

Abstract
The adaptive immune response to Francisella tularensis is dependent on the route of inoculation. Intradermal inoculation with the F. tularensis live vaccine strain (LVS) results in a robust Th1 response in the lungs, whereas intranasal inoculation produces fewer Th1 cells and instead many Th17 cells. Interestingly, bacterial loads in the lungs are similar early after inoculation by these two routes. We hypothesize that the adaptive immune response is influenced by local events in the lungs, such as the type of cells that are first infected with Francisella. Using fluorescence-activated cell sorting, we identified alveolar macrophages as the first cell type infected in the lungs of mice intranasally inoculated with F. novicida U112, LVS, or F. tularensis Schu S4. Following bacterial dissemination from the skin to the lung, interstitial macrophages or neutrophils are infected. Overall, we identified the early interactions between Francisella and the host following two different routes of inoculation.
INTRODUCTION
Immune responses following bacterial infections are influenced by the route of infection (1–3). Cytokines produced by the innate immune response are critical in shaping the adaptive immune response (reviewed in reference 4). For example, if a naive CD4+ T cell encounters antigen in the presence of interleukin 12 (IL-12), it will differentiate into a Th1 effector T cell, but if it encounters IL-6 and transforming growth factor β (TGF-β) during antigen presentation, it will differentiate into a Th17 effector T cell (4). Our previous experiments with mice using intranasal or intradermal inoculation with Francisella tularensis subsp. holarctica live vaccine strain (LVS) demonstrated striking differences in the adaptive immune response in the lungs when these two inoculation routes were compared (2). Upon either intradermal or intranasal inoculation with LVS, bacteria rapidly disseminate and are found in the spleen, liver, and lungs 24 h after inoculation (2). After 3 days, equivalent bacterial burdens are found in the spleen and lungs of mice inoculated via either route (2). Despite similar burdens early after inoculation, intradermally inoculated mice clear the infection more rapidly than intranasally inoculated mice and have an increased gamma interferon (IFN-γ) response. Intradermal inoculation leads to significantly more CD4+ and CD8+ T cells producing IFN-γ in both the spleen and lungs on day 7 postinoculation than does intranasal inoculation (2). Faster bacterial clearance in intradermally inoculated mice correlates with the increased IFN-γ-mediated immune response. IFN-γ is required for controlling F. tularensis infection, and administration of recombinant IFN-γ decreases bacterial burdens (5–7). Intranasal infection leads to an expansion in the lungs of Th17 cells, a CD4+ T cell population not found in intradermally inoculated mice (2, 8, 9). We conclude that T cell effector function is influenced by the inoculation route. Thus, it is important to understand the initiation of the immune response and identify the earliest cells infected by Francisella in the lungs.
Francisella tularensis is a facultative intracellular, Gram-negative coccobacillus. Infection with F. tularensis causes the zoonotic disease tularemia, which is endemic in regions of the United States and Europe. Three strains of Francisella are commonly used by researchers. Francisella tularensis subsp. tularensis Schu S4 is a type A strain and highly pathogenic in humans and mice. Francisella tularensis subsp. holarctica live vaccine strain (LVS) is an attenuated, type B strain and does not cause severe disease in humans (10). Murine infection with LVS closely resembles human infection (11). The 50% lethal dose (LD50) for intranasal inoculations is approximately 103 CFU, and that for intradermal inoculation is approximately 106 (12, 13). F. novicida U112 does not cause disease in immunocompetent humans but causes severe disease in mice with a course similar to that in mice inoculated with Schu S4. Intranasal inoculation with U112 or Schu S4 is typically fatal in mice before an adaptive immune response can occur, but low-dose inoculations with LVS in mice allow for the observation of the adaptive immune response to Francisella.
Due to similar bacterial burdens early after inoculation but very different adaptive immune responses for these inoculation routes, we hypothesized that the adaptive immune response to Francisella was shaped by events early after inoculation, such as what type of cell was initially infected. We therefore sought to identify host cells infected with F. tularensis early after intranasal and intradermal inoculation. We examined three strains of Francisella to determine whether all strains exhibit similar tropisms or if different strains target different cell types. Previously, we found that alveolar macrophages comprised between 50 and 80% of cells infected with U112 or LVS 24 h after intranasal inoculation (14). These experiments, however, identified more infected cells than the initial bacterial inoculum, suggesting that multiple rounds of infection had occurred. Therefore, we were interested in identifying the infected cells 4 h postinoculation, before reinfection of new cell types occurred. We found that alveolar macrophages were the primary cell type infected after intranasal inoculation and that interstitial macrophages and neutrophils were the first lung cell types infected following intradermal inoculation and bacterial dissemination. Together, our data demonstrate that the cell types initially infected with Francisella are dependent on the inoculation route and are common among the different strains of Francisella.
MATERIALS AND METHODS
Bacteria.
