antiviral effect, viral infectivity factor (Vif), an accessory protein of HIV-1, degrades APOBEC3F and APOBEC3G through a ubiquitin/proteasome-dependent pathway (reviewed in references 6 and 7). Previous studies have directly demonstrated that Vif is a prerequisite for HIV-1 replication in vitro (e.g., human primary CD4+ T lymphocytes and macrophages) (8, 9) and in vivo (e.g., humanized mouse models) (10, 11). Moreover, G-to-A mutations have been clearly observed in the proviral DNA of HIV-1-infected patients, although the frequencies of G-to-A mutations seem to vary among individuals (12–23).
It has been reported that the incorporated APOBEC3F and APOBEC3G potently impair the HIV-1 RT process even prior to the insertion of G-to-A mutations (24–26). Moreover, this inhibition is independent of these APOBEC3s' deaminase activity (26). These findings indicate that APOBEC3F and APOBEC3G potently inhibit HIV-1 replication through at least two distinct modes: (i) deaminase activity-dependent G-to-A mutations of viral DNA and (ii) impairment of viral RT independent of deaminase activity (Fig. 1). However, how much of the inhibition of HIV-1 replication by APOBEC3 proteins is attributed to each of these two modes has not been quantitatively revealed.
FIG 1.
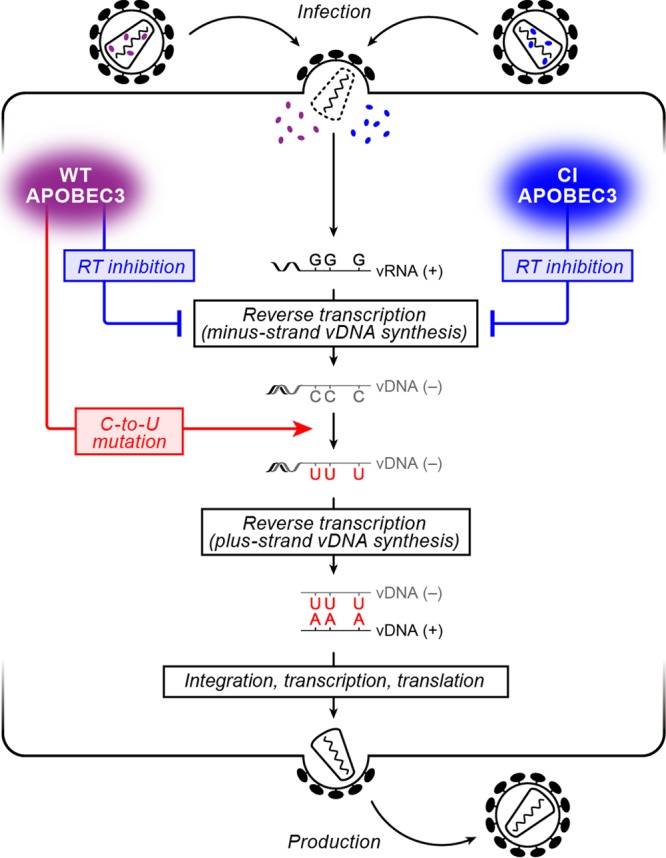
Schematic diagram of the HIV-1 life cycle and the antiviral effect of APOBEC3 proteins. (Left) Incoming WT APOBEC3 proteins (purple) cause (i) the inhibition of the viral RT process independent of their deaminase activity (blue arrow) and (ii) deaminase activity-dependent C-to-U mutations in minus-strand viral DNA (vDNA) resulting in G-to-A mutations in plus-strand vDNA (red arrow), both of which lead to the abrogation of viral replication; (Right) on the other hand, incoming CI APOBEC3 proteins (blue) cause only the inhibition of the viral RT process independent of their deaminase activity (blue arrow).
In order to quantitatively determine the relative extent of deaminase activity-dependent and -independent antiviral activity of APOBEC3 proteins, we performed a single-round infection assay using a cell culture system. 293T cells and TZM-bl cells (obtained through the NIH AIDS Research and Reference Reagent Program) (27) were maintained in Dulbecco's modified Eagle medium (Sigma) containing 10% fetal calf serum (FCS) and antibiotics. Flag-tagged wild-type (WT) APOBEC3F (GenBank accession number NM_145298.5) and WT APOBEC3G (GenBank accession number NM_021822.3) expression plasmids are based on pcDNA3.1 (Life Technologies).
