

Abstract
Streptococcus pneumoniae, a Gram-positive bacterial pathogen, causes pneumonia, meningitis, and septicemia. Innate immune responses are critical for the control and pathology of pneumococcal infections. It has been demonstrated that S. pneumoniae induces the production of type I interferons (IFNs) by host cells and that type I IFNs regulate resistance and chemokine responses to S. pneumoniae infection in an autocrine/paracrine manner. In this study, we examined the effects of type I IFNs on macrophage proinflammatory cytokine production in response to S. pneumoniae. The production of interleukin-18 (IL-18), but not other cytokines tested, was significantly decreased by the absence or blockade of the IFN-α/β receptor, suggesting that type I IFN signaling is necessary for IL-18 production. Type I IFN signaling was also required for S. pneumoniae-induced activation of caspase-1, a cysteine protease that plays a central role in maturation and secretion of IL-18. Earlier studies proposed that the AIM2 and NLRP3 inflammasomes mediate caspase-1 activation in response to S. pneumoniae. From our results, the AIM2 inflammasome rather than the NLRP3 inflammasome seemed to require type I IFN signaling for its optimal activation. Consistently, AIM2, but not NLRP3, was upregulated in S. pneumoniae-infected macrophages in a manner dependent on the IFN-α/β receptor. Furthermore, type I IFN signaling was found to contribute to IL-18 production in pneumococcal pneumonia in vivo. Taken together, these results suggest that type I IFNs regulate S. pneumoniae-induced activation of the AIM2 inflammasome by upregulating AIM2 expression. This study revealed a novel role for type I IFNs in innate responses to S. pneumoniae.
INTRODUCTION
The Gram-positive bacterial pathogen Streptococcus pneumoniae is a leading cause of pneumonia, meningitis, and septicemia and is responsible for significant mortality and morbidity worldwide (1). Due to the severe disease burden and mortality, the incidence of drug-resistant S. pneumoniae, and an increase in the number of immunocompromised patients, it is increasingly important to understand the pathogenic processes of pneumococcal diseases in order to develop novel therapeutic modalities and effective vaccines. The polysaccharide capsule and pneumolysin (PLY) are critical virulence factors of S. pneumoniae (1–3). PLY is a member of cholesterol-dependent cytolysins and forms ring- or arc-shaped pores on cholesterol-containing membranes. As PLY lacks a signal sequence for secretion, it has been hypothesized that the cytolysin is released during the breakdown of the pneumococcal cell wall. PLY plays multiple roles in the pathogenicity of S. pneumoniae, including disruption of tissue barriers, inhibition of ciliary beating on epithelial cells and bactericidal activity of neutrophils, and possible subversion of complement-mediated opsonization (3, 4). It has also been demonstrated that PLY is involved in the activation of innate immune responses to S. pneumoniae, which generate inflammation and/or contribute to host defense (3, 5–7).
Pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs) and Nod-like receptors (NLRs), play a central role in innate immunity (8, 9). PRRs sense specific pathogen-derived molecular structures and, in turn, provide signals that activate innate defense mechanisms. Pneumococcal components and products are sensed by several PRRs, leading to the induction of inflammatory mediators, some of which contribute to host defense against S. pneumoniae. TLR2, TLR9, and TLR4 have been reported to participate in the production of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), in response to S. pneumoniae and pneumococcal uptake (6, 10–12). NLR family, pyrin domain containing 3 (NLRP3) and absent in melanoma 2 (AIM2) are cytoplasmic PRRs, which sense various stimuli and cytosolic DNA, respectively (13–15). After stimulation with specific agonists, NLRP3 and AIM2 form inflammasomes which activate caspase-1, which, in turn, processes pro-IL-1β and pro-IL-18 into their bioactive forms. Recent studies have suggested that the AIM2 and NLRP3 inflammasomes mediate S. pneumoniae-induced activation of caspase-1 and secretion of bioactive IL-1β and IL-18 (16–19). Moreover, S. pneumoniae induces the production of type I interferons (IFNs) through stimulator of interferon genes (STING), an adaptor of cytosolic DNA sensors (16, 20). Thus, S. pneumoniae appears to be recognized by at least one intracellular DNA sensor. Several groups have proposed that PLY mediates the delivery of pneumococcal DNA into the cytoplasm of host cells by forming pores in the cell membrane (11, 16, 17, 20). Especially during pneumococcal infection of macrophages, PLY is thought to disrupt the membrane of S. pneumoniae-containing phagosomes, in which engulfed bacteria are autolyzed or killed, thereby mediating the release of pneumococcal components, including DNA, into the cytoplasm (11, 16, 17). Strikingly, the induction of bioactive IL-1β and IL-18 and type I IFNs by S. pneumoniae critically depends on phagocytosis of bacteria and the expression of intact PLY (7, 16, 17). A previous study showed that recognition of S. pneumoniae by NOD2, a cytosolic sensor for muramyl dipeptide, was dependent on bacterial uptake and PLY (21, 22). This also suggests that PLY mediates cytoplasmic delivery of pneumococcal components.