Francisella novicida U112 was obtained from Colin Manoil (University of Washington). F. tularensis subsp. holarctica live vaccine strain (LVS; ATCC 29684) was obtained from the American Type Culture Collection (Manassas, VA). F. tularensis subsp. tularensis Schu S4 (NR-643) was obtained from BEI Resources (Manassas, VA). Bacteria were grown on chocolate agar supplemented with 1% IsoVitaleX (Becton, Dickinson) at 37°C. Bacterial inoculations were prepared by removing bacteria from a lawn grown on chocolate agar and resuspended in sterile phosphate-buffered saline (PBS) at an optical density at 600 nm (OD600) of 1 (equivalent to 1 × 107 CFU/μl). To achieve the desired inoculation dose, appropriate dilutions were made using sterile PBS. Viable bacteria in each preparation were quantified by serial dilution and plating on chocolate agar. All experiments using Schu S4 were performed at the Duke University NIAID-Regional Biocontainment Laboratory (RBL) under biosafety level 3 (BSL3) containment.
Mice.
C57Bl/6J (B6) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were housed under specific-pathogen-free conditions at the University of North Carolina at Chapel Hill, the RBL, or the University of Arizona in accordance with their respective Institutional Animal Care and Use committees. Female mice used for experiments were between 7 and 12 weeks of age.
Inoculation of mice.
For intranasal bacterial inoculations, mice were anesthetized with 575 mg/kg (of body weight) of tribromoethanol (Avertin; Sigma) administered intraperitoneally. Mice were then intranasally inoculated with 1 × 104 CFU of U112, LVS, or Schu S4 suspended in 50 μl of PBS. For intradermal inoculations, mice were inoculated with 5 × 105 CFU of U112 or LVS in 25 μl in the tail. The inoculum was divided between three injection sites along the tail.
Single-cell suspension of mouse lung.
Following humane euthanasia, lungs were aseptically removed after perfusion with PBS and digested into a single-cell suspension as previously described (15). For intranasally inoculated mice, 50 μg/ml of gentamicin (Sigma) was added to the digestion mix to kill extracellular bacteria. Red blood cells were lysed using ammonium chloride-potassium lysis buffer (Gibco) and washed with RMPI 1640 supplemented with 10% fetal calf serum (Atlas), l-glutamine, sodium pyruvate, and β-mercaptoethanol. The total number of viable cells was determined using a hemocytometer by trypan blue exclusion.
Bead enrichment of CD45+ cells.
Lung single-cell suspensions were stained for 20 min on ice with CD45-allophycocyanin (CD45-APC; clone 30–F11; Biolegend). After washing the cells to remove unbound antibody, IMag anti-APC magnetic particles (BD) were used to enrich CD45-APC-positive cells according to the manufacturer's instructions. CD45 enrichment was determined by flow cytometry. Enriched eukaryotic cells were directly plated on chocolate agar containing 10 μg/ml of ampicillin (Sigma), and the CFU were counted 72 h later.
Identification of infected lung populations.
Lung cells in a single-cell suspension after intranasal inoculation with Francisella had Fc receptors blocked with 2.4G2 to prevent nonspecific staining and were then stained with F4/80 phycoerythrin (PE; clone BM8; eBioscience), CD11b Pacific blue (clone M1/70; Biolegend), and CD11c APC (clone N418; eBioscience). Lung cells from intradermally inoculated mice had Fc receptors blocked with 2.4G2 and were then stained with F4/80 PE, CD11b Pacific blue, CD11c APC, and GR-1 Pacific orange (clone RB6-8C5; Invitrogen). The cells were sorted using a Reflection cell sorter (iCyt/Sony; UNC) or FACSAria II (Becton Dickinson Immunocytometry Systems [BDIS]; RBL and University of Arizona) into four populations based on surface marker expression (Table 1) using the gating scheme shown in Fig. 1. Sorted populations were plated directly on chocolate agar containing 10 μg/ml of ampicillin without lysis, and bacterial CFU were counted 24 to 72 h later to enumerate the infected cells.
TABLE 1.
Identification of lung cell types infected by Francisella
| Cell type | Surface markersa |
|---|---|
| Alveolar macrophages | F4/80high, CD11chigh, CD11bmid, DEC-205mid |
| Interstitial macrophages | F4/80high, CD11cvar, CD11bhigh |
| Dendritic cells | F4/80low, CD11chigh, CD11blow |
| Neutrophils | F4/80low,CD11bhigh, GR-1high |
| Other | F4/80low, CD11clow, CD11bvar |
| Alveolar macrophages (17) | F4/80pos, CD11cneg, CD11bpos, DEC-205neg |
| Airway dendritic cells (17) | F4/80var, CD11cpos, CD11bvar, GR-1var, DEC-205pos |
| Alveolar macrophages (23) | F4/80low, CD11chigh, CD11bneg, DEC-205mid |
mid, medium level; var, variable level; pos, positive expression; neg, not expressed.
FIG 1.
