

Abstract
Streptococcus agalactiae is the causative agent of septicemia and meningitis in fish. Previous studies have shown that hyaluronidase (Hyl) is an important virulence factor in many Gram-positive bacteria. To investigate the role of S. agalactiae Hyl during interaction with macrophages, we inactivated the gene encoding extracellular hyaluronidase, hylB, in a clinical Hyl+ isolate. The isogenic hylb mutant (Δhylb) displayed reduced survival in macrophages compared to the wild type and stimulated a significantly higher release of proinflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor alpha (TNF-α), than the wild type in macrophages as well as in mice. Furthermore, only Hyl+ strains could grow utilizing hyaluronic acid (HA) as the sole carbon source, suggesting that Hyl permits the organism to utilize host HA as an energy source. Fifty percent lethal dose (LD50) determinations in zebrafish demonstrated that the hylb mutant was highly attenuated relative to the wild-type strain. Experimental infection of BALB/c mice revealed that bacterial loads in the blood, spleen, and brain at 16 h postinfection were significantly reduced in the ΔhylB mutant compared to those in wild-type-infected mice. In conclusion, hyaluronidase has a strong influence on the intracellular survival of S. agalactiae and proinflammatory cytokine expression, suggesting that it plays a key role in S. agalactiae pathogenicity.
INTRODUCTION
Streptococcus agalactiae, also known as group B streptococcus (GBS), is a Gram-positive pathogen with a broad host range. S. agalactiae is the major pathogen causing neonatal pneumonia and meningitis in humans (1, 2) and mastitis in dairy cows (3, 4). In the last decade, there has been an increase in reports of S. agalactiae infection in fish with septicemia and meningitis resulting in a disturbing case fatality rate of 100% (5–7).
The ability of S. agalactiae to establish infection in the host suggests that the organism is able to subvert the host's immune system. It is known that when S. agalactiae cells are engulfed by professional phagocytic cells, such as macrophages and neutrophils, the organism can remain viable for a long period of time (8–10). The survival of S. agalactiae in macrophages may protect the organism from clearance of more active antimicrobial molecules in the blood and thus may be important in establishing bacteremia and subsequent meningitis (10). It is reported that some major virulence factors of S. agalactiae, such as the polysaccharide capsule (11), β-hemolysin (12), and the two-component system CovS/CovR (13), may be relevant to intracellular survival.
Our previous study observed that expression of the hylB gene that encodes the S. agalactiae hyaluronidase (Hyl) was upregulated through interaction with murine macrophages in a selective capture of transcribed sequences (SCOTS) approach (14). Hyaluronidase (Hyl) is an endoglycosidase that cleaves glycosaminoglycan chains, thereby degrading hyaluronic acid (HA) (15). Since HA is a major constituent of the ground substance of most connective tissues, particularly the skin, Hyl may be an essential component in enabling the spread of the pathogens from an initial site of infection. Hyaluronidase production has been reported in some Streptococcus species (16). To date, numerous suggestions have been made as to the role of hyaluronidase in the infection process with Streptococcus spp. Streptococcus dysgalactiae hyaluronidase may have an important role in promoting dissemination of the producing organism into host tissue (17). The enzyme from Streptococcus uberis has been suggested to inhibit proliferation of a mammary epithelial cell line (18). In Streptococcus pneumoniae, hyaluronidase is thought to function as a “spreading factor” by degrading host HA (19). Starr and Engleberg (20) present evidence that extracellular hyaluronidase of group A streptococcus (GAS) facilitates the spread of large molecules but not bacteria. In S. agalactiae, much of the information on the role of hyaluronidase is speculative, with little or no data to substantiate the proposed roles.
Recent studies showed that HA plays an important role in the production of proinflammatory cytokines (21, 22). HA, a main carbohydrate component of the extracellular matrix, is the principal ligand of the CD44 glycoprotein that is found on the surface of many cell types, including lymphocytes, macrophages, and epithelial cells (23). HA is normally a glycosaminoglycan of high molecular weight. At sites of inflammation, low-molecular-weight (LMW) HA accumulates, probably due to the presence of tissue-derived hyaluronidase (HYAL). HYAL is a cell surface glycosylphosphatidylinositol-anchored protein, which in cooperation with CD44 catalyzes the initial reaction that cleaves the large HA polymers. Binding of LMW HA to CD44 can induce expression of cytokines (24–26). Thus, HYAL provides a proinflammatory effect by this mechanism. However, until recently, very little was known of the ability of hyaluronidase from a microbial source to trigger the production of cytokines.
In this study, we have investigated the function and virulence of the hyaluronidase from S. agalactiae isolated from fish by comparisons of a hylB deletion mutant with the wild-type strain. We have shown that hyaluronidase plays a role in survival within murine RAW264.7 macrophages and in stimulation of proinflammatory cytokine secretion. In addition, we present evidence indicating that hyaluronidase also plays a nutritional role.
MATERIALS AND METHODS
Ethics statement.
Animal experiments were carried out according to animal welfare standards and approved by the Ethical Committee for Animal Experiments of Nanjing Agricultural University, China. All animal experiments complied with the guidelines of the Animal Welfare Council of China.
Cell lines, bacterial strains, plasmids, and growth conditions.
RAW264.7 cells (ATCC) were maintained in Dulbecco's modified Eagle medium (DMEM) with high glucose (Gibco, Invitrogen Corp., Carlsbad, CA) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) (Invitrogen). Cells were used from passages 5 to 20.
The bacterial strains and plasmids used in this study are listed in Table 1. S. agalactiae strain GD201008-001, β-hemolysin or cytolysin positive, which belongs to serotype Ia and multilocus sequence type (MLST) ST-7, was isolated from farmed tilapia with meningoencephalitis in the Guangdong Province of China in 2010 (27). The bacterial strain was grown in Todd-Hewitt broth (THB) (Difco Laboratories, Detroit, MI) or plated on THB agar containing 5% (vol/vol) sheep blood. Escherichia coli strain DH5α was cultured in Luria-Bertani (LB) broth or on an LB plate. When necessary, antibiotics were added to the plate or broth at the following concentrations: spectinomycin (Spc) (Sigma Co., St. Louis, MO), 100 μg/ml for S. agalactiae and 50 μg/ml for E. coli; ampicillin (Sigma), 100 μg/ml for E. coli.
TABLE 1.
Characteristics of the bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| S. agalactiae | ||
| GD201008-001 serotype Ia strain | Virulent strain isolated from tilapia with meningoencephalitis in China | Collected in our laboratory |
| GD201008-001 ΔhylB mutant | hylB gene deletion mutant of GD201008-001 | This study |
| ΔhylB complemented strain | GD201008-001 ΔhylB complemented with vector pSET2::hylB; Spcr | This study |
| E. coli DH5α | Production of recombinant plasmids | Invitrogen |
| Plasmids | ||
| pSET4S | Thermosensitive suicide vector for gene replacement in Streptococcus replication of pG+host3 and pUC19; lacZ′ Spcr | 28 |
| pSET2 | E. coli-Streptococcus shuttle vector; Spcr | 29 |
| pSET4S::hylB | Recombinant vector with pSET4S background, designed for knockout of hylB; Spcr | This study |
| pSET2::hylB | Complementation vector with pSET2 background, containing promoter followed by full-length hylB ORF | This study |
Inactivation of the hylB gene.