Type I IFNs are produced during viral infections and are responsible for the first line of defense against viruses (23). Type I IFNs signal through a common receptor, the IFN-α/β receptor (IFNAR), which is a heterodimer of IFNAR1 and IFNAR2, resulting in the activation of the Janus kinase/signal transducer and activator of transcription pathway; this leads to the expression of IFN-inducible genes, some of which have antiviral effects. For the past decade, a number of studies have suggested that the production of type I IFNs is induced not only by viruses but also by bacterial pathogens (24). In bacterial infections, the role of type I IFNs varies with the type of pathogen; they can be both beneficial and detrimental to the host. Type I IFNs appear to contribute to, have no effect on, or impair host defense against S. pneumoniae, probably depending on the experimental design (20, 25–27). In pneumococcal intranasal or intratracheal infection models, genetic ablation of IFNAR1 resulted in increased nasopharyngeal colonization with S. pneumoniae in one study (20) but did not influence the clearance of bacteria from the lung and upper respiratory tract in other studies (26, 27). In the latter studies, IFNAR1 deficiency facilitated pneumococcal clearance in influenza virus-S. pneumoniae coinfection models (26, 27). In addition, a recent study showed that the transition of S. pneumoniae from the lung into the bloodstream was enhanced in the absence of IFNAR1 (25).
It has also been shown that type I IFNs positively or negatively regulate the production of chemokines, such as C-C motif chemokine 5 (CCL5; also known as RANTES), C-X-C motif chemokine 1 (CXCL1; also known as KC), CXCL2 (also known as Mip-2), and CCL2 (also known as MCP-1), in response to S. pneumoniae or a combination of influenza virus and S. pneumoniae in an autocrine and/or paracrine manner (16, 26, 27). This may, in part, account for the complicated effects of type I IFNs on host defense against pneumococcal infections. In addition to chemokines, proinflammatory cytokines are involved in host defense against S. pneumoniae (28–33), and hence, examining the effects of type I IFNs on cytokine production would be helpful to understand the exact role of type I IFNs in pneumococcal diseases. In this study, we found that type I IFN signaling is required for the production of IL-18 upon infection of macrophages with S. pneumoniae. Caspase-1 activation in response to S. pneumoniae was also dependent on IFNAR1. The AIM2 inflammasome rather than the NLRP3 inflammasome seemed to require type I IFN signaling for its optimal activation, which is most likely because of upregulation of AIM2 that is downstream of type I IFN signaling. Furthermore, type I IFN signaling was found to contribute to IL-18 production in pneumococcal pneumonia in vivo. Taken together, the results revealed a novel role for type I IFNs in innate responses to S. pneumoniae.
MATERIALS AND METHODS
Mice.
Female C57BL/6 mice were purchased from Japan SLC (Hamamatsu, Japan). IFNAR1−/− mice were kindly provided by Michel Aguet (Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland). NLRP3−/− mice were generated by Jürg Tschopp (University of Lausanne). Mice were maintained under specific-pathogen-free conditions and used at 8 to 10 weeks of age. All of the experimental procedures performed on mice were approved by the Animal Ethics and Research Committee of Kyoto University Graduate School of Medicine, Kyoto, Japan.
Bacterial strain.
Streptococcus pneumoniae D39 (serotype 2) was purchased from the National Collection of Type Cultures (NCTC 7466; Central Public Health Laboratory, London, United Kingdom). Pneumococci were grown overnight on blood agar plates at 37°C and 5% CO2. Colonies were inoculated into Todd-Hewitt broth (BD Biosciences, San Jose, CA) supplemented with 0.5% yeast extract, grown until midlogarithmic phase (optical density at 600 nm [OD600] = 0.5), and centrifuged at 6,000 × g for 15 min. The bacterial pellet was suspended in phosphate-buffered saline (PBS) and stocked at −80°C. The concentration was determined by colony counting on blood agar plates.
Preparation of macrophages and in vitro infection with S. pneumoniae.
Mice were injected intraperitoneally (i.p.) with 3% thioglycolate broth (Eiken Chemical, Tokyo, Japan), and peritoneal exudate cells (PECs) were collected 3 days later. After being washed with RPMI 1640 medium (Nacalai Tesque, Kyoto, Japan), PECs were suspended with RPMI 1640 supplemented with 10% fetal calf serum (FCS) and incubated in 48-well microplates at a density of 2.5 × 105 cells/well at 37°C plus 5% CO2, and nonadherent cells were removed after 2 h. More than 95% of the adherent PECs were F4/80+ macrophages as determined by flow cytometry. An immortalized AIM2-deficient macrophage cell line was kindly gifted from E. S. Alnemri (34). Macrophages were infected with S. pneumoniae at a multiplicity of infection (MOI) of 10 for 8 h, then 100 μg/ml of gentamicin (Wako Pure Chemical Industries, Osaka, Japan) was added to the cultures, and the cultures were incubated for an additional 16 h. Anti-mouse IFNAR1 blocking MAb (Biolegend, San Diego, CA) and purified mouse IgG1 (Biolegend) were added to the cultures at the same time as S. pneumoniae. Ultrapure Escherichia coli O111:B4 lipopolysaccharide (LPS) and nigericin were purchased from Invivogen (San Diego, CA) and Sigma-Aldrich (St. Louis, MO), respectively.
ELISA.
Levels of cytokines in culture supernatants were determined by two-site sandwich enzyme-linked immunosorbent assay (ELISA). ELISA kits for mouse IL-1β, IL-6, TNF-α, and IL-12p40 were purchased from eBioscience (San Diego, CA). A mouse IL-18 ELISA kit was purchased from Medical & Biological Laboratories (Nagoya, Japan). A mouse IL-1α ELISA kit was purchased from BD Biosciences.
Western blot analysis.