Regarding the deaminase activity of APOBEC3 proteins, it has been reported that the glutamate (E) in the catalytic site of APOBEC3 protein is involved in the proton shuttling during deamination reaction (28). Also, it has been already revealed that APOBEC3F E251 and APOBEC3G E259 are the catalytic glutamates based on mutagenesis experiments (APOBEC3F E251Q and APOBEC3G E259Q, respectively) (26, 29) and their crystal structures (30, 31). Upon these findings, we constructed catalytically inactive (CI) mutants, Flag-tagged APOBEC3F E251Q and APOBEC3G E259Q expression plasmids. These plasmids were constructed by using the GENEART site-directed mutagenesis system (Life Technologies), with WT plasmids as the template and the following primers: APOBEC3F E251Q forward, 5′-ACC CAT TGT CAT GCA CAG AGG TGC TTC CTC TCT-3′; APOBEC3F E251Q reverse, 5′-AGA GAG GAA GCA CCT CTG TGC ATG ACA ATG GGT-3′; APOBEC3G E259Q forward, 5′-GAA GGC CGC CAT GCA CAG CTG TGC TTC CTG GAC-3′; APOBEC3G E259Q reverse, 5′-GTC CAG GAA GCA CAG CTG TGC ATG GCG GCC TTC-3′. The sequences of the constructed plasmids were confirmed by using an ABI Prism 3130xl genetic analyzer (Applied Biosystems), and the data were analyzed by Sequencher v5.1 software (Hitachi).
To investigate the antiviral effect of WT and CI APOBEC3 proteins, 1 μg of a vif-deficient HIV-1-producing plasmid (pNL4-3Δvif; based on HIV-1 strain NL4-3 [GenBank accession number M19921.2]) (32) was cotransfected with the Flag-tagged plasmid expressing either WT APOBEC3F, CI APOBEC3F, WT APOBEC3G, or CI APOBEC3G at 5 different doses (0, 11, 33, 100, or 300 ng) into 293T cells by using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. At 48 h posttransfection, the culture supernatant was harvested, centrifuged, and then filtered through a 0.45-μm-pore-size filter (Millipore) to produce virus solution. To detect a viral protein (p24CA) and virion-incorporated Flag-tagged APOBEC3 proteins, SDS-PAGE/Western blotting was performed as previously described (11, 33–36). Briefly, 1 ml of the virus solution was ultracentrifuged at 100,000 × g for 1 h at 4°C using a TL-100 instrument (Beckman). The pellet was lysed with 1× SDS buffer, and the levels of viral protein (p24CA) and virion-incorporated Flag-tagged APOBEC3 proteins were detected by SDS-PAGE/Western blotting. For the Western blotting, anti-p24CA (ViroStat) and anti-Flag (clone M2; Sigma) antibodies were used. To measure the infectivity of virus solution, the TZM-bl assay was performed as previously described (11, 33–36). Briefly, 100 μl of the virus solution was inoculated into TZM-bl cells, and the β-galactosidase activity was measured by using the Galacto-Star mammalian reporter gene assay system (Roche) and a 1420 ALBOSX multilabel counter instrument (PerkinElmer) according to the manufacturer's protocol. To validate that the CI mutants used in this study (APOBEC3F E251Q and APOBEC3G E259Q) were defective in their deaminase activities, 100 μl of the virus solutions was treated with DNase I (TaKaRa) at 37°C for 2 h for the removal of the transfected plasmids and then was inoculated into TZM-bl cells. DNA was extracted by using the DNeasy blood and tissue kit (Qiagen), and the viral DNA was amplified by PCR using the following primers: forward, 5′-GTT TGG AAA GGA CCA GCA AA-3′; reverse, 5′-GCC CAA GTA TCC CCG TAA GT-3′. PCR was performed by using PrimeSTAR GXL DNA polymerase (TaKaRa) according to the manufacturer's protocol in the following sequence of conditions: (i) 98°C for 2 min; (ii) 98°C for 10 s; (iii) 55°C for 15 s; (iv) 68°C for 45 s, repeat steps ii to iv for 30 cycles; (v) 68°C for 5 min. The resultant 774 bp of the viral DNA fragment was cloned into pCR-Blunt II-TOPO vector (Life Technologies) by using the Zero Blunt TOPO PCR cloning kit (Life Technologies) according to the manufacturer's protocol. The sequencing PCR was performed by using M13 sequencing primers and an ABI Prism 3130xl genetic analyzer (Applied Biosystems), and the data were analyzed by Sequencher v5.1 software (Hitachi) and Hypermut v2.0 (http://www.hiv.lanl.gov/content/sequence/HYPERMUT/hypermut.html) (37).
As shown in Fig. 2A, APOBEC3 expression did not affect the level of released HIV-1 particles. In addition, APOBEC3 proteins were incorporated into the released virions in dose-dependent manners, and the levels of WT APOBEC3 proteins in the released virions were comparable to those of CI mutants (Fig. 2A). Next, we assessed the infectivity of the harvested viruses by TZM-bl assay (11, 33–36) and found that both WT and CI APOBEC3 proteins attenuated HIV-1 infectivity in dose-dependent manners (Fig. 2B). Consistent with previous reports (38–40), the anti-HIV-1 effect of WT APOBEC3G was greater than that of WT APOBEC3F (Fig. 2B). On the other hand, it was of interest that the antiviral effect of CI APOBEC3G (APOBEC3G E259Q) was smaller than that of CI APOBEC3F (APOBEC3F E251Q) (Fig. 2B). We then confirmed that the CI mutants did not insert G-to-A mutations at all, while WT APOBEC3 proteins elicited G-to-A mutations in dose-dependent manners (see Fig. 4A and B). These results strongly suggest that WT APOBEC3 proteins exert both G-to-A mutation-mediated (deaminase activity-dependent) and RT inhibition-mediated (i.e., deaminase activity-independent) anti-HIV-1 effects, whereas CI mutants exert only the latter effect (Fig. 1).