To knock out the gene hylB from S. agalactiae GD201008-001, a thermosensitive suicide vector, pSET4s::hylB, carrying the left arm and right arm, was constructed. The two arms were amplified from the chromosomal DNA of GD201008-001 by using the primer pairs ΔhylB-LF/ΔhylB-LRA and ΔhylB-RFA/ΔhylB-RR (Table 2). These arms, with an overlap of 24 bp, were used as a template to generate a PCR fragment comprising the flanking sequences of the hylB genomic locus using the primer pair ΔhylB-LF/ΔhylB-RR. The PCR fragment was digested with BamHI/EcoRI and cloned into the pSET4S vector digested with the same restriction enzymes to generate the hylB knockout vector, pSET4S::hylB. The recombinant plasmid pSET4s::hylB was electrotransformed into GD2010008-001 competent cells, and the strains were selected on Spc plates as described previously (28). The suspected double-crossover ΔhylB mutant strain was verified by PCR and quantitative real-time reverse transcription-PCR (qRT-PCR) with a series of specific primers listed in Table 2 and Table 3.
TABLE 2.
Primers used for construction and confirmation of S. agalactiae GD201008-001
| Primer | Sequence (5′→3′)a | Restriction site | Function |
|---|---|---|---|
| hylB-LF | CCGGAATTCTGATAAGAAAAGTCAATGGAC | EcoRI | Left arm of hylB; amplifies flank sequence located in coding region of hylB upstream (632 bp) |
| hylB-LRA | AGGTTTTTTATGTACGGCTAGTCTGTTGAAGCTGTAACTGAT | ||
| hylB-RFA | ATCAGTTACAGCTTCAACAGACTAGCCGTACATAAAAAACCT | Right arm of hylB; amplifies flank sequence located in coding region of hylB upstream (676 bp) | |
| hylB-RR | CGCGGATCCGAAGGACCAGGTGAAAAATTC | BamHI | |
| hylB-F | CCGTCTCTTGGGAAAAT | Fragment for hylB ORF; used to confirm deletion of hylBb | |
| hylB-R | CATGGTTTGAGGTTGGT | ||
| ChylB-F | CCGGAATTCAGGCAATTTACTATCAAAGTAGCAC | EcoRI | Fragment for complementation of hylB; amplifies structural gene portion of hylB, including its own promoter (3,382 bp) |
| ChylB-R | ATACTGCAGGATCTTCGTTACGCTATCGATTCTA | PstI |
Restriction sites are underlined.
The wild-type or complemented strain will produce a 2,334-bp fragment by PCR, but the mutant strain will not.
TABLE 3.
Primers used for qRT-PCR
| Primer | Sequence (5′→3′) | Function |
|---|---|---|
| hylB-up-F | CCTGAATTTACGCCAGGTCAT | Fragment of hylB upstream ORF |
| hylB-up-R | AAACTTAGCAGAAAAGAAAGG | |
| hylB-down-F | GGAGCTGGTTTCATCGGTTCA | Fragment of hylB upstream ORF |
| hylB-down-R | GAGTTGTCGTTGTGGCTTTCA | |
| hylB-self-F | AGAGTCTGGCTTGGCTTCATC | Fragment for hylB ORF |
| hylB-self-R | TACGGAAATATCCTGCATCAG | |
| 16s rRNA -F | CGACGATACATAGCCGACCT | Fragment for 16S rRNA |
| 16s rRNA -R | CCGTCACTTGGTAGATTTTCC | |
| IL-1β-F | TCCAGGATGAGGACATGAGCAC | Fragment for mouse IL-1β gene |
| IL-1β-R | GAACGTCACACACCAGCAGGTTA | |
| IL-6-F | CCACTTCACAAGTCGGAGGCTTA | Fragment for mouse IL-6 gene |
| IL-6-R | GCAAGTGCATCATCGTTGTTCATAC | |
| TNF-α-F | AAGCCTGTAGCCCACGTCGTA | Fragment for mouse TNF-α gene |
| TNF-α-R | GGCACCACTAGTTGGTTGTCTTTG | |
| β-actin-F | TGACAGGATGCAGAAGGAGA | Fragment for mouse β-actin gene |
| β-actin-R | GCTGGAAGGTGGACAGTGAG |
The construction of the pSET2::hylB complementation vector has been described previously (29). The complementation vector pSET2::hylB was constructed using the primers ChylB-F and ChylB-R (Table 2) to amplify a 3,300-bp fragment containing the open reading frame (ORF) coding for the HylB protein from the GD201008-001 genome. The complementation vector was electroporated into the ΔhylB strain and screened on THB agar for spectinomycin resistance (Spcr). The pSET2 empty vector was transformed into the wild-type strain and the ΔhylB mutant, which serves as a control.
Real-time PCR for quantitation of hyaluronidase.
Quantitative real-time reverse transcription-PCR (qRT-PCR) was carried out as described previously (30). S. agalactiae cultures were grown for 3 h in THB at 37°C, and RNA was isolated with an E.Z.N.A. bacterial RNA isolation kit (Omega, Beijing, China). The RNA was treated with DNase I (Promega, Madison, WI) to exclude genomic DNA contamination. The cDNA synthesis was performed using the PrimeScript RT reagent kit (TaKaRa, Dalian, China) according to the manufacturer's instructions. The mRNA levels were measured using two-step relative qRT-PCR. The 16S rRNA housekeeping gene was amplified as an internal control. The specific primers used for the qRT-PCR assays are listed in Table 3. The SYBR green PCR was performed using the SYBR Premix Ex Taq kit (TaKaRa) according to the manufacturer's instructions. Reactions were carried out in triplicate. An ABI 7500 RT-PCR system (Applied Biosystems) was used for relative qRT-PCR. The comparative cycle threshold (2−ΔΔCT) method (31) was used to analyze the mRNA levels.
Hyaluronic acid degradation assays.
Assays to detect degradation of hyaluronic acid were performed by using the plating method of Smith and Willett (32). The basic medium was prepared from sterile brain heart infusion broth (BBL) containing 1% (wt/vol) noble agar. The medium was autoclaved at 115°C for 15 min and cooled to 46°C before an aqueous 10-mg/ml solution of filter-sterilized hyaluronic acid was added to a final concentration of 400 μg/ml. A 5% (wt/vol) filter-sterilized solution of bovine serum albumin fraction V was then added with constant stirring to give a final concentration of 1% (wt/vol) in the medium. Bacteria were grown to an approximate optical density at 600 nm (OD600) of 0.6, and a 5-μl solution of bacteria was incubated on the plates at 37°C. After overnight incubation, each plate was flooded with 2 mol/liter acetic acid for 10 min. Degradation of the hyaluronic acid was detected as a transparent ring resulting from acetic acid precipitation of a complex of albumin and nondegraded hyaluronic acid. Assays were conducted at least three times.
Growth assays.
Growth assays to compare the reproductive abilities of bacteria were performed by using a chemically defined medium (CDM) without carbohydrate (33). Where appropriate, medium was supplemented with the commercially available carbohydrates glucose (12 mM) and human hyaluronic acid (5 mg/ml). An aqueous 10-mg/ml solution of filter-sterilized hyaluronic acid was added to the CDM. Overnight cultures were prepared for all strains (wild-type, hylB mutant, and complemented strains). The growth densities of these cultures (based on OD600 readings) were then equalized by dilution adjustments. The OD600 was measured every 2 h until the bacterial growth began to decrease. CDM supplemented with glucose served as a positive control, indicating that the bacteria were viable and that the mutant strains had no general defect in growth relative to the parent strain.
LD50 determination in zebrafish.
The care and maintenance of zebrafish used in this study followed established protocols (Pearl River Fishery Research Institute, Chinese Academy of Fishery Science). Challenge of zebrafish with S. agalactiae strain GD201008-001 was performed as described previously (34). Prior to the inoculation of the fish, cultures were collected in late log phase and washed twice in phosphate-buffered saline (PBS) (pH 7.4). Zebrafish were anesthetized with tricaine methanesulfonate (MS-222) (Hangzhou Animal Medicine Factory, China) at a concentration of 90 mg/liter. Several groups of 15 zebrafish were intraperitoneally (i.p.) injected with 10-fold serially diluted suspensions of bacteria (102 to 106 CFU) in sterile PBS. Control fish were injected with sterile PBS. Survival rates were recorded daily and over a period of 7 days postinfection. The 50% lethal dose (LD50) values were calculated by the Reed and Muench method (35).
