

Abstract
Clostridium perfringens enterotoxin causes the gastrointestinal (GI) symptoms of C. perfringens type A food poisoning and CPE-associated non-food-borne human GI diseases. It is well established that CPE induces fluid accumulation and severe tissue damage in ligated small intestinal loops of rabbits and other animals. However, a previous study had also reported that CPE binds to rabbit colonic cells yet does not significantly affect rabbit colonic loops. To the contrary, the current study determined that treatment with 50 or 100 μg/ml of CPE causes significant histologic lesions and luminal fluid accumulation in rabbit colonic loops. Interestingly, a CPE-neutralizing monoclonal antibody blocked the development of CPE-induced histologic damage but not luminal fluid accumulation in these loops. Similar luminal fluid accumulation, without significant histologic damage, also occurred after treatment of colonic loops with heat-inactivated CPE, antibody alone, or bovine serum albumin (BSA), indicating that increased osmolarity was causing or contributing to fluid accumulation in CPE-treated colonic loops. Comparative studies revealed the similar development of histologic damage and luminal fluid accumulation in both small intestinal loops and colonic loops after as little as a 1-h treatment with 50 μg/ml of CPE. Consistent with the CPE sensitivity of the small intestine and colon, Western blotting detected CPE binding and large-complex formation in both organs. In addition, Western blotting demonstrated the presence of the high-affinity CPE receptors claudin-3 and -4 in both organs of rabbits, consistent with the observed toxin binding. Collectively, these results offer support for the possible involvement of the colon in CPE-mediated GI disease.
INTRODUCTION
Substantial experimental and epidemiologic evidence has implicated Clostridium perfringens enterotoxin (CPE) as the toxin responsible for causing the gastrointestinal (GI) symptoms of C. perfringens type A food poisoning (1, 2). This food poisoning is currently the second most common bacterial food-borne illness in the United States, where an estimated one million cases occur annually (3). In addition, CPE production is essential for CPE-producing type A strains to cause ∼5 to 10% of all cases of non-food-borne human GI illnesses, including antibiotic-associated diarrhea (2, 4). These bacteria are also responsible for some GI infections in domestic animals (4).
During GI disease, sporulating C. perfringens cells produce CPE in the intestines (1). This enterotoxin, which is an ∼35-kDa single polypeptide with a unique amino acid sequence (5), belongs structurally to the aerolysin family of pore-forming toxins (6, 7). CPE action begins with its binding to receptors, which include certain members of the claudin protein family (8–10). Claudins are ∼20- to 27-kDa protein components of the mammalian tight junctions in epithelia and endothelia, where they serve important structural and functional roles (11). Once bound to a claudin receptor, e.g., claudin-3 or claudin-4 (8, 12), CPE becomes localized on the membrane surface in a prepore complex named CH-1, for CPE hexamer-1. CH-1 is ∼450 kDa in mass but runs anomalously as an ∼155-kDa species on SDS-PAGE (13). The six CPE proteins present in CH-1 are then thought to extend β-hairpins into the membranes to create an active pore (14). Formation of this pore triggers a calcium influx into intestinal cells, which kills those cells in a toxin dose-dependent manner; i.e., low CPE doses induce classical caspase-3-mediated apoptosis, while high CPE doses cause oncosis (15, 16).
Previous studies using rabbit small intestinal loop models showed that as little as 50 μg/ml of CPE causes significant lesions in the small intestine, where substantial damage starts at the villus tips (17–20). This damage then progresses down the entire villus into the crypts, producing necrosis of the epithelium and lamina propria, as well as villus blunting and edema. The histologic lesions caused by CPE in the small intestine are considered to be a major contributor to the development of the substantial intestinal fluid and electrolyte losses that manifest as diarrhea during CPE-associated food-borne or non-food-borne GI disease (1, 4, 18).
To our knowledge, only a single previous study has examined whether CPE also affects the colon in vivo (21). That earlier study concluded that CPE causes minimal or no damage in rabbit colonic loops, despite high levels of enterotoxin binding to this organ. This putative detection of CPE binding, in the absence of damage, to the rabbit colon was interesting since it is generally accepted that once bound to cells, CPE quickly forms a pore that leads to the development of cell death (1). If those previous findings that bound CPE cannot damage the rabbit colon are correct, it could suggest the existence of an unrecognized postbinding step(s) in CPE action. In addition, for understanding the pathology of CPE-induced GI disease, it is critical to determine whether or not the colon is responsive to this enterotoxin. Therefore, the current study sought to reevaluate the question of whether CPE binds to and damages the rabbit colon.
MATERIALS AND METHODS
Purification of CPE.
C. perfringens strain T-1 was isolated from retail turkey meat in Pittsburgh, PA (22). This type A strain carries the cpe and cpa genes, encoding (respectively) the CPE or alpha toxin, but does not possess (not shown) the tpeL, cpb2, or pfoA gene, which would encode TpeL, beta2 toxin, or perfringolysin O, respectively. In the Duncan-Strong sporulation medium used for CPE purification, CPE is the only known toxin produced by strain T-1 (data not shown). CPE was purified to electrophoretic homogeneity from strain T-1 using described previously techniques (23).
