

Abstract
Meningitis and meningoencephalitis caused by Escherichia coli are associated with high rates of mortality and neurological sequelae. A high prevalence of neurological disorders has been observed in geriatric populations at risk of hypovitaminosis D. Vitamin D has potent effects on human immunity, including induction of antimicrobial peptides (AMPs) and suppression of T-cell proliferation, but its influence on microglial cells is unknown. The purpose of the present study was to determine the effects of vitamin D deficiency on the phagocytosis rate, intracellular killing, and immune response of murine microglial cultures after stimulation with the Toll-like receptor (TLR) agonists tripalmitoyl-S-glyceryl-cysteine (TLR1/2), poly(I·C) (TLR3), lipopolysaccharide (TLR4), and CpG oligodeoxynucleotide (TLR9). Upon stimulation with high concentrations of TLR agonists, the release of tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) was decreased in vitamin D-deficient compared to that in vitamin D-sufficient microglial cultures. Phagocytosis of E. coli K1 after stimulation of microglial cells with high concentrations of TLR3, -4, and -9 agonists and intracellular killing of E. coli K1 after stimulation with high concentrations of all TLR agonists were lower in vitamin D-deficient microglial cells than in the respective control cells. Our observations suggest that vitamin D deficiency may impair the resistance of the brain against bacterial infections.
INTRODUCTION
Vitamin D deficiency is associated not only with osteoporosis but also with various health events not related to the musculoskeletal system (1, 2). Hypovitaminosis D is very common in older persons, affecting more than 50% of American and European elderly persons who live at home and more than 80% of those who live in nursing homes (3, 4, 5). Brain, prostate, breast, and colon tissues, among others, as well as immune cells express vitamin D receptors (VDR) and respond to 1,25-dihydroxyvitamin D [1,25(OH)2D], the active form of vitamin D (6).
The active form of vitamin D is present in the brain. The central nervous system (CNS) contains all the enzymatic material required to locally produce its own active form of vitamin D (7, 8). Vitamin D acts as an auto- or paracrine hormone of the neurosteroid type (7, 8), binding locally to VDR expressed in neural and glial cells of the temporal, orbital, and cingulate cortices but also in the thalamus, the amygdaloid complex, and the spinal cord (9, 10). Directly or indirectly, 1,25-dihydroxyvitamin D controls more than 200 genes, including genes responsible for the regulation of cellular proliferation, differentiation, apoptosis, and angiogenesis (11). Vitamin D signaling has emerged as a key regulator of immunity in humans (12). Studies of the innate immune response to pathogens such as Mycobacterium tuberculosis have shown that pathogen recognition receptor (PRR)-mediated activation of localized vitamin D metabolism and signaling is a key event associated with resistance to infection (13). Vitamin D, acting in an intracrine fashion, is able to induce expression of antibacterial proteins. The net effect of these actions is to support increased bacterial killing in a variety of cell types (14).
Death in the acute phase of the disease and neurological as well as neuropsychological sequelae are frequent complications of bacterial CNS infections. Escherichia coli is a Gram-negative bacillus causing local infections in the urinary tract, abdomen, and lungs. Systemic spread of these infections is frequent, leading to sepsis and meningitis, and is associated with high rates of mortality and morbidity in infants and immunocompromised and elderly persons despite advances in antimicrobial chemotherapy (15). The presence of the capsule K1 confers invasiveness to the strains and enables them to penetrate the blood-brain barrier in vivo (16, 17).
Mammals have two main forms of immune defense against infectious agents that are sequentially activated: innate (phagocytosis and antigen presentation) and adaptive immunity (T and B lymphocyte function). Both innate immunity and adaptive immunity contribute to the host defense against bacteria. The brain shows a well-organized innate immune reaction in response to bacteria in blood and cerebrospinal fluid (18–20). Microglia, the resident phagocytes of the CNS, constitute the first line of defense when bacteria have entered the brain (21, 22). Microglial cells express Toll-like receptors (TLRs) that can recognize pathogen-associated molecular patterns (PAMPs) and thereby play an important role as regulators of the local innate immune response (23, 24). With their ability to produce proinflammatory cytokines and chemokines and to present antigens, microglia together with perivascular and meningeal macrophages attract circulating immune cells to the site of infection (21, 25).
Previous studies have shown that activation of microglia occurs in both cerebral and systemic infections, most likely as a mechanism to increase the resistance of the brain against invading pathogens (19, 20). Stimulation of microglial cells with TLR agonists can increase the phagocytic and intracellular killing properties of microglial cells (26). Recent studies described the key mechanisms associated with vitamin D metabolism and signaling for both innate and adaptive immune responses (14, 27). In particular, studies of the interaction between vitamin D and the immune system have highlighted the importance of the local conversion of the precursor 25-hydroxyvitamin D (25-OHD) to active 1,25-dihydroxyvitamin D as a mechanism for maintaining antibacterial activity in humans. The aims of this study were (i) to investigate how vitamin D deficiency influences the ability of microglial cells to phagocytose and intracellularly kill bacteria at rest and after stimulation by TLR agonists and (ii) to compare the cytokine and nitric oxide (NO) release of microglial cells prepared from vitamin D-deficient and vitamin D-sufficient mice after stimulation with TLR agonists.
MATERIALS AND METHODS
Primary mouse microglial cell cultures.
Primary cultures of microglial cells were prepared from brains of newborn C57BL/6 mice (postnatal day 0 to day 2 [p0–p2]), whose parents were fed a vitamin D-deficient or vitamin D-sufficient diet. To obtain low vitamin D concentrations in the circulation of the newborns, beginning 6 weeks before breeding, their parents were fed a diet with a low vitamin D concentration (vitamin D concentration under detection level and normal calcium and phosphate concentrations) (Ssniff, Experimentalfutter für Labortiere, Soest, Germany) (Table 1). After 6 weeks, the 25-hydroxyvitamin D3 concentrations in serum were measured for the parent mice using liquid chromatography and tandem mass spectroscopy (LC-MS/MS). Serum samples were measured with the MassChrom 25-OH-vitamin D3/D2 LC-MS/MS kit (Chromsystems, Munich, Germany). The measurements were performed on an AB Sciex API 4000 LC-MS/MS system (AB Sciex, Darmstadt, Germany). The high-performance liquid chromatography (HPLC) component was from Shimadzu (Duisburg, Germany). The assay was adapted to a 50-μl sample volume. The limit of quantification was estimated at approximately 3 ng/ml for the adapted assay.
TABLE 1.