Experimental infection of mice.
Female BALB/c mice (5 to 7 weeks of age) were purchased from the Experimental Animal Center, Yangzhou University. Previous studies had shown that the bacterial strain GD201008-001 was highly virulent to BALB/c mice by intraperitoneal administration, with LD50 values of less than 10 CFU (14). The mice infected with a predetermined dose of 5 × 102 CFU (50-fold greater than the LD50 in mice infected with the GD201008-001 wild-type strain) of the wild-type, mutant, and complemented strains. Control mice were injected with sterile PBS. For CFU organ burden determinations, groups of five mice were sacrificed at 16 h postinfection to collect the spleen, brain, and blood. The organs were harvested, homogenized, and plated onto THB agar plates for bacterial cell counting to determine tissue colonization. Experiments were repeated at least three times to ensure reproducibility. For time-to-death studies, groups of 10 mice were infected with matched inocula and monitored for mortality.
Determination of intracellular growth efficiency.
RAW264.7 macrophages were grown in DMEM containing 10% FBS in 24-well tissue culture plates at a concentration of 4 × 105macrophages/well. S. agalactiae GD201008-001 wild-type, mutant, and complemented strains were grown in THB for 3 h at 37°C, harvested by centrifugation at 5,000 × g for 10 min, washed three times in sterile 10 mM PBS, and resuspended in fresh serum-free DMEM without antibiotics. The cell monolayers were infected at a multiplicity of infection (MOI) of 1:1 for 1 h at 37°C. Extracellular bacteria were removed by washing macrophages three times in PBS, and the medium was replaced with 1% FBS–DMEM containing 100 μg/ml gentamicin and 5 μg/ml penicillin G; the cells remained in this medium for the duration of the assay. Cells were cultured throughout all experiments at 37°C in 5% CO2. To measure the intracellular bacterial survival rate, cells were sampled at 1 h after the addition of antibiotics (time 0) and subsequently at 2, 4, 6, 8, 12, and 24 h. Infected cells were washed three times with PBS and then incubated for 15 min at 37°C with 0.02% Triton X-100 to lyse the macrophages and release intracellular bacteria. Various dilutions of the lysates were plated on THB agar plates and incubated overnight at 37°C. Colonies were counted to determine the number of intracellular bacteria. To analyze the number of bacteria surviving over time compared to the initial number of intracellular bacteria, the relative CFU (rCFU) was calculated as follows: rCFU = CFU at time point x/CFU at time point 0 (13).
LDH cytotoxicity assay.
Bacterial cell-mediated cytotoxicity of RAW264.7 macrophages was detected by measuring lactate dehydrogenase (LDH) activity present in the culture supernatant. For LDH measurements, the CytoTox 96 nonradioactive cytotoxicity assay (Promega) was used according to the manufacturer's instructions. Briefly, S. agalactiae cells were grown and diluted as described above. Cells grown in 96-well plates were infected with 100-μl aliquots of a bacterial suspension (MOI of 1, 10, or 100). The plates were centrifuged at 800 × g for 10 min to bring the bacteria to the surface of each monolayer and were then incubated for 4 h at 37°C with 5% CO2. Maximum release was achieved by lysis of monolayers with Triton X-100 at a final concentration of 1% (vol/vol). The amount of enzyme was determined using a microplate reader (absorbance at 492 nm). The LDH released by uninfected cells and bacteria alone was designated spontaneous release. Cytotoxicity was calculated as follows: % cytotoxicity = (test LDH release − spontaneous release)/(maximal release − spontaneous release).
Cytokine assays.
RAW264.7 macrophages were infected with the bacteria at an MOI of 10:1 for 1 h. To measure the cytokine expression of the infected cells, cells were sampled at 3, 6, 9, 12, 15, and 24 h after the addition of antibiotics. Then the infected cells were treated with 0.02% Triton X-100 for 15 min at 37°C to lyse the macrophages. Uninfected RAW264.7 macrophages in medium served as controls.
Spleen tissues were removed from mice 5, 10, and 15 h after injection with 5 × 102 CFU of S. agalactiae and from PBS-treated mice. Tissues were then homogenized in 1 ml of lysis buffer (Qiagen, West Sussex, United Kingdom). The homogenized tissues were then centrifuged at 2,000 × g for 10 min, and the supernatants were sterilized with a Millipore filter (0.45-μm pore size).
Total RNA was then isolated from the above supernatants of the spleens and RAW264.7 macrophages using an AllPrep RNA microkit (Qiagen). The cDNA synthesis was performed using the PrimeScript RT reagent kit (TaKaRa) according to the manufacturer's instructions. The mRNA levels were measured using two-step relative qRT-PCR. The β-actin housekeeping gene was amplified as an internal control. The sequences of the primers for tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), IL-6, and β-actin are listed in Table 3. Real-time PCR was performed using a SYBR Premix Ex Taq kit (TaKaRa) and ABI 7500 RT-PCR system. The comparative cycle threshold (2−ΔΔCT) method was used to analyze the mRNA levels.
To determine whether differences in mRNA levels reflect differences in secreted protein levels, we also used commercial enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN) to examine the levels of TNF-α, IL-1β, and IL-6 released from the infected RAW264.7 macrophages. RAW264.7 macrophages were infected with bacteria as described above. After incubation for 3, 6, 9, 12, 15, or 24 h, the culture supernatants were collected from the infected macrophages and used to measure the cytokine levels according to the manufacturer's instructions.
Statistical analyses.
In all experiments, data points were plotted using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA). Data are presented as mean values ± standard errors of the means (SEM). Two-way analysis of variance (ANOVA) was used for the analysis of qRT-PCR results. The animal infection and intracellular growth analyses were performed using the nonparametric Mann-Whitney U test. A P value of <0.05 was considered significant.
Nucleotide sequence accession number.
The genome sequence of S. agalactiae strain GD201008-001 has been deposited in the GenBank database under accession no. CP003810.
RESULTS
Inactivation of the hylB gene.
To determine the function of hylB in S. agalactiae, a hylB mutant (ΔhylB) was constructed by homologous replacement in strain GD201008-001 (Fig. 1A). A complemented strain (CΔhylB) was constructed by introduction of the hylB gene into the ΔhylB mutant using a complementation vector. The gene knockout mutant and complemented strains were identified by PCR with specific primers (Fig. 1B). The qRT-PCR analysis indicated that hylB mRNA expression could not be detected in the hylB mutant, whereas by complementation of the mutant strain, hylB mRNA was detected (Fig. 1C). Moreover, inactivation of hylB gene did not have an effect either upstream or downstream of the deletion. The results indicated that the hylB mutant and complemented strains were successfully constructed.
FIG 1.

Confirmation of the S. agalactiae GD201008-001 hylB mutant and complemented strains. (A) Schematic of the strategy for producing the hylB mutant by allelic exchange mutagenesis. (B) Multiple-PCR analysis of the wild-type (WT), mutant (ΔhylB), and complemented (CΔhylB) strains. The primer combinations used in the PCR were as follows: lanes 1, 2, and 3, hylB-LF/hylB-RR; lanes 4, 5, and 6, hylB-F/hylB-R. Genomic DNA from the following strains was used as the template: the wild-type strain (lanes 1 and 4), the ΔhylB mutant strain (lanes 2 and 5), and the CΔhylB complemented strain (lanes 3 and 6). The 5-kb DNA ladder marker (M) is shown on the left. (C) mRNA expression levels of hylB upstream, downstream, and hylB self ORF in the S. agalactiae GD201008-001 wild-type, mutant, and complemented strains in vitro. The values of the hylB-associated genes in the wild-type strain were normalized to 1.0. The relative changes in gene expression ratios of selected genes were normalized to the expression of a single housekeeping gene (16S rRNA gene) and calculated as described by the 2−ΔΔCT method. Error bars indicate SEM from three independent experiments.
