

Abstract
EspC is a non-locus of enterocyte effacement (LEE)-encoded autotransporter produced by enteropathogenic Escherichia coli (EPEC) that is secreted to the extracellular milieu by a type V secretion system and then translocated into epithelial cells by the type III secretion system. Here, we show that this efficient EspC delivery into the cell leads to a cytopathic effect characterized by cell rounding and cell detachment. Thus, EspC is the main protein involved in epithelial cell cytotoxicity detected during EPEC adhesion and pedestal formation assays. The cell detachment phenotype is triggered by cytoskeletal and focal adhesion disruption. EspC-producing EPEC is able to cleave fodrin, paxillin, and focal adhesion kinase (FAK), but these effects are not observed when cells are infected with an espC isogenic mutant. Recovery of these phenotypes by complementing the mutant with the espC gene but not with the espC gene mutated in the serine protease motif highlights the role of the protease activity of EspC in the cell detachment phenotype. In vitro assays using purified proteins showed that EspC, but not EspC with an S256I substitution [EspCS256I], directly cleaves these cytoskeletal and focal adhesion proteins. Kinetics of protein degradation indicated that EspC-producing EPEC first cleaves fodrin (within the 11th and 9th repetitive units at the Q1219 and D938 residues, respectively), and this event sequentially triggers paxillin degradation, FAK dephosphorylation, and FAK degradation. Thus, cytoskeletal and focal adhesion protein cleavage leads to the cell rounding and cell detachment promoted by EspC-producing EPEC.
INTRODUCTION
Enteropathogenic Escherichia coli (EPEC) infection is a leading cause of infantile diarrhea that can be severe and lethal in developing countries (1). The hallmark of EPEC infection is a histopathological lesion formed at the mucosal intestinal surface that resembles a pedestal-like structure known as the attaching and effacing (A/E) lesion (2). The genes responsible for the A/E phenotype are located in a 35.6-kb pathogenicity island termed the locus of enterocyte effacement (LEE). Other pathogens that share this pathogenicity island are known as A/E pathogens, including enterohemorrhagic E. coli (EHEC), rabbit EPEC (REPEC), and Citrobacter rodentium, which infects mice (3, 4). The LEE contains diverse genes encoding secreted proteins for the type III secretion system (T3SS), termed EPEC-secreted proteins (Esp), and its own translocated intimin receptor (Tir) (5). Tir is injected by the translocation apparatus directly into the cell and is inserted in the membrane, exposing an extracellular domain that is recognized by intimin, an EPEC-membrane adhesin. Intimin-Tir interaction leads to intimate adherence and pedestal formation beneath adherent bacteria (6). During infection, other effector proteins are translocated into the cell (EspF, EspG, EspH, Map, and EspZ), and they interfere with different aspects of cell physiology (7, 8). Notably, there are several identified non-LEE-encoded effectors in A/E pathogens that are translocated by the T3SS, including Cif, NleA/EspI, TccP/EspFU, EspJ, NleB, NleC, and NleD (9–16). The outcomes of infection as seen in biopsy specimens from patients include moderate to severe damage at the intestinal mucosa, irregular atrophy of surface epithelium, and subnuclear vacuolization of crypt epithelium (17). At the structural level the biopsy specimens showed intracellular damage characterized by dilatation of rough endoplasmic reticulum, mitochondrial changes, and cytoplasmic pallor (18).
A second pathogenicity island of EPEC that encodes EspC, an autotransporter protein, has been identified in pathogenic EPEC1 strains. A recent study showed that espC is one of the most prevalent genes among those encoding autotransporter proteins in both typical and atypical EPEC strains (19). Unlike proteins secreted by the T3SS, EspC secretion is mediated by the type V secretion system (T5SS) (20, 21). EspC shows the three classical domains (signal sequence, passenger domain, and translocation unit) of autotransporter proteins and has a conserved serine protease motif (22). Even thought EspC is a non-LEE-encoded protein, the regulation of espC is coupled to the global regulator Ler encoded in LEE that controls virulence gene expression during EPEC pathogenesis, including genes encoding the T3SS, secreted Esp proteins, Tir, and intimin (23, 24). Previously, it was shown that during epithelial cell infection by EPEC, the first protein found in the supernatant is EspC (60 min), and 30 min later T3SS translocator proteins are secreted to the supernatant (25). We have shown that EspC is not efficiently internalized under nonphysiological conditions (i.e., as a purified protein) because no receptor is involved in its uptake and because no intracellular traffic is required. However, the physiologically secreted EspC by EPEC, which is enhanced in tissue culture medium and by cell contact, is efficiently internalized during the interaction of EPEC and epithelial cells (26). Recently, we also showed that during EPEC infection, EspC is secreted from the bacteria by the type V secretion system (T5SS), and then it is efficiently translocated into the epithelial cells by the type III secretion system (T3SS) translocon (27).
We have found that purified EspC, once inside the cells, has enterotoxic and cytotoxic activities on rat jejunum preparations mounted in Ussing chambers and on cultured epithelial cells, respectively (20, 28). Our group has also found that EspC causes cytotoxic effects, including cytoskeletal damage that depends on the internalization of EspC and its functional serine protease motif (28). Here, we examined the role of EspC during the infection of epithelial cells by EPEC by characterizing its cellular targets and the relationship with cytotoxicity, which is commonly observed in adhesion and pedestal formation assays.
MATERIALS AND METHODS
Bacterial strains and purification of recombinant proteins.
Characteristics of the strains used in this study are listed in Table 1. All strains were routinely grown in Luria-Bertani (LB) broth or minimum essential medium (MEM) (without supplements) aerobically at 37°C. EPEC cultures were activated for 3 h as previously described (29).
TABLE 1.
Bacterial strains and plasmid used in this study
| Strain | Genotype and/or description | Reference |
|---|---|---|
| E2348/69 | Prototype EPEC isolate (O127:H6), espC+ | 20, 46 |
| ΔespC strain | E2348/69 ΔespC, EspC mutant | 21 |
| REPEC | REPEC O103:K-H2, LEE+ espC mutant | 47 |
| ΔespC(pespC) strain | E2348/69 ΔespC(pJLM174), complemented espC mutant | 27 |
| ΔespC(pespCS256I) strain | E2348/69 ΔespC(pespCS256I), transformed mutant with espC mutated in serine 256 | 27 |
| REPEC(pespC) | REPEC O103:K-H2, LEE+ espC mutant transformed with espC | 27 |
| CVD452 | E2348/69 ΔescN, T3SS mutant | 48 |
| HB101 | Nonpathogenic strain, K-12/B hybrid | 49 |
| HB101(pJLM174) | Minimal espC clone, cloned in pBAD30 | 20 |
| HB101(pespCS256I) | espC clone mutated in the residue S256 | 27 |
| BL21(pGEX-3X18531) | Clone encoding GST-fodrin | 50 |
To purify recombinant proteins, strain HB101(pJLM174) or HB101(pespCS256I) (where pespCS256I is a plasmid carrying the espC gene that encodes a change from S to I at amino acid position 256) was grown overnight in LB medium plus arabinose (0.2%, wt/vol) and ampicillin (100 μg/ml) at 37°C with shaking. Supernatants were obtained by centrifugation at 7,000 × g for 15 min, filter sterilized through 0.22-μm-pore-size filters (Corning, Cambridge, MA), and concentrated 100-fold in an Ultrafree Centrifugal Filter device with a 100-kDa cutoff (Millipore, Bedford, MA). Recombinant EspC and the EspC protein with the substitution S256I (EspCS256I) were filter sterilized again (28), aliquoted, and quantified by the Bradford method (30).
Tissue culture cells and bacterial infection.
The human epithelial cell line HEp-2 (ATCC CCL23) or rabbit kidney RK13 (ATCC CCL-37) line was cultured in MEM supplemented with 10% fetal calf serum (FCS) (HyClone, Logan, UT), 1% nonessential amino acids, 5 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were normally harvested with 10 mM EDTA and 0.25% trypsin (Gibco-BRL, Grand Island, NY) in phosphate-buffered saline (PBS; pH 7.4), resuspended in the appropriate volume of supplemented MEM, and incubated at 37°C in a humidified atmosphere of 5% CO2.