Composition of the low-vitamin D3 diet (E15312-244; ssniff Spezialdiäten GmbH)
| Ingredient | Unit | Value |
|---|---|---|
| Main ingredients | ||
| Casein | % | 24.000 |
| Corn starch, pregelatinized | % | 30.000 |
| Maltodextrin | % | 19.570 |
| Dextrose | % | 10.000 |
| Cellulose powder | % | 5.000 |
| l-Cystine | % | 0.200 |
| Vitamin premix without vitamin D3 | % | 1.000 |
| Mineral premix | % | 6.000 |
| Choline Cl (50%) | % | 0.200 |
| Dye (red or blue) | % | 0.030 |
| Soybean oil | % | 4.000 |
| Proximate contents | ||
| Crude protein | % | 20.8 |
| Crude fat | % | 4.2 |
| Crude fiber | % | 5.0 |
| Crude ash | % | 5.3 |
| Starch | % | 28.9 |
| Dextrin | % | 19.4 |
| Sugar | % | 10.8 |
| Calcium | % | 0.90 |
| Phosphorus | % | 0.63 |
| Vitamin A | IU/kg | 15,000 |
| Vitamin D3 | IU/kg | <100 |
| Energy (Atwater) | MJ/kg | 15.2 |
| Protein | kcal% | 23 |
| Fat | kcal% | 10 |
| Carbohydrates | kcal% | 67 |
After removal of the meninges, brains were homogenized, and the homogenates were treated with trypsin (Sigma-Aldrich, Taufkirchen, Germany) for 10 min to isolate the cells and then treated with DNase (Sigma-Aldrich), centrifuged, and suspended in Dulbecco's modified Eagle medium (DMEM; Gibco, Karlsruhe, Germany) supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 U/ml of penicillin, and 100 μg/ml of streptomycin. Cells were plated at a density of two brains per T75 culture flask (Corning Costar, Wiesbaden, Germany) and incubated at 37°C with 5% CO2. Culture medium was changed twice a week. After 14 days of culture, microglial cells were isolated from the mixed glial (astrocytes and microglia) cultures by shaking, and the cells of the supernatants were plated in 96-well plates at a density of 50,000 cells/well. To test for the purity of the cultures, Griffonia simplicifolia isolectin B4 and Iba staining was performed. For this purpose, microglial cells were plated on poly-l-lysine-coated coverslips, fixed in 4% formaldehyde, treated with Triton X-100 (0.1% in phosphate-buffered saline [PBS]) for 30 min, and then incubated with biotinylated isolectin B4 (5 μg/ml diluted in PBS, containing 1% [wt/vol] bovine serum albumin; Sigma, Taufkirchen, Germany) and with primary anti-Iba-1 antibody (Wako, Japan) at a dilution of 1:400 for 90 min. Anti-rabbit biotinylated secondary antibody was added at a dilution of 1:200 (GE Healthcare, Amersham, Germany) for Iba-1 staining for 1 h. Afterwards, cells were treated with avidin-biotin complex (Vector, Burlingame, CA) for 30 min, and diaminobenzidine was used for visualization, resulting in a brown staining of the somata of microglial cells. The stainings showed that the cultures contained >98% microglia.
Microglial stimulation with the TLR agonists Pam3CSK4, poly(I·C), LPS, and CpG.
Microglial cells from vitamin D-sufficient C57BL/6 and vitamin D-deficient C57BL/6 newborn mice plated in 96-well plates were cultured in medium containing 10% FCS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin for 20 h. Microglial cultures were then exposed for 24 h to one of the TLR agonists. Tripalmitoyl-S-glyceryl-cysteine (Pam3CSK4; 910.5 Da; EMC Microcollections, Tübingen, Germany) was used as a specific agonist of TLR1/2 at concentrations of 1, 0.1, 0.01, 0.001, and 0.0001 μg/ml. Poly(I·C) (1.5 to 8 kb; Invivogen, San Diego, CA) was used as a TLR3 agonist at concentrations of 10, 1, 0.1, 0.01, and 0.001 μg/ml. For activation of TLR4, microglial cells were exposed to lipopolysaccharide (LPS) from E. coli serotype O26:B6 (Sigma, Taufkirchen, Germany) at concentrations of 1, 0.1, 0.01, 0.001, and 0.0001 μg/ml. CpG oligodesoxynucleotide 1668 (TCC ATG ACG TTC CTG ATG CT; 6,383 Da; TIB Molbiol, Berlin, Germany) was used as a specific ligand of TLR9 at concentrations of 10, 1, 0.1, 0.01, and 0.001 μg/ml. In our experimental setting, we used the concentrations of stimulators according to previously published results in murine microglial cultures to ensure that all compounds were tested at comparable concentrations in terms of microglia (26, 28) as well as macrophage and splenocyte stimulation (29, 30). In the experiments comparing NO release upon activation of microglia, gamma interferon (IFN-γ; 100 U/ml) was added as a costimulant to augment NO production. Control groups with unstimulated microglial cells were included in all experiments. Supernatants from stimulated microglial cultures and unstimulated controls were collected after 24 h of incubation and stored at −20°C until measurement of cytokine and chemokine levels.
Flow cytometric analysis of microglial cells.
To assess the surface markers of cultured microglial cells, microglia from vitamin D-sufficient and vitamin D-deficient C57BL/6 newborn mice were seeded into 12-well plates at 2 × 105 cells/well and incubated overnight. After 24 h, microglial cells were preincubated with medium or one TLR agonists in the highest concentration used for phagocytosis (Pam3CSK4, 1 μg/ml, and CpG, 10 μg/ml) for 24 h. Incubations were performed in DMEM supplemented with 10% FCS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin at 5% CO2 at 37°C. After incubation, the culture medium was removed, and microglial cells were washed once with prewarmed medium and twice with prewarmed PBS (37°C). For detachment from the culture dish surface, cells were exposed to a 0.05% trypsin–0.02% EDTA solution (Biochrom, Berlin, Germany). The enzymatic reaction was stopped by adding culture medium, and cells were carefully scraped off to be collected in microcentrifuge tubes. Afterwards, cells were centrifuged at 800 × g for 12 min at 4°C and resuspended in 1 ml of FACS buffer (PBS supplemented with 2% FCS, 0.01 M EDTA [pH 8.0], and 0.1% NaN3). After another centrifugation step, cells were resuspended in 40 μl of flow FACS buffer, containing rat anti-mouse CD16/CD32 antibody (Fc Block, clone 93; BioLegend, San Diego, CA; working concentration, 1:100). Following incubation on ice for 10 min, 40 μl of FACS buffer containing the appropriate fluorescence-coupled antibodies were added; antibodies were as follows: allophycocyanin (APC)-conjugated anti-mouse CD45 antibody (clone 30–F11; BioLegend; working concentration, 1:400), eFluor-conjugated anti-mouse CD11b antibody (clone M1/70; eBioscience, San Diego, CA; working concentration, 1:200), and phycoerythrin (PE)-conjugated anti-mouse F4/80 antibody (clone A3-1; BioLegend; working concentration, 1:200). Cells were incubated for 30 min on ice in the dark. Finally, cells were washed again with 1 ml of FACS buffer, centrifuged at 800 × g for 12 min at 4°C, and resuspended in 300 μl of FACS buffer to be acquired on a FACSCantoII flow cytometer (BD Biosciences, Heidelberg, Germany). Microglial cells were gated (10,000 events) and characterized according to their forward-scatter (FSC) and side-scatter (SSC) characteristics. Only cells positive for the pan-populational marker protein CD11b were analyzed for their additional expression of the pan-populational microglial marker protein CD45. The data were analyzed using FlowJo (Tree Star, Ashland, OR) software.
Measurement of microglial cell viability.
Microglial cell viability was determined using the WST-1 cell proliferation reagent (Roche Applied Science, Mannheim, Germany). The assay is based on the cleavage of a tetrazolium salt by active mitochondria, producing a soluble formazan. This conversion occurs only in viable cells. Cells were incubated with the WST-1 reagent for 3 h. Then, the formazan dye formed was quantified by measuring the optical density at 490 nm using a Genios multiplate reader (Tecan, Crailsheim, Germany). The absorbance was directly correlated with the metabolic activity of the cells. As a negative control, 1.0% Triton X-100 (Sigma, Taufkirchen, Germany) was applied, causing cell lysis.
Bacterial strain.
An E. coli strain with the capsule K1, isolated from a child with neonatal meningitis (the strain was a gift from G. Zysk, Institute of Medical Microbiology, Düsseldorf, Germany), was used. E. coli was grown in tryptic soy broth, centrifuged, and then suspended in DMEM supplemented with 10% FCS. The bacterial inoculum was determined as the number of CFU added to each well by quantitative plating on sheep blood agar plates.
Phagocytosis assay.
After stimulation, microglial cells from either vitamin D-sufficient or vitamin D-deficient C57BL/6 mice were coincubated with E. coli K1 (6 × 106 CFU/well) for 90 min at 37°C and 5% CO2. After coincubation with E. coli, microglial cells were washed twice with PBS and incubated for 1 h with DMEM containing gentamicin (100 μg/ml; Sigma-Aldrich, Taufkirchen, Germany) to destroy extracellular bacteria. After incubation with the antibiotic, microglial cells were washed twice with PBS and lysed with 100 μl of distilled water. The ingested E. coli CFU were counted by quantitative plating of 1:10 dilutions on sheep blood agar plates (31, 32).
Intracellular survival assay.