Growth assays.
A previous report has shown that S. pneumoniae can utilize HA as the sole carbon source for growth (36); therefore, we hypothesized that HA also provides a carbon source for S. agalactiae growth. Our study demonstrated that S. agalactiae strain GD201008-001 cultured on HA (5 mg/ml) was capable of growing to a maximal OD600 that did not differ significantly from that observed when grown on glucose (12 mM), although bacteria grown on HA exhibited a significant growth delay (Fig. 2). The time taken to reach an OD600 of 0.6 when grown on HA was longer than when grown on glucose, presumably as HA must be degraded before being utilized for growth. The ΔhylB strain grown on HA showed a significant growth defect compared with the wild-type strain, whereas the growth of the complemented strain was almost restored to the level of the wild-type strain (Fig. 2). Furthermore, compared with the wild-type and complemented strains, the hylB mutant did not exhibit detectable degradation of hyaluronic acid by the plate assay (Fig. 3). These data suggested that S. agalactiae adapted slowly to growth in the medium with HA as the sole carbon source, but once adapted, efficient degradation of HA occurred. Furthermore, growth of the ΔhylB strain on HA was negligible, while complementation of HylB restored growth of the mutant, providing further confirmation of the successful generation of the mutant and complemented strains.
FIG 2.

Growth assays of the wild-type (WT), hylB mutant (ΔhylB), and complemented (CΔhylB) strains. Strains were grown in chemically defined medium (CDM) supplemented with 5 mg/ml hyaluronic acid (HA) or 12 mM glucose. OD600 values are the means ± SEM from three independent experiments.
FIG 3.
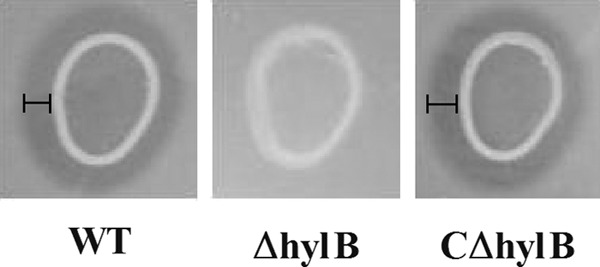
Variation in the ability of the wild-type (WT), hylB mutant (ΔhylB), and complemented (CΔhylB) strains of S. agalactiae to degrade hyaluronic acid. Degradation of the hyaluronic acid was detected as a clear zone (labeled areas) resulting from acetic acid precipitation of a complex of albumin and nondegraded hyaluronic acid. Assays were conducted at least twice.
S. agalactiae infection in zebrafish.
To investigate the role of hylB in S. agalactiae virulence, zebrafish were injected i.p. with the wild-type, mutant, or complemented strain. The mortality of zebrafish was recorded daily and followed over a period of 7 days following infection. As shown in Table 4, the LD50 value of the ΔhylB mutant (7.94 × 102 CFU) was 6-fold higher than that of the wild-type strain (1.31 × 102 CFU), indicating a significant reduction in the virulence of the mutant. Also, the LD50 of the complemented strain (2.37 × 102 CFU) was restored to a level 3-fold higher than that of the mutant strain. The results suggested that hyaluronidase may be an important virulence factor of S. agalactiae GD201008-001 in zebrafish.
TABLE 4.
LD50 of the wild-type S. agalactiae GD201008-001 strain, hylB mutant, and complemented strain in zebrafish
| Strain | No. of dead animals/total no. tested at challenge dose (CFU) of: |
LD50 (CFU) | ||||
|---|---|---|---|---|---|---|
| 1.0 × 105 | 1.0 × 104 | 1.0 × 103 | 1.0 × 102 | 50 | ||
| WT | 15/15 | 13/15 | 12/15 | 7/15 | 4/15 | 1.31 × 102 |
| ΔhylB mutant | 15/15 | 11/15 | 8/15 | 3/15 | 1/15 | 7.94 × 102 |
| CΔhylB complemented strain | 15/15 | 12/15 | 10/15 | 6/15 | 3/15 | 2.37 × 102 |
S. agalactiae infection in mice.
The mortality of mice was recorded. To better understand the effects of hylB during systemic infection, we harvested spleens, blood, and brains of mice infected with the wild-type, mutant, or complemented strain. Mice infected with 5 × 102 CFU were examined over the following 16 h for signs of systemic infection. S. agalactiae colonizations in spleens and brains at 16 h postinfection, as well as blood bacteremia, were significantly reduced with the ΔhylB mutant, with an approximate decrease of 6-fold in the spleen and 3-fold in both the blood and brain compared to those in the animals infected with the wild-type strain (Fig. 4). Certainly, reduction in CFU does not distinguish between reduced replication and reduced survival. However, in spite of this, these data indicated an important role for the hylB gene during in vivo replication and/or survival within lymphoid organs in a systemic S. agalactiae infection model. Furthermore, these observations implicate the hylB gene as a key element required by S. agalactiae to penetrate the blood-brain barrier (BBB) and cause meningitis in mice.
FIG 4.

Bacterial loads in different tissues from mice infected intraperitoneally with the S. agalactiae GD201008-001 wild-type (WT), mutant (ΔhylB), and complemented (CΔhylb) strains. Bacterial loads in blood (A) are expressed as CFU/ml, and those in the brain (B) and spleen (C) are expressed as CFU/g of tissue. Results are expressed as means ± SEM from at least three infected mice at 16 h postinfection. Asterisks indicate significant differences (P < 0.05) in the numbers of CFU observed between the mutant and wild-type strains.
The time-to-death analysis showed that 16 h after infection, death began to occur in the mice challenged with wild-type and complemented strains, and all of the infected mice died by hour 18. However, the animals infected with the hylB mutant showed a delay in time to death. Death occurred 20 h postinfection, and all mice were dead by hour 24. Before their death, mice infected with either Hyl+ or Hyl− strains exhibited similar clinical symptoms, such as depression, swollen eyes, rough hair coat, and lethargy.
Effect of S. agalactiae hyaluronidase on RAW264.7 macrophages.
We observed that replication and/or survival of the S. agalactiae ΔhylB mutant strain in the tissues of mice was significantly reduced compared with that of the wild-type strain. Following invasion, resistance to macrophage killing is an essential step in bacterium pathogenesis. To elucidate the role of hylB in this context, we investigated the survival capacity of the wild-type, mutant, and complemented strains in RAW264.7 macrophages. Intracellular bacterial counts and total survival within RAW264.7 macrophages were quantified by CFU determination. Compared with the wild-type and complemented strains, the ΔhylB mutant exhibited reduced resistance to killing after ingestion by macrophages in a time-dependent manner (Fig. 5A). The ΔhylB mutant displayed impaired survival in macrophages compared to the wild-type strain. Similar numbers of intracellular viable wild-type and Hyl-deficient bacteria were measured in RAW264.7 macrophages at 1 h postinfection, demonstrating that the reduced number of the ΔhylB mutant cells in RAW264.7 macrophages was not due to diminished bacterial uptake by macrophages (Fig. 5B).
FIG 5.