PECs were cultured on 12-well plates at a density of 5 × 105 cells/well in RPMI 1640 plus FCS at 37°C for 2 h. Then nonadherent cells were washed out, and the medium was replaced with Opti-MEM (Life Technologies, Carlsbad, CA). Adherent cells were infected with S. pneumoniae at an MOI of 10. Supernatants were collected 24 h after infection, and the cells were lysed with radioimmunoprecipitation assay buffer (Nacalai Tesque). The supernatants were concentrated 20-fold using 20% (wt/vol) trichloroacetate (Nacalai Tesque). The precipitates and cell lysates were subjected to SDS-PAGE and subsequently transferred to polyvinylidene difluoride membranes. The membranes were immunoblotted with anti-caspase-1 Ab (Santa Cruz Biotechnology, Santa Cruz, CA), anti-IL-1β Ab (R&D Systems, Minneapolis, MN), anti-IL-18 MAb (Medical & Biological Laboratories), or anti-β-actin MAb (Sigma-Aldrich).
Quantification of S. pneumoniae organisms associating with and phagocytosed by macrophages.
The number of S. pneumoniae organisms associating with or phagocytosed by macrophages was determined as previously described (17). Adherent macrophages were infected with S. pneumoniae at an MOI of 10 for 4, 6, or 8 h. To enumerate S. pneumoniae organisms associating with macrophages, cells were washed with chilled PBS three times to remove nonassociating bacteria and lysed in PBS containing 0.1% Triton X-100. The cell lysates were diluted with PBS and plated on blood agar plates. CFU were counted after overnight incubation. To enumerate the phagocytosed bacteria, macrophages were infected with S. pneumoniae as described above and additionally cultured for 30 min in the presence of 100 μg/ml of gentamicin. The cells were then washed, and the bacteria were counted. To monitor the survival of S. pneumoniae inside macrophages, the numbers of intracellular bacteria were determined every 0.5 h from 1 h to 3 h after gentamicin addition.
Quantitative RT-PCR analysis.
Total cellular RNA of adherent PECs was extracted by using NucleoSpin RNA Cleanup (MACHEREY-NAGEL, Düren, Germany), and quantitative real-time RT-PCR was carried out using the ABI Prism 7000 (Life Technologies) and Express Two-Step SYBR GreenER (Life Technologies), according to the manufacturer's instructions. Primers for quantitative real-time RT-PCR were as follows: for β-actin, 5′-GCC CTG AGG CTC TTT TCC AG-3′ (forward) and 5′-TGC CAC AGG ATT CCA TAC CC-3′ (reverse); for AIM2, 5′-TTG TAT CTA GGC TGA TCC TGG GAC-3′ (forward) and 5′-ACC TGC ACT TTG AAT CAG GTG GTC-3′ (reverse); for NLRP3, 5′-AGA GCC TAC AGT TGG GTG AAA TG-3′ (forward) and 5′-CCA CGC CTA CCA GGA AAT CTC-3′ (reverse); and for IL-6, 5′-TTC CAT CCA GTT GCC TTC TTG-3′ (forward) and 5′-GAA GGC CGT GGT TGT CACC-3′ (reverse).
Intranasal infection of mice.
Mice were anesthetized with pentobarbital (Nacalai Tesque) and inoculated with 5 × 107 bacteria in 30 μl of PBS. Bronchoalveolar lavage fluid (BALF) was collected 12 h after S. pneumoniae infection. BALF samples from noninfected mice were collected as a negative control. The collected BALF samples were first centrifuged at 200 × g for 3 min at 4°C, and the supernatants were transferred to new tubes for cytokine detection. The pellets were washed and the total cell number was determined. A total of 106 cells from each sample were incubated with anti-mouse CD16/32 MAb (Biolegend) for 30 min at 4°C. Then the cells were stained with phycoerythrin (PE)-conjugated anti-mouse F4/80 MAb (Biolegend) and fluorescein isothiocyanate (FITC)-conjugated anti-Gr-1 MAb (Biolegend) for 30 min at 4°C and analyzed on a FACSCalibur flow cytometer (BD Biosciences). The proportions of F4/80+ macrophages and F4/80− Gr-1+ neutrophils were calculated.
Statistical analysis.
For comparisons between two groups, Student's t test was used. Multigroup comparisons of mean values were made according to analysis of variance (ANOVA) and the Bonferroni post hoc test. Statistical significance was determined as a P value of <0.05.
RESULTS
Type I IFN signaling is required for IL-18 production in response to S. pneumoniae.
To determine whether type I IFNs regulate the production of proinflammatory cytokines, thioglycolate-elicited macrophages from wild-type (WT) or IFNAR1-deficient mice were infected with S. pneumoniae for 24 h, and cytokine levels in the culture supernatants were determined. In the absence of IFNAR1, S. pneumoniae-induced production of IL-18 was markedly reduced, while that of IL-1β was marginally reduced (Fig. 1A and B). The production of IL-1α, TNF-α, IL-6, and IL-12p40 was not affected by IFNAR1 deficiency (Fig. 1C to F). Blockade of IFNAR1 using an anti-IFNAR1 MAb resulted in a significant reduction in the production of IL-18 but not of IL-1β and IL-6 (Fig. 1G to I). Therefore, it appears that type I IFN signaling is involved in the production of IL-18 during S. pneumoniae infection. WT and IFNAR1-deficient macrophages produced comparable levels of IL-18 and IL-1β after stimulation with LPS plus nigericin, which activates the NLRP3 inflammasome, suggesting that secretion of these cytokines can occur without type I IFN signaling in this situation (Fig. 1J and K).
FIG 1.