TLR-stimulated [Pam3CSK4, 1 μg/ml; poly(I·C), 10 μg/ml; LPS, 1 μg/ml; and CpG, 10 μg/ml] or unstimulated microglial cells from vitamin D-sufficient and vitamin D-deficient C57BL/6 mice were incubated with E. coli K1 for 90 min to monitor survival inside microglia. Thereafter, cells were washed twice with PBS and incubated in culture medium containing gentamicin (100 μg/ml) for various times. At various points (90, 180, and 300 min), the monolayers were washed with PBS and lysed with distilled water. The amounts of intracellular bacteria were determined by quantitative plating of serial 1:10 dilutions on sheep blood agar plates (n ≥ 15, performed on at least 2 days).
Nitrite assay.
NO release was quantified by measurement of nitrite, one of its stable reaction products, in the supernatants of microglial cultures using the Griess reaction. Aliquots (100 μl) of the supernatant were mixed with 100 μl of Griess reagent [equal volumes of 1% sulfonilamide in 30% acetate and 0.1% N-(1-naphthyl)ethylenediamine in 60% acetate] in a 96-well plate. After 10 min, the optical density at 570 nm was measured with a Genios multiplate reader (Tecan, Crailsheim, Germany). Concentrations were calculated by comparison of the absorptions with a standard curve (28).
Cytokine and chemokine release.
Tumor necrosis factor alpha (TNF-α), interleukin (IL-6), and chemokine (C-X-C motif) ligand 1 (CXCL1) (also termed KC, the mouse equivalent of growth-regulated oncogene α [GRO-α]) levels were determined using DuoSet enzyme-linked immunosorbent assay (ELISA) development kits (R&D Systems, Wiesbaden, Germany). Procedures were performed according to the manufacturer's instructions. The color reaction was quantified at 450 nm in a microplate reader (Bio-Rad, Munich, Germany). The limit of detection was 7.5 pg/ml for TNF-α, IL-6, and CXCL1.
Statistics.
Descriptive values of serum vitamin D3 concentrations are shown as means and individual measurements and were analyzed using unpaired t test. The cytokine, chemokine, and nitrite release data are reported as medians and interquartile ranges (Q25 and Q75). Phagocytosis data are also shown as medians and corresponding interquartile ranges (Q25 and Q75). SAS version 9.3 (SAS Institute, NC) was used to perform statistical analysis. Statistical testing for the effect of concentration, the group membership, and their interaction were done according to a factorial analysis of variance (ANOVA) for the logarithms of the outcomes (phagocytosis of E. coli K1 and cytokine and chemokine release), adjusting for the random effect of the mice. The significance level was set to 5%, and adjustments for multiple comparisons were performed with the Bonferroni method. Descriptive values of cell viability test, intracellular survival assays, and NO release are shown as means and SE and were analyzed by one-way ANOVA and corrected for repeated testing with the Bonferroni multiple-comparison test. For visualization, GraphPad Prism software (GraphPad Software, San Diego, CA) was used.
RESULTS
Feeding with diet containing low vitamin D concentration resulted in decreased serum 25-OH vitamin D3 concentrations.
Six weeks of feeding a diet containing low vitamin D concentrations in the parent mice resulted in serum 25-OH vitamin D3 concentrations significantly lower than those in mice fed with a diet containing normal amounts of vitamin D (7.3 ± 1.6 versus 31.7 ± 1.2 ng/ml; P < 0.001) (Fig. 1).
FIG 1.
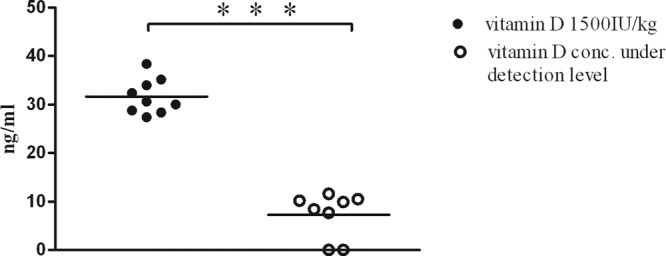
25-OH D3 concentrations in mouse serum. Six weeks after mice were fed a diet containing a low vitamin D concentration (vitamin D concentration under detection level), the serum 25-OH D3 concentrations were significantly lower than in mice fed a diet containing normal amounts of vitamin D (1,500 IU/kg) (P < 0.0001). Each symbol represents the serum concentration of an individual mouse. The values for both mice with the lowest 25-OH D3 values in the vitamin D-deficient group were below the limit of quantification. For statistical analysis, unpaired t test was used. ***, P < 0.001.
TLR agonist stimulation did not influence the expression of CD11b and CD45 on microglial cells.
Microglial cells from vitamin D-sufficient and vitamin D-deficient C57BL/6 mice were gated for the pan-populational surface marker protein CD11b (Fig. 2A). All CD11b-positive microglia additionally were positive for the pan-populational marker CD45 (Fig. 2B and C). No significant changes in the percentage of CD45-positive microglial cells were found between medium- and TLR agonist-treated microglial cells. After stimulation of microglial cells with different TLR agonists, the induction marker F4/80 was expressed on the surface of CD11b+ CD45+ microglia in vitamin D-sufficient and -deficient microglia (Fig. 2C). Levels of expression of the induction marker F4/80 on the surface of CD11b+ CD45+ microglia 24 h after stimulation were not significantly different between vitamin D-sufficient and vitamin D-deficient microglial cells (Fig. 2D).
FIG 2.

Flow cytometric analysis of microglial cell surface marker proteins. Vitamin D-sufficient and vitamin D-deficient microglial cells were left undisturbed under medium condition or stimulation with Pam3CSK4 (10 μg/ml) or CpG (10 μg/ml) for 24 h. (A) Expression of the pan-populational surface marker protein CD11b. CD11b-positive vitamin D-sufficient and vitamin D-deficient microglial cells showed positivity for the pan-populational surface marker protein CD45, as exemplified for medium (B) and Pam3CSK4 stimulation (C). Levels of expression of the induction marker F4/80 on the surface of CD11b+ CD45+ microglia 24 h after stimulation were not significantly different between vitamin D-sufficient and vitamin D-deficient microglial cells (D). Only stimulated microglial cells expressed F4/80 (C and D). Data are provided as means ± SE (n = 4 per condition from two independent experiments) and were analyzed using one-way ANOVA followed by Bonferroni's correction for multiple comparisons.
Cell viability.
The TLR agonist concentrations used in our study were not toxic in accordance with our previously published data (28). Stimulation of microglial cells with the highest dose of TLR1/2, -3, -4, and -9 agonists did not result in a loss of cell viability (Fig. 3) in all treatment groups compared with the unstimulated group.
FIG 3.
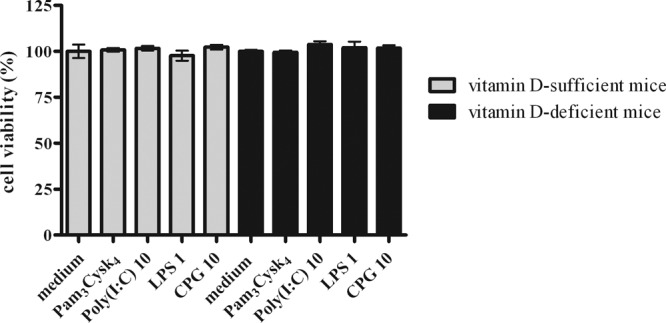
Cell viability of vitamin D-sufficient and vitamin D-deficient microglial cells after treatment with Pam3CSK4 at 1 μg/ml, poly(I·C) at 10 μg/ml, LPS at 1 μg/ml, and CpG at 10 μg/ml (n = 4), shown as means ± SE. Treatment with all TLR agonists did not result in a loss of microglial viability in both groups compared to the viability of untreated cells. Values are as follows: medium, vitamin D sufficient, 100% ± 7.5%, and vitamin D deficient, 100% ± 1.5%; Pam3CSK4 at 1 μg/ml, vitamin D sufficient, 100.8% ± 1.9%, and vitamin D deficient, 99.5% ± 1.9%; poly(I·C) at 10 μg/ml, vitamin D sufficient, 101.7% ± 2.4%, and vitamin D deficient, 103.8% ± 3.2%; LPS at 1 μg/ml, vitamin D sufficient, 97.7% ± 5.4%, and vitamin D deficient, 102.1% ± 6.4%; and CpG at 10 μg/ml, vitamin D sufficient, 102.% ± 2.6%, and vitamin D deficient, 101.7% ± 3.1%. Data were analyzed using one-way ANOVA followed by Bonferroni's correction for multiple comparisons. No significant differences were noted.