Intracellular growth of S. agalactiae in RAW264.7 macrophages. (A) Intracellular survival of wild-type S. agalactiae GD201008-001 in RAW264.7 macrophages compared to that of the ΔhylB mutant and complemented strains infected at an MOI of 1. Phagocytosis was allowed to proceed for 1 h before the addition of antibiotics for 1 h. This initial antibiotic treatment was extended for different times up to 24 h. The relative number of CFU (rCFU) was estimated by plating out the lysates of infected macrophages and counting the number of CFU at each time point. Asterisks indicate the time points when the intracellular bacteria survival rates elicited by the ΔhylB mutant were significantly lower (P < 0.05) than those produced by wild-type infection. (B) RAW264.7 macrophages were infected with different numbers of cells of the S. agalactiae GD201008-001 wild-type (WT), mutant (ΔhylB), and complemented (CΔhylb) strains. At 1 h postinfection, cells were extensively washed to remove extracellular bacteria after antibiotic treatment and were lysed; CFU were then measured. The data shown are means ± SEM from three independent experiments.
The cytotoxic effect of hyaluronidase on RAW264.7 macrophages was investigated to determine its role in macrophage injury. The LDH assay was carried out with RAW264.7 macrophages at MOI of 1, 10, and 100 for 4 h, and the data obtained in the experiment showed that, similar to the wild-type and complemented strains, the ΔhylB mutant produced no significant injury (Fig. 6).
FIG 6.

Effect of bacterial cell-mediated cytotoxicity as measured by LDH cytotoxicity assays. RAW264.7 macrophages were infected with S. agalactiae GD201008-001 wild-type (WT), mutant (ΔhylB), and complemented (CΔhylb) strains at the indicated multiplicities of infection (MOI) for 4 h to measure the LDH release into the supernatant. “Control” corresponds to maximum lysis achieved using 2% Triton X-100. The data shown are means ± SEM from three independent experiments.
Hyaluronidase attenuates the cytokine output of S. agalactiae-infected mice or cultured macrophages.
Previous studies have determined that cytokines can act, in an autocrine fashion, to render macrophages less permissive for bacterial growth (37). Thus, we hypothesized that the reduced intracellular survival of the ΔhylB mutant within macrophages might be due, at least in part, to the high levels of cytokines produced in response to infection by the mutant. To address this hypothesis, we investigated the levels of IL-1β, IL-6, and TNF-α in response to infection with the wild-type, ΔhylB mutant, and complemented strains in RAW264.7 macrophages. The ΔhylB mutant caused a marked enhancement of mRNA levels of IL-1β, IL-6, and TNF-α in RAW264.7 cells compared to those of the wild-type and complemented strains (Fig. 7). The altered phenotype of the mutant was most marked in terms of IL-6 production, the difference between the wild-type and mutant strains being almost 5-fold at all time points (Fig. 7B). The results showed that cytokine expression by RAW264.7 macrophages may be inhibited by hyaluronidase following S. agalactiae infection. Furthermore, infection of macrophages with the three S. agalactiae strains led to significantly increased expression of IL-1β, IL-6, and TNF-α compared with that detected in the uninfected controls (Fig. 7B). However, among the different cytokines, IL-6 showed the highest mRNA levels in the infected macrophages, and IL-1β and TNF-α exhibited relatively low levels (Fig. 7A and C). Furthermore, we performed ELISA and demonstrated the association between cytokine production and protein level in RAW264.7 macrophages (see Fig. S1 in the supplemental material). The findings suggest the presence of a good correlation between mRNA expression and secreted protein levels.
FIG 7.

Intracellular S. agalactiae-induced cytokine expression in RAW264.7 macrophages. RAW264.7 macrophages were infected with S. agalactiae GD201008-001 wild-type (WT), mutant (ΔhylB), and complemented (CΔhylB) strains at an MOI of 10:1 at 1 h. Extracellular bacteria were killed by antibiotics, and cells were harvested at different time points. The expression levels of IL-1β, IL-6, and TNF-α were measured by quantitative RT-PCR. Uninfected cells served as the control. Levels of IL-1β, IL-6, and TNF-α mRNA were normalized to mRNA levels of β-actin and then were expressed as n-fold increases with respect to the control. The data shown are means ± SEM from no less than six independent experiments. Asterisks indicate the time points when the cytokine levels elicited by the ΔhylB mutant were significantly greater (P < 0.05) than those produced by wild-type infection.
Cultured murine RAW264.7 macrophages may not be sufficient to accurately represent host animals to evaluate cytokine production. Therefore, we investigated the induction of cytokine expression in BALB/c mice at 5, 10, and 15 h postinfection. Compared with the wild-type and complemented strains, infection with the ΔhylB mutant also significantly increased accumulation of IL-1β, IL-6, and TNF-α mRNA in the spleens at all the time points assessed (Fig. 8). Furthermore, compared with uninfected mice, dramatic increases in IL-1β, IL-6, and TNF-α expression levels were found following infection with the ΔhylB mutant at 10 h postinfection. Sustained high levels of the three cytokines were still present at the end of the detection period. A time-dependent increase in cytokine expression levels was also observed in the wild-type and complemented strains. These results provide further evidence that hyaluronidase from S. agalactiae inhibits proinflammatory cytokine expression.
FIG 8.

Cytokine expression in the spleens of BALB/c mice injected intraperitoneally with 5 × 102 CFU of S. agalactiae GD201008-001 wild-type (WT), mutant (ΔhylB), and complemented (CΔhylB) strains. Supernatants from spleen homogenates were collected at different time points after infection and assayed for IL-1β (A), IL-6 (B), and TNF-α (C) by quantitative RT-PCR. Uninfected mice served as the control. Five mice per group were sacrificed at each time point. Levels of IL-1β, IL-6, and TNF-α mRNA were normalized to mRNA levels of β-actin and then were expressed as n-fold increases with respect to the control. The data shown are means ± SEM from no less than six independent experiments. Asterisks indicate the time points when the cytokine levels elicited by the ΔhylB mutant were significantly greater (P < 0.05) than those produced by wild-type infection.
DISCUSSION
Many pathogenic bacteria produce hyaluronidases, which serve as virulence factors (16). Since hyaluronan is a major constituent of the ground substance of most connective tissues, hyaluronidase may be an essential factor in enabling the spread of the pathogens from an initial site of infection (38). As observed in other bacteria, hyaluronidase in S. agalactiae has been assumed to facilitate the spread of the organism through the tissues of the infected host (15). The mechanism of the enzymatic degradation of hyaluronan in S. agalactiae has been determined (39). However, the role of hyaluronidase in the pathological processes of S. agalactiae is relatively unclear. The purpose of the present study was to examine the role of hyaluronidase in S. agalactiae pathogenesis and to attempt to demonstrate some novel functions for this enzyme.
Intracellular survival within phagocytes may contribute to disease progression by reducing the effectiveness of phagocyte clearance. In addition, some pathogens actively exploit phagocytes to facilitate movement between host tissues, and it is possible that a related process may underlie S. agalactiae dissemination (8, 9). It has been reported that some virulence factors and regulatory genes of S. agalactiae affect survival of the bacterium in professional phagocytes (13, 40, 41). The ability to sense the environment and mount an appropriate adaptive transcriptional response may be of crucial importance for S. agalactiae colonization and pathogenicity. Our previous study showed that the expression of hylB was upregulated by the interaction of S. agalactiae with murine macrophages (14). The present study demonstrated that inactivation of hylB reduced the numbers of intracellular viable S. agalactiae cells (Fig. 5A), indicating that hyaluronidase plays a key role in bacterial intracellular survival in macrophages. However, further study showed that the reduction in the numbers of the S. agalactiae ΔhylB mutant cells in RAW264.7 macrophages was independent of diminished bacterial uptake by macrophages (Fig. 5B) or an increased cytotoxic effect of the bacterium on macrophages (Fig. 6). These observations raise the question of how hyaluronidase influences the interaction between S. agalactiae and macrophages. Once inside a macrophage, bacteria must acquire nutrients for growth. Hyaluronate lyase is an important surface enzyme of many streptococcal species and presumably provides energy and a carbon source for bacterial cells. Disaccharides are the major end products of HA degradation, and these can be transported and metabolized intracellularly to supply the required nutrients (as a carbon source) for a pathogen as it replicates and spreads (16). By analogy with what is observed in other bacteria (32, 42), our study showed that S. agalactiae could grow utilizing HA as its sole source of carbon. The inactivation of hylB resulted in severe defects of bacterial growth in the medium of HA as the sole carbon source, while complementation of HylB restored growth of the mutant (Fig. 2). These data indicated that hyaluronidase-mediated degradation of HA may play an important role in the growth of S. agalactiae under glucose deficiency. Therefore, hyaluronidase may be beneficial for survival of S. agalactiae in macrophages.