Sample preparation.
Purified native CPE (25 μg, 50 μg, or 100 μg) was dissolved in 1 ml of Hanks' balanced salt solution (HBSS) containing Ca2+ and Mg2+ (Cellgro). A 1-ml sample of HBSS buffer alone was used as a negative control. To address whether CPE was the active agent inducing fluid accumulation and histologic damage in the rabbit intestinal loop models, several control experiments were performed. Before their addition to colonic loops, selected samples containing 50 μg/ml of CPE were preincubated for 1 h at 37°C with 1.5 mg of monoclonal antibody (MAb) 3C9 (MAb3), which neutralizes CPE by blocking receptor binding, or with the same amount of a nonneutralizing CPE monoclonal antibody (MAb1). Both CPE MAbs had been created and characterized in a previous study (24). Similarly, prior to their addition to colonic or small intestinal loops, some 50-μg/ml CPE samples were inactivated by boiling for 10 min. Inactivation of boiled CPE was confirmed by lack of cytotoxicity on Caco-2 cells (data not shown). Finally, a 50-μg/ml aliquot of bovine serum albumin (BSA) (Sigma) was inoculated into selected colonic loops.
Rabbit small intestinal and colonic loop models.
After premedication with acepromazine, xylazine, and burprenorphine, fasted young adult (male or female) New Zealand White rabbits (Charles River, CA) were anesthetized with ketamine. Anesthesia was then maintained with inhalatory isofluorane. A midline laparotomy was performed to expose the small intestine and/or colon. After washing with saline solution injected into the lumen, the exposed intestinal segments were gently massaged before preparation of colonic or small intestinal loops. Two-centimeter lengths of each intestinal section were isolated by ligation, leaving an empty segment between the loops. To eliminate a possible ischemic component to the toxin-induced intestinal damage, care was taken to avoid overdistending the bowel loops or interfering with the blood supply. The serosal surface of the loops was kept wet during surgery by frequent soaking with sterile normal saline solution. The sequence of inocula was varied between animals, and loop location did not affect outcomes (data not shown). Each inoculum was tested in duplicate in each animal. After injection of the inoculum, the abdominal incision was closed by separate muscle and skin sutures, and the animals were then kept deeply anesthetized throughout the specified experimental period.
Measurement of fluid accumulation and histologic damage in intestinal loops.
Following the specified treatment period, rabbits were euthanized by an overdose of sodium barbiturate (Beuthanasia; Schering-Plough Animal Health, Kenilworth, NJ). After the abdominal cavity was opened, the small intestinal and colonic loops were excised in the same order as inoculated. The length and weight of each loop were then determined. The loops were then cut open to remove fluid before the loops were reweighed. The difference in weight before and after fluid removal was used to calculate the loop weight-to-length ratio (g/cm) of fluid secretion.
For histology, tissues were fixed by immersion in 10% buffered (pH 7.4) formalin for 24 to 48 h and then dehydrated through graded alcohol solutions to xylene before being embedded in paraffin wax. Thick sections (4 μm) were cut and stained with hematoxylin and eosin using standard procedures. The stained tissue sections were then examined by a pathologist in a blinded fashion. Using a subjective quantitative scoring system, the degree of damage was scored using a scale of 0 to 5. In this scoring, 0 indicates no histologic damage, while 1, 2, 3, 4, and 5 indicate increasingly severe damage. Histologic parameters considered during the evaluation of the small intestine and colon included epithelial desquamation, epithelial necrosis, lamina propria necrosis, inflammation, hemorrhage, and submucosal edema. In addition, villus blunting was considered for the evaluation of the small intestine. All procedures involving animals were reviewed and approved by the University of California, Davis, Committee for Animal Care and Use (permit 16546).
Analysis of CPE binding and large-complex formation in rabbit small intestine or colon.
To assess CPE binding and large-complex formation in small intestinal or colonic specimens that had been collected from rabbit loops challenged with 50 μg/ml or 100 μg/ml of CPE, 100-mg tissue specimens from the small intestines or colons of rabbits were suspended in 1 ml of radioimmunoprecipitation assay (RIPA) buffer (Fisher Scientific) containing protease inhibitor cocktail (Roche) plus 1 μl of Benzonase (EMD Novagen). The suspension was then homogenized using a Qsonica sonicator, with an output of 25% and three 10-s pulses. A 25-μl aliquot of supernatant from each homogenate was added to 25 μl of 2× Laemmli buffer and centrifuged for 5 min at 13,000 rpm. These sample supernatants were then electrophoresed on an SDS-containing 4% polyacrylamide gel. After transfer of the separated proteins onto a nitrocellulose membrane, the blot was subjected to Western blotting for detection of bound CPE species using rabbit polyclonal CPE antiserum (25) and the Clean-Blot IP detection reagent (with horseradish peroxidase [HRP]). Equivalent volumes of similarly homogenized small intestinal and colonic tissue from rabbit loops that had received only HBSS buffer were used as negative controls.
Detection of claudin-3 or claudin-4 in rabbit small intestine or colon.