HEp-2 cells grown in 60-mm petri dishes were infected with activated cultures of EPEC (multiplicity of infection [MOI] of 10) or the indicated isogenic mutant alone or with purified EspC for the indicated time. EPEC-infected HEp-2 cells were incubated in the presence of d-mannose (1%) and the appropriate antibiotic. Cells were delicately washed three times with ice-cold PBS, pH 7.4, and scraped into a buffer consisting of Tris-HCl (0.25 M), pH 7.5, phenylmethylsulfonyl fluoride (PMSF) (50 μg/ml), aprotinin (0.5 μg/ml), and EDTA (0.5 μM). Then cells were lysed by three freeze-thaw cycles (5-min incubation in a dry ice-ethanol bath and 3-min incubation in a thermoblock at 37°C). Protein concentrations were estimated by the method of Bradford. Equivalent amounts were boiled for 7 min, analyzed on sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE), and electrotransferred to nitrocellulose membranes for immunoblot analyses essentially as previously described (26), using anti-fodrin (MAB1622; Chemicon International), anti-FAK (C20) (sc-558; Santa Cruz Biotechnology), rabbit anti-phospho-FAK (Tyr397) [pFAK(Tyr397)] (sc-11765-R; Santa Cruz Biotechnology), and anti-paxillin (H-114) (sc-5574; Santa Cruz Biotechnology) antibodies.
Confocal microscopy.
HEp-2 or RK13 cells were seeded into eight-well LabTek slides (VWR, Bridgeport, NJ) at a density of 3 × 104 cells/well. Before infection with activated EPEC or REPEC cultures, cells were washed three times with warmed PBS (pH 7.4) and incubated at 37°C in MEM (without supplements) for 30 min. Infections were carried out at the indicated times in the presence of 1% d-mannose (Research Organics, Inc.). Infected HEp-2 cells were washed with PBS, fixed with 2% formalin-PBS, washed, permeabilized by the addition of 0.1% Triton X-100–PBS, and stained with 0.05 μg of tetramethyl rhodamine isothiocyanate (TRITC)-phalloidin/ml and with a polyclonal rabbit anti-EspC, as previously described (28), followed by an anti-rabbit fluorescein-labeled antibody. Slides were mounted on Gelvatol, covered with glass coverslips, and examined under a Leica TCS SP8 confocal microscope.
The total number of epithelial cells attached to the glass substrate was determined by counting their nuclei, and the percentage of detached cells was calculated as the inverse of attached cells by using the total number of epithelial cells from untreated cells as a reference (100%). Cell rounding was identified by cell morphology using as a reference the ratio nucleus/cytoplasm (globular cells) detected by optical sections. Cell rounding was quantified in three optical fields by comparing the distended cells versus the rounded cells.
Immunoprecipitation and degradation assays.
HEp-2 cells grown in 60-mm petri dishes were delicately washed three times with ice-cold PBS, pH 7.4, and scraped into radioimmunoprecipitation assay (RIPA) buffer (1% Nonidet P-40, 50 mM Tris-HCl, pH 7.5. 150 mM NaCl, 0.02% SDS). Cells were collected in Eppendorf tubes at 4°C. Cells were lysed by sonication for 10 s in ice water using a Soniprep sonicator at 30% amplitude. The supernatant was centrifuged at 14,000 × g for 15 min at 4°C.
Five micrograms of polyclonal anti-FAK (C20) (sc-558; Santa Cruz Biotechnology) or anti-paxillin (H-114) (sc-5574; Santa Cruz Biotechnology) antibody and protein A-agarose suspension (Roche Diagnostics, Mannheim, Germany) was incubated with cell lysates overnight at 4°C to precipitate the protein-antibody complex. The beads were washed three times with RIPA buffer, and the immunocomplexes were resuspended in 525 μl of reaction buffer (250 mM Tris-HCl, pH 7.5, 1 mM dithiothreitol [DTT]).
Two micrograms of immunoprecipitated FAK or paxillin was mixed with an equal volume of 2× digestion buffer (0.3 mM CaCl2, 10 mM DTT) containing 20 μg/ml of EspC or EspCS256I. Reactions were carried out at 37°C for different time periods (30 s and 1, 2, 5, 10, 15, and 30 min) and then stopped by the addition of 4× SDS sample buffer. Samples were boiled, and proteins were separated by SDS-PAGE and analyzed by immunoblotting using anti-paxillin antibody (H-114; 1:1,000) or anti-FAK antibody (C20 or A17; 1:1,000). Membranes were developed by using horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody (Zymed) as indicated by the manufacturer. HRP was detected with enhanced chemiluminescence (ECL) reagent from Amersham.
Statistical analysis.
Results are expressed as means and standard deviations. Statistical significance was determined by a two-tailed Student t test. A P value of <0.005 is considered significant.
RESULTS
EspC internalization causes cytotoxicity after 3 h of the epithelial cell-EPEC interaction.
To investigate the role of EspC in the cytotoxicity detected during the interaction of epithelial cells with EPEC, HEp-2 cells were infected with EPEC strain E2348/69, and the cytotoxic effect was compared with that of an espC isogenic mutant (ΔespC strain) and a complemented mutant [ΔespC(pespC) strain]. After 3 h of infection, cells were immunostained with anti-EspC antibodies, and the actin cytoskeleton was stained with rhodamine-phalloidin. The EspC-producing EPEC E2348/69 strain was able to secrete and deliver EspC inside the cells (as analyzed by confocal microscopy in 12 optical sections), which led to cell rounding and cell detachment (Fig. 1A), while an EPEC strain mutated in the espC gene (ΔespC strain) was unable to cause these cytotoxic effects but was capable of producing several pedestal-like structures. Furthermore, complementation of the ΔespC mutant with the espC gene [ΔespC(pespC) strain] restored the capacity to cause cytotoxicity, and the effects were similar to those seen with the wild-type strain. To further demonstrate that EspC activity is responsible for the cytotoxic effects, ΔespC bacteria were complemented with an espC gene containing a mutation in the serine protease motif [ΔespC(pespCS256I) strain], which thus lacks serine protease activity, or with the addition of exogenous EspC during the infection (EPEC ΔespC + EspC). The strain complemented with the espC gene encoding the substitution at S256 [ΔespC(pespCS256I) strain] was unable to cause cytotoxicity, and several pedestal structures and actin cytoskeleton fibers were observed. In contrast, the ΔespC strain, as we demonstrated before, was able to internalize exogenous EspC (27) and also caused cytotoxic effects similar to those observed in cells treated with the wild-type EPEC (Fig. 1A). The effects of EspC on cell rounding (Fig. 1B) and cell detachment (Fig. 1C) were quantified for all tested strains and compared among them. These data and their statistical analyses showed a correlation with the morphological damage described above (Fig. 1A and B). Together, our results show that physiologically internalized EspC induces cytotoxicity of human cells via its serine protease motif.
FIG 1.

EspC is required to cause the cytotoxic effect caused by EPEC. (A) HEp-2 cells were infected by either EPEC (strain E2348/69), EPEC ΔespC (mutant ΔespC), EPEC ΔespC(pespC) [complemented ΔespC(pespC) strain], EPEC ΔespC(pespCS256I) [complemented strain with espC gene mutated in the serine protease motif ΔespC(pespCS256I)], or EPEC ΔespC supplemented with purified EspC (ΔespC + EspC strain) for 3 h at an MOI of 10. Infected cells were fixed and permeabilized. Cells were immunostained with anti-EspC antibody, followed by a secondary antibody conjugated to fluorescein isothiocyanate, and the actin cytoskeleton was detected with rhodamine-phalloidin. Untreated cells were used as negative controls. Slides were observed by confocal microscopy, and in the case of cells treated with EPEC ΔespC(pespCS256I) only a middle optical section is shown to demonstrate that EspCS256I is internalized as the native protein. (B and C) Quantification of cell rounding and cell detachment. After cell treatments as described in panel A, the numbers of rounded cells and detached cells were quantified as described in Materials and Methods. Results are means plus standard deviations of three independent experiments. A P value of <0.005 was considered significant. ***, P < 0.001 (E2348/69 versus ΔespC or ΔescN strain); +++, P < 0.001 (ΔespC strain versus ΔespC + EspC strain); ++, P < 0.005 [ΔespC strain versus ΔespC(pespC) or ΔescN strain].