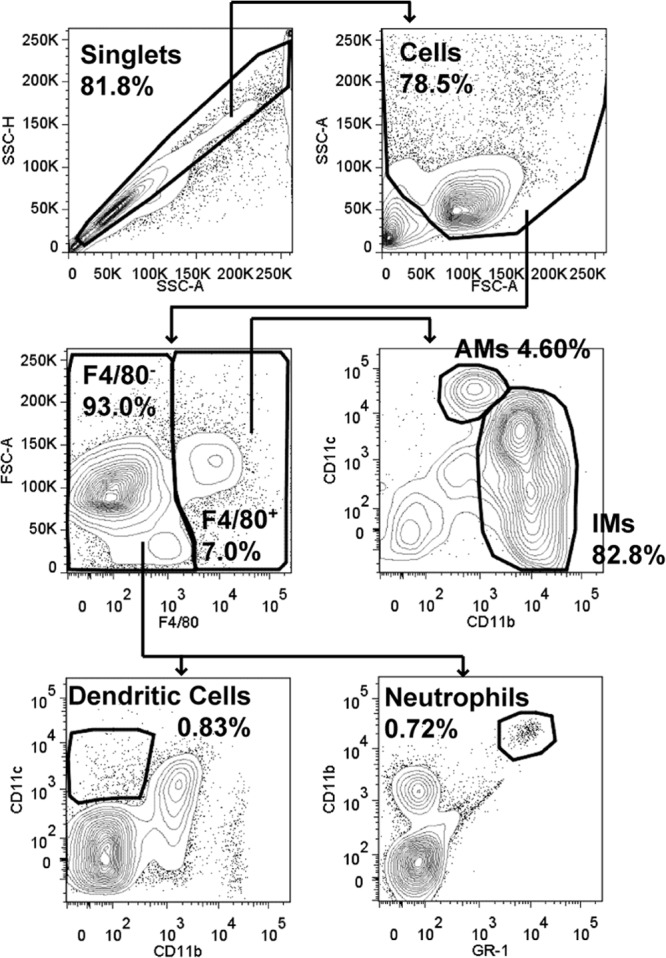
Lung gating scheme. Single cells were discriminated from doublets by plotting side scatter height (SSC-H) versus side scatter area (SSC-A). Cells were selected by plotting SSC-A versus forward scatter area (FSC-A). F4/80− and F4/80+ cells were gated on by plotting FSC-A versus F4/80. From the F4/80+ gate, alveolar macrophages (AMs) were discriminated from interstitial macrophages (IMs) by plotting CD11c versus CD11b. Of the F4/80− cells, dendritic cells were identified by plotting CD11c versus CD11b and neutrophils were identified by plotting CD11b versus GR-1. For each gate, the percentage of the parent gate is indicated in bold (for example, AMs are 4.6% of the cells within the F4/80+ gate).
Statistical analysis.
A Kruskal-Wallis test was used to determine whether the distribution of infected cells was significantly different and not due to random sampling. GraphPad Prism (v.5.04) was used for analysis.
RESULTS
LVS infects myeloid cells after intranasal inoculation.
The adaptive immune response to LVS is influenced by the route of infection despite similar bacterial burdens, and therefore antigen load, early after inoculation (2). We hypothesized that the adaptive immune response in the lungs was shaped by the cell type(s) infected immediately after intranasal inoculation or after bacterial dissemination to the lungs after intradermal inoculation. We therefore sought to identify which cell populations were infected following inoculation. A previous study reported that a variety of lung cell types are infected with Francisella 24 and 72 h after intranasal inoculation (16). Although the majority of infected cells 24 h after intranasal inoculation with green fluorescent protein (GFP)-expressing Francisella strains were myeloid, alveolar type II epithelial cells were also identified as an infected cell type by flow cytometry (16). Therefore, our initial investigation into identifying the first infected lung cells sought to determine whether cells initially targeted by LVS were of the myeloid or nonmyeloid lineage. We chose to use 4 h after inoculation so that Francisella had sufficient time to reach and infect the cells it initially targets but not time for multiple rounds of reinfection. An intranasal inoculum dose of 1 × 104 CFU yielded approximately 100 infected cells out of 1 × 107 host lung cells at 4 h postinoculation for all strains, giving us confidence that we were identifying the initially infected cells.
B6 mice were intranasally inoculated with 1 × 104 CFU of LVS and euthanized 4 h later. Lung tissue was digested into a single-cell suspension in the presence of gentamicin to kill extracellular bacteria. Cells were then stained with anti-CD45-APC, and anti-APC magnetic beads were used to positively select for myeloid cells. Figure 2A shows representative flow cytometry histograms of CD45 staining within the preenrichment, negative selection (CD45−), and positive selection (CD45+) samples. Eukaryotic cells were directly plated on chocolate agar, and the colonies within the CD45− and CD45+ pools were counted. A total of 99% of the resulting LVS colonies were on the CD45+ plates, indicating that LVS initially targets myeloid cells for infection (Fig. 2B). Although we did not repeat these experiments using U112 or Schu S4, we predicted similar results between strains and indeed did observe that all strains targeted the same cell types after intranasal inoculation (see below).
FIG 2.

LVS infects myeloid cells following intranasal inoculation. B6 mice were intranasally inoculated with 1 × 104 CFU of LVS. Four hours postinfection, mice were sacrificed and lungs were removed and digested into a single-cell suspension. Cells were stained with CD45-APC, and then CD45+ cells were enriched using magnetic beads. (A) Representative flow cytometry analysis of CD45 enrichment. (B) CD45+ and CD45− populations were directly plated on chocolate agar, and the colonies were counted 72 h later. We counted 123 total CFU among 4 mice. Data are weighted by the total number of CFU and presented as the percentage of CFU within a population from 4 infected mice in 2 independent experiments.
Alveolar macrophages are the dominant infected cell type after intranasal inoculation in all Francisella strains.