After stimulation with high concentrations of TLR3, -4, and -9 agonists, microglial cells prepared from vitamin D-deficient mice ingested significantly fewer E. coli bacteria than did microglial cells from vitamin D-sufficient mice.
The rates of phagocytosis of E. coli by microglia prepared from vitamin D-deficient mice and microglial cells from vitamin D-sufficient mice were compared quantitatively after coincubation of microglia and bacteria for 90 min in unstimulated cultures and in microglial cultures that had been previously stimulated with TLR1/2, TLR3, TLR4, or TLR9 agonists (n = 21 wells per concentration for TLR1/2, TLR4, and TLR9 and n = 18 for TLR3). In phagocytosis assays involving E. coli K1, unstimulated cells ingested bacteria at a low rate (cultures of microglia from vitamin D-sufficient mice, median, 50 [20, 80] CFU/ml; cultures of microglia from vitamin D-deficient mice, median, 40 [20, 100] CFU/well). The numbers of ingested E. coli bacteria 90 min after stimulation with different TLR agonists showed an increase of ingested bacteria in a dose-dependent manner in microglia from both vitamin D-sufficient and vitamin D-deficient mice. The dose-dependent stimulation of microglia by TLR agonists is in accordance with data previously published by us (28) and others reporting stimulation with TLR agonists in macrophages and splenocytes (29, 30). Except for the TLR1/2 agonist (Pam3CSK4), the group-concentration interaction between the two groups (vitamin D-sufficient microglia and vitamin D-deficient microglia) and the phagocytosis rate were positive (P ≤ 0.05) for all TLR agonists (Table 2). Prestimulation with the highest concentrations of TLR3, TLR4, or TLR9 agonists significantly increased the phagocytosis rate of E. coli in vitamin D-sufficient microglia compared to that in the vitamin D-deficient microglia (P value [adjusted], <0.05 [Fig. 4]). After stimulation with TLR1/2, vitamin D-sufficient microglia cells showed a slightly higher phagocytosis rate than vitamin D-deficient microglia. The difference did not reach statistical significance. At the other concentrations of TLR3, -4, and -9 studied, there were no significant differences between the vitamin D-sufficient and vitamin D-deficient microglial cells.
TABLE 2.
The group-concentration interactions between vitamin D-sufficient and vitamin D-deficient microglia and cytokines/chemokines and phagocytosis rate for all TLR agonists studieda
| Stimulus | Cytokine, chemokine, or infection process | Concn significance | Group-concn interaction significance |
|---|---|---|---|
| LPS | IL-6 | + | + |
| TNF-α | + | + | |
| CXCL1 | + | + | |
| Phagocytosis | + | + | |
| Pam3CSK4 | IL-6 | + | + |
| TNF-α | + | + | |
| CXCL1 | + | + | |
| Phagocytosis | + | − | |
| Poly(I·C) | IL-6 | + | + |
| TNF-α | + | + | |
| CXCL1 | + | − | |
| Phagocytosis | + | + | |
| CpG | IL-6 | + | + |
| TNF-α | + | − | |
| CXCL1 | + | − | |
| Phagocytosis | + | + |
The release of the cytokines and chemokines and the phagocytosis rate after incubation of microglial cells with different TLR agonists were in both groups concentration dependent for all TLR agonists (P ≤ 0.05), indicating that the tested compounds stimulated microglial cells in a dose-dependent manner. Except for Pam3CSK4, the group-concentration interaction between stimulation of microglia and phagocytosis rate was found for all TLR agonists, indicating that vitamin D deficiency significantly impaired phagocytosis (P ≤ 0.05; two-way ANOVA followed by Bonferroni's correction for multiple comparisons). The cytokines and chemokines also showed a significant group-concentration interaction, documenting the influence of low vitamin D on cytokine and chemokine release. For CXCL1 after stimulation with poly(I·C) and CpG and for TNF-α after stimulation with CpG, however, no significant group-concentration interaction was found. Date were analyzed using two-way ANOVA followed by Bonferroni's multiple-comparison test.
FIG 4.

Phagocytosis of E. coli K1 by microglial cells after 24 h of prestimulation with various TLR agonists [Pam3CSK4, poly(I·C), LPS, and CpG] at different concentrations. The number of ingested bacteria was determined by quantitative plating of the cell lysates on blood agar plates. Data are shown as recovered bacterial CFU per ml (median and 25% and 75% interquartile ranges). Prestimulation with TLR agonists increased the number of bacteria ingested by microglia in comparison to unstimulated cells in both groups. After stimulation with high concentrations of TLR3, -4, and -9 agonists, vitamin D-deficient microglial cells ingested significantly fewer E. coli than did vitamin D-sufficient microglial cells. Data were analyzed using two-way ANOVA followed by Bonferroni's correction for multiple comparisons. Filled black circles, vitamin D-sufficient microglial cells; open grey circles, vitamin D-deficient microglial cells. *, P < 0.05.
Intracellular killing of E. coli K1 in vitamin D-deficient microglial cells was decreased compared to that in vitamin D-sufficient microglial cells.
After demonstrating the enhanced ability of vitamin D-sufficient microglial cells to phagocytose bacteria upon stimulation with TLR agonists, we studied whether stimulated microglial cells from vitamin D-deficient or from vitamin D-sufficient C57BL/6 mice killed more ingested bacteria. We performed intracellular survival assays with the highest concentrations of TLR agonists used in the phagocytosis studies. The numbers of surviving intracellular bacteria after 90 min of phagocytosis and 60 min of gentamicin treatment and the numbers of killed bacteria at 300 min after the end of phagocytosis are shown in Table 3. For each group, the absolute amount of killed bacteria was calculated as the difference between each individual value at 300 min and the mean of ingested bacteria of the respective group at 60 min of gentamicin exposure. In both groups, the amounts of intracellularly surviving bacteria decreased throughout the experiment. Stimulation of vitamin D-sufficient microglia with TLR agonists led to an increased number of killed bacteria in comparison to those for unstimulated cells and stimulated vitamin D-deficient microglial cells (P < 0.001).
TABLE 3.
Intracellular killing of E. coli K1 by vitamin D-sufficient and vitamin D-deficient microglial cellsa
| Stimulus (μg/ml) | Phagocytosed E. coli K1 at 90 min (CFU/ml) |
Intracellularly killed E. coli K1 after 300 min (CFU/ml) |
||
|---|---|---|---|---|
| D+ | D− | D+ | D− | |
| Medium | 429.3 (105) | 410.7 (103) | 375.1 (20.41) | 333.3 (21.3)*** |
| Pam3CSK4 (1) | 16,253 (3,922) | 9,133 (2,113) | 15,033 (496.7) | 7,207 (753.8)*** |
| Poly (I·C) (10) | 4,160 (606.4) | 1,519 (280.8) | 3,603 (130.5) | 1,213 (80.2)*** |
| LPS (1) | 21,133 (4,111) | 7,800 (1,730) | 18,120 (754.9) | 6,271 (520.3)*** |
| CPG (10) | 11,393 (2,259) | 5,333 (687.8) | 9,340 (293.2) | 4,559 (245.6)*** |
Intracellular killing of E. coli K1 by microglial cells was decreased in vitamin D-deficient (D−) microglial cells after stimulation with high concentrations of TLR1/2, -3, -4, and -9 agonists. Unstimulated and stimulated vitamin D-sufficient (D+) and vitamin D-deficient microglial cells were incubated with bacteria for 90 min. Then, gentamicin was added for 60 and 300 min to ensure killing of extracellularly located bacteria. For each group, phagocytosis was expressed as the number of recovered bacteria after 60 min of exposure to gentamicin. The absolute number of killed bacteria at 300 min was calculated as the difference between the number of intracellular bacteria at 60 and 300 min. Data are shown as means (SE in parentheses) from at least two independent experiments (n ≥ 15 wells/group per time point) and were analyzed by one-way ANOVA and corrected for repeated testing using the Bonferroni procedure for multiple comparisons. ***, P < 0.001.