EspC causes actin cytoskeleton disruption triggering cell detachment.
To determine the timing for the cell-EPEC interaction to cause cytoskeleton disruption and cell detachment, HEp-2 cells were infected with EPEC or EPEC ΔespC for different times and then treated with an antibiotic (gentamicin) for 1 h to kill the bacteria. After this treatment, infected cells were incubated for 8 h in fresh medium and then fixed and stained with rhodamine-phalloidin. Confocal microscopy analyses showed that 2 h of EPEC-epithelial cell interaction was not sufficient to cause cytotoxic effects, and most of the cells were similar to those treated with the mutant EPEC ΔespC, in which actin stress fibers and pedestal-like structures were clearly observed (Fig. 2). However, 3 h of EPEC-epithelial cell interaction was sufficient to produce cell toxicity, observed as cytoskeleton contraction and cell detachment. These cytotoxic effects were not observed in cells treated with EPEC ΔespC under the same conditions, and their cytoskeletons were preserved, allowing formation of the actin-rich pedestal structures induced by attached bacteria (Fig. 2). Interestingly, after 3 h, cell detachment depended on the interaction time. After 4 h of EPEC infection most cells were detached, whereas cells infected with the EPEC ΔespC strain showed an increased quantity of pedestal structures, and some cells started to undergo cell damage, probably induced by other bacterial effectors. Finally, after 5 h of EPEC infection, cells were almost absent on the glass substrate, while it was still possible to observe cells that had been infected with the EPEC ΔespC strain (Fig. 2).
FIG 2.
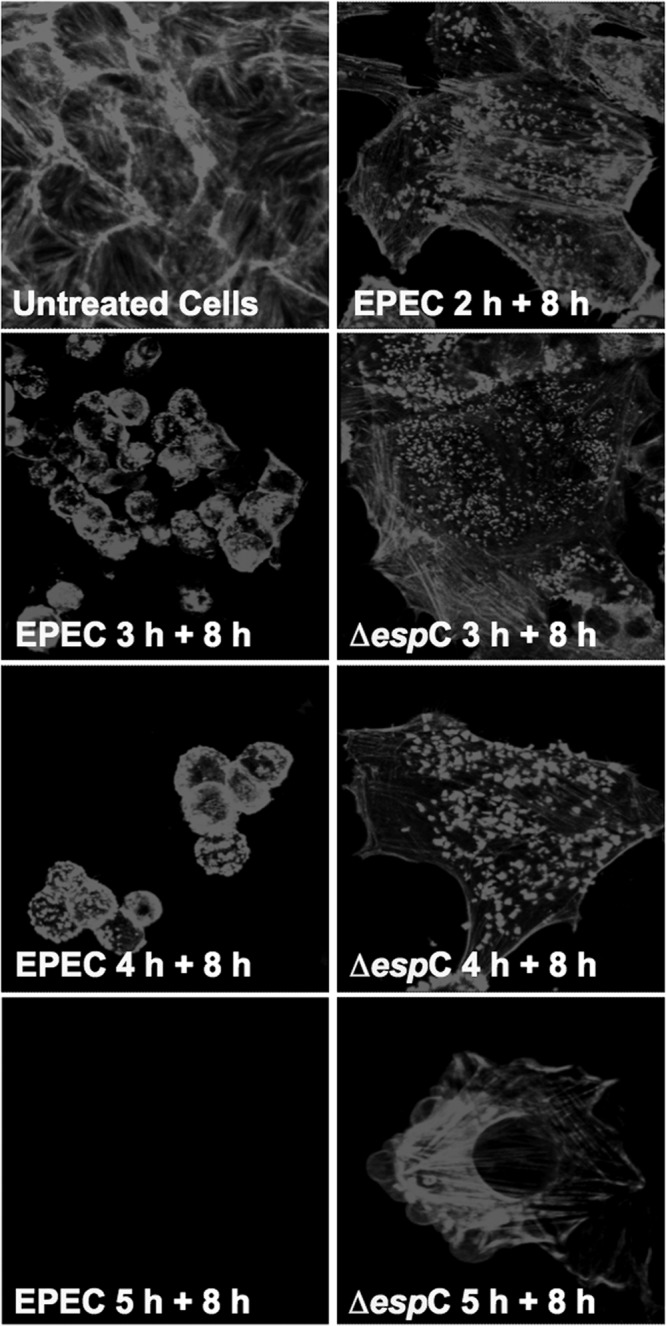
EspC is the main toxic effector that causes cytotoxicity after 3 h of EPEC infection. HEp-2 cells were infected with either EPEC or EPEC ΔespC (ΔespC) at different times (2, 3, 4, or 5 h). After the infection, cells were treated with gentamicin to kill bacteria and incubated for 8 h. Infected cells were fixed and permeabilized. Actin cytoskeleton was stained with rhodamine-phalloidin. Untreated cells were used as negative controls. Slides were observed by confocal microscopy.
To further demonstrate that EspC is responsible for the cytoskeleton disruption and cell detachment induced by EPEC after 3 h of interaction with epithelial cells, we transformed an EPEC strain that infects rabbits (REPEC) with the espC gene from EPEC E2348/69. REPEC does not harbor the espC gene, but it does carry a T3SS, which is required for EspC internalization (27). HEp-2 cells were alternatively infected with wild-type REPEC or REPEC(pespC). After 3 h of infection, REPEC(pespC) clearly caused cytoskeleton disruption and cell detachment, while the wild-type REPEC strain did not show these effects (Fig. 3). Cytoskeleton disruption and cell detachment were more evident with time. After 4 h most of the cells were detached, and those that remained were observed as rounded cells; after 5 h of interaction almost no cells were observed. However, in cells infected with the wild-type REPEC, no clear cytotoxicity was observed at 3 or 4 h, and a slight effect was observed on the cell cytoskeleton at 5 h of interaction (Fig. 3).
FIG 3.
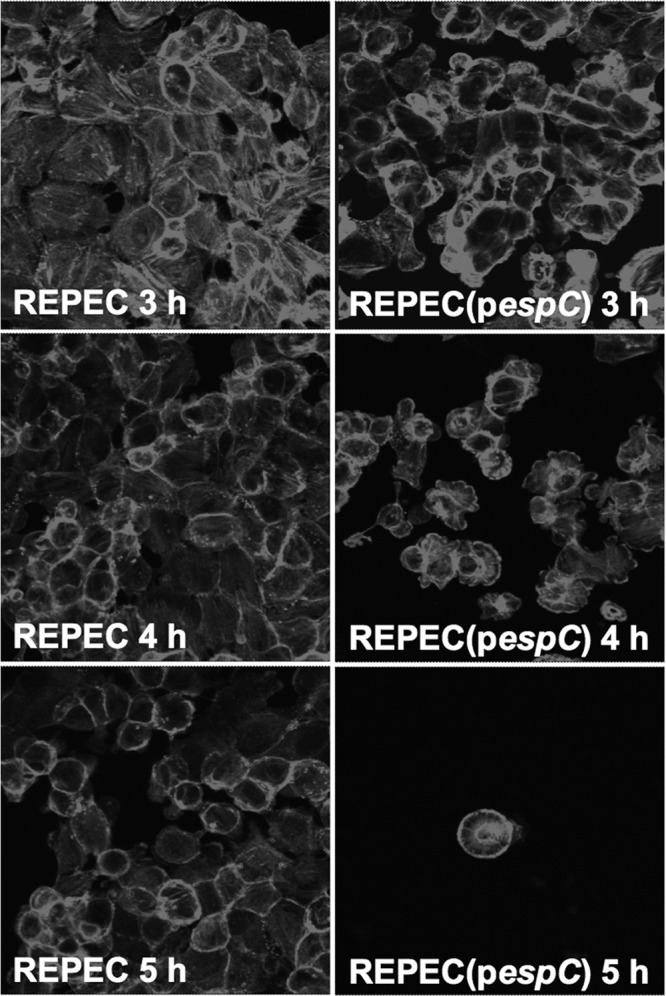
EspC increases the cytotoxicity of rabbit EPEC (REPEC). HEp-2 cells were infected with either REPEC or REPEC transformed with the espC gene [REPEC(pespC)] for 3, 4, or 5 h. Infected cells were fixed and permeabilized. Actin cytoskeleton was stained with rhodamine-phalloidin. Slides were observed by confocal microscopy.