Of the myeloid cells in the lungs, we predicted that alveolar macrophages, interstitial macrophages, and dendritic cells were the cell types most likely to be initially infected with Francisella after intranasal inoculation. To identify infected cells in the lung early after intranasal inoculation, B6 mice were intranasally inoculated with 1 × 104 CFU of U112, LVS, or Schu S4 and euthanized 4 h postinoculation. Lung tissue was digested in the presence of gentamicin to kill extracellular bacteria. The lung single-cell suspensions were stained for F4/80, CD11c, and CD11b, and cell populations were sorted based on expression of these surface markers (Table 1 and Fig. 1). Sorted eukaryotic cells were plated directly on chocolate agar without lysis of the cells, and the resulting colonies were counted. Data from multiple mice were combined for each Francisella strain, and a weighted average was used to identify which cell type made up the majority of infected cells (Fig. 3). Approximately 90% of infected cells were alveolar macrophages for each Francisella strain, indicating that these cells were initially targeted by Francisella after intranasal inoculation. The remaining 10% of infected cells consisted of a mixture of interstitial macrophages, dendritic cells, and others. The results were consistent across individual mice, although more variability was observed in the minor infected cell populations (interstitial macrophages, dendritic cells, and others) (Table 2). In individual mice, alveolar macrophages constituted 86 to 96% of infected cells after U112 inoculation, 71 to 93% of infected cells after LVS inoculation, and 93 to 96% of infected cells after Schu S4 inoculation (Table 2). Furthermore, determination of number of LVS CFU per 105 sorted cells showed that the alveolar macrophage population contained at least 7-fold more CFU per 105 sorted cells than did interstitial macrophages, dendritic cells, or others (Fig. 4). Together, these data indicate that alveolar macrophages are the dominant first infected cell type immediately after intranasal inoculation with each of the three distinct strains of Francisella.
FIG 3.
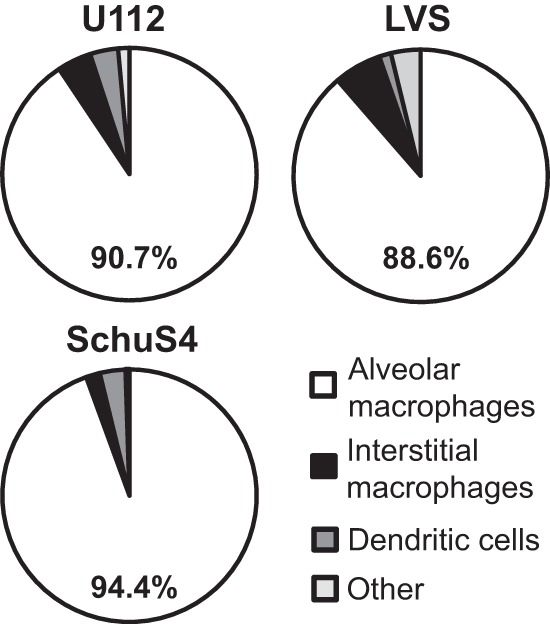
Alveolar macrophages are the primary infected cell type in the lungs after intranasal inoculation with Francisella. B6 mice were intranasally inoculated with 1 × 104 CFU of U112, LVS, or Schu S4. Four hours postinoculation, mice were sacrificed and lungs were removed and digested into a single-cell suspension and stained for sorting. Alveolar macrophages, interstitial macrophages, dendritic cells, and other cell populations were sorted and directly plated on chocolate agar. Resulting colonies were counted 24 to 72 h later. Data are weighted by the total number of CFU and presented as the percentage of CFU within a population from 2 mice (U112; 139 total CFU), 6 mice (LVS; 132 total CFU), or 3 mice (Schu S4; 398 total CFU) from 1 (U112), 3 (LVS), or 2 (Schu S4) independent experiments. A Kruskal-Wallis test was used to determine whether the distribution of infected cells was significantly different. Significant differences were as follows: for U112, not significant (P = 0.1767); for LVS, P ≤ 0.01; and for Schu S4, P ≤ 0.05.
TABLE 2.
Mean percentage of infected cells in the lung 4 h after intranasal inoculation
| Bacterial strain | % of infected cells, mean ± SD (range) |
|||
|---|---|---|---|---|
| Alveolar macrophages | Interstitial macrophages | Dendritic cells | Other | |
| F. novicida U112 (n = 2) | 90.82 ± 6.65 (86.11–95.52) | 4.32 ± 0.16 (4.17–4.48) | 3.51 ± 3.47 (0.0–6.94) | 1.41 ± 1.40 (0.0–2.78) |
| F. tularensis LVS (n = 6) | 87.86 ± 7.72 (71.43–92.86) | 8.13 ± 10.6 (0.0–28.57) | 1.67 ± 4.08 (0.0–10) | 3.72 ± 4.81 (0.0–9.52) |
| F. tularensis Schu S4 (n = 3) | 94.27 ± 1.27 (93.33–95.71) | 3.45 ± 2.96 (0.83–6.67) | 1.90 ± 2.69 (0.0–4.98) | 0.38 ± 0.36 (0.0–0.71) |
FIG 4.