To evaluate the expression of CPE receptors claudin-3 and claudin-4 in the rabbit small intestine and colon, 100-mg tissue specimens from the small intestines or colons of healthy rabbits were homogenized and processed as described above. Those samples were then loaded onto 12% SDS-polyacrylamide gels, followed by transfer onto a nitrocellulose membrane and subsequent Western blotting. For claudin-3 and claudin-4 detection, the membrane was incubated with a rabbit claudin-3 or claudin-4 polyclonal antibody (Invitrogen), followed by detection with the Clean-Blot IP detection reagent system (HRP) (Thermo Scientific). This claudin-3 polyclonal antibody does not cross-react with claudin-4 on Western blots (data not shown).
Statistical analyses.
Histologic damage and fluid accumulation differences were analyzed using the Freidman test, the Kruskal-Wallis test, and Student t test, each performed with GraphPad Prism 6.
RESULTS
Histologic damage develops in rabbit colonic loops after CPE treatment.
CPE concentrations measured in feces from diseased people range up to, and occasionally exceed, ∼100 μg/ml (26, 27). Therefore, an initial experiment treated colonic loops for 6 h with three pathophysiologically relevant toxin doses, i.e., 25, 50, or 100 μg/ml of CPE. After this 6-h challenge, colonic loops treated with the 50- or 100-μg/ml dose of CPE had developed significant histologic lesions that were absent from similar colonic loops treated with HBSS buffer (Fig. 1A). Mild histologic damage was also noted after a 6-h challenge of colonic loops with the 25-μg/ml CPE dose, although those effects did not reach statistical significance (P > 0.05) compared against buffer-treated colonic loops.
FIG 1.
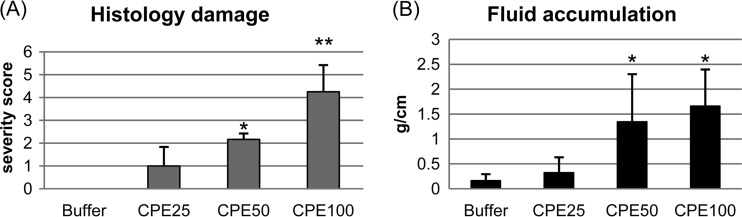
Histologic damage and luminal fluid accumulation in colonic loops treated with CPE (25 μg/ml, 50 μg/ml, or 100 μg/ml) or HBSS buffer alone. (A) Colonic histopathology damage after a 6-h treatment with the indicated CPE doses. Results shown are the mean ± standard deviation (SD) for six rabbits. Histologic damage severity was based on a five-point scale, with 5 representing the most severe damage (see Materials and Methods). * or ** denotes a significant difference compared to the buffer control (P < 0.05 or P < 0.001, respectively). (B) Luminal fluid accumulation (g/cm) after a 6-h treatment with the specified CPE dose. Results shown are the mean ± SD for six rabbits. * denotes a significant difference compared to the buffer control (P < 0.05).
To explore the time course of CPE-induced histologic lesion development in the colon, a brief (1-h) challenge of rabbit colonic loops was also performed with the 50-μg/ml CPE dose. The development of statistically significant colonic damage was detected even after this short-term CPE treatment period (Fig. 2A). However, the histologic lesions present after this 1-h CPE treatment were significantly (P < 0.05) less than those noted following a 6-h treatment with the same CPE dose.
FIG 2.

Comparison of CPE effects in small intestinal versus colonic loops. (A) Comparison of histologic lesions between small intestinal and colonic loops treated with 50 μg/ml of CPE. After a 1-h challenge, both the CPE-treated colonic loops and small intestinal (SI) loops showed histologic lesions that were absent from loops treated with buffer. This damage became even stronger after a 6-h treatment with this CPE dose. Values shown are the mean ± SD for 4 to 6 rabbits. * or ** indicates a significant difference (P < 0.05 or P < 0.001, respectively) relative to the buffer control. Differences between CPE-treated small intestine and colon were not statistically significant after either a 1- or 6-h CPE treatment (P > 0.05). (B) Comparison of luminal fluid accumulation (g/cm) between SI and colonic loops treated with 50 μg/ml of CPE. After a 1-h challenge, both CPE-treated colonic loops and SI loops had accumulated more luminal fluid than similar loops treated with buffer. For the colon, this CPE-induced fluid accumulation increased further by 6 h of treatment. These differences were significant at a P value of <0.05 or <0.001, indicated by * or **, respectively. The differences in fluid accumulation between the CPE-treated SI and colonic loops were significant (P < 0.05) for either the 1- or 6-h CPE treatment. Values shown are the mean ± SD for 4 to 6 rabbits.
To compare the development of CPE-induced damage in the rabbit colon versus the small intestine, similar studies were repeated using rabbit small intestinal loops treated with a 50-μg/ml CPE dose. With this toxin challenge, statistically significant small intestinal lesions developed within 1 h (Fig. 2A). This small intestinal damage became significantly (P < 0.05) more severe by 6 h. At equivalent time points, more CPE-induced damage was observed in the small intestinal loops than in the colonic loops, although those differences did not reach statistical significance (P > 0.05).