EspC-producing EPEC is able to cleave epithelial fodrin and generates two subproducts of degradation.
We have previously shown that purified EspC is able to cleave recombinant glutathione S-transferase (GST)-fodrin; therefore we tested if an EspC-producing EPEC strain is able to cleave epithelial fodrin from HEp-2 cells. HEp-2 cells were infected for 2 or 6 h with EPEC, EPEC ΔespC, EPEC ΔespC(pespC), or EPEC ΔespC(pespCS256I). As we showed above, at 2 h cytoskeleton disruption and cell detachment were not detected, but at 6 h these effects were clearly observed (Fig. 4A). After the infection times, cells were lysed and analyzed by immunoblotting using anti-fodrin antibodies. As expected, at 2 h of EPEC infection, the fodrin protein band was detected unbroken, as observed in untreated cells or in cells infected with EPEC ΔespC (Fig. 4A). However, at 6 h of EPEC infection, the fodrin protein band drastically decreased, and a subproduct of fodrin degradation was detected at around 90 to 120 kDa. This was not detected in cells infected with EPEC ΔespC but was recovered when cells were infected with the complemented EPEC ΔespC(pespC) strain. Interestingly, the cleavage of fodrin was not detected when cells were infected with the EPEC ΔespC strain complemented with espC mutated in the serine protease motif [EPEC ΔespC(pespCS256I)] (Fig. 4A).
FIG 4.

EspC cleaves fodrin (alpha II spectrin) within the 11th and 9th repetitive units. (A) Cleavage of epithelial fodrin by EPEC, EPEC ΔespC, EPEC ΔespC(pespC), or EPEC ΔespC(pespCS256I). HEp-2 cells were infected with the strains indicated above for 2 or 6 h. Infected cells were lysed, and the proteins were analyzed by immunoblotting using anti-fodrin antibody. The reaction was visualized using HRP-labeled rabbit anti-mouse antibodies and developed using Western blotting chemiluminescence reagent. The subproducts of fodrin degradation are indicated with asterisks, and the original fodrin is indicated with an arrow. Samples for all panels are identified at the top of each lane. (B) Cleavage of GST-fodrin by EspC. Purified GST-fodrin (2.5 μg) was incubated with EspC (1 μg) for 4 h and then separated by SDS-PAGE. Subproducts of GST-fodrin degradation by EspC containing the amino terminus (43 and 34 kDa) were sequenced. Sequencing of the 43-kDa subproduct showed that the first cleavage site is localized within the 11th repeat unit, between Q1219 and L1220, while the 34-kDa subproduct showed that the second cleavage site is localized within the 9th repeat unit, between D938 and L939.
As we have previously shown, EspC is able to cleave GST-fodrin in vitro (28), which is a 109-kDa recombinant protein containing repeats 8 to 14 of α-fodrin fused to GST; therefore we decided to perform this degradation assay using purified proteins in order to investigate the cleavage sites of EspC on fodrin. We previously reported that according to the GST-fodrin structure/sequence and the subproducts of fodrin generated (after 1 h, two subproducts of 72 and 43 kDa; after 4 h, two additional subproducts derived from the 72-kDa band were detected as 45 and 34 kDa) since only the 72- and 45-kDa bands were detected with anti-GST antibodies, the N-terminal (28). Thus, we decided to determine the N-terminal amino acid sequence of the 43- and 34-kDa subproducts from recombinant GST-fodrin since these fragments do not contain the GST N-terminal amino acids. N-terminal amino acid sequences from these fragments were LAEERSQLGS for the 43-kDa subproduct and LSAYGSSIQA for the 34-kDa subproduct (University of California, San Diego, Division of Biology Protein Sequencer Facility). These data and the sequence of GST-fodrin (Fig. 4B) indicate that EspC cleavage sites in human fodrin occur within fodrin's 11th and 9th repetitive units. In the 11th repetitive unit, the cleavage site occurs between Q1219 and L1220 in helix C close to a CaM binding site, where a Pet cleavage site is (31), and the second cleavage in the 9th repetitive unit occurs between D938 and L939. According to our analysis in silico, in the native protein, the first cleavage correspond to a 108-kDa protein, which matches the subproduct detected in HEp-2 cells seen between 90 and 120 kDa (Fig. 4A).
EspC-producing EPEC also cleaves other proteins involved in cell detachment: FAK and paxillin.
Since cell detachment could also be induced by disruption of focal adhesion, we sought evidence for FAK and paxillin degradation by EspC. HEp-2 cells were infected for 1, 2, or 6 h with EPEC, EPEC ΔespC, EPEC ΔespC(pespC), or EPEC ΔespC(pespCS256I); as negative controls, cells were untreated or infected with EPEC ΔescN (mutant in the T3SS, which is unable to translocate EspC into the cells). After infection, cells were lysed and analyzed by immunoblotting using anti-FAK antibodies. As expected, the EPEC strain was unable to cleave FAK at 1 or 2 h of interaction with epithelial cells, and the FAK protein band was observed, as in untreated cells. However, at 6 h of infection, the FAK band disappeared, indicating its degradation by the EPEC strain. Interestingly, FAK was not degraded when epithelial cells were infected with the mutant in espC (ΔespC strain). Furthermore, the complemented strain [EPECΔespC(pespC)] was able to cleave FAK, similar to the wild-type EPEC, but the complemented strain with the serine protease motif mutant [EPEC ΔespC(pespCS256I)] was unable to cleave FAK. As with fodrin, FAK is cleaved by the serine protease motif of EspC (Fig. 5A).
FIG 5.
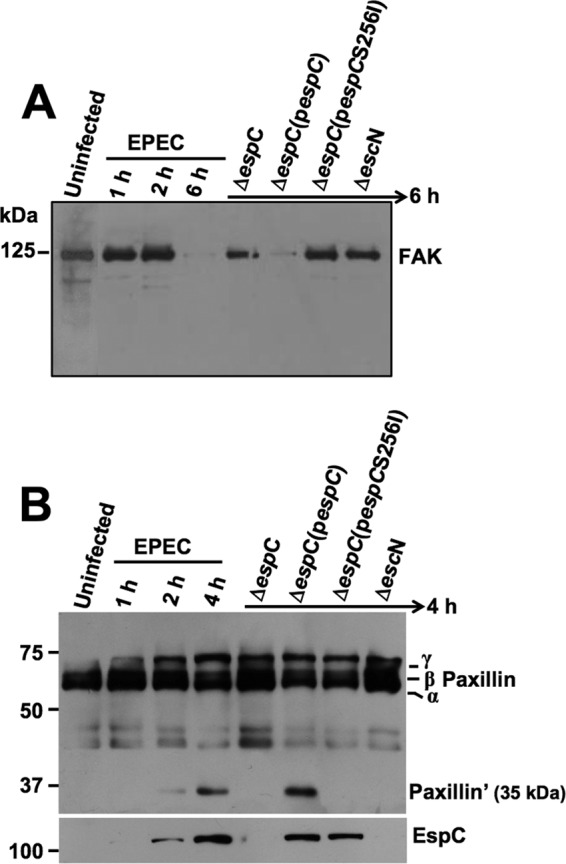
EspC causes FAK and paxillin degradation. (A) FAK degradation. HEp-2 cells were infected with the strains as indicated at the top of each lane and for the indicated times. Infected cells were lysed, and proteins were analyzed by immunoblotting using anti-FAK antibody. The reaction was visualized using HRP-labeled rabbit anti-mouse antibody and developed using Western blotting chemiluminescence reagent. (B) Paxillin degradation. HEp-2 cells were infected with the strains as indicated at the top of each lane and for the indicated times. Infected cells were lysed, and the proteins were analyzed by immunoblotting using anti-paxillin as described for panel A. Note EspC detection in the cytoplasmic fraction using anti-EspC antibodies (in the same blot), indicating EspC translocation into the cells (lower panel).