In recent years, the zebrafish has been used as an ideal experimental model of infections with several streptococcal species such as Streptococcus iniae, Streptococcus pyogenes (43, 44), and S. agalactiae (34). Our data obtained using the zebrafish model indicated a 6-fold reduction in virulence of the hylB mutant compared with the wild type (Table 4). Considering that BALB/c mice are highly susceptible to S. agalactiae GD2010008-001 (14), we used this mouse model to further assess the pathogenicity of this enzyme. The time-to-death analysis showed that 100% of the mice challenged with wild-type or complemented strains died by hour 18, whereas all mice infected with the mutant died by hour 24, indicating that the inactivation of hylB resulted in a significant delay in time to death. The investigation for bacterial loads showed that the bacterial replication and/or survival in the blood, spleen, and brain was significantly reduced for the hylB mutant compared to wild-type-infected animals (Fig. 4). Recent publications suggest that hyaluronidase plays a role in bacterial dissemination and invasion and also in breaching the BBB (45, 46). The observation that the deletion of hyaluronidase resulted in a significant decrease in the CFU counts in the blood and brain of infected mice (Fig. 4) leads us to hypothesize that hyaluronidase plays an important role in the development of meningitis caused by S. agalactiae. The molecular mechanism of the involvement of the hylB gene of S. agalactiae in the penetration of BBB represents a critical focus of further work. Collectively, these data suggest that hyaluronidase is an important S. agalactiae virulence factor.
Previous studies have shown that S. agalactiae virulence factors stimulate the release of various proinflammatory cytokines from human mononuclear cells and murine macrophages (40, 47). However, in the present study, we demonstrated that the S. agalactiae hyaluronidase limited the secretion of proinflammatory cytokines IL-1β, IL-6, and TNF-α from infected RAW264.7 macrophages and in infected mice (Fig. 7; see Fig. S1 in the supplemental material). Indeed, high levels of proinflammatory cytokines inhibit bacterial survival in macrophages (48, 49). Furthermore, these cytokines modulate both local and systemic inflammatory responses and help to recruit and activate leukocytes, thereby increasing bactericidal activity (50, 51). Thus, limitation of the secretion of these cytokines can greatly enhance bacterial growth in vivo. This conclusion may explain our finding that hyaluronidase contributes to the ability of S. agalactiae to survive in mice and macrophages.
A large amount of evidence indicates that bacterial virulence factors have opposing effects on the expression of proinflammatory cytokines. Costa et al. (52) revealed that β-hemolysin from S. agalactiae triggered the production of high levels of IL-1β by activating the NOD-like receptor family pyrin domain containing 3 (NLRP3) sensor. On the other hand, Vossenkamper et al. (53) showed that the type three secretion system (T3SS) of enteropathogenic Escherichia coli (EPEC) inhibits NF-κB signal transduction events, which results in decreasing release of proinflammatory cytokines by infected dendritic cells. Our findings indicated that hyaluronidase of S. agalactiae suppresses the release of proinflammatory cytokines. The actual mechanism involved in cytokine regulation is not clear. We cannot exclude the possibility that hyaluronidase alters the expression of other virulence determinants or regulators, which in turn reduce the production and secretion of the proinflammatory cytokines in S. agalactiae. The CovS/CovR system is known to be an important transcriptional regulator in pathogenic streptococci, regulating approximately 7% of the S. agalactiae genome (40, 54). It has been reported that one S. agalactiae strain carrying a cov mutation showed a marked increase in the release of proinflammatory cytokines (55).
Recent reports have shown that tissue-derived hyaluronidase can cleave HA into fragments that trigger the production of proinflammatory cytokines (54, 55). However, our study indicated that the inactivation of hyaluronidase from S. agalactiae stimulated a significantly higher release of proinflammatory cytokines than that from the wild type in macrophages as well as in mice (Fig. 8), suggesting a different function from tissue-derived hyaluronidase. It can be speculated that this is due to variations in HA metabolism catalyzed by hyaluronidases from different sources. HA catabolism depends on individual hyaluronidase activities under pathological conditions and the products generated during catabolic pathway with contrasting biological activities. The vertebrate enzymes are hydrolases, while the enzymes from microbial source are eliminases (30). Previous studies indicated that hyaluronidase from S. agalactiae cleaves HA into disaccharides that are utilized for bacterial growth (39). Therefore, elimination of HA via hyaluronidase from S. agalactiae might inhibit the interaction of HA with its receptor CD44, thus interfering with the expression of cytokines.
The complex underlying mechanism and regulation of the expression levels and the release patterns of various proinflammatory cytokines from macrophages in response to bacterial stimulation remain to be fully elucidated. In this study, we showed that a significantly higher level of IL-6 was released from RAW264.7 macrophages infected with the wild-type or hylB mutant forms of S. agalactiae at every time point investigated, relative to the release of IL-1β and TNF-α (Fig. 7; see Fig. S1 in the supplemental material). These in vitro findings correlated with the observations in mice of a significantly higher release of proinflammatory cytokines, especially IL-6, after infection with S. agalactiae (Fig. 8). These findings are in accordance with previous reports that during most invasive bacterial infections, the immune system of the host facilitates activation and recruitment of neutrophils and macrophages and the production of proinflammatory cytokines, most notably the release of high levels of IL-6 (56–58). The marked upregulation of IL-6 secretion from macrophages in response to S. agalactiae described here has not previously been reported. As IL-6 is secreted by and has effects on a wide range of cell types, it is now believed to be important for maintaining the homeostatic balance within tissues (59). Furthermore, the release of the three proinflammatory cytokines from macrophages showed different regular patterns. In the case of IL-1β and TNF-α, expression levels reached a maximum at 12 h and 6 h, respectively, and then declined, whereas IL-6 production exhibited a gradually rising trend (Fig. 7; see Fig. S1). These data suggest that S. agalactiae has various effects on the cytokine output of infected macrophages.
In conclusion, the results presented herein provide insights into novel roles of S. agalactiae hyaluronidase in bacterial survival within macrophages and release of proinflammatory cytokines. Understanding this mechanism will provide unique insights into the pathogen and facilitate the development of new control strategies for S. agalactiae infection.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Program for New Century Excellent Talents in University (NCET-07-0440).
Footnotes
Published ahead of print 7 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00022-14.