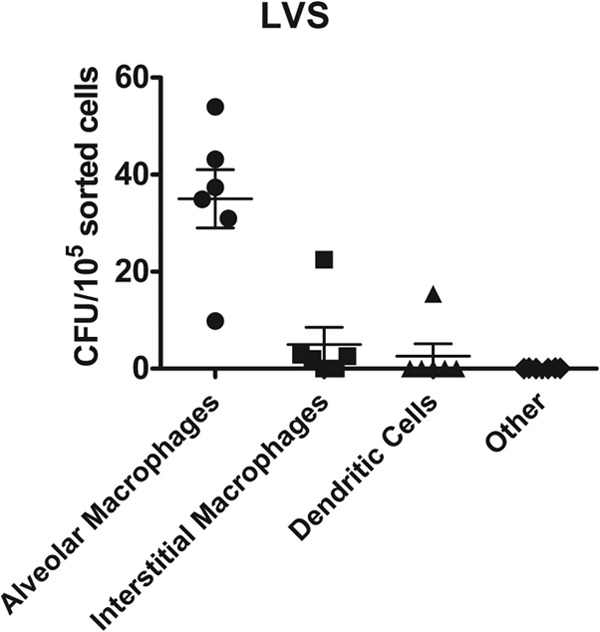
Alveolar macrophages are infected with LVS at the highest frequency. B6 mice were intranasally inoculated with 1 × 104 CFU of LVS. Four hours postinoculation, mice were sacrificed and lungs were removed and digested into a single-cell suspension and stained for sorting. Alveolar macrophages, interstitial macrophages, dendritic cells, and other cell populations were sorted and directly plated on chocolate agar. The total number of sorted cells for each population was recorded during the sort. Resulting colonies were counted 72 h later. Data are represented as the number of CFU per 105 sorted cells for each population from 6 mice in 3 independent experiments.
Interstitial macrophages and neutrophils are the dominant infected cell types in the lungs after intradermal inoculation.
We observed very different adaptive immune responses in the lungs after intranasal versus intradermal inoculation (2) and therefore hypothesized that different innate immune events occurred early after infection. One possibility was different infected cell types in the lungs depending on the route of infection, particularly since one inoculation route introduced bacteria directly into the lungs, whereas intradermal inoculation required bacteria to disseminate from the skin to the lungs. We therefore sought to identify the early infected cell type(s) in the lungs after intradermal inoculation and subsequent bacterial dissemination to the lungs. Mice were intradermally inoculated with 5 × 105 CFU of U112 or LVS. Pilot experiments determined that euthanizing mice 48 h postinoculation allowed enough time for bacteria to disseminate from the skin to the lungs and that 48 h was the earliest time point that bacteria could be reproducibly found in the lungs. Lung single-cell suspensions were stained and sorted as previously described. In contrast to the case with intranasal inoculation, interstitial macrophages and neutrophils together comprise >90% of all cells infected with Francisella after intradermal inoculation and bacterial dissemination to the lungs (Fig. 5 and Table 3). Importantly, alveolar macrophages were not appreciably infected with Francisella in the lungs following bacterial dissemination from the skin (<2% of infected cells). Additionally, when we calculated the number of CFU per 105 sorted cells for each population, we found that interstitial macrophages and neutrophils were both infected at a rate over 30 times greater than alveolar macrophages for both LVS and U112 inoculation (Fig. 6). We detected less than 500 infected cells in the mice intradermally inoculated with LVS. Although we detected more infected cells (100 to 6,000) in the mice intradermally inoculated with U112, the percentage of each infected cell population was similar to findings for LVS, even with variability in the number of infected cells, allowing us to be confident that we were observing early infection events in the lungs following bacterial dissemination. These results indicate that pulmonary interstitial macrophages and neutrophils are infected with Francisella in the lungs after intradermal inoculation. Furthermore, these results indicate that different cell types are infected with Francisella in the lungs depending on the inoculation route and support our hypothesis that the differences observed in the adaptive immune response are a result of different infected cell types.
FIG 5.
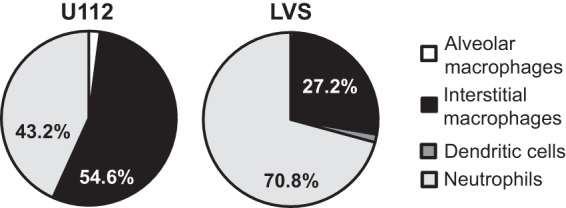
Interstitial macrophages and neutrophils are the primary cell types infected with U112 or LVS in the lungs after intradermal inoculation. B6 mice were intradermally inoculated with 5 × 105 CFU of U112 or LVS in 50 μl of PBS at the base of the tail. Forty-eight hours postinoculation, mice were sacrificed and lungs were removed and digested into a single-cell suspension and stained for sorting. Alveolar macrophages, interstitial macrophages, dendritic cells, and neutrophil populations were sorted and directly plated on chocolate agar. Resulting colonies were counted 24 to 72 h later. Data are weighted by the total number of CFU and presented as the percentage of CFU within a population from 4 mice (U112; 9,344 total CFU) or 2 mice (LVS; 537 total CFU) from 1 experiment per strain. A Kruskal-Wallis test was used to determine whether the distribution of infected cells was significantly different. The distributions were not significantly different for U112 (P = 0.1184) or LVS (P = 0.1116).
TABLE 3.