On the other hand, HEp-2 cells were infected for 1, 2, or 4 h with EPEC, EPEC ΔespC, and EPEC ΔespC(pespC), and lysed cells were analyzed by immunoblotting using anti-paxillin antibodies (Fig. 5B). Paxillin was also cleaved during EPEC infection, and a 35-kDa subproduct (paxillin′) was detected after 4 h; in contrast the mutant ΔespC strain was unable to cleave paxillin, and the profile of paxillin bands was similar to that in untreated cells. Complementation of the mutant with the espC gene [ΔespC(pespC) strain] restored the ability to cleave paxillin, and the subproduct of 35 kDa was also observed. The complementation strain with the serine protease motif mutant [ΔespC(pespCS256I) strain], however, was unable to cleave paxillin. All the EspC- or EspCS256I-producing EPEC strains were able to translocate these proteins into the cells, as detected in the cytoplasmic fractions (Fig. 5B).
Kinetics of FAK and paxillin cleavage by EspC-producing EPEC.
Since cell detachment occurs between 3 and 4 h of interaction of epithelial cells and EPEC, we hypothesized that FAK and paxillin cleavage must occur before this phenotype appears. Thus, we determined the kinetics of FAK and paxillin cleavage by EPEC, EPEC ΔespC, and EPEC ΔespC(pespCS256I). HEp-2 cells were infected at different times (1, 2, 3, 4, and 6 h) with the strains indicated above, and lysed cells were analyzed by immunoblotting using anti-FAK or anti-paxillin antibodies (Fig. 6). The EPEC strain was able to cleave FAK in a time-dependent manner, starting at 2 h of infection until 6 h when the FAK band disappeared (Fig. 6A). However, FAK was not cleaved at any time when cells were infected with the EPEC ΔespC or EPEC ΔespC(pespCS256I) strain. On the other hand, when immunoblots were probed with the anti-paxillin antibodies, EPEC also caused the cleavage of paxillin in a time-dependent manner starting at 2 h, but a period of 6 h was not enough to completely cleave the paxillin isoforms. However, a distinct paxillin subproduct of 35 kDa (indicated as paxillin′) was clearly observed beginning at 2 h of infection and up to 6 h of infection (Fig. 6). As with FAK, paxillin bands were not cleaved at any time when cells were infected with the EPEC ΔespC or EPEC ΔespC(pespCS256I) strain; therefore, no subproduct of paxillin degradation was seen (Fig. 6B).
FIG 6.

Kinetics of FAK and paxillin cleavage by EspC-producing EPEC strains. HEp-2 cells were infected with the strains indicated at the top of each lane and for the indicated times. Infected cells were lysed, and the proteins were analyzed by immunoblotting using anti-FAK (A) or anti-paxillin (B) antibodies. The reaction was visualized using HRP-labeled rabbit anti-mouse antibodies and developed using Western blotting chemiluminescence reagent. Paxillin′ indicates a subproduct from paxillin cleavage.
Purified EspC is able to cleave immunoprecipitated FAK and paxillin.
Even though EspC-producing EPEC was able to degrade FAK and paxillin, this effect could either be induced by EspC through activating host enzymes or result from direct cleavage by EspC. Therefore, we tested if purified EspC is able to cleave purified FAK or paxillin. Immunoprecipitated FAK or paxillin was incubated with purified EspC at different times, and proteins were analyzed by immunoblotting using anti-FAK, anti-paxillin, or anti-EspC antibodies. EspC was able to cleave FAK from the first 30 s of EspC-FAK interaction. Interestingly, FAK cleavage was easily detected by the generation of subproducts identified using antibodies against the carboxy terminus or amino terminus of FAK (Fig. 7). The amino-terminal subproduct of around 90 kDa was clearly detected after 30 s until it disappeared at 15 min of EspC-FAK interaction, while the carboxy-terminal subproduct of around 78 kDa increased from 30 s until 5 min, when it was similar to the 90-kDa amino-terminal subproduct at 30 s. Unlike the 90-kDa subproduct, the 78-kDa subproduct was not further degraded at 15 min. These data and the fact that FAK subproducts of degradation during the cell infection assays were not observed indicate that FAK is quickly and completely degraded after its first cleavage by EspC (Fig. 7). As expected, the purified EspC mutant (EspCS256I) was unable to cleave FAK; the subproducts of FAK degradation were not observed, and the protein band was intact, as in untreated cells (control).
FIG 7.
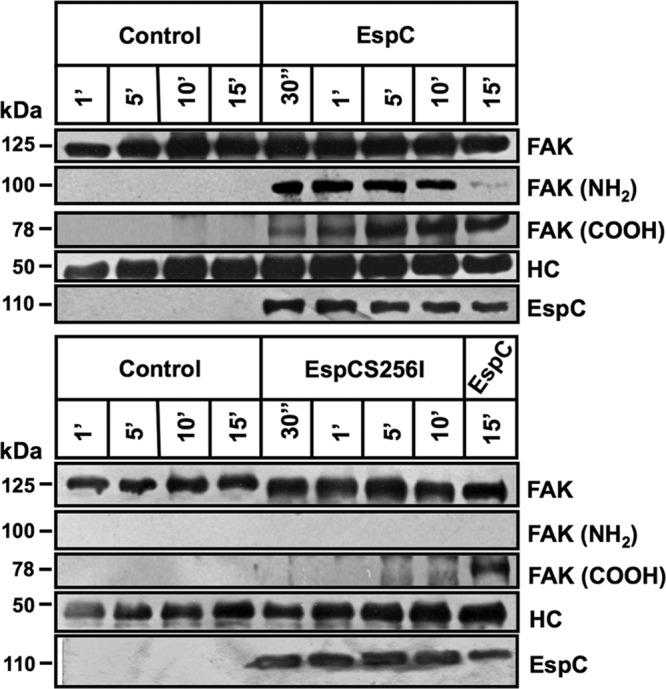
Purified EspC directly cleaves immunoprecipitated FAK. HEp-2 cells were lysed, and protein extracts were incubated with primary antibody anti-FAK plus protein A-agarose overnight. Immunoprecipitated FAK was incubated with purified EspC or EspCS256I for 30 s or 1, 5, 10, or 15 min. Reactions were stopped with Laemmli buffer, and products were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and analyzed by immunoblotting using anti-FAK (full-length protein), anti-FAK-NH2, anti-FAK-COOH, and anti-EspC as primary antibodies and HRP-conjugated anti-isotype secondary antibody. The reaction was developed using Western blotting chemiluminescence reagent. HC, heavy chains of antibodies used for the immunoprecipitation (visualized as a loading control).