REFERENCES
- 1.Rajagopal L. 2009. Understanding the regulation of group B streptococcal virulence factors. Future Microbiol. 4:201–221. 10.2217/17460913.4.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaiwarith R, Jullaket W, Bunchoo M, Nuntachit N, Sirisanthana T, Supparatpinyo K. 2011. Streptococcus agalactiae in adults at Chiang Mai University Hospital: a retrospective study. BMC Infect. Dis. 11:149. 10.1186/1471-2334-11-149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keefe GP. 1997. Streptococcus agalactiae mastitis: a review. Can. Vet. J. 38:429–437 [PMC free article] [PubMed] [Google Scholar]
- 4.Richards VP, Lang P, Bitar PD, Lefebure T, Schukken YH, Zadoks RN, Stanhope MJ. 2011. Comparative genomics and the role of lateral gene transfer in the evolution of bovine adapted Streptococcus agalactiae. Infect. Genet. Evol. 11:1263–1275. 10.1016/j.meegid.2011.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pridgeon JW, Klesius PH. 2011. Development and efficacy of a novobiocin-resistant Streptococcus iniae as a novel vaccine in Nile tilapia (Oreochromis niloticus). Vaccine 29:5986–5993. 10.1016/j.vaccine.2011.06.036 [DOI] [PubMed] [Google Scholar]
- 6.Ferguson HW, Morales JA, Ostland VE. 1994. Streptococcosis in aquarium fish. Dis. Aquat. Organ. 19:1–6. 10.3354/dao019001 [DOI] [Google Scholar]
- 7.Evans JJ, Klesius PH, Shoemaker CA. 2004. Efficacy of Streptococcus agalactiae (group B) vaccine in tilapia (Oreochromis niloticus) by intraperitoneal and bath immersion administration. Vaccine 22:3769–3773. 10.1016/j.vaccine.2004.03.012 [DOI] [PubMed] [Google Scholar]
- 8.Cornacchione P, Scaringi L, Fettucciari K, Rosati E, Sabatini R, Orefici G, von Hunolstein C, Modesti A, Modica A, Minelli F, Marconi P. 1998. Group B streptococci persist inside macrophages. Immunology 93:86–95. 10.1046/j.1365-2567.1998.00402.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valenti-Weigand P, Benkel P, Rohde M, Chhatwal GS. 1996. Entry and intracellular survival of group B streptococci in J774 macrophages. Infect. Immun. 64:2467–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan EL, Chhatwal GS, Rohde M. 2006. Reduced ability of penicillin to eradicate ingested group A streptococci from epithelial cells: clinical and pathogenetic implications. Clin. Infect. Dis. 43:1398–1406. 10.1086/508773 [DOI] [PubMed] [Google Scholar]
- 11.Lemire P, Houde M, Lecours MP, Fittipaldi N, Segura M. 2012. Role of capsular polysaccharide in group B Streptococcus interactions with dendritic cells. Microbes Infect. 14:1064–1076. 10.1016/j.micinf.2012.05.015 [DOI] [PubMed] [Google Scholar]
- 12.Liu GY, Doran KS, Lawrence T, Turkson N, Puliti M, Tissi L, Nizet V. 2004. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc. Natl. Acad. Sci. U. S. A. 101:14491–14496. 10.1073/pnas.0406143101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cumley NJ, Smith LM, Anthony M, May RC. 2012. The CovS/CovR acid response regulator is required for intracellular survival of group B Streptococcus in macrophages. Infect. Immun. 80:1650–1661. 10.1128/IAI.05443-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo C, Chen R, Kalhoro DH, Wang Z, Liu G, Liu LUCY. 2014. Identification of genes preferentially expressed by highly virulent piscine Streptococcus agalactiae upon interaction with macrophages. PLoS One 9:e87980. 10.1371/journal.pone.0087980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchard DG, Lin B, Willingham TR, Baker JR. 1994. Characterization of the group B streptococcal hyaluronate lyase. Arch. Biochem. Biophys. 315:431–437. 10.1006/abbi.1994.1521 [DOI] [PubMed] [Google Scholar]
- 16.Hynes WL, Walton SL. 2000. Hyaluronidases of Gram-positive bacteria. FEMS Microbiol. Lett. 183:201–207. 10.1111/j.1574-6968.2000.tb08958.x [DOI] [PubMed] [Google Scholar]
- 17.Calvinho LF, Almeida RA, Oliver SP. 1998. Potential virulence factors of Streptococcus dysgalactiae associated with bovine mastitis. Vet. Microbiol. 61:93–110. 10.1016/S0378-1135(98)00172-2 [DOI] [PubMed] [Google Scholar]
- 18.Matthews KR, Rejman JJ, Turner JD, Oliver SP. 1994. Proliferation of a bovine mammary epithelial-cell line in the presence of bacterial virulence factors. J. Dairy Sci. 77:2959–2964. 10.3168/jds.S0022-0302(94)77237-4 [DOI] [PubMed] [Google Scholar]
- 19.Berry AM, Lock RA, Thomas SM, Rajan DP, Hansman D, Paton JC. 1994. Cloning and nucleotide sequence of the Streptococcus pneumoniae hyaluronidase gene and purification of the enzyme from recombinant Escherichia coli. Infect. Immun. 62:1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Starr CR, Engleberg NC. 2006. Role of hyaluronidase in subcutaneous spread and growth of group A streptococcus. Infect. Immun. 74:40–48. 10.1128/IAI.74.1.40-48.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campo GM, Avenoso A, D'Ascola A, Scuruchi M, Prestipino V, Calatroni A, Campo S. 2012. Hyaluronan in part mediates IL-1beta-induced inflammation in mouse chondrocytes by up-regulating CD44 receptors. Gene 494:24–35. 10.1016/j.gene.2011.11.064 [DOI] [PubMed] [Google Scholar]
- 22.Turley EA, Noble PW, Bourguignon LY. 2002. Signaling properties of hyaluronan receptors. J. Biol. Chem. 277:4589–4592. 10.1074/jbc.R100038200 [DOI] [PubMed] [Google Scholar]
- 23.Pure E, Cuff CA. 2001. A crucial role for CD44 in inflammation. Trends Mol. Med. 7:213–221. 10.1016/S1471-4914(01)01963-3 [DOI] [PubMed] [Google Scholar]
- 24.Noble PW, Lake FR, Henson PM, Riches DW. 1993. Hyaluronate activation of CD44 induces insulin-like growth factor-1 expression by a tumor necrosis factor-alpha-dependent mechanism in murine macrophages. J. Clin. Invest. 91:2368–2377. 10.1172/JCI116469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harada H, Takahashi M. 2007. CD44-dependent intracellular and extracellular catabolism of hyaluronic acid by hyaluronidase-1 and -2. J. Biol. Chem. 282:5597–5607. 10.1074/jbc.M608358200 [DOI] [PubMed] [Google Scholar]
- 26.Lepperdinger G, Strobl B, Kreil G. 1998. HYAL2, a human gene expressed in many cells, encodes a lysosomal hyaluronidase with a novel type of specificity. J. Biol. Chem. 273:22466–22470. 10.1074/jbc.273.35.22466 [DOI] [PubMed] [Google Scholar]
- 27.Liu G, Zhang W, Lu C. 2012. Complete genome sequence of Streptococcus agalactiae GD201008-001, isolated in China from tilapia with meningoencephalitis. J. Bacteriol. 194:6653. 10.1128/JB.01788-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takamatsu D, Osaki M, Sekizaki T. 2001. Thermosensitive suicide vectors for gene replacement in Streptococcus suis. Plasmid 46:140–148. 10.1006/plas.2001.1532 [DOI] [PubMed] [Google Scholar]
- 29.Takamatsu D, Osaki M, Sekizaki T. 2001. Construction and characterization of Streptococcus suis-Escherichia coli shuttle cloning vectors. Plasmid 45:101–113. 10.1006/plas.2000.1510 [DOI] [PubMed] [Google Scholar]