FIG 2.

Antiviral ability of APOBEC3F and APOBEC3G proteins. (A) Western blotting of the released HIV-1 particles. The level of HIV-1 particles released into the culture supernatant was analyzed by Western blotting, and representative results are shown. (B) Quantification of antiviral activity of APOBEC3F and APOBEC3G by experimental and mathematical analyses. The infectivity of released HIV-1 particles was quantified by TZM-bl assay. The assay was performed in triplicate. The averages of experimental data are shown as dots with standard deviations represented by bars, and a graph of the best-fit model is superimposed.
FIG 4.

Mutation signatures and in silico analysis. (A and B) Summary of viral DNA sequence analyzed in this study. (A) Raw sequence data (the sequences are available upon request). (B) Summarized table. a, The number in parenthesis represents the amount of transfected APOBEC3-expressing plasmid; b, the numbers of analyzed nucleotides and clones (in parenthesis) are shown; c, hypermutation index (38, 43) is calculated as follows: (no. of G-to-A mutations − no. of A-to-G mutations)/sequence length (bp) × 1,000; d, the number and the percentage (in parenthesis) of dinucleotide contexts of G-to-A mutation sites in total G-to-A mutations are shown. ND, not determined, probably due to a high level of G-to-A hypermutations mediated by APOBEC3G in the primer-binding region of template DNA that resulted in the failure of the PCR process under this condition (53, 54). (C) In silico analyses. The effects of G-to-A mutations mediated by APOBEC3F (GA to AA, red line) or APOBEC3G (GG to AG, blue line) on the proportion of proviral DNA with stop codons (S, y axis) were estimated with equation 9 as described in the text.
In order to quantitatively estimate the contribution of these two mechanisms of action (i.e., RT inhibition and G-to-A mutation) in restricting HIV-1 replication, we built a mathematical model from the experimental data. Because RT inhibition and G-to-A mutation are independently caused by WT APOBEC3 proteins, their effect as a whole is represented as the union of two independent events. Here, we estimated the combination of each antiviral effect by Bliss independence, which is a major index for the evaluation of drug combination effects (41, 42). We define x as the amount of transfected APOBEC3 expression plasmid (ng), and fRT(x) and fMu(x) are the unaffected fractions of viral infectivity by RT inhibition and G-to-A mutation, respectively. Then, the total effect of the two antiviral activities, fWT(x), is assumed to be the product of the individual effect of the two antiviral activities as follows:
| (1) |
We analyzed the experimental data of viral infectivity at the 4 different doses (Fig. 2A), using the phenomenological model, equation 1. Furthermore, we assume that RT inhibition and G-to-A mutation are dose-dependently described by simple Hill functions of x, and then we have
| (2) |
and
| (3) |
respectively. Here, hRT and hMu are the Hill coefficients, and KRT and KMu are the amounts of transfected APOBEC3 expression plasmid (ng) required to decrease the viral infectivity by 50%. Using equations 2 and 3, the viral infectivities affected by WT and CI APOBEC3 are calculated by
| (4) |
and
| (5) |
respectively. To estimate the parameters hRT, hMu, KRT, and KMu, we fitted the above models fWT(x) and fCI(x) (i.e., equations 4 and 5) to 9 sets of the corresponding experimental measurement of HIV-1 infectivity by using a nonlinear least-squares method, which minimizes the sum of squared residuals (SSR) as follows:
| (6) |
where EWT(xi) and ECI(xi) are the experimental data of the infectivity of viruses released from the cells cotransfected with different doses of WT or CI APOBEC3 expression plasmid xi (i = 1, 2, 3, 4, and 5 correspond to xi = 0, 11, 33, 100, and 300 ng). The principle axis method in Mathematica 8.0 (Wolfram) was used for searching the optimal parameters. The experiments shown in Fig. 2B were performed in triplicate for each WT and CI dose. By pairing these two sets of three values, we obtained 3 × 3 = 9 combinations between WT and CI values. For each combination, we estimated h and K values by fitting (i.e., we obtained 9 h values and 9 K values for APOBEC3F and APOBEC3G, respectively). To calculate the confidence intervals for the estimated parameters, we used a nonparametric bootstrap method with 20,000 resamples of 9 KMu values of APOBEC3F and APOBEC3G, respectively. Using the mean values of the estimated parameters, fMu(x) predicts the anti-HIV-1 effect of APOBEC3F and APOBEC3G solely mediated by G-to-A mutations (Fig. 2B, represented in red lines). Intriguingly, these analyses revealed that the G-to-A mutation-mediated anti-HIV-1 effect of APOBEC3G (hMu of APOBEC3G, 1.93 ± 0.18; KMu of APOBEC3G, 5.38 ± 0.49; 95% confidence interval, 5.09 to 5.69) was significantly greater than that of APOBEC3F (hMu of APOBEC3F, 1.41 ± 0.39; KMu of APOBEC3F, 40.8 ± 21.3; 95% confidence interval, 28.96 to 54.95) (Fig. 2B; KMus between APOBEC3F and APOBEC3G, P = 0.038 by the nonparametric bootstrap method). In addition, sequencing analysis revealed that the hypermutation index (38, 43), which represents the level of G-to-A mutation in viral DNA, of APOBEC3G was higher than that of APOBEC3F (see Fig. 4B). Because the levels of APOBEC3F and APOBEC3G in the released virions were comparable (Fig. 2A), these results suggest that APOBEC3G more frequently inserts G-to-A mutations and, therefore, more potently attenuates viral infection than APOBEC3F does.