Qualitatively, colonic loops treated for 1 h with 50 μg/ml of CPE exhibited mild to moderate necrosis and desquamation of superficial epithelial cells with only minimal lamina propria necrosis (Fig. 3A). By 6 h of this CPE treatment, colonic damage consisted of severe mucosal necrosis characterized by loss of superficial epithelium and necrosis of lamina propria (Fig. 3A). After equivalent CPE treatment, pathology similar to that observed in the colon developed in the small intestine, along with moderate and severe villus blunting after 1 and 6 h of challenge, respectively (Fig. 3B).
FIG 3.

Comparison of histologic damage between CPE-treated small intestinal and colonic loops. Tissues were processed by histology and stained using hematoxylin and eosin. Sections of treated or control tissues were then photomicrographed at a final magnification of ×200. Shown are representative photomicrographs after 1- or 6-h buffer or CPE treatments performed in colonic (A) or small intestinal (B) loops. In both the colonic and small intestinal loops, CPE produced microscopic lesions, characterized mainly by severe mucosal necrosis, that were absent from matching buffer-treated loops. In addition, small intestinal loops exhibited severe villus blunting. Results shown are representative of histologic examination for six rabbits.
Luminal fluid accumulation increases in rabbit colon loops challenged with CPE.
After a 6-h treatment with 50 or 100 μg/ml of CPE, the lumen of colonic loops had accumulated significantly more fluid than the lumen of similar loops receiving HBSS buffer alone (Fig. 1B). A 6-h challenge with a 25-μg/ml dose of CPE also caused more fluid accumulation in colonic loops than did treatment with buffer (Fig. 1B), but this effect did not reach statistical significance (P > 0.05).
The time course of CPE-induced fluid accumulation in the colon was explored using a more brief (1-h) challenge of rabbit colonic loops with a 50-μg/ml CPE dose. Significant colonic fluid accumulation was evident even after this short CPE treatment period (Fig. 2B). However, luminal fluid accumulation after a 1-h CPE treatment was significantly (P < 0.05) less than fluid levels measured following a 6-h treatment with the same CPE dose.
To compare the effects of CPE treatment on fluid accumulation in the rabbit colon versus small intestine, rabbit small intestinal loops were challenged for 1 h or 6 h with 50 μg/ml of CPE. In these studies, statistically significant small intestinal fluid accumulation developed within 1 h of toxin challenge (Fig. 2B), with this effect then becoming significantly (P < 0.05) stronger by 6 h of CPE treatment. At equivalent time points, significantly more CPE-induced fluid accumulation was measured in colonic loops than in small intestinal loops after either a 1- or 6-h toxin treatment (P < 0.05).
Effects of CPE monoclonal antibodies on CPE-induced histologic damage and fluid accumulation in rabbit colon.
To evaluate whether the significant damage and luminal fluid accumulation in colonic loops caused by treatment with 50- or 100-μg/ml doses of CPE (Fig. 1 and 2) were due to active CPE, additional studies were performed using colonic loops treated for 6 h with CPE (50 μg/ml), CPE (50 μg/ml) preincubated with MAb3, a neutralizing monoclonal antibody that blocks CPE binding to receptors (24), or CPE (50 μg/ml) preincubated with nonneutralizing CPE monoclonal antibody MAb1.
These analyses showed that colonic loops treated for 6 h with samples containing 50 μg/ml of CPE or 50 μg/ml of CPE preincubated with MAb 1 developed significant histologic damage compared to HBSS buffer-treated colonic loops (Fig. 4A). In contrast, colonic loops similarly challenged with 50 μg/ml of CPE preincubated with MAb 3 did not develop significant histologic damage (Fig. 4A).
FIG 4.

Effects of MAbs and other treatments on CPE activities in colonic loops. Colonic loops were challenged for 6 h with CPE (50 μg/ml), heat-inactivated CPE (50 μg/ml), CPE (50 μg/ml) plus CPE-neutralizing MAb3, CPE (50 μg/ml) plus CPE-nonneutralizing MAb1, BSA, or buffer. (A) Histologic damage. Relative to buffer-treated or any other colonic loops, significant (*, P < 0.05) damage developed only in those colonic loops treated with CPE (50 μg/ml) or CPE (50 μg/ml) plus MAb1. Results shown are the mean ± SD from 4 to 6 rabbits. (B) Fluid accumulation (g/cm). Relative to buffer-treated loops, all loops except those receiving MAb3 or BSA showed significant (P < 0.05) fluid accumulation. However, relative to loops treated with CPE (50 μg/ml), there were no statistically significant (P < 0.05) differences in fluid accumulation for any samples tested for this figure, except those receiving buffer alone. Results shown are the mean ± SD from 4 to 6 rabbits.
At the microscopic level, histologic changes in colonic loops treated for 6 h with either 50 μg/ml of CPE or 50 μg/ml of CPE preincubated with MAb 1 included severe mucosal necrosis characterized by a loss of superficial epithelium, necrosis, and hemorrhage of the lamina propria (Fig. 5). In contrast, colonic loops challenged with buffer or CPE (50 μg/ml) that had been preincubated with CPE-neutralizing MAb3 showed only very minor or no histologic changes (Fig. 5).
FIG 5.