On the other hand, EspC was also able to degrade purified paxillin (it was immunoprecipitated from HEp-2 cells using antibodies that recognize all isoforms). At 5 min of EspC-paxillin interaction, a slight difference in the paxillin isoform pattern was detected in comparison to that of untreated cells (control cells). This increased at 10 min, and paxillin degradation was clearly observed at 15 min; α-paxillin was the protein band mainly degraded. EspCS256I was unable to cleave paxillin, and the protein bands were similar to those in untreated cells (Fig. 8). It is clear that the kinetics of paxillin degradation was different when purified paxillin was used since the degradation was fast, occurring before 15 min; therefore the subproduct of 35 kDa is not seen (Fig. 8) because further degradation of it occurs in vitro using purified proteins.
FIG 8.

Purified EspC directly cleaves immunoprecipitated paxillin. HEp-2 cells were lysed, and protein extracts were incubated with the primary antibody anti-paxillin plus protein A-agarose overnight. Immunoprecipitated paxillin was incubated with purified EspC or EspCS256I for 5, 10, and 15 min. Reactions were stopped with Laemmli buffer, and products were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and analyzed by immunoblotting using anti-paxillin as a primary antibody and HRP-conjugated anti-isotype secondary antibody. The reaction was developed using Western blotting chemiluminescence reagent. HC, heavy chains of antibodies used for the immunoprecipitation (visualized as a loading control).
EspC from EPEC initially cleaves fodrin and then focal adhesion proteins to cause cell detachment.
Since both fodrin and focal adhesion proteins are involved in cell detachment, we decided to investigate which of these events, the cleavage of fodrin or that of FAK (cytoskeleton disruption or focal adhesion disruption), triggered the cell rounding before the cell detachment. By using the same population of cells and probing the same immunoblots sequentially with anti-fodrin, anti-FAK or anti-phosphorylated FAK, and anti-actin (as a loading control), we determined the kinetics of protein degradation of FAK and fodrin and, at the same time, for phosphorylated FAK. As shown in Fig. 9, during EPEC infection, the first detected event is the degradation of fodrin, followed by dephosphorylation and degradation of FAK. The kinetics of fodrin or FAK degradation was not observed when HEp-2 cells were treated with EPEC ΔespC or a laboratory E. coli avirulent strain, HB101. Densitometry analyses showed that, indeed, fodrin degradation preceded FAK degradation (Fig. 9, lower panels). After the first hour of infection, fodrin degradation occurred faster (from 90% to around 40% at 2 h of infection) than FAK degradation (from 95% to around 90% at 2 h). Then, fodrin decay was linear until 3 h (to 8%), while the linear FAK decay started at 2 h of infection until 4 h (to 15%). Finally, fodrin was completely degraded at 4 h, while FAK degradation was prolonged until 6 h when 8% of FAK still remained.
FIG 9.

EspC-producing EPEC initially cleaves fodrin and then causes focal adhesion disruption by dephosphorylation and cleavage of FAK. HEp-2 cells were infected with EPEC (E2348/69), EPEC ΔespC, and HB101 (MOI 10) at different times. Cells were lysed, and protein extracts were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Protein degradation was analyzed by immunoblotting using anti-FAK, anti-fodrin, anti-pFAK (Tyr 397), and anti-actin as primary antibodies and HRP-conjugated anti-isotype secondary antibody. The reaction was developed using Western blotting chemiluminescence reagent. Lower panels show densitometry data of protein bands from three independent experiments, which were performed to compare the decay kinetics of fodrin and FAK.
DISCUSSION
Epithelial cell death is a well-known consequence of EPEC infection (32–37). Furthermore, in human EPEC infection cases, pathological evidence of intestinal damage has been found (17, 18, 38). In the present study, we have investigated the role of EspC on cell detachment during EPEC infection. We previously showed that EspC is an important non-LEE-encoded enterotoxin (20) that is also capable of causing cytotoxicity in epithelial cells (28). EspC cytotoxic effects depend on EspC internalization and its serine protease motif (27, 28). Here, we found that EspC is sufficient to cause cell rounding, cell detachment, and cell death. These effects are consequences of fodrin and focal adhesion protein degradation produced by the serine protease motif of EspC. The sequence of these events is initiated by fodrin degradation, followed by paxillin degradation, FAK dephosphorylation, and FAK degradation. EspC first cleaves fodrin within its 11th repetitive unit between Q1219 and L1220 in helix C, close to a CaM binding site, and immediately EspC also causes cleavage in the 9th repetitive unit between D938 and L939; these cleavages trigger the cell detachment induced by EPEC, including the focal adhesion disruption.
We have previously shown that purified EspC is able to cause cytotoxicity in epithelial cells, but a considerable amount of EspC is needed to cause cell damage (28). However, we have recently found that EspC is efficiently secreted and delivered into epithelial cells by concerted cooperation of the T5SS and T3SS (27). Thus, here we showed that the efficient delivery of EspC by the T3SS is sufficient to cause cytotoxicity in epithelial cells. EspC-caused cytotoxicity is quite effective since an EPEC MOI of 10 for 3 h was enough to produce cell detachment, an effect that resembles the cytotoxicity seen in the adhesion pattern or fluorescent actin staining (FAS) assays (39, 40). Remarkably, an espC isogenic mutant was unable to cause these cytotoxic effects even though the mutant was able to cause A/E lesions and inject other virulence factors, and the complementation of this mutant with the espC gene restored the ability of the bacteria to cause cytotoxicity. This recovery was also complemented when the isogenic mutant was interacted with epithelial cells and purified EspC; exogenous EspC was translocated by the T3SS into the cells (27). However, when the isogenic mutant was transformed with the espC gene containing a site-directed mutation (espCS256I) in the serine protease motif, it was unable to complement the cytotoxic phenotype, supporting the role of this motif in cytotoxicity caused by EspC and our previous observations using purified protein (28).
The cytotoxic effects caused by an EspC-producing EPEC strain were characterized by cytoskeleton contraction, cell rounding, and cell detachment. The kinetics of cell damage after EPEC infection for different times, followed by gentamicin treatment for 1 h and incubation for 8 h, showed that EspC-caused toxicity depends on the time of EspC translocation since the effects are seen after 2 h of EPEC-epithelial cell interaction; at 3 h, most of the cells were rounded or detached, and this effect was progressive; at 5 h almost all cells were detached. However, these effects were not seen in the first hours after cells were infected with the espC isogenic mutant, but were present at 5 h, when perhaps other EPEC effectors are involved. Thus, by using an espC mutant it is possible to have a good model for pedestal formation since several pedestal-like structures were observed without cytotoxic effects over a long period (more than 10 h). Interestingly, the cell detachment effect caused by EspC-producing EPEC was mimicked by rabbit EPEC (REPEC), which does not harbor the espC gene but does have a T3SS that allows EspC translocation when it is transformed with the espC gene (27); thus REPEC(pespC) was able to cause cytotoxicity at the same infection times as the human EPEC wild-type strain.
We have previously shown that purified EspC is able to cleave recombinant fodrin in two sites within the molecule (28). Here, we showed that EspC-producing EPEC is able to cleave epithelial fodrin but not the espC isogenic mutant or the mutant complemented with the espC gene mutated in the serine protease motif. These data indicate that fodrin is an intracellular target of EspC. Since fodrin is a cytoskeletal protein, this explains the cytoskeletal disruption induced by EPEC that leads to cell detachment and cell death. However, other EPEC target proteins can also be involved in cell detachment, such as those involved in focal adhesion. Indeed, Shifrin et al. showed that EPEC induces cell detachment, modification of focal adhesion, and dephosphorylation of FAK and that these events are dependent on a functional T3SS in the infecting EPEC (41), but the EPEC effector responsible for these effects was not reported. Here, we showed that EspC-producing EPEC, but not an espC isogenic mutant, is able to cleave two main focal adhesion proteins, FAK and paxillin, and that the serine motif of EspC was essential to complement these phenotypes, as well as the T3SS, which allows EspC translocation. Interestingly, paxillin and FAK cleavage by EspC-producing EPEC preceded the cell detachment that started around 2 h after infection and increased with time until total degradation occurred, at around 6 h. In addition, our data suggest that paxillin cleavage occurs before FAK cleavage but that FAK is more susceptible than paxillin to degradation by the serine protease motif of EspC. Since fodrin and FAK are also cleaved during cell death by caspase and calpain (reviewed in reference 42), which might be induced by EspC, we showed here that purified EspC, but not EspCS256I, is able to cleave immunoprecipitated paxillin and FAK in vitro. Fortunately, the EspC-FAK interaction in vitro allowed us to detect amino- and carboxy-terminal degradation subproducts by using antibodies against either of these ends, suggesting that further FAK degradation in the cells could be occurring by means of other host proteases. It is worth mentioning that EspC is the most prominent protein secreted by EPEC before the T3SS translocator proteins (25), and unlike other cytotoxic LEE effectors, such as EspF, Map, and EspH (32, 43, 44), EspC is not subject to the T3SS timing for its secretion (45). On the other hand, the paxillin 35-kDa subproduct detected during EPEC infection, which appears to be derived from β-paxillin, was not detected during the EspC-paxillin interaction in vitro, where the main degraded isoform was α-paxillin. This suggests that the availability (or exposure) of paxillin is important for its proteolytic processing.