Mean percentage of infected cells in the lung 48 h after intradermal inoculation
| Bacterial strain | % of infected cells, mean ± SD (range) |
|||
|---|---|---|---|---|
| Alveolar macrophages | Interstitial macrophages | Dendritic cells | Neutrophils | |
| F. novicida U112 (n = 4) | 1.21 ± 1.84 (0.0–3.89) | 62.89 ± 26.82 (38.46–100) | 0.036 ± 0.044 (0.0–0.089) | 35.86 ± 26.75 (0.0–61.54) |
| F. tularensis LVS (n = 2) | 1.26 ± 1.47 (0.22–2.30) | 32.91 ± 11.97 (24.44–41.38) | 4.13 ± 5.53 (0.22–8.05) | 61.69 ± 18.98 (48.28–75.11) |
FIG 6.
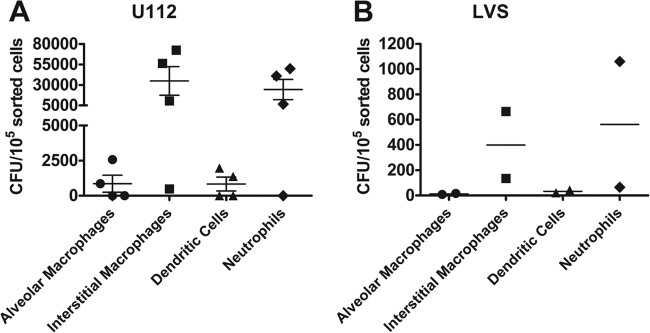
Interstitial macrophages and neutrophils are infected with U112 and LVS at the highest frequencies. B6 mice were intranasally inoculated with 1 × 104 CFU of U112 or LVS. Four hours postinoculation, mice were sacrificed and lungs were removed and digested into a single-cell suspension and stained for sorting. Alveolar macrophages, interstitial macrophages, dendritic cells, and other cell populations were sorted and directly plated on chocolate agar. The total number of sorted cells for each population was recorded during the sort. Resulting colonies were counted 24 or 72 h later. Data are represented as the number of CFU per 105 sorted cells for each population from 4 (U112) or 2 (LVS) mice in 1 experiment per strain.
DISCUSSION
Francisella is capable of infecting a variety of cell types upon inoculation (14, 16–18). The early interactions between the host and pathogen set the stage for the adaptive immune response. We and others have shown that the route of inoculation influences the type of adaptive immune response that develops (2, 3). We were particularly interested in the early interactions between Francisella and the host following intranasal and intradermal inoculations because of differential adaptive immune responses in the lungs. Intranasal and intradermal inoculations with LVS lead to similar bacterial burdens early after inoculation, yet the adaptive immune responses are very different (2). We hypothesized that the cell type infected with Francisella immediately after inoculation shaped the adaptive immune response. We therefore sought to identify the cells that were infected with Francisella after inoculation that were likely responsible for shaping subsequent adaptive immunity.
We identified infected cells by sorting individual populations using fluorescence-activated cell sorting. This technique only identified host cells infected with live Francisella, since our experimental readout was colonies grown on agar. We had to intranasally inoculate mice with 1 × 104 CFU to have detectable infected cells after sorting. This inoculum dose is 20-fold higher than our typical LVS intranasal inoculation dose. We believe that the higher dose increased the number of infected cells without altering the distribution of infected cell types because nearly all of the infected cells were alveolar macrophages.
All three strains of Francisella predominantly infected alveolar macrophages following intranasal inoculation. Alveolar macrophages are the resident macrophages of the airway and interact with inhaled antigens. It is therefore not surprising that inhalation of Francisella leads to infection of alveolar macrophages. Other pathogens, like Mycobacterium tuberculosis, Mycoplasma pulmonis, and Legionella pneumophila, target alveolar macrophages upon infection as well (19–22). Experiments found that LVS infects pulmonary dendritic cells 1 h after intratracheal inoculation with 5 × 104 CFU using flow cytometry to detect carboxyfluorescein succinimidyl ester (CFSE)-labeled bacteria inside host cells (17). A potential explanation for the seemingly disparate results between the two experiments is the use of different surface markers to define airway dendritic cells and alveolar macrophages. Surface markers used to define lung cellular populations by our group and others are shown in Table 1. The markers described by Bosio and Dow (17) for alveolar macrophages best fit with our definition of interstitial macrophages, and their definition for airway dendritic cells best fits with our definition of alveolar macrophages (Table 1). More recently, Guth et al. reported that alveolar macrophages express middle levels of DEC-205 and CD11c, giving this macrophage cell subset a more dendritic-cell-like surface phenotype (23). We also observed mid-level expression of DEC-205 of alveolar macrophages. Therefore, the cell populations are likely the same based on surface marker phenotype.
While we did not pursue experiments to determine the underlying mechanisms responsible for bacterial dissemination, we identified interstitial macrophages and neutrophils as the dominant infected cell types in the lungs after intradermal inoculation and bacterial dissemination. We carefully timed the lung harvest after intradermal inoculation so that we were identifying infected cells soon after bacteria disseminated to the lungs. These results indicate that not only does the route of infection shape the adaptive immune response but also two different types of innate cells are initially infected with Francisella in the lungs, which we predict helps shape the different downstream adaptive responses. In the lungs, the differences in infected cell types between the two different inoculation routes may simply be spatial. Alveolar macrophages are located primarily in the alveoli. This spatial location would put them in direct contact with Francisella after an intranasal inoculation but out of the way during bacterial dissemination from the skin.