Histology after the treatments of colonic loops for Fig. 4. CPE (50 μg/ml) or CPE (50 μg/ml) plus MAb1 produced similar microscopic lesions, characterized mainly by severe mucosal necrosis and occasional congestion/hemorrhage of the lamina propria. Heat-inactivated CPE (50 μg/ml), CPE (50 μg/ml) plus MAb3, BSA, or buffer produced no histologic lesions.
While those studies (Fig. 4A and 5) confirmed that active CPE was responsible for the damage observed in CPE-treated colonic loops, the results regarding luminal fluid accumulation in colonic loops were more complicated (Fig. 4B). Compared to treatment with HBSS buffer, all three CPE-containing treatments caused a significant increase in luminal fluid accumulation in rabbit colon loops. However, there were no statistically significant differences (P > 0.05) in fluid accumulation levels between those three CPE-containing samples, even though samples containing CPE preincubated with MAb3 did not exhibit histologic damage.
Further analysis of luminal fluid accumulation responses in rabbit colonic loops.
The increased luminal fluid accumulation noted in colonic loops after challenge with 50 μg/ml of CPE that had been preincubated with sufficient amounts of MAb3 to block the development of significant histologic damage could indicate that (i) colonic luminal fluid accumulation caused by CPE treatment is due to some other active contaminant in the CPE preparation, (ii) colonic luminal fluid accumulation requires active CPE but at lower levels than necessary to cause histologic damage, or (iii) colonic luminal fluid accumulation caused by CPE treatment is an osmotic effect that does not require active CPE (or other active factors).
To help distinguish between those possibilities, additional studies were performed. When rabbit colonic loops were treated for 6 h with 50 μg/ml of heat-inactivated CPE (Fig. 4B and 5), significant loop fluid accumulation was still observed even though this inactivated toxin caused no significant histologic damage (Fig. 4A). This result, coupled with the finding that fluid accumulation still occurred when using CPE that had been preincubated with sufficient amounts of MAb3 to block histologic damage, suggested that addition of any protein might suffice to induce colonic loop fluid accumulation. This hypothesis was confirmed when it was determined that addition of MAb1 or MAb3 alone (no CPE) or even BSA alone (no CPE) (Fig. 4B and 5), at a molar equivalent of only a 25-μg/ml dose of CPE, induced rabbit colonic fluid accumulation levels in rabbit colonic loops that were not significantly different from the luminal fluid accumulation levels measured after a 6-h challenge of colonic loops with 50 μg/ml of CPE.
CPE can bind and form large complexes in rabbit colon.
Receptor binding and formation of the CH-1 large CPE complex are considered to be essential early steps in CPE action on Caco-2 cells (1, 13, 28). Despite its true size of ∼450 kDa, the CH-1 complex migrates anomalously in SDS-PAGE as a smeary ∼155-kDa species on the SDS-containing 4% polyacrylamide gels necessary to analyze this large protein species (13). Previous studies had demonstrated that CPE binds and forms a similarly migrating CH-1-like large complex in mouse internal organs, including the small intestine (29).
Therefore, the current study analyzed whether CPE also binds and forms a large CPE complex in the rabbit colon. CPE Western blot analysis of tissue homogenates from colonic loops challenged with 50 or 100 μg/ml of enterotoxin clearly demonstrated CPE binding and the formation of a large CPE complex (Fig. 6A). Similar to the CH-1-like CPE complex formed in Caco-2 cells (13, 28) or mouse intestine (29), the CPE large complex formed in colonic tissues migrated on SDS-containing 4% polyacrylamide gels with an apparent size of ∼155 kDa. A similarly migrating large complex was also detected in CPE-treated rabbit small intestine (Fig. 6A). As expected, control loops treated with HBSS buffer alone (no CPE) did not show any CPE binding or large-complex formation.
FIG 6.

Western blot analyses of rabbit colon. (A) CPE Western blot analyses of homogenates from colonic or small intestinal loops challenged for 6 h with 50 or 100 μg/ml of CPE. In both the small intestine and colon, CPE bound and formed a large complex that migrates as a 155-kDa CPE species, similar to the CH-1-like CPE complex formed in mouse small intestine or in Caco-2 cells (25, 29). (B) Claudin-3 (upper panel) or claudin-4 (lower panel) Western blot analyses of homogenates from small intestines and colons from four control rabbits. Note that the CPE receptors claudin-3 and claudin-4 are clearly produced in both colon and small intestine in all four tested rabbits.
Rabbit colon contents of claudin-3 and claudin-4 CPE receptors.
Because CPE bound to the rabbit colon in the preceding experiment (Fig. 6A), Western blot studies were performed to determine whether this organ contains the high-affinity CPE receptors claudin-3 and claudin-4 as potential contributors to this binding. The results obtained detected both of these CPE receptors in the colon and the small intestine (Fig. 6B). However, some animal-to-animal variations were noted in the relative abundances of these two claudins in the small intestine and colon (Fig. 6B).
DISCUSSION
To fully understand CPE-mediated GI disease, it is important to identify which organs are affected by this enterotoxin in vivo. In 1982, McDonel and Demers (21) reported that CPE induces diarrhea via its effects on the small intestine but not the colon. This conclusion was based upon results from their experiments using CPE-treated colonic loops, where no significant histologic damage or fluid accumulation was detected in those loops, although CPE treatment did cause some luminal mucus and protein accumulation (21).