Since both the cytoskeletal fodrin protein and focal adhesion FAK protein are involved in cell detachment, we showed here that fodrin cleavage triggers paxillin degradation, FAK dephosphorylation, and FAK degradation. Therefore, EspC is a cytoskeletal toxin that cleaves fodrin, and this event is followed by adhesion focal disruption, events that result in cell rounding and cell detachment. Thus, the focal adhesion disruption phenotypes produced by EPEC previously detected (41) appear to be a consequence of fodrin degradation by EspC.
In summary, we have demonstrated that during EPEC infection, EspC is secreted by the T5SS, and then it is efficiently translocated into host cells by the T3SS (27). Once inside the cells, EspC is able to cleave fodrin in two sites (within the 11th repetitive unit between Q1219 and L1220 and then within the 9th repetitive unit between D938 and L939), which leads to focal adhesion disruption including paxillin degradation and FAK dephosphorylation and degradation. This cascade of events results in the cell rounding and cell detachment that lead to cell death.
ACKNOWLEDGMENTS
This work was supported by grants from Consejo Nacional de Ciencia y Tecnología (CONACYT; 44660-M and 128490) to F.N.-G.
We thank Lucia Chavez-Dueñas and Jazmin Huerta for their technical help and Oscar Amezquita for reproduction of some preliminary results.
Footnotes
Published ahead of print 18 March 2014
REFERENCES
- 1. Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140. 10.1038/nrmicro818 [DOI] [PubMed] [Google Scholar]
- 2. Moon HW, Whipp SC, Argenzio RA, Levine MM, Giannella RA. 1983. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 41:1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frankel G, Phillips AD, Rosenshine I, Dougan G, Kaper JB, Knutton S. 1998. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol. Microbiol. 30:911–921. 10.1046/j.1365-2958.1998.01144.x [DOI] [PubMed] [Google Scholar]
- 4. Goosney DL, DeVinney R, Pfuetzner RA, Frey EA, Strynadka NC, Finlay BB. 2000. Enteropathogenic E. coli translocated intimin receptor, Tir, interacts directly with alpha-actinin. Curr. Biol. 10:735–738. 10.1016/S0960-9822(00)00543-1 [DOI] [PubMed] [Google Scholar]
- 5. McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92:1664–1668. 10.1073/pnas.92.5.1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB. 1997. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91:511–520. 10.1016/S0092-8674(00)80437-7 [DOI] [PubMed] [Google Scholar]
- 7. Elliott SJ, Krejany EO, Mellies JL, Robins-Browne RM, Sasakawa C, Kaper JB. 2001. EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect. Immun. 69:4027–4033. 10.1128/IAI.69.6.4027-4033.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McNamara BP, Koutsouris A, O'Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. 2001. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J. Clin. Invest. 107:621–629. 10.1172/JCI11138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campellone KG, Robbins D, Leong JM. 2004. EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. Cell 7:217–228. 10.1016/j.devcel.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 10. Garmendia J, Phillips AD, Carlier MF, Chong Y, Schuller S, Marches O, Dahan S, Oswald E, Shaw RK, Knutton S, Frankel G. 2004. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell Microbiol. 6:1167–1183. 10.1111/j.1462-5822.2004.00459.x [DOI] [PubMed] [Google Scholar]
- 11. Dahan S, Wiles S, La Ragione RM, Best A, Woodward MJ, Stevens MP, Shaw RK, Chong Y, Knutton S, Phillips A, Frankel G. 2005. EspJ is a prophage-carried type III effector protein of attaching and effacing pathogens that modulates infection dynamics. Infect. Immun. 73:679–686. 10.1128/IAI.73.2.679-686.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gruenheid S, Sekirov I, Thomas NA, Deng W, O'Donnell P, Goode D, Li Y, Frey EA, Brown NF, Metalnikov P, Pawson T, Ashman K, Finlay BB. 2004. Identification and characterization of NleA, a non-LEE-encoded type III translocated virulence factor of enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 51:1233–1249. 10.1046/j.1365-2958.2003.03911.x [DOI] [PubMed] [Google Scholar]
- 13. Marches O, Ledger TN, Boury M, Ohara M, Tu X, Goffaux F, Mainil J, Rosenshine I, Sugai M, De Rycke J, Oswald E. 2003. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol. Microbiol. 50:1553–1567. 10.1046/j.1365-2958.2003.03821.x [DOI] [PubMed] [Google Scholar]
- 14. Mundy R, Petrovska L, Smollett K, Simpson N, Wilson RK, Yu J, Tu X, Rosenshine I, Clare S, Dougan G, Frankel G. 2004. Identification of a novel Citrobacter rodentium type III secreted protein, EspI, and roles of this and other secreted proteins in infection. Infect. Immun. 72:2288–2302. 10.1128/IAI.72.4.2288-2302.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kelly M, Hart E, Mundy R, Marches O, Wiles S, Badea L, Luck S, Tauschek M, Frankel G, Robins-Browne RM, Hartland EL. 2006. Essential role of the type III secretion system effector NleB in colonization of mice by Citrobacter rodentium. Infect. Immun. 74:2328–2337. 10.1128/IAI.74.4.2328-2337.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marches O, Wiles S, Dziva F, La Ragione RM, Schuller S, Best A, Phillips AD, Hartland EL, Woodward MJ, Stevens MP, Frankel G. 2005. Characterization of two non-locus of enterocyte effacement-encoded type III-translocated effectors, NleC and NleD, in attaching and effacing pathogens. Infect. Immun. 73:8411–8417. 10.1128/IAI.73.12.8411-8417.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rothbaum RJ, Giannella RA, Partin JC. 1982. Diarrhea caused by adherent enteropathogenic E. coli. J. Pediatr. 101:486. 10.1016/S0022-3476(82)80105-4 [DOI] [PubMed] [Google Scholar]
- 18. Rothbaum RJ, Partin JC, Saalfield K, McAdams AJ. 1983. An ultrastructural study of enteropathogenic Escherichia coli infection in human infants. Ultrastruct. Pathol. 4:291–304. 10.3109/01913128309140582 [DOI] [PubMed] [Google Scholar]
- 19. Abreu AG, Bueris V, Porangaba TM, Sircili MP, Navarro-Garcia F, Elias WP. 2013. Autotransporter protein-encoding genes of diarrheagenic Escherichia coli are found in both typical and atypical enteropathogenic E. coli strains. Appl. Environ. Microbiol. 79:411–414. 10.1128/AEM.02635-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mellies JL, Navarro-Garcia F, Okeke I, Frederickson J, Nataro JP, Kaper JB. 2001. espC pathogenicity island of enteropathogenic Escherichia coli encodes an enterotoxin. Infect. Immun. 69:315–324. 10.1128/IAI.69.1.315-324.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stein M, Kenny B, Stein MA, Finlay BB. 1996. Characterization of EspC, a 110-kilodalton protein secreted by enteropathogenic Escherichia coli which is homologous to members of the immunoglobulin A protease-like family of secreted proteins. J. Bacteriol. 178:6546–6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Navarro-Garcia F, Sonnested M, Teter K. 2010. Host-Toxin Interactions Involving EspC and Pet, Two Serine Protease Autotransporters of the Enterobacteriaceae. Toxins (Basel) 2:1134–1147. 10.3390/toxins2051134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elliott SJ, Sperandio V, Giron JA, Shin S, Mellies JL, Wainwright L, Hutcheson SW, McDaniel TK, Kaper JB. 2000. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 68:6115–6126. 10.1128/IAI.68.11.6115-6126.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mellies JL, Elliott SJ, Sperandio V, Donnenberg MS, Kaper JB. 1999. The Per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol. Microbiol. 33:296–306. 10.1046/j.1365-2958.1999.01473.x [DOI] [PubMed] [Google Scholar]