Type I IFN signaling is required for IL-18 production in response to S. pneumoniae. (A to F) Adherent PECs from C57BL/6 (WT) and IFNAR1−/− mice were left uninfected or infected with S. pneumoniae D39 at an MOI of 10. Gentamicin (final concentration of 100 μg/ml) was added to the cultures 8 h after infection, and culture supernatants were collected after an additional 16 h of incubation (24 h after infection). Levels of IL-18 (A), IL-1β (B), IL-1α (C), TNF-α (D), IL-6 (E), and IL-12p40 (F) in the culture supernatants were determined by ELISA. (G to I) Anti-IFNAR1 blocking MAb (10 μg/well) or isotype control Ab (10 μg/well) was added to the cultures at the same time as S. pneumoniae. Levels of IL-18 (G), IL-1β (H), and IL-6 (I) were determined by ELISA. (J and K) Adherent PECs were stimulated with LPS (10 ng/ml) for 3 h and sequentially with nigericin (5 μM). After 21 h, culture supernatants were collected to assess the levels of IL-18 (J) and IL-1β (K) by ELISA. All of the experiments were repeated at least two times, with similar results. Data are presented as the means and standard deviations of triplicate assays. Statistical significance was determined by one-way ANOVA followed by Bonferroni's test. *, P < 0.05; n.s., not significant.
Type I IFN signaling contributes to S. pneumoniae-induced caspase-1 activation.
IL-18 and IL-1β require cleavage by caspase-1 to be secreted in their active forms. We examined the role of type I IFNs in caspase-1 activation in macrophages infected with S. pneumoniae. Infection of macrophages with this bacterium induced the secretion of the p10 fragment of active caspase-1, which was significantly reduced in the absence of IFNAR1 (Fig. 2). Thus, it is suggested that type I IFN signaling contributes to induction of caspase-1 activation by S. pneumoniae. Consistent with previous observations that type I IFNs suppress the expression of pro-IL-1β (35), the protein level of pro-IL-1β was higher in IFNAR1-deficient macrophages than in WT macrophages (Fig. 2). On the other hand, protein levels of pro-IL-18 were comparable between WT and IFNAR1-deficient macrophages. This may explain why S. pneumoniae-induced IL-1β production was only modestly affected by deficiency of IFNAR1, despite a significant reduction in IL-18 production and caspase-1 activation.
FIG 2.

Type I IFN signaling contributes to S. pneumoniae (S. p)-induced caspase-1 activation. Adherent PECs from WT and IFNAR1−/− mice were left uninfected or infected with S. pneumoniae D39 at an MOI of 10 for 24 h as described for Fig. 1, and culture supernatants and cell lysates were then collected. The culture supernatants were concentrated by TCAtrichloroacetic acid precipitation. The precipitates and cell lysates were subjected to Western blot analysis using antibodies specific for caspase-1, IL-1β, IL-18, or β-actin. Filled and open triangles indicate molecular mass markers of 47 kDa and 9 kDa, respectively. All of the experiments were repeated three times, with similar results.
Type I IFN signaling is dispensable for uptake and killing of S. pneumoniae by macrophages.
We have previously shown that the production of IL-18 and the activation of caspase-1 in response to S. pneumoniae depend on phagocytosis of pneumococci by host macrophages (17). To know whether type I IFNs are involved in pneumococcal uptake and killing, WT and IFNAR1-deficient macrophages were tested for the ability to ingest and kill S. pneumoniae. IFNAR1-deficient macrophages showed normal uptake and killing of S. pneumoniae (Fig. 3), suggesting that IFNAR1 deficiency does not affect phagocytosis and killing of this pathogen by macrophages.
FIG 3.
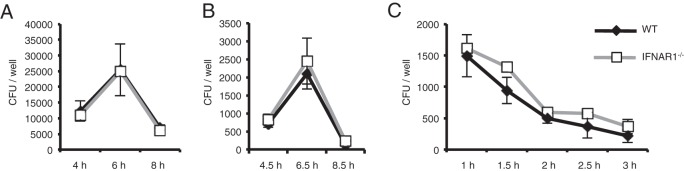
Type I IFN signaling is dispensable for uptake and killing of S. pneumoniae by macrophages. Adherent PECs were infected with S. pneumoniae D39 at an MOI of 10. (A) Bacteria associating with macrophages were counted at 4, 6, and 8 h after infection as described in Materials and Methods. (B) Phagocytosed bacteria were counted. Non-cell-associated bacteria were removed by washing at 4, 6, and 8 h after infection, and macrophages were cultured for an additional 30 min in the presence of 100 μg/ml of gentamicin. The bacteria in cell lysates were then counted. (C) The viability of phagocytosed bacteria was determined. Gentamicin was added to the cultures 4 h after infection, and the cells were additionally incubated for the indicated times. The bacteria in cell lysates were then counted. All of the experiments were repeated two times, with similar results. Data are presented as the means and standard deviations of triplicate assays.
Type I IFN signaling is required for induction of AIM2-mediated IL-18 production by S. pneumoniae.
S. pneumoniae induces the production of IL-18 and activation of caspase-1 through the AIM2 and NLRP3 inflammasomes (16–19). Thus, we examined which kind of inflammasome requires type I IFN signaling for optimal activation upon S. pneumoniae infection. IFNAR1 blockade significantly reduced the production of IL-18, but not of IL-1β, by NLRP3-deficient macrophages infected with S. pneumoniae (Fig. 4). This indicates that NLRP3-independent production of IL-18 requires type I IFN signaling. On the other hand, AIM2-deficient macrophages hardly secreted IL-18 and IL-1β after infection with S. pneumoniae (Fig. 4), and thus, it was difficult to test the effect of inhibition of type I IFN signaling on IL-18 production in the absence of AIM2. AIM2-deficient macrophages could secrete a large amount of IL-1β in response to LPS plus nigericin used as a positive control (data not shown). Based on these results, we concluded that upon S. pneumoniae infection, AIM2 rather than NLRP3 mediates the production of IL-18 in a manner dependent on type I IFN signaling.