NO release upon stimulation with agonists of TLR1/2, -3, -4, and -9 did not differ in vitamin D-deficient mice and vitamin D-sufficient mice microglia.
A mechanism by which vitamin D may be able to exert antimicrobial activity is bacterial killing using the NO pathway (33). As expected from previously published data (28), high concentrations of the TLR agonists Pam3CSK4 (TLR1/2), LPS (TLR4), and CpG (TLR9) induced NO release in microglial cells. To study the possible mechanism by which vitamin D deficiency can influence the intracellular killing of E. coli, we measured the NO release at the highest TLR agonist concentrations studied, at which we also found the maximum phagocytosis rate. We could not find a significant difference in NO release between vitamin D-sufficient and vitamin D-deficient microglial cells (Fig. 5). There was only a tendency of vitamin D-deficient microglia to release less NO than vitamin D-sufficient microglial cells.
FIG 5.
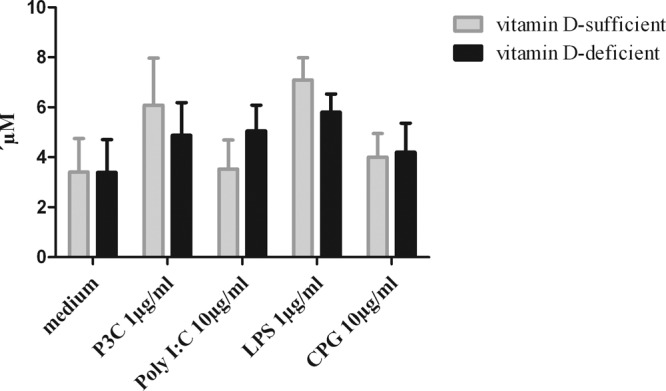
NO release in primary mouse microglial cells upon stimulation with high concentrations of TLR1/2, -3, -4, and -9 agonists. NO release after stimulation with high concentrations of the agonists did not differ between vitamin D-deficient and vitamin D-sufficient microglia. Nitric oxide concentrations in the supernatants are given as means plus SE. Data were analyzed using one-way ANOVA followed by Bonferroni's correction for multiple comparisons. No significant differences were noted.
Stimulation of vitamin D-deficient microglia by TLR agonists led to decreased TNF-α and IL-6 release compared to that of vitamin D-sufficient microglia.
To confirm microglial activation by the TLR agonists, we measured the concentrations of TNF-α, IL-6, and CXCL1 (KC) in the supernatants of microglial cultures (n = 21 per concentration of TLR1/2, -4, and -9 agonists and n = 18 per concentration of the TLR3 agonist) after stimulation with different concentrations of Pam3CSK4, poly(I·C), LPS, and CpG (Fig. 6). In all experiments, a group of unstimulated cells was included for comparison. The release of the cytokines and chemokines after incubation of microglial cells with different TLR agonists in both vitamin D-deficient and vitamin D-sufficient microglial cells was concentration dependent for all TLR agonists (P value [adjusted], ≤0.05 [Table 2]), indicating that the tested concentrations were effective in activating microglial cells.
FIG 6.

Cytokine and chemokine release of unstimulated and stimulated vitamin D-sufficient and vitamin D-deficient microglial cells. IL-6 (circles), TNF-α (triangles), and CXCL1 (squares) concentrations in the supernatants of microglial cell cultures after 24 h of stimulation with Pam3CSK4 (P3C), poly(I·C), LPS, and CpG are shown. Stimulation of vitamin D-deficient microglia by TLR agonists induced decreased TNF-α and IL-6 release compared to that of vitamin D-sufficient microglia but no differences in CXCL1 release. Data are given as median and interquartile ranges (25% and 75%). Data were analyzed using two-way ANOVA followed by Bonferroni's correction for multiple comparisons. Filled black symbols, vitamin D-sufficient microglial cells; open grey symbols, vitamin D-deficient microglial cells. *, P < 0.05.
The group-concentration interactions between vitamin D-sufficient and vitamin D-deficient microglial cells and cytokines and chemokines for all TLR agonists are summarized in Table 2. Stimulation of vitamin D-sufficient microglia with different TLR agonists generally resulted in a stronger release of cytokines and chemokines than stimulation of vitamin D-deficient microglia. This was true for IL-6 and TNF-α and partly for CXCL1, although the concentrations of this chemokine were low at all TLR agonist concentrations studied (Fig. 6).
Incubation of microglia with Pam3CSK4 (TLR1/2) at concentrations of 1, 0.1, and 0.01 μg/ml resulted in significantly decreased levels of IL-6 in vitamin D-deficient microglia compared to that in vitamin D-sufficient microglia (P < 0.05 [Fig. 6A]). Pam3CSK4 (TLR1/2) at 0.001 and 0.0001 μg/ml also induced the release of smaller amounts of IL-6 in vitamin D-deficient microglia than in vitamin D-sufficient microglia, but these differences failed to reach statistical significance. The differences in TNF-α release between vitamin D-deficient and vitamin D-sufficient microglia after treatment of microglia with Pam3CSK4 at the individual concentrations were not statistically significant as a consequence of the correction for repeated testing (Fig. 6A).
Treatment of microglia with 10, 1, 0.1, 0.001, and 0.0001 μg/ml of poly(I·C) (TLR3) resulted in significant differences in IL-6 and TNF-α release between vitamin D-deficient and -sufficient microglia (P < 0.05 [Fig. 6B]). The stimulation of vitamin D-sufficient microglia with LPS (TLR4) at concentrations of 1, 0.1, 0.01, and 0.001 μg/ml induced a significantly higher release of IL-6 than for vitamin D-deficient microglia (Fig. 6C). The TNF-α release after stimulation of microglia with LPS at 1 μg/ml was significantly decreased in vitamin D-deficient microglia. The results after stimulation with LPS concentrations of 0.1, 0.01, 0.001, and 0.0001 μg/ml are presented in Fig. 6C.
The stimulation of microglia with CpG (TLR9) at concentrations of 10, 1, and 0.1 μg/ml induced a significantly higher release of IL-6 (Fig. 6D) and, for concentrations of 1 and 0.1 μg/ml, also significantly larger amounts of TNF-α release (P < 0.05 [Fig. 6D]) in vitamin D-sufficient microglia.
DISCUSSION
Clinical and experimental observations support the hypothesis that insufficient supply with vitamin D may lead to the dysregulation of the human immune response and may therefore be an underlying cause of infectious diseases and immune disorders (34, 35). The concentration of the biologically active compound vitamin D, 1,25(OH)2D, is dependent on the serum vitamin D status (14). To assess the role of vitamin D deficiency in an in vitro experimental model of bacterial CNS infection, we analyzed the influence of vitamin D deficiency on the immune response of microglial cells after stimulation with different TLR agonists.
“Nonclassical” effects of vitamin D have been recognized for many years, but only in recent years have these effects been accepted as important components of vitamin D action. These nonclassical responses to 1,25(OH)2D include antiproliferative or anticancer effects (36) promoting innate antimicrobial responses (34) as well as effects on hypertension (37) and immunomodulation (27, 38). A central feature of many of these nonclassical actions of vitamin D is that the synthesis of active 1,25(OH)2D appears to occur in a cell-specific manner: enzyme 25-OHD-1α-hydroxylase (encoded by the gene CYP27B1) is expressed by many extrarenal tissues, including those of the immune system (27, 39). The influence of vitamin D on the course of CNS infections is not known.