Since that work by McDonel and Demers (21), several subsequent findings have suggested the need for reassessing whether CPE can damage the colon. First, an epidemiologic study of a severe C. perfringens type A food poisoning outbreak in Oklahoma concluded that, under medical conditions leading to reduced intestinal motility, CPE-positive type A strains can damage the colon, producing a potentially lethal necrotizing colitis (30). Second, in vitro treatment of human colonic tissue with only 10 μg/ml of CPE caused slight morphologic and fluid transport changes, although the extent of those effects did not reach statistical significance (31). Finally, it is now well established that purified CPE is cytotoxic for several colonic cell lines, including Caco-2 cells (10, 25, 28).
Because of this uncertainty in the literature, the current study revisited whether CPE affects the colon in vivo. Using rabbit colonic loops, both 50- and 100-μg/ml doses of the enterotoxin were found to induce significant histologic lesions and luminal fluid accumulation. An explanation for the discrepancy between the results of this study and those of the previous study (21) regarding the ability of CPE to induce damage and fluid accumulation is not obvious. It is not attributable to differences in CPE treatment times, since McDonel and Demers (21) challenged colonic loops for 1 h with high-dose CPE, which was a sufficient time in the current study to observe the development of significant colonic damage. Consistent with our finding, another previous study reported mild, although not statistically significant, in vitro damage to human colonic tissue after a 1-h treatment with only 10 μg/ml of CPE (31). The difference in results between the current study and that of McDonel and Demers (21) also is not due to use of higher CPE treatment doses in the current study, since the earlier study employed much higher CPE doses (supraphysiologic at ∼400 μg/ml) than our study. The colonic lesions and fluid accumulation induced by electrophoretically pure CPE in the current study could be neutralized by a monoclonal antibody (24), confirming CPE involvement in the observed colonic damage. Further supporting CPE causation of colonic damage, the T-1 strain used as the source for CPE in our study produces CPE but no other known toxins when grown in the Duncan-Strong medium used for CPE purification. Finally, the amino acid sequence of CPE produced by strain T-1 is identical to the CPE sequence encoded by NCTC8239, the strain used by McDonel and Demers for CPE purification (data not shown). In addition, the CPEs produced by T-1 and NCTC8239 have identical activities on Caco-2 cells (data not shown). Therefore, the different conclusion about CPE-induced colonic damage reached by our study was not due to use of a CPE variant with more activity than the CPE produced by NCTC8239.
However, there is a possible explanation for the different conclusions of the two studies regarding CPE activity in the rabbit colon. The early study by McDonel and Demers (21) never demonstrated any small intestinal activity for their CPE preparation. In the absence of any positive control, it is conceivable that the CPE preparation used in the earlier study possessed weak or no activity.
Results from the current study also demonstrated that after similar CPE treatments, the rabbit small intestine developed histologic lesions, consistent with previous reports (20). The current study also determined that the rabbit colon and small intestine exhibit similar time frames of significant lesion development after treatment with 50 μg/ml of CPE. In addition, the CPE sensitivities of the organs appears to be similar, since a 1-h treatment with 10 to 25 μg/ml of this toxin causes minimal, statistically insignificant damage to the colon (this study and reference 31) or small intestine (20), while both organs develop significant lesions after a 1-h treatment with 50 μg/ml of CPE (this study). Their similar CPE responsiveness suggests that both the small intestine and colon may be targets during CPE-mediated GI disease.
The extensive damage observed in both CPE-treated rabbit small intestine and colon during the current study correlated with the formation of similar CH-1-like large complexes in both organs. Detection of a CH-1-like complex in the CPE-treated colon is likely to be significant for pathogenesis, since the CH-1 large complex is thought to contain the CPE pore and to be responsible for the cytotoxicity that results in CPE-induced intestinal tissue damage (1).
Understanding the contribution of histologic damage to the observed development of luminal fluid accumulation in the CPE-treated colon proved to be complicated, since inactivated CPE or CPE neutralized by MAb3, neither of which induced significant histologic damage, caused luminal fluid accumulation similar to that caused by the same dose of active CPE in rabbit colonic loops. Those observations suggested that osmotic effects contribute to CPE-induced luminal fluid accumulation, which was supported by the similar luminal fluid accumulation cause by active CPE versus MAbs or even BSA alone, even though the MAbs or BSA caused no histologic damage. These observations are consistent with previous studies describing osmotic diarrhea involving the colon. In those studies, increased osmotic pressure in the colon from other agents induced diarrhea, with this effect comprising the basis for several commonly used laxatives (32–35).
Even if histologic lesions are not required for causing fluid transport alterations in the CPE-treated colon, this damage still could have pathologic importance. The C. perfringens type A food poisoning outbreak in Oklahoma was exceptionally severe, with several deaths in nonelderly people (30). As mentioned, it has been postulated that reduced intestinal motility, due to medication side effects, may prolong contact between CPE and the intestines (30). In this regard, it is notable that in the Oklahoma outbreak investigation (30) colonic necrosis was observed in several people whose small intestines were unremarkable. Therefore, damage from prolonged contact between the colon and CPE may have facilitated entry of the enterotoxin into the bloodstreams of these people, so it could then damage their internal organs and contribute to death. Similar enterotoxemia was experimentally demonstrated recently in mice intestinally challenged with CPE (29).