FIG 4.
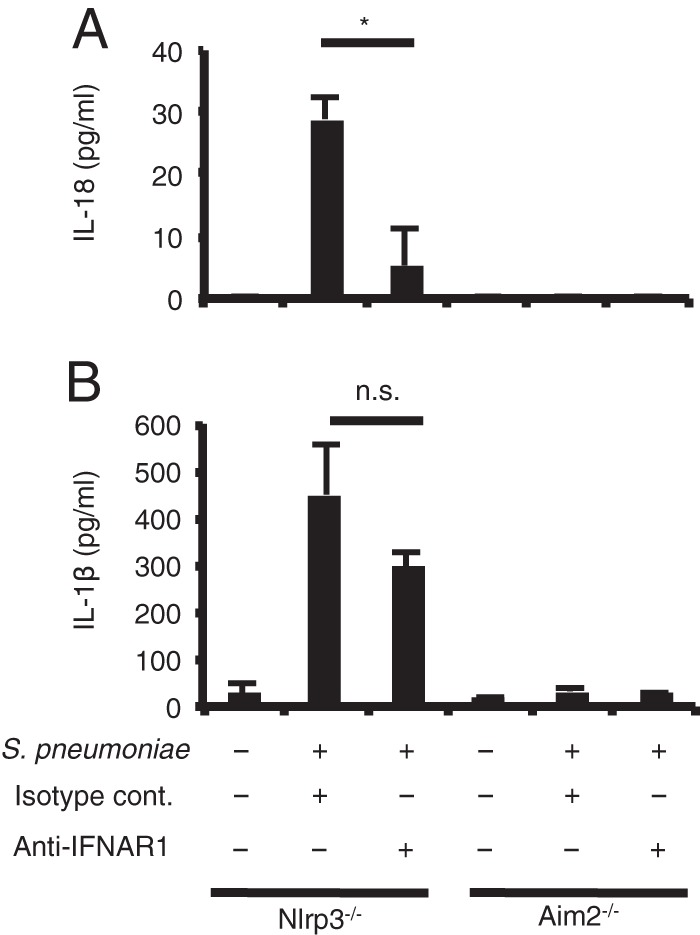
Type I IFN signaling promotes AIM2-mediated IL-18 induction by S. pneumoniae. Adherent PECs from NLRP3−/− mice and immortalized AIM2−/− macrophages were left uninfected or infected with S. pneumoniae D39 at an MOI of 10. Gentamicin (final concentration 100 μg/ml) was added to cultures 8 h after infection, and culture supernatants were collected after an additional 16 h of incubation (24 h after infection). Anti-IFNAR1 blocking MAb (10 μg/ml) or isotype control Ab (10 μg/ml) was added to the cultures at the same time as S. pneumoniae. Levels of IL-18 (A) and IL-1β (B) were determined by ELISA. All of the experiments were repeated two times, with similar results. Data are presented as the means and standard deviations of triplicate assays. Statistical significance was determined by one-way ANOVA followed by Bonferroni's test. *, P < 0.05; n.s., not significant.
Type I IFN signaling is required for upregulation of AIM2 in S. pneumoniae infection.
It has been demonstrated that stimulation of macrophages with TLR ligands or proinflammatory cytokines, such as TNF-α, leads to upregulation of NLRP3 through the activation of NF-κB, which is critical for NLRP3 inflammasome activation (36, 37). On the other hand, AIM2 is an IFN-inducible protein (13). Thus, we determined the expression levels of NLRP3 and AIM2 in S. pneumoniae-infected macrophages. The expression of AIM2, NLRP3, and IL-6 was strongly increased following infection with S. pneumoniae (Fig. 5). In IFNAR1-deficient macrophages, the increase in AIM2 expression was impaired (Fig. 5A), whereas expression levels of NLRP3 and IL-6 were almost comparable to those in WT macrophages (Fig. 5B and C). Therefore, type I IFN signaling appears to be required for upregulation of AIM2 during S. pneumoniae infection.
FIG 5.
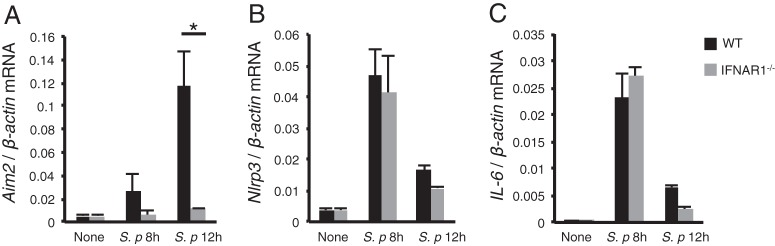
Type I IFN signaling is required for upregulation of AIM2 during S. pneumoniae infection. Adherent PECs from WT and IFNAR1−/− mice were infected with S. pneumoniae D39 at an MOI of 10 for 8 h or 12 h, and the expression of Aim2 (A), Nlrp3 (B), and Il6 (C) was analyzed by real-time RT-PCR. The target mRNA concentrations were normalized to β-actin mRNA concentrations. All of the experiments were repeated three times, with similar results. Data are presented as the means and standard deviations of triplicate assays. Tests for statistical significance were performed by using the Student t test. *, P < 0.05.