Furthermore, to quantitatively estimate the relative contribution of G-to-A mutation and RT inhibition to the anti-HIV-1 effect, we defined the relative contribution rates of G-to-A mutation and RT inhibition to the total antiviral effects, RCMu(x) and RCRT(x), as follows:
| (7) |
| (8) |
where fRT(x) − fWT(x) means the amplification of antiviral effects by adding the G-to-A mutation to the condition of preexisting RT inhibition, and fRT(x) − fWT(x) + fMu(x) − fWT(x) is the sum of each amplification of the antiviral effects by G-to-A mutation and RT inhibition (Fig. 3A). In other words, RCMu(x) represents the contribution of the G-to-A mutation to the total antiviral effects relative to that of the RT inhibition. Calculating RCMu(x) and RCRT(x) in WT APOBEC3F and WT APOBEC3G with equations 7 and 8 shows that RCRT(x) in WT APOBEC3F (30.2% ± 4.18%; 95% confidence interval, 17.07% to 45.38%) was greater than that in WT APOBEC3G (0.66% ± 0.69%; 95% confidence interval, 0.42% to 0.93%) (Fig. 3B; P = 0.043 by the nonparametric bootstrap method).
FIG 3.
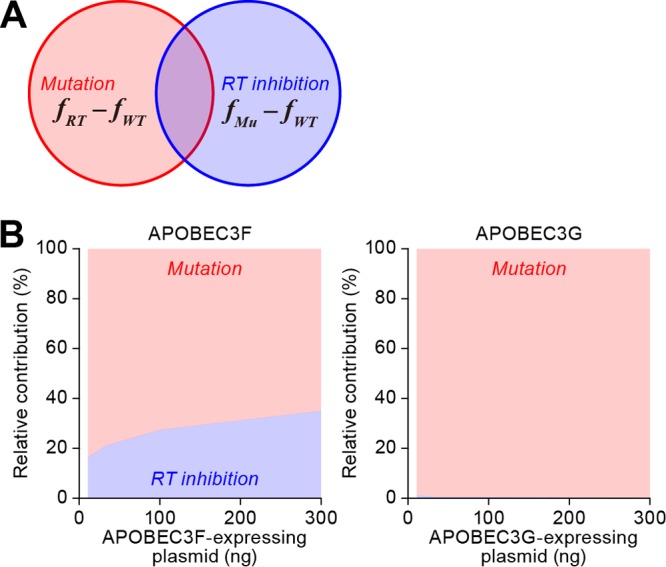
Quantification of mutation-dependent and -independent antiviral effects by APOBEC3F and APOBEC3G. (A) A Venn diagram of the relative contribution rates of deaminase activity-dependent G-to-A mutations [Mutation, RCMu(x)] and deaminase activity-independent RT inhibition [RT inhibition, RCRT(x)] to the total antiviral effect of APOBEC3 proteins. (B) Relative contribution rate of the different modes of antiviral activities with APOBEC3F and APOBEC3G. RCMu(x) (red area) and RCRT(x) (blue area) of APOBEC3F and APOBEC3G are estimated as described in the text. RCRT(x) in APOBEC3F, 11 ng, 26.5% ± 36.7%; 33 ng, 27.9% ± 30.6%; 100 ng, 30.6% ± 20.2%; 300 ng, 36.0% ± 12.9%. RCRT(x) in APOBEC3G, 11 ng, 1.64% ± 1.02%; 33 ng, 0.64% ± 0.41%; 100 ng, 0.26% ± 0.17%; 300 ng, 0.11% ± 0.08%.