Finally, Western blot analyses demonstrated the presence of claudin-3 and -4 in the rabbit small intestine and colon. Since these two claudins are both high-affinity CPE receptors (8, 10, 12, 25), their presence in the colon helps to explain the binding of CPE to the colon detected in this study and the previous study by McDonel and Demers (21). Furthermore, since binding is considered the essential first step in CPE action (1), the presence of these high-affinity CPE receptors likely facilitates CH-1-like CPE complex formation, and the development of histologic damage, in colonic loops. The presence of these claudin CPE receptors in the rabbit colon also likely holds relevance for understanding the CPE responsiveness of the human colon, where these two claudins are also present (36, 37).
In summary, the current findings support CPE effects on the colon as a potential contributor to CPE-mediated GI disease. This information will broaden understanding of CPE-mediated GI disease pathology and could impact future therapeutic development.
ACKNOWLEDGMENTS
This research was supported by National Institute of Allergy and Infectious Diseases grant AI19844-32.
We thank Jackie Parker and Jim Cravotta for excellent technical assistance.
Footnotes
Published ahead of print 18 March 2014
REFERENCES
- 1. McClane BA, Robertson SL, Li J. 2013. Clostridium perfringens, p 465–89 In Doyle MP, Buchanan RL. (ed), Food Microbiology: fundamentals and frontiers, 4th ed. ASM Press, Washington, DC [Google Scholar]
- 2. Sarker MR, Carman RJ, McClane BA. 1999. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33:946–958. 10.1046/j.1365-2958.1999.01534.x [DOI] [PubMed] [Google Scholar]
- 3. Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M, Roy S, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17:7–15. 10.3201/eid1701.P11101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins TD. 2006. The enterotoxic clostridia, p 688–752 In Dworkin M, Falkow S, Rosenburg E, Schleifer H, Stackebrandt E. (ed), The prokaryotes, 3rd ed. Springer Press, New York, NY [Google Scholar]
- 5. Czeczulin JR, Hanna PC, McClane BA. 1993. Cloning, nucleotide sequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect. Immun. 61:3429–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kitadokoro K, Nishimura K, Kamitani S, Fukui-Miyazaki A, Toshima H, Abe H, Kamata Y, Sugita-Konishi Y, Yamamoto S, Karatani H, Horiguchi Y. 2011. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J. Biol. Chem. 286:19549–19555. 10.1074/jbc.M111.228478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Briggs DC, Naylor CE, Smedley JG, III, Lukoyanova N, Robertson S, Moss DS, McClane BA, Basak AK. 2011. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J. Mol. Biol. 413:138–149. 10.1016/j.jmb.2011.07.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. 1997. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J. Biol. Chem. 272:26652–26658. 10.1074/jbc.272.42.26652 [DOI] [PubMed] [Google Scholar]
- 9. Gao Z, McClane BA. 2012. Use of Clostridium perfringens enterotoxin and the enterotoxin receptor-binding domain (C-CPE) for cancer treatment: opportunities and challenges. J. Toxicol. 2012:981626. 10.1155/2012/981626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shrestha A, McClane BA. 2013. Human claudin-8 and -14 are receptors capable of conveying the cytotoxic effects of Clostridium perfringens enterotoxin. mBio 4:e00594–12. 10.1128/mBio.00594-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson JM, Van Itallie CM. 2009. Physiology and function of the tight junction. Cold Spring Harbor Persp. Biol. 1:a002584. 10.1101/cshperspect.a002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katahira J, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. 1997. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 136:1239–1247. 10.1083/jcb.136.6.1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smedley JG, III, Uzal FA, McClane BA. 2007. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 75:2381–2390. 10.1128/IAI.01737-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen J, Theoret JR, Shrestha A, Smedley JG, III, McClane BA. 2012. Cysteine scanning mutagenesis supports the importance of Clostridium perfringens enterotoxin amino acids 80 to 106 for membrane insertion and pore formation. Infect. Immun. 80:4078–4088. 10.1128/IAI.00069-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chakrabarti G, McClane BA. 2005. The importance of calcium influx, calpain, and calmodulin for the activation of Caco-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 7:129–146 [DOI] [PubMed] [Google Scholar]
- 16. Chakrabarti G, Zhou X, McClane BA. 2003. Death pathways activated in Caco-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 71:4260–4270. 10.1128/IAI.71.8.4260-4270.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McDonel JL. 1986. Toxins of Clostridium perfringens types A, B, C, D, and E, p 477–517 In Dorner F, Drews H. (ed), Pharmacology of bacterial toxins. Pergamon Press, Oxford, United Kingdom [Google Scholar]