Type I IFN signaling facilitates IL-18 production in pneumococcal pneumonia in vivo.
Finally, we examined the involvement of type I IFN signaling in cytokine production in pneumococcal pneumonia in vivo. Levels of IL-18, IL-1β, and IL-6 in BALF were significantly elevated after intranasal infection with S. pneumoniae (Fig. 6A to C). Although the concentrations of IL-1β and IL-6 were not reduced by the absence of IFNAR1 (Fig. 6B and C), IL-18 levels were significantly lower in IFNAR1-deficient mice than in WT mice (Fig. 6A). There was no significant difference between WT and IFNAR1-deficient mice in the number of monocytes/macrophages and neutrophils, which are possible sources of IL-18, in BALF after pneumococcal infection (Fig. 6D and E). In conclusion, consistent with the above in vitro observations, type I IFN signaling appears to be important for IL-18 production in S. pneumoniae infection in vivo.
FIG 6.

Type I IFN signaling facilitates IL-18 production in pneumococcal pneumonia in vivo. WT and IFNAR1−/− mice were left uninfected or intranasally challenged with S. pneumoniae D39, BALF was collected from the mice 12 h after infection, and the levels of IL-18 (A), IL-1β (B), and IL-6 (C) were measured by ELISA. The numbers of macrophages (D) and neutrophils (E) in each BALF sample were determined by cell counting and flow cytometric analysis. Tests for statistical significance were performed by using one-way ANOVA followed by Bonferroni's test. *, P < 0.05.
DISCUSSION
Type I IFNs are well-known antiviral mediators (23). They also have anti-inflammatory effects and are widely used to control autoimmune diseases. In addition, type I IFNs have recently been suggested to have functions in bacterial infections (24). In earlier studies, type I IFN signaling had positive or negative effects on host resistance and chemokine responses to S. pneumoniae (16, 20, 25–27). In our present study, we found that type I IFN signaling contributes to the activation of caspase-1 and production of IL-18 in response to S. pneumoniae. Considering the importance of this inflammatory response in the control and pathology of pneumococcal infections, our findings would be helpful to better understand host-pathogen interactions in pneumococcal diseases.
Type I IFN signaling has been reported to contribute to AIM2 inflammasome activation induced by Francisella tularensis, Listeria monocytogenes, or nonvirulent mycobacteria (38–41). We previously showed that L. monocytogenes induced the upregulation of AIM2 in an IFNAR1-dependent manner (42). One study suggested that upon F. tularensis infection, type I IFN signaling leads to upregulation of AIM2, thereby enhancing the activation of the AIM2 inflammasome (40), whereas another study proposed type I IFN signaling enhances F. tularensis-induced AIM2 inflammasome activation by promoting the killing of engulfed bacteria to generate the cytosolic DNA rather than by upregulating AIM2 expression (38). We observed that the lack of IFNAR1 led to an impaired upregulation of AIM2, but not to defective uptake and killing of pneumococci, in S. pneumoniae-infected macrophages. Judging from the results, it is most likely that type I IFNs enhance the activation of the AIM2 inflammasome through upregulation of AIM2 during S. pneumoniae infection.
A previous study has suggested that type I IFNs suppress the expression of pro-IL-1β (35). In agreement with this, the protein level of pro-IL-1β was higher in IFNAR1-deficient macrophages than in WT macrophages after S. pneumoniae infection. One may speculate that an increase in the amount of pro-IL-1β leads to an increase in its cleaved form when in the presence of active caspase-1. However, IL-1β production in response to S. pneumoniae was not enhanced but marginally reduced in the absence of IFNAR1, which is probably a reflection of diminished caspase-1 activation under this condition. The elevation of IL-1β in BALF after S. pneumoniae infection was not affected by deficiency of IFNAR1, despite a significant reduction in BALF IL-18 levels in the absence of IFNAR1. This might be due to an increase in pro-IL-1β expression in the absence of IFNAR1. Another possibility is that IL-1β might be produced by an inflammasome-independent mechanism, which does not require type I IFN signaling, in pneumococcal pneumonia. Indeed, in our recent experiments, BALF IL-18 levels, but not BALF IL-1β levels, were elevated in a caspase-1-dependent manner after intranasal infection with S. pneumoniae (R. Fang and K. Tsuchiya, unpublished data). Taken together, our results show that type I IFN signaling appears to differentially regulate the production of IL-18 and IL-1β upon S. pneumoniae infection.
IL-18 and IL-1β have been reported to be protective against pneumococcal pneumonia (28, 29, 32, 33, 43). In earlier studies, IL-18 was shown to promote the clearance of S. pneumoniae in a nasopharyngeal colonization model and pneumonia models (29, 43). Thus, the type I IFN enhancement of AIM2-mediated IL-18 production may play a protective role in host defense against pneumococcal respiratory tract infections. Consistently, both IFNAR and IL-18 have been shown to contribute to the clearance of S. pneumoniae from the nasopharynx in colonization models (20, 43). However, several studies have suggested that in some situations, type I IFN signaling impairs host defense against pneumococcal pneumonia. Importantly, type I IFN signaling not only regulates the AIM2 inflammasome but also has effects on other innate immune responses to S. pneumoniae, including the inhibition of the production of chemokines involved in optimal protection (26, 27). It is therefore conceivable that type I IFNs may exacerbate pneumococcal pneumonia through affecting chemokine production and/or other mechanisms despite their role in enhancing AIM2 inflammasome activation. Alternatively, enhanced activation of the AIM2 inflammasome in the lung may be detrimental to the host, for example, because of increased cell death due to pyroptosis, among other reasons (14, 15). In contrast to pneumonia models, deficiency or blockade of IL-18 ameliorated pathology in pneumococcal meningitis models, with or without reduced bacterial outgrowth in the brain (44, 45), suggesting that IL-18 plays a host-detrimental role in S. pneumoniae meningitis. IL-18 is an IFN-γ-inducing cytokine. It was proposed that IL-18-stimulated IFN-γ production by NK cells contributes to pathogenesis in pneumococcal meningitis (46). Accordingly, IL-18 is a key cytokine when considering pathology of and host defense against S. pneumoniae infections, and thus, type I IFN signaling may influence the outcome of pneumococcal diseases by enhancing inflammasome activation and IL-18 production.