Summarizing these mathematical analyses (Fig. 3B), the relative contribution of the deaminase activity-dependent antiviral effect of APOBEC3G [RCMu(x) = 99.3% ± 0.69%] was 150-fold greater than its deaminase-independent (i.e., RT inhibition-dependent) antiviral effect [RCRT(x) = 0.66% ± 0.69%]. This indicates that APOBEC3G strongly depends on its catalytic activity in inhibiting HIV-1 replication. On the other hand, the relative contribution of deaminase activity to the antiviral effect of APOBEC3F [RCMu(x) = 69.8% ± 4.18%] was only 2.3-fold greater than that of its RT inhibition [RCRT(x) = 30.2% ± 4.18%]. These findings suggest that APOBEC3G depends more on its deaminase-dependent antiviral effect than APOBEC3F does. In this regard, it is known that APOBEC3F induces mainly GA-to-AA mutations, while APOBEC3G induces more of GG-to-AG mutations (italics and underlining indicate the mutation) (7, 44). Consistent with previous observations (7, 44), we observed that WT APOBEC3F and WT APOBEC3G inserted GA-to-AA and GG-to-AG mutations, respectively, in dose-dependent manners (Fig. 4B). For a detailed assessment of the impact of G-to-A mutations caused by APOBEC3F and APOBEC3G, we performed in silico analyses. If there are L lethal mutation sites (i.e., TGG [tryptophan] to TAG [stop codon]) in a full-genome proviral DNA sequence, which is constructed by F sites in total, we can calculate the proportion S of proviral DNA with at least one stop codon after the proviral DNA suffers from N times of APOBEC3-mediated mutations per full-length proviral DNA as follows:
| (9) |
To perform this in silico simulation, we obtained 1,596 full-genome HIV-1 sequences registered in the Los Alamos HIV sequence database (http://www.hiv.lanl.gov). In the 1,596 data sets (containing 13,799,872 bases), the following motifs were picked up by using the seqinR package for the R statistical computing environment v2.15.2 (http://www.r-project.org): the number of APOBEC3F-targeted motifs (FAPOBEC3F, the number of GA sites), 1,134,377; the number of APOBEC3F-targeted motifs directly leading to lethal mutation (LAPOBEC3F, the number of TGGA with TGG as a codon, leading to TGAA), 59,788; the number of APOBEC3G-targeted motifs (FAPOBEC3G, the number of GG sites), 929,607; the number of APOBEC3G-targeted motifs directly leading to lethal mutation (LAPOBEC3G, the number of TGG with TGG as a codon, leading to TAG), 151,234. Calculating SAPOBEC3F and SAPOBEC3G with equation 9 shows that APOBEC3G-mediated mutations (GG to AG) quickly become lethal (Fig. 4C; 3.90 mutations with 50% being lethal, 13.0 mutations with 90% being lethal). On the other hand, the APOBEC3F-mediated mutation (GA to AA) less frequently became lethal (Fig. 4C, 12.80 mutations with 50% being lethal, 42.53 mutations with 90% being lethal). Taken together, these findings well explain the differences in the profiles of antiviral activities between these APOBEC3 proteins, which were observed in our experimental and mathematical investigations (Fig. 2B and 3). They strongly suggest that APOBEC3G impairs HIV-1 replication mostly dependent on its deaminase activity. In addition, APOBEC3G is more effective than APOBEC3F because of its (i) higher G-to-A mutation frequency than APOBEC3F (indicated by hypermutation index in Fig. 4B) and (ii) mutation signature readily resulting in lethal mutation (GG-to-AG mutations; Fig. 4C). On the other hand, APOBEC3F suppresses HIV-1 replication with the combination of G-to-A mutations and RT inhibition. In our in silico simulation (Fig. 4C), we assumed that only termination mutations (L) result in the abrogation of viral replication. However, there are other mutations which can result in the failure of viral replication, such as (i) long-terminal repeat, (ii) polypurine tracts, (iii) tRNA-binding regions, (iv) initiation codons (i.e., ATG), and (v) nonsynonymous mutations in viral genes. Therefore, as previously described (44), it should be noted that our simulation (Fig. 4C) may underestimate the antiviral effect of G-to-A mutations caused by APOBEC3.
Although we observed the dose-dependent increase of G-to-A mutations by WT APOBEC3F and APOBEC3G (Fig. 4B), we detected some amplicons without G-to-A mutations (Fig. 4A). This raises a possibility that a portion of viruses released from the cells transfected with APOBEC3-expressing plasmid may not package APOBEC3 proteins. On the other hand, it is known that the viral clones suffered from APOBEC3-mediated G-to-A mutations are less efficiently amplified by PCR than the clones without any mutations, because the primer-binding sites can be also mutated (44). Therefore, the nonmutated amplicons detected (Fig. 4A) may be due to a technical bias on PCR rather than the presence of virions without packaged APOBEC3 proteins. Moreover, a biochemical analysis has previously estimated that approximately 4 to 9 APOBEC3 molecules are packaged per vif-deficient HIV-1 particle (45). However, it seems technically difficult to quantify the absolute number of APOBEC3 molecules per virion. To fully reveal this issue, further investigations will be needed.