- 18. Sherman S, Klein E, McClane BA. 1994. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J. Diarrheal Dis. Res. 12:200–207 [PubMed] [Google Scholar]
- 19. Smedley JG, III, Saputo J, Parker JC, Fernandez-Miyakawa ME, Robertson SL, McClane BA, Uzal FA. 2008. Noncytotoxic Clostridium perfringens enterotoxin (CPE) variants localize CPE intestinal binding and demonstrate a relationship between CPE-induced cytotoxicity and enterotoxicity. Infect. Immun. 76:3793–3800. 10.1128/IAI.00460-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uzal FA, McClane BA. 2012. Animal models to study the pathogenesis of enterotoxigenic Clostridium perfringens infections. Microbes Infect. 14:1009–1016. 10.1016/j.micinf.2012.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDonel JL, Demers GW. 1982. In vivo effects of enterotoxin from Clostridium perfringens type A in rabbit colon: binding vs. biologic activity. J. Infect. Dis. 145:490–494. 10.1093/infdis/145.4.490 [DOI] [PubMed] [Google Scholar]
- 22. Wen Q, McClane BA. 2004. Detection of enterotoxigenic Clostridium perfringens type A isolates in American retail foods. Appl. Environ. Microbiol. 70:2685–2691. 10.1128/AEM.70.5.2685-2691.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McDonel JL, McClane BA. 1988. Production, purification and assay of Clostridium perfringens enterotoxin. Method Enzymol. 165:94–103. 10.1016/S0076-6879(88)65018-X [DOI] [PubMed] [Google Scholar]
- 24. Wnek AP, Strouse RJ, McClane BA. 1985. Production and characterization of monoclonal antibodies against Clostridium perfringens type A enterotoxin. Infect. Immun. 50:442–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robertson SL, Smedley JG, III, Singh U, Chakrabarti G, Van Itallie CM, Anderson JM, McClane BA. 2007. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 9:2734–2755. 10.1111/j.1462-5822.2007.00994.x [DOI] [PubMed] [Google Scholar]
- 26. Bartholomew BA, Stringer MF, Watson GN, Gilbert RJ. 1985. Development and application of an enzyme-linked immunosorbent assay for Clostridium perfringens type A enterotoxin. J. Clin. Pathol. 38:222–228. 10.1136/jcp.38.2.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berry P, Rodhouse J, Hughes S, Bartholomew B, Gilbert RJ. 1988. Evaluation of ELISA, RPLA, and Vero cell assays for detecting Clostridium perfringens enterotoxin in faecal specimens. J. Clin. Pathol. 41:458–461. 10.1136/jcp.41.4.458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh U, Van Itallie CM, Mitic LL, Anderson JM, McClane BA. 2000. Caco-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 275:18407–18417. 10.1074/jbc.M001530200 [DOI] [PubMed] [Google Scholar]
- 29. Caserta JA, Robertson SL, Saputo J, Shrestha A, McClane BA, Uzal FA. 2011. Development and application of a mouse interstinal loop model to study the in vivo action of Clostridium perfringens enterotoxin. Infect. Immun. 79:3020–3027. 10.1128/IAI.01342-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bos J, Smithee L, McClane BA, Distefano RF, Uzal F, Songer JG, Mallonee S, Crutcher JM. 2005. Fatal necrotizing enteritis following a foodborne outbreak of enterotoxigenic Clostridium perfringens type A infection. Clin. Infect. Dis. 15:78–83. 10.1086/429829 [DOI] [PubMed] [Google Scholar]
- 31. Fernandez Miyakawa ME, Pistone Creydt V, Uzal FA, McClane BA, Ibarra C. 2005. Clostridium perfringens enterotoxin damages the human intestine in vitro. Infect. Immun. 73:8407–8410. 10.1128/IAI.73.12.8407-8410.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gorkiewicz G, Thallinger GG, Trajanoski S, Lackner S, Stocker G, Hinterleitner T, Gully C, Hogenauer C. 2013. Alterations in the colonic microbiota in response to osmotic diarrhea. PLoS One 8:e55817. 10.1371/journal.pone.0055817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clausen MR, Jørgensen J, Mortensen PB. 1998. Comparison of diarrhea induced by ingestion of fructooligosaccharide Idolax and disaccharide lactulose: role of osmolarity versus fermentation of malabsorbed carbohydrate. Dig. Dis. Sci. 43:2696–2707. 10.1023/A:1026659512786 [DOI] [PubMed] [Google Scholar]
- 34. Hammer HF, Santa Ana CA, Schiller LR, Fordtran JS. 1989. Studies of osmotic diarrhea induced in normal subjects by ingestion of polyethylene glycol and lactulose. J. Clin. Invest. 84:1056–1062. 10.1172/JCI114267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N, Bruewer M. 2009. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 296:G1140–G1149. 10.1152/ajpgi.90534.2008 [DOI] [PubMed] [Google Scholar]
- 36. Hewitt KJ, Agarwal R, Morin PJ. 2006. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer 6:186. 10.1186/1471-2407-6-186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu Z, Ding L, Lu Q, Chen YH. 2013. Claudins in intestines: Distribution and functional significance in health and diseases. Tissue Barriers 1:e24978. 10.4161/tisb.24978 [DOI] [PMC free article] [PubMed] [Google Scholar]