- 25. Kenny B, Finlay BB. 1995. Protein secretion by enteropathogenic Escherichia coli is essential for transducing signals to epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 92:7991–7995. 10.1073/pnas.92.17.7991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vidal JE, Navarro-Garcia F. 2006. Efficient translocation of EspC into epithelial cells depends on enteropathogenic Escherichia coli and host cell contact. Infect. Immun. 74:2293–2303. 10.1128/IAI.74.4.2293-2303.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vidal JE, Navarro-Garcia F. 2008. EspC translocation into epithelial cells by enteropathogenic Escherichia coli requires a concerted participation of type V and III secretion systems. Cell Microbiol. 10:1975–1986. 10.1111/j.1462-5822.2008.01181.x [DOI] [PubMed] [Google Scholar]
- 28. Navarro-Garcia F, Canizalez-Roman A, Sui BQ, Nataro JP, Azamar Y. 2004. The serine protease motif of EspC from enteropathogenic Escherichia coli produces epithelial damage by a mechanism different from that of Pet toxin from enteroaggregative E. coli. Infect. Immun. 72:3609–3621. 10.1128/IAI.72.6.3609-3621.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosenshine I, Ruschkowski S, Finlay BB. 1996. Expression of attaching/effacing activity by enteropathogenic Escherichia coli depends on growth phase, temperature, and protein synthesis upon contact with epithelial cells. Infect. Immun. 64:966–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 31. Canizalez-Roman A, Navarro-Garcia F. 2003. Fodrin CaM-binding domain cleavage by Pet from enteroaggregative Escherichia coli leads to actin cytoskeletal disruption. Mol. Microbiol. 48:947–958. 10.1046/j.1365-2958.2003.03492.x [DOI] [PubMed] [Google Scholar]
- 32. Crane JK, McNamara BP, Donnenberg MS. 2001. Role of EspF in host cell death induced by enteropathogenic Escherichia coli. Cell Microbiol. 3:197–211. 10.1046/j.1462-5822.2001.00103.x [DOI] [PubMed] [Google Scholar]
- 33. Crane JK, Majumdar S, Pickhardt DF., III 1999. Host cell death due to enteropathogenic Escherichia coli has features of apoptosis. Infect. Immun. 67:2575–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barnett Foster D, Abul-Milh M, Huesca M, Lingwood CA. 2000. Enterohemorrhagic Escherichia coli induces apoptosis which augment bacterial binding and phosphatidylethanolamine exposure on the plasma membrane outer leaflet. Infect. Immun. 68:3108–3115. 10.1128/IAI.68.6.3108-3115.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abul-Milh M, Wu Y, Lau B, Lingwood CA, Barnett Foster D. 2001. Induction of epithelial cell death including apoptosis by enteropathogenic Escherichia coli expressing bundle-forming pili. Infect. Immun. 69:7356–7364. 10.1128/IAI.69.12.7356-7364.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crane JK, Vezina CM. 2005. Externalization of host cell protein kinase C during enteropathogenic Escherichia coli infection. Cell Death Differ. 12:115–127. 10.1038/sj.cdd.4401531 [DOI] [PubMed] [Google Scholar]
- 37. Flynn AN, Buret AG. 2008. Caspases-3, -8, and -9 are required for induction of epithelial cell apoptosis by enteropathogenic E. coli but are dispensable for increased paracellular permeability. Microb. Pathog. 44:311–319. 10.1016/j.micpath.2007.10.007 [DOI] [PubMed] [Google Scholar]
- 38. Fagundes-Neto U, Freymuller E, Gandolfi Schimitz L, Scaletsky I. 1996. Nutritional impact and ultrastructural intestinal alterations in severe infections due to enteropathogenic Escherichia coli strains in infants. J. Am. Coll. Nutr. 15:180–185. 10.1080/07315724.1996.10718586 [DOI] [PubMed] [Google Scholar]
- 39. Nataro JP, Kaper JB, Robins-Browne R, Prado V, Vial P, Levine MM. 1987. Patterns of adherence of diarrheagenic Escherichia coli to HEp-2 cells. Pediatr. Infect. Dis. J. 6:829–831. 10.1097/00006454-198709000-00008 [DOI] [PubMed] [Google Scholar]
- 40. Knutton S, McConnell MM, Rowe B, McNeish AS. 1989. Adhesion and ultrastructural properties of human enterotoxigenic Escherichia coli producing colonization factor antigens III and IV. Infect. Immun. 57:3364–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shifrin Y, Kirschner J, Geiger B, Rosenshine I. 2002. Enteropathogenic Escherichia coli induces modification of the focal adhesions of infected host cells. Cell Microbiol. 4:235–243. 10.1046/j.1462-5822.2002.00188.x [DOI] [PubMed] [Google Scholar]
- 42. Wang KK. 2000. Calpain and caspase: can you tell the difference? Trends Neurosci. 23:20–26. 10.1016/S0166-2236(99)01479-4 [DOI] [PubMed] [Google Scholar]
- 43. Simovitch M, Sason H, Cohen S, Zahavi EE, Melamed-Book N, Weiss A, Aroeti B, Rosenshine I. 2010. EspM inhibits pedestal formation by enterohaemorrhagic Escherichia coli and enteropathogenic E. coli and disrupts the architecture of a polarized epithelial monolayer. Cell Microbiol. 12:489–505. 10.1111/j.1462-5822.2009.01410.x [DOI] [PubMed] [Google Scholar]
- 44. Wong AR, Raymond B, Collins JW, Crepin VF, Frankel G. 2012. The enteropathogenic E. coli effector EspH promotes actin pedestal formation and elongation via WASP-interacting protein (WIP). Cell Microbiol. 14:1051–1070. 10.1111/j.1462-5822.2012.01778.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mills E, Baruch K, Charpentier X, Kobi S, Rosenshine I. 2008. Real-time analysis of effector translocation by the type III secretion system of enteropathogenic Escherichia coli. Cell Host Microbe 3:104–113. 10.1016/j.chom.2007.11.007 [DOI] [PubMed] [Google Scholar]
- 46. Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR, Sotman S. 1978. Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i:1119–1122 [DOI] [PubMed] [Google Scholar]
- 47. Camguilhem R, Milon A. 1989. Biotypes and O serogroups of Escherichia coli involved in intestinal infections of weaned rabbits: clues to diagnosis of pathogenic strains. J. Clin. Microbiol. 27:743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jarvis KG, Giron JA, Jerse AE, McDaniel TK, Donnenberg MS, Kaper JB. 1995. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. U. S. A. 92:7996–8000. 10.1073/pnas.92.17.7996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boyer HW, Roulland-Dussoix D. 1969. A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41:459–472. 10.1016/0022-2836(69)90288-5 [DOI] [PubMed] [Google Scholar]
- 50. Stabach PR, Cianci CD, Glantz SB, Zhang Z, Morrow JS. 1997. Site-directed mutagenesis of alpha II spectrin at codon 1175 modulates its mu-calpain susceptibility. Biochemistry 36:57–65. 10.1021/bi962034i [DOI] [PubMed] [Google Scholar]