In 1986, Rook and colleagues found in cultured human monocytes that active vitamin D, 1,25(OH)2D, can inhibit the growth of Mycobacterium tuberculosis (13). At that time, the physiological significance of this finding was unclear. Subsequent studies showed that monocytes not only are able to phagocytose mycobacteria but also can initiate a cellular response to M. tuberculosis because they express pathogen recognition receptors (PRRs) that bind pathogen-associated molecular patterns (PAMPs) expressed by M. tuberculosis (40). To clarify the innate immune responses to M. tuberculosis, Liu and colleagues (41) used DNA arrays that characterized changes in gene expression following activation of macrophage TLR1/2 receptors by one of the putative PRRs of M. tuberculosis. In that study, local synthesis of 1,25(OH)2D by monocytes was an integral part of the normal innate immune function of these cells: this activated vitamin D bound to the monocyte VDR and then was able to act as a transcription factor leading to the induction of cathelicidin, a potent antimicrobial protein (42), and the promotion of phagocytosis and intracellular killing (41, 43, 44).
Induction of antibacterial activity by vitamin D is not restricted to monocytes and macrophages but has also been reported for a variety of cell types, including bronchial epithelial cells (45), myeloid cell lines (46), and decidual (47) and trophoblastic (48) cells of the placenta. Our experiments were performed with mouse microglial cells. Although the innate antibacterial activity of vitamin D has been thought to be restricted to primates that express the promoter vitamin D response element (VDRE) required for vitamin D-mediated transcriptional regulation of antibacterial proteins (44), some mouse antimicrobial molecules such as angiogenin-4 (Ang4) also appear to be influenced by vitamin D (49). Although the function of Ang4 appears to be restricted to specific areas of the gastrointestinal tract, this observation underlines the versatility of vitamin D as a promoter of antibacterial activity in different tissues and animal species (34). In our model using vitamin D-deficient mouse microglial cells, we also were able to demonstrate the antibacterial activity of vitamin D (decreased phagocytosis rate and intracellular killing of E. coli in vitamin D-deficient microglial cells).
Activated microglia can influence innate immunity (21). In our previous work we have shown that stimulation of TLR1/2, -3, -4, and -9 with pathogen-derived compounds, such as bacterial products, or with endogenous ligands, such as fibronectin for TLR4, can increase bacterial phagocytosis and intracellular killing of bacteria by murine microglial cells (31, 32). Here, we demonstrate that vitamin D deficiency can decrease the ability of microglia to phagocytose and intacellularly kill a pathogenic E. coli strain after stimulation with high doses of TLR1/2, -3, -4, and -9 agonists. This suggests that vitamin D deficiency may affect the cellular innate immune response of the CNS by decreasing phagocytosis and intracellular killing of bacteria invading the brain. For the TLR1/2 agonist and at lower TLR3, -4, and -9 agonist concentrations used in our study, the effect of vitamin D deficiency was mild, and the differences in phagocytosis rate between the vitamin D-sufficient microglial cells and vitamin D-deficient microglial cells were found to be not statistically significant when individual concentrations were compared as a consequence of correction for repeated testing. Once bacteria are engulfed, they are incorporated into phagolysosomes and exposed to reactive oxygen species and other bacteriotoxic compounds, which eventually results in bacterial lysis. The stimulation of microglia with TLR agonists is dose dependent (reference 28 and this study), and the efficacy of the phagocytic activity of reactive microglia depends not only on their ability to ingest bacteria but also on the pathogen's ability to modulate phagocyte signaling (49, 50). This may be an additional reason why we found small differences in the phagocytosis rate between vitamin D-deficient and -sufficient microglia which failed to reach statistical significance at many individual concentrations tested.
Another mechanism by which vitamin D may be able to exert antimicrobial activity is via nonspecific bacterial killing. This is best illustrated by studies of the relationship between vitamin D and reactive oxygen species (ROS) which can act as bacteriocides. Initial analysis of M. tuberculosis killing in response to 1,25(OH)2D suggested that this probably did not involve ROS (13). However, macrophages infected with M. tuberculosis in the presence of 1,25(OH)2D produced high levels of superoxide anions (51). More recently, attention has focused on nitric oxide (NO), which is produced by macrophages as part of their innate immune response to infection. NO can function as an ROS and thereby exert bactericidal effects (33). The NO pathway appears to play a pivotal role in the murine response to M. tuberculosis infection. In our study, we did not find any differences between vitamin D-deficient and vitamin D-sufficient microglial cells with respect to NO release after stimulation of these cells with high concentrations of TLR agonists. In accordance with our observation, the relationship between vitamin D and NO appears to be uncertain (34). In neuronal cells, 1,25(OH)2D suppressed the NO-generating enzyme inducible nitric oxide synthase (iNOS) (52). Macrophages infected with the intracellular protozoan Leismania major showed decreased NO production and bacterial killing when treated with 1,25(OH)2D (53). In this instance, it was suggested that vitamin D may promote rather than suppress L. major infection. Conversely, rats on a low-vitamin D diet showed a global elevation of 3-nitrotyrosine in comparison to control and high-vitamin D-treated animals, suggesting an increase nitrosative stress in the vitamin D-deficient brain (54).
Vitamin D not only stimulates innate immunity but also modulates adaptive immunity to minimize inflammation and autoimmune diseases (38). The active vitamin D, 1,25(OH)2D, which is produced in monocytes or macrophages, is released to act locally on activated T lymphocytes, which regulate the synthesis of cytokines, such as interleukin 2 (IL-2) (55), IL-4, IL-10, and gamma interferon (IFN-γ) (56), and activated B lymphocytes, which synthesize immunoglobulins (57, 58).
One of the main functions of activated microglia is to produce a whole array of soluble factors, including cytokines and chemokines with potent immuno- and neuroregulatory effects. Several signals of intrinsic origin or infectious nature have been identified to trigger this process (59, 60). In our study, the stimulation of vitamin D-deficient microglia for 24 h by TLR agonists induced decreased TNF-α and IL-6 release compared to that of vitamin D-sufficient microglia, but no strong difference in CXCL1 release was noted. Microglial responses after stimulation with compounds from Gram-positive and Gram-negative bacteria are not uniform. Microglial cytokines and chemokines can be regulated by different factors, and their induction is independently organized in macrophage-like cells, resulting in a divergent release of TNF-α, IL-6, and CXCL1 after stimulation with different bacterial compounds, under different environmental conditions such as decreased or normal vitamin D supply (25). Although not all differences at individual concentrations were statistically significant, the vitamin D-deficient microglia, with the partial exception of CXCL1, generally released smaller amounts of cytokines and chemokines upon TLR agonist stimulation than their vitamin D-sufficient counterparts.
In conclusion, stimulation by various TLR agonists can increase the phagocytosis and intracellular killing of E. coli by microglial cells. This process can be negatively influenced under vitamin D deficiency conditions, resulting in decreased rates of phagocytosis and intracellular killing of the bacteria. The vitamin D deficiency affects the shaping of the microglial chemokine release during inflammation and probably impairs the effectiveness of the local immune system of the CNS against invading pathogens.
ACKNOWLEDGMENTS
This work was supported by Robert-Bosch-Stiftung grant 32.5.1141.0042.0 (M. Djukic), Sparkasse Göttingen, and Evangelisches Krankenhaus Göttingen-Weende.
Helpful advice from Richard Lukacin and Oliver Midasch, Chromsystems, is gratefully acknowledged.