APOBEC3-associated mutation patterns (i.e., GG-to-AG and/or GA-to-AA mutations) have been observed in HIV-1-infected CD4+ T lymphocytes (38, 39, 46) and macrophages (38) as well as in infected humanized mouse models (10, 11) and individuals (14, 22), strongly suggesting that some APOBEC3 proteins, at least APOBEC3F and APOBEC3G, are closely associated with HIV-1 infection, replication, and pathogenesis. In addition, APOBEC3F and APOBEC3G are highly associated with single-strand nucleic acids, including viral RNA and DNA, which is a prerequisite for their packaging into the virions and exerting their antiviral activity in the newly infected cells (47–49). APOBEC3F and APOBEC3G catalyze C-to-U mutations by directly binding to minus-strand viral DNA, and the binding of APOBEC3 to minus-strand viral DNA results in the inhibition of the viral RT process, which is independent of APOBEC3's catalytic activity (47, 48). Therefore, it is technically unable to prepare the APOBEC3 mutant causing G-to-A mutations without RT inhibition and to experimentally elucidate the sole effect of deaminase activity of APOBEC3 proteins on HIV-1 replication. By applying mathematical modeling to the experimental data, here we quantitatively estimated the sole effect of G-to-A mutations by APOBEC3 proteins, which cannot be determined through conventional experimental methods (Fig. 2B; the sole effects of G-to-A mutations by APOBEC3F and APOBEC3G are represented in red lines). Moreover, since the combination of the two different antiviral mechanisms has some synergistic or additive effects to inhibit viral replication, the combination of deaminase activity-dependent and -independent effects cannot be estimated by linearly summing the experimental measurement of their individual antiviral effect. Similar problems occur in evaluating the nonlinear effects of two-drug combinations, and, therefore, mathematical models such as Bliss independence are useful and necessary for estimating the combination effects by using single-drug effects. In this study, we applied Bliss independence to evaluate the antiviral effects of G-to-A mutation and RT inhibition in consideration of the nonlinear effects of two different antiviral mechanisms of action.
So far, our experimental and mathematical approach has quantitatively revealed the replication dynamics of retroviruses (50, 51) and enteroviruses (52). To the best of our knowledge, here we quantitatively determined the antiviral effect of cellular proteins, human APOBEC3, using the experimental data for the first time. The synergistic experimental and mathematical strategy is a powerful approach to quantitatively investigate the dynamics of virus infections in a way that is impossible by conventional experimental strategies.
ACKNOWLEDGMENTS
We thank Keiko Okano (Research Administration Office, Kyoto University) for proofreading the manuscript.
This study was supported in part by grants from the following: the Aihara Innovative Mathematical Modeling Project, the Japan Society for the Promotion of Science (JSPS), through the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program), initiated by the Council for Science and Technology Policy of Japan (to Kazuyuki Aihara, Shingo Iwami, and Kei Sato); Takeda Science Foundation (to Kei Sato); Sumitomo Foundation Research Grant (to Kei Sato); Senshin Medical Research Foundation (to Kei Sato); Kyushu University Interdisciplinary Programs in Education and Projects in Research Development (to Shingo Iwami); Grant-in-Aid for Young Scientists B25800092 (to Shingo Iwami) from JSPS; Grants-in-Aid for Scientific Research B24390112 and S22220007 (to Yoshio Koyanagi) from JSPS; Grant-in-Aid for Scientific Research on Innovative Areas 24115008 (to Yoshio Koyanagi) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; and Research on HIV/AIDS (to Yoshio Koyanagi) from the Ministry of Health, Labor and Welfare of Japan.