- 30.Ju CX, Gu HW, Lu CP. 2012. Characterization and functional analysis of atl, a novel gene encoding autolysin in Streptococcus suis. J. Bacteriol. 194:1464–1473. 10.1128/JB.06231-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 32.Smith RF, Willett NP. 1968. Rapid plate method for screening hyaluronidase and chondroitin sulfatase-producing microorganisms. Appl. Microbiol. 16:1434–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kloosterman TG, Bijlsma JJ, Kok J, Kuipers OP. 2006. To have neighbour's fare: extending the molecular toolbox for Streptococcus pneumoniae. Microbiology 152:351–359. 10.1099/mic.0.28521-0 [DOI] [PubMed] [Google Scholar]
- 34.Patterson H, Saralahti A, Parikka M, Dramsi S, Trieu-Cuot P, Poyart C, Rounioja S, Ramet M. 2012. Adult zebrafish model of bacterial meningitis in Streptococcus agalactiae infection. Dev. Comp. Immunol. 38:447–455. 10.1016/j.dci.2012.07.007 [DOI] [PubMed] [Google Scholar]
- 35.Stanic M. 1963. A simplification of the estimation of the 50 percent endpoints according to the Reed and Muench method. Pathol. Microbiol. (Basel) 26:298–302 (In German.) [PubMed] [Google Scholar]
- 36.Marion C, Stewart JM, Tazi MF, Burnaugh AM, Linke CM, Woodiga SA, King SJ. 2012. Streptococcus pneumoniae can utilize multiple sources of hyaluronic acid for growth. Infect. Immun. 80:1390–1398. 10.1128/IAI.05756-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McHugh SL, Newton CA, Yamamoto Y, Klein TW, Friedman H. 2000. Tumor necrosis factor induces resistance of macrophages to Legionella pneumophila infection. Proc. Soc. Exp. Biol. Med. 224:191–196. 10.1046/j.1525-1373.2000.22420.x [DOI] [PubMed] [Google Scholar]
- 38.Girish KS, Kemparaju K. 2007. The magic glue hyaluronan and its eraser hyaluronidase: a biological overview. Life Sci. 80:1921–1943. 10.1016/j.lfs.2007.02.037 [DOI] [PubMed] [Google Scholar]
- 39.Li S, Jedrzejas MJ. 2001. Hyaluronan binding and degradation by Streptococcus agalactiae hyaluronate lyase. J. Biol. Chem. 276:41407–41416. 10.1074/jbc.M106634200 [DOI] [PubMed] [Google Scholar]
- 40.Sagar A, Klemm C, Hartjes L, Mauerer S, van Zandbergen G, Spellerberg B. 2013. The β-hemolysin and intracellular survival of Streptococcus agalactiae in human macrophages. PLoS One 8:e60160. 10.1371/journal.pone.0060160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sendi P, Johansson L, Dahesh S, Van Sorge NM, Darenberg J, Norgren M, Sjolin J, Nizet V, Norrby-Teglund A. 2009. Bacterial phenotype variants in group B streptococcal toxic shock syndrome. Emerg. Infect. Dis. 15:223–232. 10.3201/eid1502.080990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costagliola C, DelPrete A, Winkler NR, Carpineto P, Ciancaglini M, Piccolomini R, Mastropasqua L. 1996. The ability of bacteria to use Na-hyaluronate as a nutrient. Acta Ophthalmol. Scand. 74:566–568 [DOI] [PubMed] [Google Scholar]
- 43.Lowe BA, Miller JD, Neely MN. 2007. Analysis of the polysaccharide capsule of the systemic pathogen Streptococcus iniae and its implications in virulence. Infect. Immun. 75:1255–1264. 10.1128/IAI.01484-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neely MN, Pfeifer JD, Caparon M. 2002. Streptococcus-zebrafish model of bacterial pathogenesis. Infect. Immun. 70:3904–3914. 10.1128/IAI.70.7.3904-3914.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kostyukova NN, Volkova MO, Ivanova VV, Kvetnaya AS. 1995. A study of pathogenic factors of Streptococcus pneumoniae strains causing meningitis. FEMS Immunol. Med. Microbiol. 10:133–137. 10.1111/j.1574-695X.1995.tb00022.x [DOI] [PubMed] [Google Scholar]
- 46.Zwijnenburg PJ, van der Poll T, Florquin S, van Deventer SJ, Roord JJ, van Furth AM. 2001. Experimental pneumococcal meningitis in mice: a model of intranasal infection. J. Infect. Dis. 183:1143–1146. 10.1086/319271 [DOI] [PubMed] [Google Scholar]
- 47.Vallejo JG, Baker CJ, Edwards MS. 1996. Roles of the bacterial cell wall and capsule in induction of tumor necrosis factor alpha by type III group B streptococci. Infect. Immun. 64:5042–5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy-Simandle K, Stewart CR, Dao J, DebRoy S, Rossier O, Bryce PJ, Cianciotto NP. 2011. Legionella pneumophila type II secretion dampens the cytokine response of infected macrophages and epithelia. Infect. Immun. 79:1984–1997. 10.1128/IAI.01077-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. 2005. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 434:1138–1143. 10.1038/nature03491 [DOI] [PubMed] [Google Scholar]
- 50.Medzhitov R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819–826. 10.1038/nature06246 [DOI] [PubMed] [Google Scholar]
- 51.Sims JE, Smith DE. 2010. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10:89–102. 10.1038/nri2691 [DOI] [PubMed] [Google Scholar]
- 52.Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, Midiri A, Galbo R, Trieu-Cuot P, Papasergi S, Teti G, Henneke P, Mancuso G, Golenbock DT, Beninati C. 2012. Activation of the NLRP3 inflammasome by group B streptococci. J. Immunol. 188:1953–1960. 10.4049/jimmunol.1102543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vossenkamper A, Marches O, Fairclough PD, Warnes G, Stagg AJ, Lindsay JO, Evans PC, le Luong A, Croft NM, Naik S, Frankel G, MacDonald TT. 2010. Inhibition of NF-kappaB signaling in human dendritic cells by the enteropathogenic Escherichia coli effector protein NleE. J. Immunol. 185:4118–4127. 10.4049/jimmunol.1000500 [DOI] [PubMed] [Google Scholar]
- 54.Lamy MC, Zouine M, Fert J, Vergassola M, Couve E, Pellegrini E, Glaser P, Kunst F, Msadek T, Trieu-Cuot P, Poyart C. 2004. CovS/CovR of group B streptococcus: a two-component global regulatory system involved in virulence. Mol. Microbiol. 54:1250–1268. 10.1111/j.1365-2958.2004.04365.x [DOI] [PubMed] [Google Scholar]
- 55.Patras KA, Wang NY, Fletcher EM, Cavaco CK, Jimenez A, Garg M, Fierer J, Sheen TR, Rajagopal L, Doran KS. 2013. Group B Streptococcus CovR regulation modulates host immune signalling pathways to promote vaginal colonization. Cell. Microbiol. 15:1154–1167. 10.1111/cmi.12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fan H, Williams DL, Zingarelli B, Breuel KF, Teti G, Tempel GE, Spicher K, Boulay G, Birnbaumer L, Halushka PV, Cook JA. 2007. Differential regulation of lipopolysaccharide and Gram-positive bacteria induced cytokine and chemokine production in macrophages by Galpha(i) proteins. Immunology 122:116–123. 10.1111/j.1365-2567.2007.02619.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurtz SL, Foreman O, Bosio CM, Anver MR, Elkins KL. 2013. Interleukin-6 is essential for primary resistance to Francisella tularensis live vaccine strain infection. Infect. Immun. 81:585–597. 10.1128/IAI.01249-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buckner MMC, Antunes LCM, Gill N, Russell SL, Shames SR, Finlay BB. 2013. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits macrophage colonization by Salmonella enterica serovar Typhimurium. PLoS One 8:e69759. 10.1371/journal.pone.0069759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schett G. 2011. Effects of inflammatory and anti-inflammatory cytokines on the bone. Eur. J. Clin. Invest. 41:1361–1366. 10.1111/j.1365-2362.2011.02545.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.