Footnotes
Published ahead of print 31 March 2014
REFERENCES
- 1.Kalueff AV, Tuohimaa P. 2007. Neurosteroid hormone vitamin D and its utility in clinical nutrition. Curr. Opin. Clin. Nutr. Metab. Care 10:12–19. 10.1097/MCO.0b013e328010ca18 [DOI] [PubMed] [Google Scholar]
- 2.Zittermann A. 2003. Vitamin D in preventive medicine: are we ignoring the evidence? Br. J. Nutr. 89:552–572. 10.1079/BJN2003837 [DOI] [PubMed] [Google Scholar]
- 3.Gloth FM, III, Gundberg CM, Hollis BW, Haddad JG, Jr, Tobin JD. 1995. Vitamin D deficiency in homebound elderly persons. JAMA 21:1683–1686 [DOI] [PubMed] [Google Scholar]
- 4.Holick MF. 2006. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin. Proc. 81:353–373. 10.4065/81.3.353 [DOI] [PubMed] [Google Scholar]
- 5.Flicker L, MacInnis RJ, Stein MS, Scherer SC, Mead KE, Nowson CA, Thomas J, Lowndes C, Hopper JL, Wark JD. 2005. Should older people in residential care receive vitamin D to prevent falls? Results of a randomized trial. J. Am. Geriatr. Soc. 53:1881–1888. 10.1111/j.1532-5415.2005.00468.x [DOI] [PubMed] [Google Scholar]
- 6.Dusso AS, Brown AJ, Slatopolsky E. 2005. Vitamin D. Am. J. Physiol. Renal Physiol. 289:F8–F28. 10.1152/ajprenal.00336.2004 [DOI] [PubMed] [Google Scholar]
- 7.Garcion E, Wion-Barbot N, Montero-Menei CN, Berger F, Wion D. 2002. New clues about vitamin D functions in the nervous system. Trends Endocrinol. Metab. 13:100–105. 10.1016/S1043-2760(01)00547-1 [DOI] [PubMed] [Google Scholar]
- 8.McGrath J, Feron F, Eyles D, Mackay-Sim A. 2001. Vitamin D: the neglected neurosteroid? Trends Neurosci. 24:570–572. 10.1016/S0166-2236(00)01949-4 [DOI] [PubMed] [Google Scholar]
- 9.Prüfer K, Veenstra TD, Jirikowski GF, Kumar R. 1999. Distribution of 1,25-dihydroxyvitamin D 3 receptor immunoreactivity in the rat brain and spinal cord. J. Chem. Neuroanat. 16:135–145. 10.1016/S0891-0618(99)00002-2 [DOI] [PubMed] [Google Scholar]
- 10.Langub MC, Herman JP, Malluche HH, Koszewski NJ. 2001. Evidence of functional vitamin D receptors in rat hippocampus. Neuroscience 104:49–56. 10.1016/S0306-4522(01)00049-5 [DOI] [PubMed] [Google Scholar]
- 11.Nagpal S, Na S, Rathnachalam R. 2005. Noncalcemic actions of vitamin D receptor ligands. Endocr. Rev. 26:662–687. 10.1210/er.2004-0002 [DOI] [PubMed] [Google Scholar]
- 12.Olliver M, Spelmink L, Hiew J, Meyer-Hoffert U, Henriques-Normark B, Bergman P. 6 August 2013. Immunomodulatory effects of vitamin D on innate and adaptive immune responses to Streptococcus pneumoniae. J. Infect. Dis. 10.1093/infdis/jit355 [DOI] [PubMed] [Google Scholar]
- 13.Rook GA, Steele J, Fraher L, Barker S, Karmali R, O'Riordan J, Stanford J. 1986. Vitamin D3, gamma interferon, and control of proliferation of Mycobacterium tuberculosis by human monocytes. Immunology 57:159–163 [PMC free article] [PubMed] [Google Scholar]
- 14.Hewison M. 2012. Vitamin D and immune function: an overview. Proc. Nutr. Soc. 71:50–61. 10.1017/S0029665111001650 [DOI] [PubMed] [Google Scholar]
- 15.Russo TA, Johnson JR. 2000. Proposal for a new inclusive designation for extraintestinal pathogenic isolates of Escherichia coli: ExPEC. J. Infect. Dis. 181:1753–1754. 10.1086/315418 [DOI] [PubMed] [Google Scholar]
- 16.Silver RP, Aaronson W, Vann WF. 1988. The K1 capsular polysaccharide of Escherichia coli. Rev. Infect. Dis. 10(Suppl 2):S282–S286. 10.1093/cid/10.Supplement_2.S282 [DOI] [PubMed] [Google Scholar]
- 17.Xie Y, Kim KJ, Kim KS. 2004. Current concepts on Escherichia coli K1 translocation of the blood-brain barrier. FEMS Immunol. Med. Microbiol. 42:271–279. 10.1016/j.femsim.2004.09.001 [DOI] [PubMed] [Google Scholar]
- 18.Aravalli RN, Peterson PK, Lokensgard JR. 2007. Toll-like receptors in defense and damage of the central nervous system. J. Neuroimmune Pharmacol. 2:297–312. 10.1007/s11481-007-9071-5 [DOI] [PubMed] [Google Scholar]
- 19.Laflamme N, Rivest S. 1999. Effects of systemic immunogenic insults and circulating proinflammatory cytokines on the transcription of the inhibitory factor κB alpha within specific cellular populations of the rat brain. J. Neurochem. 73:309–321. 10.1046/j.1471-4159.1999.0730309.x [DOI] [PubMed] [Google Scholar]
- 20.Xu J, Ling EA. 1994. Upregulation and induction of surface antigens with special reference to MHC class II expression in microglia in postnatal rat brain following intravenous or intraperitoneal injections of lipopolysaccharide. J. Anat. 184:285–296 [PMC free article] [PubMed] [Google Scholar]
- 21.Hanisch UK, Kettenmann H. 2007. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10:1387–1394. 10.1038/nn1997 [DOI] [PubMed] [Google Scholar]
- 22.Nau R, Bruck W. 2002. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 25:38–45. 10.1016/S0166-2236(00)02024-5 [DOI] [PubMed] [Google Scholar]
- 23.Simard AR, Rivest S. 2005. Do pathogen exposure and innate immunity cause brain diseases? Neurol. Res. 27:717–725. 10.1179/016164105X49526 [DOI] [PubMed] [Google Scholar]
- 24.Takeda K, Kaisho T, Akira S. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. 10.1146/annurev.immunol.21.120601.141126 [DOI] [PubMed] [Google Scholar]
- 25.Häusler KG, Prinz M, Nolte C, Weber JR, Schumann RR, Kettenmann H, Hanisch UK. 2002. Interferon-gamma differentially modulates the release of cytokines and chemokines in lipopolysaccharide and pneumococcal cell wall-stimulated mouse microglia and macrophages. Eur. J. Neurosci. 16:2113–2122. 10.1046/j.1460-9568.2002.02287.x [DOI] [PubMed] [Google Scholar]
- 26.Ribes S, Ebert S, Regen T, Agarwal A, Tauber SC, Czesnik D, Spreer A, Bunkowski S, Eiffert H, Hanisch UK, Hammerschmidt S, Nau R. 2010. Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infect. Immun. 78:865–871. 10.1128/IAI.01110-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hewison M. 2011. Vitamin D and innate and adaptive immunity. Vitam. Horm. 86:23–62. 10.1016/B978-0-12-386960-9.00002-2 [DOI] [PubMed] [Google Scholar]
- 28.Ebert S, Gerber J, Bader S, Mühlhauser F, Brechtel K, Mitchell TJ, Nau R. 2005. Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J. Neuroimmunol. 159:87–96. 10.1016/j.jneuroim.2004.10.005 [DOI] [PubMed] [Google Scholar]
- 29.Walk RM, Elliot ST, Blanco FC, Snyder JA, Jacobi AM, Rose SD, Behlke MA, Salem AK, Vukmanovic S, Sandler AD. 2012. T-cell activation is enhanced by targeting IL-10 cytokine production in toll-like receptor-stimulated macrophages. ImmunoTargets Ther. 1:13–23. 10.2147/ITT.S32615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li P, Neubig RR, Zingarelli B, Borg K, Halushka PV, Cook JA, Fan H. 2012. Toll-like receptor-induced inflammatory cytokines are suppressed by gain of function or overexpression of Gα(i2) protein. Inflammation 35:1611–1617. 10.1007/s10753-012-9476-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ribes S, Ebert S, Czesnik D, Regen T, Zeug A, Bukowski S, Mildner A, Eiffert H, Hanisch UK, Hammerschmidt S, Nau R. 2009. Toll-like receptor prestimulation increases phagocytosis of Escherichia coli DH5alpha and Escherichia coli K1 strains by murine microglial cells. Infect. Immun. 77:557–564. 10.1128/IAI.00903-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ribes S, Adam N, Ebert S, Regen T, Bunkowski S, Hanisch UK, Nau R. 2010. The viral TLR3 agonist poly(I:C) stimulates phagocytosis and intracellular killing of Escherichia coli by microglial cells. Neurosci. Lett. 482:17–20. 10.1016/j.neulet.2010.06.078 [DOI] [PubMed] [Google Scholar]