Footnotes
Published ahead of print 12 March 2014
REFERENCES
- 1.Freed EO, Martin MA. 2007. HIVs and their replication, p 2107–2185 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Malim MH, Bieniasz PD. 2012. HIV restriction factors and mechanisms of evasion. Cold Spring Harb. Perspect. Med. 2:a006940. 10.1101/cshperspect.a006940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liddament MT, Brown WL, Schumacher AJ, Harris RS. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 14:1385–1391. 10.1016/j.cub.2004.06.050 [DOI] [PubMed] [Google Scholar]
- 4.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 23:2451–2458. 10.1038/sj.emboj.7600246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. 10.1038/nature00939 [DOI] [PubMed] [Google Scholar]
- 6.Izumi T, Shirakawa K, Takaori-Kondo A. 2008. Cytidine deaminases as a weapon against retroviruses and a new target for antiviral therapy. Mini Rev. Med. Chem. 8:231–238. 10.2174/138955708783744047 [DOI] [PubMed] [Google Scholar]
- 7.Harris RS, Liddament MT. 2004. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4:868–877. 10.1038/nri1489 [DOI] [PubMed] [Google Scholar]
- 8.Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J. 1992. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 66:6489–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Schwedler U, Song J, Aiken C, Trono D. 1993. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 67:4945–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krisko JF, Martinez-Torres F, Foster JL, Garcia JV. 2013. HIV restriction by APOBEC3 in humanized mice. PLoS Pathog. 9:e1003242. 10.1371/journal.ppat.1003242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato K, Izumi T, Misawa N, Kobayashi T, Yamashita Y, Ohmichi M, Ito M, Takaori-Kondo A, Koyanagi Y. 2010. Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice. J. Virol. 84:9546–9556. 10.1128/JVI.00823-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgibbon JE, Mazar S, Dubin DT. 1993. A new type of G→A hypermutation affecting human immunodeficiency virus. AIDS Res. Hum. Retroviruses 9:833–838. 10.1089/aid.1993.9.833 [DOI] [PubMed] [Google Scholar]
- 13.Gandhi SK, Siliciano JD, Bailey JR, Siliciano RF, Blankson JN. 2008. Role of APOBEC3G/F-mediated hypermutation in the control of human immunodeficiency virus type 1 in elite suppressors. J. Virol. 82:3125–3130. 10.1128/JVI.01533-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janini M, Rogers M, Birx DR, McCutchan FE. 2001. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4+ T cells. J. Virol. 75:7973–7986. 10.1128/JVI.75.17.7973-7986.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kieffer TL, Kwon P, Nettles RE, Han Y, Ray SC, Siliciano RF. 2005. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J. Virol. 79:1975–1980. 10.1128/JVI.79.3.1975-1980.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Land AM, Ball TB, Luo M, Pilon R, Sandstrom P, Embree JE, Wachihi C, Kimani J, Plummer FA. 2008. Human immunodeficiency virus (HIV) type 1 proviral hypermutation correlates with CD4 count in HIV-infected women from Kenya. J. Virol. 82:8172–8182. 10.1128/JVI.01115-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Kappes JC, Conway JA, Price RW, Shaw GM, Hahn BH. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 65:3973–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, Mallal S. 2006. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J. Virol. 80:9259–9269. 10.1128/JVI.00888-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piantadosi A, Humes D, Chohan B, McClelland RS, Overbaugh J. 2009. Analysis of the percentage of human immunodeficiency virus type 1 sequences that are hypermutated and markers of disease progression in a longitudinal cohort, including one individual with a partially defective Vif. J. Virol. 83:7805–7814. 10.1128/JVI.00280-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulenga NK, Sarr AD, Hamel D, Sankale JL, Mboup S, Kanki PJ. 2008. The level of APOBEC3G (hA3G)-related G-to-A mutations does not correlate with viral load in HIV type 1-infected individuals. AIDS Res. Hum. Retroviruses 24:1285–1290. 10.1089/aid.2008.0072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vartanian JP, Henry M, Wain-Hobson S. 2002. Sustained G→A hypermutation during reverse transcription of an entire human immunodeficiency virus type 1 strain Vau group O genome. J. Gen. Virol. 83:801–805 [DOI] [PubMed] [Google Scholar]
- 22.Vartanian JP, Meyerhans A, Asjo B, Wain-Hobson S. 1991. Selection, recombination, and G→A hypermutation of human immunodeficiency virus type 1 genomes. J. Virol. 65:1779–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, Daniels M, Ferrari G, Haynes BF, McMichael A, Shaw GM, Hahn BH, Korber B, Seoighe C. 2009. HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog. 5:e1000414. 10.1371/journal.ppat.1000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bishop KN, Holmes RK, Malim MH. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 80:8450–8458. 10.1128/JVI.00839-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. 2008. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4:e1000231. 10.1371/journal.ppat.1000231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holmes RK, Koning FA, Bishop KN, Malim MH. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 282:2587–2595. 10.1074/jbc.M607298200 [DOI] [PubMed] [Google Scholar]
- 27.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905. 10.1128/AAC.46.6.1896-1905.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiu YL, Greene WC. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317–353. 10.1146/annurev.immunol.26.021607.090350 [DOI] [PubMed] [Google Scholar]