In this study, we found that type I IFN signaling regulates caspase-1 activation and IL-18 production during S. pneumoniae infection. Our results suggest a novel role for type I IFNs in an inflammasome-mediated response to S. pneumoniae, providing an insight into the cytokine network that is involved in the control and pathology of pneumococcal infections.
ACKNOWLEDGMENTS
We thank Michel Aguet, EPFL School of Life Sciences, for giving us permission to use the IFNAR1−/− mice and Jürg Tschopp, University of Lausanne, for permission to use the NLRP3−/− mice. We thank Emad S. Alnemri, Thomas Jefferson University, for providing the AIM2−/− macrophage cell line; Shigekazu Nagata, Kyoto University, for providing the IFNAR1−/− mice; and Hiroko Tsutsui, Hyogo College of Medicine, for providing the NLRP3−/− mice.
This work was supported by JSPS KAKENHI grant numbers 23590504 and 24590522.
Footnotes
Published ahead of print 18 March 2014
REFERENCES
- 1. Tuomanen EI, Austrian R, Masure HR. 1995. Pathogenesis of pneumococcal infection. N. Engl. J. Med. 332:1280–1284. 10.1056/NEJM199505113321907 [DOI] [PubMed] [Google Scholar]
- 2. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301. 10.1038/nrmicro1871 [DOI] [PubMed] [Google Scholar]
- 3. Marriott HM, Mitchell TJ, Dockrell DH. 2008. Pneumolysin: a double-edged sword during the host-pathogen interaction. Curr. Mol. Med. 8:497–509. 10.2174/156652408785747924 [DOI] [PubMed] [Google Scholar]
- 4. Paton JC, Rowan-Kelly B, Ferrante A. 1984. Activation of human complement by the pneumococcal toxin pneumolysin. Infect. Immun. 43:1085–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Houldsworth S, Andrew PW, Mitchell TJ. 1994. Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect. Immun. 62:1501–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U. S. A. 100:1966–1971. 10.1073/pnas.0435928100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shoma S, Tsuchiya K, Kawamura I, Nomura T, Hara H, Uchiyama R, Daim S, Mitsuyama M. 2008. Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infect. Immun. 76:1547–1557. 10.1128/IAI.01269-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30:16–34. 10.3109/08830185.2010.529976 [DOI] [PubMed] [Google Scholar]
- 9. Opitz B, van Laak V, Eitel J, Suttorp N. 2010. Innate immune recognition in infectious and noninfectious diseases of the lung. Am. J. Respir. Crit. Care Med. 181:1294–1309. 10.1164/rccm.200909-1427SO [DOI] [PubMed] [Google Scholar]
- 10. Albiger B, Dahlberg S, Sandgren A, Wartha F, Beiter K, Katsuragi H, Akira S, Normark S, Henriques-Normark B. 2007. Toll-like receptor 9 acts at an early stage in host defence against pneumococcal infection. Cell. Microbiol. 9:633–644. 10.1111/j.1462-5822.2006.00814.x [DOI] [PubMed] [Google Scholar]
- 11. Koppe U, Suttorp N, Opitz B. 2012. Recognition of Streptococcus pneumoniae by the innate immune system. Cell. Microbiol. 14:460–466. 10.1111/j.1462-5822.2011.01746.x [DOI] [PubMed] [Google Scholar]
- 12. Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. 1999. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 163:1–5 [PubMed] [Google Scholar]
- 13. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. 2009. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513. 10.1038/nature07710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Latz E. 2010. The inflammasomes: mechanisms of activation and function. Curr. Opin. Immunol. 22:28–33. 10.1016/j.coi.2009.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuchiya K, Hara H. 2014. The inflammasome and its regulation. Crit. Rev. Immunol. 34:41–80. 10.1615/CritRevImmunol.2013008686 [DOI] [PubMed] [Google Scholar]
- 16. Koppe U, Hogner K, Doehn JM, Muller HC, Witzenrath M, Gutbier B, Bauer S, Pribyl T, Hammerschmidt S, Lohmeyer J, Suttorp N, Herold S, Opitz B. 2012. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J. Immunol. 188:811–817. 10.4049/jimmunol.1004143 [DOI] [PubMed] [Google Scholar]
- 17. Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, Yamamoto T, Fernandes-Alnemri T, Yang R, Hernandez-Cuellar E, Dewamitta SR, Xu Y, Qu H, Alnemri ES, Mitsuyama M. 2011. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J. Immunol. 187:4890–4899. 10.4049/jimmunol.1100381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Pétrilli V, Andrew PW, Kadioglu A, Lavelle EC. 2010. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 6:e1001191. 10.1371/journal.ppat.1001191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, Holmes A, Trendelenburg G, Heimesaat MM, Bereswill S, van der Linden M, Tschopp J, Mitchell TJ, Suttorp N, Opitz B. 2011. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol. 187:434–440. 10.4049/jimmunol.1003143 [DOI] [PubMed] [Google Scholar]
- 20. Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ, Fitzgerald KA, Schindler C, Prince A. 2011. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio 2:e00016-11. 10.1128/mBio.00016-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davis KM, Nakamura S, Weiser JN. 2011. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J. Clin. Invest. 121:3666–3676. 10.1172/JCI57761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Opitz B, Püschel A, Schmeck B, Hocke AC, Rosseau S, Hammerschmidt S, Schumann RR, Suttorp N, Hippenstiel S. 2004. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J. Biol. Chem. 279:36426–36432. 10.1074/jbc.M403861200 [DOI] [PubMed] [Google Scholar]