- 33.Kohchi C, Inagawa H, Nishizawa T, Soma G. 2009. ROS and innate immunity. Anticancer Res. 29:817–821 [PubMed] [Google Scholar]
- 34.Hewison M. 2011. Antibacterial effects of vitamin D. Nat. Rev. Endocrinol. 7:337–345. 10.1038/nrendo.2010.226 [DOI] [PubMed] [Google Scholar]
- 35.Bergman P, Norlin AC, Hansen S, Rekha RS, Agerberth B, Björkhem-Bergman L, Ekström L, Lindh JD, Andersson J. 2012. Vitamin D3 supplementation in patients with frequent respiratory tract infections: a randomised and double-blind intervention study. BMJ Open. 2:e001663. 10.1136/bmjopen-2012-001663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spina CS, Tangpricha V, Uskokovic M, Adorinic L, Maehr H, Holick MF. 2006. Vitamin D and cancer. Anticancer Res. 26:2515–2524 [PubMed] [Google Scholar]
- 37.Zhou C, Lu F, Cao K, Xu D, Goltzman D, Miao D. 2008. Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 74:170–179. 10.1038/ki.2008.101 [DOI] [PubMed] [Google Scholar]
- 38.Adams JS, Hewison M. 2008. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat. Clin. Pract. Endocrinol. Metab. 4:80–90. 10.1038/ncpendmet0716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, Hewison M. 2001. Extrarenal expression of 25-hydroxyvitamin D(3)-1a-hydroxylase. J. Clin. Endocrinol. Metab. 86:888–894. 10.1210/jcem.86.2.7220 [DOI] [PubMed] [Google Scholar]
- 40.Janeway CA, Jr, Medzhitov R. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. 10.1146/annurev.immunol.20.083001.084359 [DOI] [PubMed] [Google Scholar]
- 41.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zügel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773. 10.1126/science.1123933 [DOI] [PubMed] [Google Scholar]
- 42.Risso A. 2000. Leukocyte antimicrobial peptides: multifunctional effector molecules of innate immunity. J. Leukoc. Biol. 68:785–792. 10.1189/jlb.1938-3673 [DOI] [PubMed] [Google Scholar]
- 43.Zanetti M. 2004. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol. 75:39–48. 10.1189/jlb.0403147 [DOI] [PubMed] [Google Scholar]
- 44.Gombart AF, Saito T, Koeffler HP. 2009. Exaptation of an ancient Alu short interspersed element provides a highly conserved vitamin D-mediated innate immune response in humans and primates. BMC Genomics 10:321. 10.1186/1471-2164-10-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yim S, Dhawan P, Ragunath C, Christakos S, Diamond G. 2007. Induction of cathelicidin in normal and CF bronchial epithelial cells by 1,25-dihydroxyvitamin D(3). J. Cyst. Fibros. 6:403–410. 10.1016/j.jcf.2007.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gombart AF, Borregaard N, Koeffler HP. 2005. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 19:1067–1077. 10.1096/fj.04-3284com [DOI] [PubMed] [Google Scholar]
- 47.Evans KN, Nguyen L, Chan J, Innes BA, Bulmer JN, Kilby MD, Hewison M. 2006. Effects of 25-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 on cytokine production by human decidual cells. Biol. Reprod. 75:816–822. 10.1095/biolreprod.106.054056 [DOI] [PubMed] [Google Scholar]
- 48.Liu N, Kaplan AT, Low J, Nguyen L, Liu GY, Equils O, Hewison M. 2009. Vitamin D induces innate antibacterial responses in human trophoblasts via an intracrine pathway. Biol. Reprod. 80:398–406. 10.1095/biolreprod.108.073577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lagishetty V, Misharin AV, Liu NQ, Lisse TS, Chun RF, Ouyang Y, McLachlan SM, Adams JS, Hewison M. 2010. Vitamin D deficiency in mice impairs colonic antibacterial activity and predisposes to colitis. Endocrinology 151:2423–2432. 10.1210/en.2010-0089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fexby S, Bjarnsholt T, Jensen PO, Roos V, Høiby N, Givskov M, Klemm P. 2007. Biological Trojan horse: antigen 43 provides specific bacterial uptake and survival in human neutrophils. Infect. Immun. 75:30–34. 10.1128/IAI.01117-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sly LM, Lopez M, Nauseef WM, Reiner NE. 2001. 1α,25-Dihydroxyvitamin D3-induced monocyte antimycobacterial activity is regulated by phosphatidylinositol 3-kinase and mediated by the NADPH-dependent phagocyte oxidase. J. Biol. Chem. 276:35482–35493. 10.1074/jbc.M102876200 [DOI] [PubMed] [Google Scholar]
- 52.Garcion E, Nataf S, Berod A, Darcy F, Brachet P. 1997. 1,25-Dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Brain Res. Mol. Brain Res. 45:255–267. 10.1016/S0169-328X(96)00260-4 [DOI] [PubMed] [Google Scholar]
- 53.Ehrchen J, Helming L, Varga G, Pasche B, Loser K, Gunzer M, Sunderkötter C, Sorg C, Roth J, Lengeling A. 2007. Vitamin D receptor signaling contributes to susceptibility to infection with Leishmania major. FASEB J. 21:3208–3218. 10.1096/fj.06-7261com [DOI] [PubMed] [Google Scholar]
- 54.Keeney JT, Förster S, Sultana R, Brewer LD, Latimer CS, Cai J, Klein JB, Porter NM, Allan Butterfield D. 2013. Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: implications for low vitamin D-dependent age-related cognitive decline. Free Radic. Biol. Med. 18:324–334. 10.1016/j.freeradbiomed.2013.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lemire JM, Adams JS, Kermani-Arab V, Bakke AC, Sakai R, Jordan SC. 1985. 1,25-Dihydroxyvitamin D3 suppresses human T helper/inducer lymphocyte activity in vitro. J. Immunol. 134:3032–3035 [PubMed] [Google Scholar]
- 56.Abbas AK, Murphy KM, Sher A. 1996. Functional diversity of helper T lymphocytes. Nature 383:787–793. 10.1038/383787a0 [DOI] [PubMed] [Google Scholar]
- 57.Holick MF. 2007. Vitamin D deficiency. N. Engl. J. Med. 357:266–281. 10.1056/NEJMra070553 [DOI] [PubMed] [Google Scholar]
- 58.Peelen E, Knippenberg S, Muris AH, Thewissen M, Smolders J, Tervaert JW, Hupperts Damoiseaux RJ. 2011. Effects of vitamin D on the peripheral adaptive immune system: a review. Autoimmun. Rev. 10:733–743. 10.1016/j.autrev.2011.05.002 [DOI] [PubMed] [Google Scholar]
- 59.Hanisch UK. 2001. Microglia as a source and target of cytokine activities in the brain, p 79–124 In Streit WJ. (ed), Microglia in the degenerating and regenerating CNS. Springer-Verlag, New York, NY [Google Scholar]
- 60.Smith ME, van der Maesen K, Somera FP. 1998. Macrophage and microglial responses to cytokines in vitro: phagocytic activity, proteolytic enzyme release, and free radical production. J. Neurosci. Res. 54:68–78. [DOI] [PubMed] [Google Scholar]