Because alveolar macrophages were infected following intranasal inoculation with LVS, we sought to determine whether the disease course was altered in the absence of alveolar macrophages. Alveolar macrophages express high levels of CD11c and can therefore be depleted in CD11c.DOG mice upon intranasal treatment with diphtheria toxin (DT). We chose to use CD11c.DOG mice instead of other depletion strategies so that alveolar macrophages were specifically depleted while other phagocytic cells remained untouched. Alveolar macrophages can also be depleted by intranasal administration of liposomal clodronate; however, this treatment is nonspecific and depletes >90% of lung and airway antigen-presenting cells (17). Although we successfully depleted alveolar macrophages from the lungs of CD11c.DOG mice with intranasal inoculation of diphtheria toxin, this treatment caused changes to the cytokine and chemokine milieu prior to infection with LVS (unpublished data). Diphtheria toxin also changed the proportion and absolute number of lung cellular populations (unpublished data). Although we observed an increase in LVS lung bacterial burdens when CD11c.DOG mice were depleted of alveolar macrophages prior to LVS inoculation, the changes in the lungs' cytokine and chemokine milieu as well as cellular distribution made it impossible to ascribe increased bacterial burdens simply to the lack of alveolar macrophages.
Alveolar macrophages have been shown in other models to be both protective and detrimental during infection (17, 19, 24–26). CBA/J mice succumb rapidly (day 3) to Klebsiella pneumoniae in the absence of alveolar macrophages and have significantly higher bacterial burdens in the plasma and lungs, suggesting that alveolar macrophages control bacterial replication in the lungs (24, 26). B6 mice, normally resistant to Mycoplasma pulmonis, were more susceptible to infection in the absence of alveolar macrophages, indicating that alveolar macrophages are important for host defense during M. pulmonis infection (25). In contrast, during Mycobacterium tuberculosis infection, mice lacking alveolar macrophages were less susceptible to infection and had decreased mycobacterial burdens in the lungs and liver, suggesting that the presence of alveolar macrophages is detrimental during infection (19). Bosio and Dow found that depletion of alveolar macrophages with clodronate followed 18 h later by intratracheal inoculation with a lethal dose of LVS led to decreased bacterial burdens and an increase in mean time to death (17). It is possible that the difference in bacterial burdens observed in untreated and clodronate-treated mice was due to the absence of cells to infect, because nearly all phagocytic cells were reported to be depleted, leaving few cells for LVS to infect (17). While specific depletion of alveolar macrophages might address this possibility, selective depletion of alveolar macrophages without other changes in cellular composition has not been possible.
Several groups have reported that LVS vaccination prior to lethal challenge with a highly virulent, type A strain of Francisella must be administered intranasally and not intradermally in order to achieve protective immunity (27, 28). The failure of intradermally vaccinated mice to survive a lethal type A challenge suggests that the T cell response is not successfully primed via this route. We have demonstrated that the adaptive immune response is different depending on the route of LVS inoculation (2). Herein, we have shown that different cell types are initially infected with LVS, again depending on the route of inoculation. Taken together, these data suggest that alveolar macrophages could play a role in successful T cell priming (via antigen presentation and/or cytokine milieu), leading to a T cell response that is protective after secondary challenge with virulent Francisella. Alternatively, when interstitial macrophages are among the cells initially infected, an environment is established in which the T cells successfully clear the primary infection but fail to protect upon secondary challenge.
Overall, we have shown that alveolar macrophages are initially infected with Francisella in the lungs after intranasal inoculation. We also determined that interstitial macrophages and neutrophils are infected with Francisella in the lungs following bacterial dissemination from intradermal inoculation in the skin. We had previously observed a differential adaptive immune response following intranasal and intradermal inoculation, despite similar bacterial burdens early after inoculation. We predicted that there would be differences in the innate immune response in the lungs that contributed to the development of two distinct T cell responses, and this was the case; different types of cells were infected in the lungs following each inoculation route.
ACKNOWLEDGMENTS
We thank Paula Campbell of the University of Arizona Flow Cytometry Core, Lisa Bixby of the University of North Carolina Flow Cytometry Core, and John Whitesides of the Duke University Human Vaccine Institute/Regional Biocontainment Laboratory Flow Facility for their expertise and assistance with cell sorting.
Selected studies were performed in the Regional Biocontainment Laboratory at Duke University, which received partial support for construction from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) (grant UC6-AI058607). This work was supported by NIAID Southeast Regional Center of Excellence for Emerging Infections and Biodefense grant U54 AI057157.