- 29.Schumacher AJ, Hache G, Macduff DA, Brown WL, Harris RS. 2008. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 82:2652–2660. 10.1128/JVI.02391-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohn MF, Shandilya SM, Albin JS, Kouno T, Anderson BD, McDougle RM, Carpenter MA, Rathore A, Evans L, Davis AN, Zhang J, Lu Y, Somasundaran M, Matsuo H, Harris RS, Schiffer CA. 2013. Crystal structure of the DNA cytosine deaminase APOBEC3F: the catalytically active and HIV-1 Vif-binding domain. Structure 21:1042–1050. 10.1016/j.str.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen KM, Harjes E, Gross PJ, Fahmy A, Lu Y, Shindo K, Harris RS, Matsuo H. 2008. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 452:116–119. 10.1038/nature06638 [DOI] [PubMed] [Google Scholar]
- 32.Izumi T, Io K, Matsui M, Shirakawa K, Shinohara M, Nagai Y, Kawahara M, Kobayashi M, Kondoh H, Misawa N, Koyanagi Y, Uchiyama T, Takaori-Kondo A. 2010. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. U. S. A. 107:20798–20803. 10.1073/pnas.1008076107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato K, Misawa N, Fukuhara M, Iwami S, An DS, Ito M, Koyanagi Y. 2012. Vpu augments the initial burst phase of HIV-1 propagation and downregulates BST2 and CD4 in humanized mice. J. Virol. 86:5000–5013. 10.1128/JVI.07062-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi T, Ode H, Yoshida T, Sato K, Gee P, Yamamoto SP, Ebina H, Strebel K, Sato H, Koyanagi Y. 2011. Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85:932–945. 10.1128/JVI.01668-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato K, Yamamoto SP, Misawa N, Yoshida T, Miyazawa T, Koyanagi Y. 2009. Comparative study on the effect of human BST-2/tetherin on HIV-1 release in cells of various species. Retrovirology 6:53. 10.1186/1742-4690-6-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato K, Aoki J, Misawa N, Daikoku E, Sano K, Tanaka Y, Koyanagi Y. 2008. Modulation of human immunodeficiency virus type 1 infectivity through incorporation of tetraspanin proteins. J. Virol. 82:1021–1033. 10.1128/JVI.01044-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rose PP, Korber BT. 2000. Detecting hypermutations in viral sequences with an emphasis on G→A hypermutation. Bioinformatics 16:400–401. 10.1093/bioinformatics/16.4.400 [DOI] [PubMed] [Google Scholar]
- 38.Chaipan C, Smith JL, Hu WS, Pathak VK. 2013. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol. 87:444–453. 10.1128/JVI.00676-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85:11220–11234. 10.1128/JVI.05238-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zennou V, Bieniasz PD. 2006. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology 349:31–40. 10.1016/j.virol.2005.12.035 [DOI] [PubMed] [Google Scholar]
- 41.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. 2006. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2:458–466. 10.1038/nchembio817 [DOI] [PubMed] [Google Scholar]
- 42.Keith CT, Borisy AA, Stockwell BR. 2005. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 4:71–78. 10.1038/nrd1609 [DOI] [PubMed] [Google Scholar]
- 43.Kijak GH, Janini LM, Tovanabutra S, Sanders-Buell E, Arroyo MA, Robb ML, Michael NL, Birx DL, McCutchan FE. 2008. Variable contexts and levels of hypermutation in HIV-1 proviral genomes recovered from primary peripheral blood mononuclear cells. Virology 376:101–111. 10.1016/j.virol.2008.03.017 [DOI] [PubMed] [Google Scholar]
- 44.Albin JS, Harris RS. 2010. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol. Med. 12:e4. 10.1017/S1462399409001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nowarski R, Britan-Rosich E, Shiloach T, Kotler M. 2008. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat. Struct. Mol. Biol. 15:1059–1066. 10.1038/nsmb.1495 [DOI] [PubMed] [Google Scholar]
- 46.Refsland EW, Hultquist JF, Harris RS. 2012. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog. 8:e1002800. 10.1371/journal.ppat.1002800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chelico L, Pham P, Calabrese P, Goodman MF. 2006. APOBEC3G DNA deaminase acts processively 3′→5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13:392–399. 10.1038/nsmb1086 [DOI] [PubMed] [Google Scholar]
- 48.Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35:7096–7108. 10.1093/nar/gkm750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwatani Y, Takeuchi H, Strebel K, Levin JG. 2006. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 80:5992–6002. 10.1128/JVI.02680-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwami S, Sato K, De Boer RJ, Aihara K, Miura T, Koyanagi Y. 2012. Identifying viral parameters from in vitro cell cultures. Front. Microbiol. 3:319. 10.3389/fmicb.2012.00319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwami S, Holder BP, Beauchemin CA, Morita S, Tada T, Sato K, Igarashi T, Miura T. 2012. Quantification system for the viral dynamics of a highly pathogenic simian/human immunodeficiency virus based on an in vitro experiment and a mathematical model. Retrovirology 9:18. 10.1186/1742-4690-9-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuhara M, Iwami S, Sato K, Nishimura Y, Shimizu H, Aihara K, Koyanagi Y. 2013. Quantification of the dynamics of enterovirus 71 infection by experimental-mathematical investigation. J. Virol. 87:701–705. 10.1128/JVI.01453-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. 10.1016/S0092-8674(03)00423-9 [DOI] [PubMed] [Google Scholar]
- 54.Suspene R, Henry M, Guillot S, Wain-Hobson S, Vartanian JP. 2005. Recovery of APOBEC3-edited human immunodeficiency virus G→A hypermutants by differential DNA denaturation PCR. J. Gen. Virol. 86:125–129. 10.1099/vir.0.80426-0 [DOI] [PubMed] [Google Scholar]