- 23. González-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 12:125–135. 10.1038/nri3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malireddi RK, Kanneganti TD. 2013. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front. Cell. Infect. Microbiol. 3:77. 10.3389/fcimb.2013.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. LeMessurier KS, Hacker H, Chi L, Tuomanen E, Redecke V. 2013. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog. 9:e1003727. 10.1371/journal.ppat.1003727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakamura S, Davis KM, Weiser JN. 2011. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Invest. 121:3657–3665. 10.1172/JCI57762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, Deng JC. 2009. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119:1910–1920. 10.1172/JCI35412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kafka D, Ling E, Feldman G, Benharroch D, Voronov E, Givon-Lavi N, Iwakura Y, Dagan R, Apte RN, Mizrachi-Nebenzahl Y. 2008. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int. Immunol. 20:1139–1146. 10.1093/intimm/dxn071 [DOI] [PubMed] [Google Scholar]
- 29. Lauw FN, Branger J, Florquin S, Speelman P, van Deventer SJ, Akira S, van der Poll T. 2002. IL-18 improves the early antimicrobial host response to pneumococcal pneumonia. J. Immunol. 168:372–378 [DOI] [PubMed] [Google Scholar]
- 30. van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF. 1997. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J. Infect. Dis. 176:439–444. 10.1086/514062 [DOI] [PubMed] [Google Scholar]
- 31. Rijneveld AW, Florquin S, Branger J, Speelman P, Van Deventer SJ, van der Poll T. 2001. TNF-alpha compensates for the impaired host defense of IL-1 type I receptor-deficient mice during pneumococcal pneumonia. J. Immunol. 167:5240–5246 [DOI] [PubMed] [Google Scholar]
- 32. Yang H, Ko HJ, Yang JY, Kim JJ, Seo SU, Park SG, Choi SS, Seong JK, Kweon MN. 2013. Interleukin-1 promotes coagulation, which is necessary for protective immunity in the lung against Streptococcus pneumoniae infection. J. Infect. Dis. 207:50–60. 10.1093/infdis/jis651 [DOI] [PubMed] [Google Scholar]
- 33. Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, Van Furth AM. 2003. IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J. Immunol. 170:4724–4730 [DOI] [PubMed] [Google Scholar]
- 34. Wu J, Fernandes-Alnemri T, Alnemri ES. 2010. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30:693–702. 10.1007/s10875-010-9425-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. 2011. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223. 10.1016/j.immuni.2011.02.006 [DOI] [PubMed] [Google Scholar]
- 36. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. 2009. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183:787–791. 10.4049/jimmunol.0901363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franchi L, Eigenbrod T, Nunez G. 2009. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 183:792–796. 10.4049/jimmunol.0900173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. 2010. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11:385–393. 10.1038/ni.1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. 2007. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204:987–994. 10.1084/jem.20062665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. 2010. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U. S. A. 107:9771–9776. 10.1073/pnas.1003738107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VA, Vogel SN, Fitzgerald KA, Sutterwala FS, Briken V. 2013. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-beta and AIM2 inflammasome-dependent IL-1beta production via its ESX-1 secretion system. J. Immunol. 191:3514–3518. 10.4049/jimmunol.1301331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsuchiya K, Hara H, Kawamura I, Nomura T, Yamamoto T, Daim S, Dewamitta SR, Shen Y, Fang R, Mitsuyama M. 2010. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J. Immunol. 185:1186–1195. 10.4049/jimmunol.1001058 [DOI] [PubMed] [Google Scholar]
- 43. Paterson GK, Blue CE, Mitchell TJ. 2005. Role of interleukin-18 in experimental infections with Streptococcus pneumoniae. J. Med. Microbiol. 54:323–326. 10.1099/jmm.0.45873-0 [DOI] [PubMed] [Google Scholar]
- 44. Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, Pfister HW, Fontana A, Hammerschmidt S, Koedel U. 2011. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J. Immunol. 187:5440–5451. 10.4049/jimmunol.1100790 [DOI] [PubMed] [Google Scholar]
- 45. Zwijnenburg PJ, van der Poll T, Florquin S, Akira S, Takeda K, Roord JJ, van Furth AM. 2003. Interleukin-18 gene-deficient mice show enhanced defense and reduced inflammation during pneumococcal meningitis. J. Neuroimmunol. 138:31–37. 10.1016/S0165-5728(03)00088-2 [DOI] [PubMed] [Google Scholar]
- 46. Mitchell AJ, Yau B, McQuillan JA, Ball HJ, Too LK, Abtin A, Hertzog P, Leib SL, Jones CA, Gerega SK, Weninger W, Hunt NH. 2012. Inflammasome-dependent IFN-gamma drives pathogenesis in Streptococcus pneumoniae meningitis. J. Immunol. 189:4970–4980. 10.4049/jimmunol.1201687 [DOI] [PubMed] [Google Scholar]