The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 31 March 2014
REFERENCES
- 1.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. 2007. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27:203–213. 10.1016/j.immuni.2007.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolard MD, Hensley LL, Kawula TH, Frelinger JA. 2008. Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infect. Immun. 76:2651–2659. 10.1128/IAI.01412-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK. 2010. Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat. Immunol. 11:83–89. 10.1038/ni.1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nurieva RI, Chung Y. 2010. Understanding the development and function of T follicular helper cells. Cell. Mol. Immunol. 7:190–197. 10.1038/cmi.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anthony LS, Ghadirian E, Nestel FP, Kongshavn PA. 1989. The requirement for gamma interferon in resistance of mice to experimental tularemia. Microb. Pathog. 7:421–428. 10.1016/0882-4010(89)90022-3 [DOI] [PubMed] [Google Scholar]
- 6.Collazo CM, Sher A, Meierovics AI, Elkins KL. 2006. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 8:779–790. 10.1016/j.micinf.2005.09.014 [DOI] [PubMed] [Google Scholar]
- 7.Leiby DA, Fortier AH, Crawford RM, Schreiber RD, Nacy CA. 1992. In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect. Immun. 60:84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzger DW, Salmon SL, Kirimanjeswara G. 2013. Differing effects of interleukin-10 on cutaneous and pulmonary Francisella tularensis live vaccine strain infection. Infect. Immun. 81:2022–2027. 10.1128/IAI.00024-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skyberg JA, Rollins MF, Samuel JW, Sutherland MD, Belisle JT, Pascual DW. 2013. Interleukin-17 protects against the Francisella tularensis live vaccine strain but not against a virulent F. tularensis type A strain. Infect. Immun. 81:3099–3105. 10.1128/IAI.00203-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oyston PC. 2009. Francisella tularensis vaccines. Vaccine 27(Suppl 4):D48–D51. 10.1016/j.vaccine.2009.07.090 [DOI] [PubMed] [Google Scholar]
- 11.Fortier AH, Green SJ, Polsinelli T, Jones TR, Crawford RM, Leiby DA, Elkins KL, Meltzer MS, Nacy CA. 1994. Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol. Ser. 60:349–361 [PubMed] [Google Scholar]
- 12.Metzger DW, Bakshi CS, Kirimanjeswara G. 2007. Mucosal immunopathogenesis of Francisella tularensis. Ann. N. Y. Acad. Sci. 1105:266–283. 10.1196/annals.1409.007 [DOI] [PubMed] [Google Scholar]
- 13.Fortier AH, Slayter MV, Ziemba R, Meltzer MS, Nacy CA. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 59:2922–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, Kawula TH. 2008. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 76:5843–5852. 10.1128/IAI.01176-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrigan LM, Tuladhar S, Brunton JC, Woolard MD, Chen CJ, Saini D, Frothingham R, Sempowski GD, Kawula TH, Frelinger JA. 2013. Infection with Francisella tularensis LVS clpB leads to an altered yet protective immune response. Infect. Immun. 81:2028–2042. 10.1128/IAI.00207-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall JD, Craven RR, Fuller JR, Pickles RJ, Kawula TH. 2007. Francisella tularensis replicates within alveolar type II epithelial cells in vitro and in vivo following inhalation. Infect. Immun. 75:1034–1039. 10.1128/IAI.01254-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bosio CM, Dow SW. 2005. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175:6792–6801 [DOI] [PubMed] [Google Scholar]
- 18.Bosio CM, Bielefeldt-Ohmann H, Belisle JT. 2007. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J. Immunol. 178:4538–4547 [DOI] [PubMed] [Google Scholar]
- 19.Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, van Deventer SJ, van der Poll T. 2001. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J. Immunol. 166:4604–4611 [DOI] [PubMed] [Google Scholar]
- 20.Davis JK, Davidson MK, Schoeb TR, Lindsey JR. 1992. Decreased intrapulmonary killing of Mycoplasma pulmonis after short-term exposure to NO2 is associated with damaged alveolar macrophages. Am. Rev. Respir. Dis. 145:406–411. 10.1164/ajrccm/145.2_Pt_1.406 [DOI] [PubMed] [Google Scholar]
- 21.Gordon SB, Read RC. 2002. Macrophage defences against respiratory tract infections. Br. Med. Bull. 61:45–61. 10.1093/bmb/61.1.45 [DOI] [PubMed] [Google Scholar]
- 22.Nash TW, Libby DM, Horwitz MA. 1984. Interaction between the legionnaires' disease bacterium (Legionella pneumophila) and human alveolar macrophages. Influence of antibody, lymphokines, and hydrocortisone. J. Clin. Invest. 74:771–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. 2009. Lung environment determines unique phenotype of alveolar macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 296:L936–L946. 10.1152/ajplung.90625.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheung DO, Halsey K, Speert DP. 2000. Role of pulmonary alveolar macrophages in defense of the lung against Pseudomonas aeruginosa. Infect. Immun. 68:4585–4592. 10.1128/IAI.68.8.4585-4592.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hickman-Davis JM, Michalek SM, Gibbs-Erwin J, Lindsey JR. 1997. Depletion of alveolar macrophages exacerbates respiratory mycoplasmosis in mycoplasma-resistant C57BL mice but not mycoplasma-susceptible C3H mice. Infect. Immun. 65:2278–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Broug-Holub E, Toews GB, van Iwaarden JF, Strieter RM, Kunkel SL, Paine R, III, Standiford TJ. 1997. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect. Immun. 65:1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu TH, Hutt JA, Garrison KA, Berliba LS, Zhou Y, Lyons CR. 2005. Intranasal vaccination induces protective immunity against intranasal infection with virulent Francisella tularensis biovar A. Infect. Immun. 73:2644–2654. 10.1128/IAI.73.5.2644-2654.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conlan JW, Shen H, Kuolee R, Zhao X, Chen W. 2005. Aerosol-, but not intradermal-immunization with the live vaccine strain of Francisella tularensis protects mice against subsequent aerosol challenge with a highly virulent type A strain of the pathogen by an alphabeta T cell- and interferon gamma-dependent mechanism. Vaccine 23:2477–2485. 10.1016/j.vaccine.2004.10.034 [DOI] [PubMed] [Google Scholar]