

Abstract
The defining motor characteristics of Parkinson’s disease (PD) are mediated by the neurotransmitter dopamine (DA). Dopamine molecules spend most of their lifespan stored in intracellular vesicles awaiting release and very little time in the extracellular space or the cytosol. Without proper packaging of transmitter and trafficking of vesicles to the active zone, dopamine neurotransmission cannot occur. In the cytosol, dopamine is readily oxidized; excessive cytosolic dopamine oxidation may be pathogenic to nigral neurons in PD. Thus, factors that disrupt vesicular function may impair signaling and increase the vulnerability of dopamine neurons. This review outlines the many mechanisms by which disruption of vesicular function may contribute to the pathogenesis of PD. From direct inhibition of dopamine transport into vesicles by pharmacological or toxicological agents to alterations in vesicle trafficking by PD-related gene products, variations in the proper compartmentalization of dopamine can wreak havoc on a functional dopamine pathway. Findings from patient populations, imaging studies, transgenic models, and mechanistic studies will be presented to document the relationship between impaired vesicular function and vulnerability of the nigrostriatal dopamine system. Given the deleterious effects of impaired vesicular function, strategies aimed at enhancing vesicular function may be beneficial in the treatment of PD.
Keywords: Dopamine, Vesicle, Transport, VMAT2, Parkinson’s disease
Introduction
The disease James Parkinson first described in 1817 as the Shaking Palsy, which now bears his name, is the second most common neurodegenerative disorder. Parkinson’s disease (PD) is defined by its motor symptoms, tremor, rigidity, bradykinesia, but is, in fact, multifaceted and is associated with nonmotor symptoms as well, including autonomic, cognitive, and neuropsychiatric disturbances. The motor symptoms of PD are in all likelihood due to deficits in synaptic dopamine transmission, while the nonmotor symptoms may involve other neurotransmitters. PD pathology is characterized by deposition of α-synuclein-positive Lewy bodies in susceptible neurons and neuronal degeneration. However, Lewy pathology and neurodegeneration do not follow the same pattern of progression. Lewy bodies are, early on, deposited in the dorsal motor nucleus of the brainstem and spread in an ascending manner throughout the nervous system, but neurodegeneration is limited to specific areas. The dopaminergic neurons of the substantia nigra pars compacta (SNpc), which innervate the dorsal striatum, are preferentially destroyed in the course of the disease. Notably, cell loss is also observed in the locus ceruleus, in sympathetic neurons of the peripheral nervous system (releasing norepinephrine), and the dorsal raphe (releasing serotonin) [1, 2].
The key role for vesicular function in PD was first hinted at over 50 years ago. In 1952, reserpine, an alkaloid isolated from the Indian snakeroot Rauwolfia serpentine, was introduced to Western medicine and widely prescribed for its antihypertensive and antipsychotic properties. Despite the beneficial effects of reserpine, side effects include depression, gastric dysmotility, and extrapyramidal symptoms, “resembling a complete Parkinson Syndrome” [3]. While the molecular target of reserpine would not be identified for decades, investigators noted that reserpine depleted dopamine in biological tissue and caused parkinsonism in rats [4, 5]. Arvid Carlsson found that administration of the dopamine precursor, L-3,4-dihydroxyphenylalanine (levodopa), rescued the parkinsonian phenotype in rats [6]. Soon after, Ehringer and Hornykiewicz discovered that striatal dopamine deficiency was responsible for Parkinsonian motor deficits [7] and in 1961, Birkmayer and Hornykiewicz intravenously administered levodopa to a PD patient for the first time with marked success [8]. An oral form of levodopa was approved for the treatment of PD in 1970.
Storage of neurotransmitters relies on the function of synaptic vesicles; disrupting vesicular function results in decreased release and increased cytoplasmic neurotransmitter, which is toxic to neurons. The toxicity of cytoplasmic dopamine, in particular, has been studied extensively and results from oxidative stress stemming from excessive autoxidation and enzymatic deamination. Similar mechanisms of toxicity have been studied in the norepinephrine system, and to a lesser extent, the serotonin system. For the purpose of this review, we will focus on the vesicular storage of dopamine and ways it is disrupted in PD. We will both review evidence that defective vesicular function may be a determinant of PD pathogenesis and examine clinical indices of vesicular function and their potential as biomarkers for disease progression.
Synaptic Vesicle Function
Vesicles consist of spherical lipid bilayers and a vast array of proteins that mediate their various functions. Combined biochemical and genomic approaches have led to the identification of dozens of integral vesicular proteins involved in these activities, as well as biomolecules that are transiently associated with vesicles throughout their lifecycle [9–13]. The composition and size of synaptic vesicles vary with respect to their neuronal phenotype and cargo [14]. Synaptic vesicles transport and store a variety of neurotransmitters including acetylcholine, amino acids (including GABA and glycine, which are inhibitory and glutamate, which is excitatory), biogenic amines (including DA, NE, and serotonin), and neuropeptides. By concentrating neurotransmitters into discrete packages for subsequent release, synaptic vesicles are essential for neurotransmission. The vast majority of neurotransmitters within a neuron are found stored within vesicles. As such, vesicles must sequester transmitters against a concentration gradient, using specialized transporters. Within monoaminergic neurons, the type II vesicular monoamine transporter (VMAT2; SLC18A2) preferentially transports 1 cytosolic monoamine into the vesicle in exchange for release of 2 protons into the cytosol [15]. This process is dependent on an electrochemical gradient generated by the vacuolar ATPase, which transports protons into the vesicle, acidifying the vesicle lumen and providing substrate for antiport by VMAT2 [15, 16].
Formed in the Golgi apparatus, vesicles are transported to the synaptic terminal via transient interactions between cytoskeletal and vesicular proteins [17]. At the synaptic terminal, vesicles exist in distinct pools of various stages in the vesicle cycle; movement between these pools is dynamically regulated by vesicular proteins that respond to neuronal activity [18]. A subset of vesicles, comprising the readily releasable pool (RRP), dock at the active zone of the presynaptic membrane awaiting calcium influx. Calcium induces vesicles to fuse with the presynaptic membrane and release their contents by exocytic release (Fig. 1a) [18]. Closely associated with the RRP is the larger reserve pool, and collectively these make up the recycling pool. Most distal to the terminal is the resting pool [19]. From electrophysiological and morphological observations in cultured hippocampal neurons, investigators have estimated that there are 6–8 vesicles in the RRP, 17–20 in the reserve pool, and 180 in the much larger resting pool [20–23]. Docking of RRP vesicles is mediated by interactions between vesicular and plasmalemmal proteins of the SNARE complex [24], including synaptotagmins, which are calcium-sensing proteins that initiate membrane fusion during exocytosis [25, 26]. With prolonged stimulation, vesicles in the reserve pool are recruited to the RRP. Vesicles fused to the plasma membrane have several possible fates, including reuse by so-called “kiss-and-stay” (immediate reuse) and “kiss-and-run” (trafficking to the reserve pool) mechanisms [18, 27, 28]. Vesicle components remaining in the plasma membrane are recycled or degraded by endosomes.
Fig. 1.
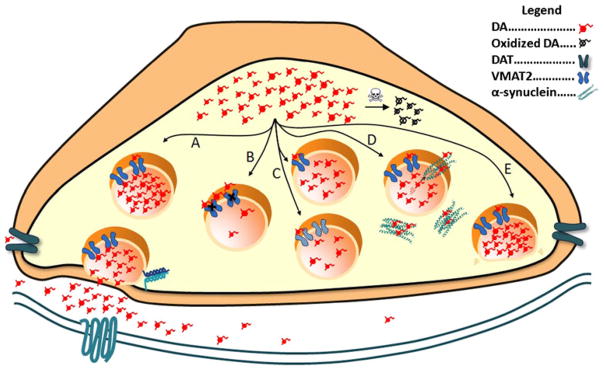
Mechanisms of vesicular disruption in Parkinson’s disease. After synthesis, dopamine is transported into vesicles. Cytoplasmic dopamine can be oxidized; in excess, this can lead to cellular injury. a, Vesicles in the readily releasable pool dock at the presynaptic membrane. In response to an action potential and calcium influx, vesicle membranes fuse to the plasma membrane and release their contents into the synaptic space. Dopamine is then reclaimed by the dopamine transporter (DAT) and then subsequently repackaged into the vesicle. b, Pharmacological inhibition of VMAT2 prevents uptake of dopamine leading to a depleted vesicle and reduced release. c, Genetic reduction of VMAT2 expression via promoter polymorphisms in humans or genetic manipulation in mice (upper vesicle) or VMAT2 function as noted in [126•] (lower vesicle) causes a reduction in vesicular filling and subsequent release. d, α-synuclein can form pores in the vesicle and cause depletion of dopamine from the vesicle or interact with cytoplasmic dopamine to form toxic species. e, Altered trafficking of the vesicle to the presynaptic membrane has been proposed to occur in the presence of overabundant α-synuclein or mutated LRRK2
Proteins, such as LRRK2 and α-synuclein, which are implicated in PD, may disrupt vesicular trafficking and function. Mutations in the LRRK2 gene (encoding the leucine-rich repeat kinase 2), account for the highest proportion of genetic cases of PD [29]. LRRK2 has a functional kinase domain with a multitude of cellular phospho-targets that may alter vesicular activities [30]. In addition, the circularized beta-propeller structure of the WD-40 domains in LRRK2 has been shown to interact with cytoskeletal and vesicular proteins. Mutations in LRRK2 may disrupt vesicular trafficking and endosomal recycling of vesicular proteins (Fig. 1e) [30].
The physiological role of α-synuclein (encoded by SNCA) remains elusive, though its genetic and pathological role in PD is well established [31–33]. Monomeric α-synuclein has been shown to reversibly bind and adjoin vesicle membranes [34]. Based on this and its presynaptic localization, it is hypothesized that its endogenous function is to regulate vesicular trafficking between the active zone and cytoplasmic pools. Combining transgenic models of α-synuclein overexpression and deletion, Scott and Roy observed that α-synuclein regulates the size of the recycling pool in cultured hippocampal neurons because overexpression decreased the recycling pool size and deletion increased its size and enhanced intersynaptic vesicular trafficking [35•]. Rats overexpressing human α-synuclein have reduced dopamine vesicle density and correlative reductions in motor activity [36•]. In the context of pathogenesis, fibrilization of α-synuclein forms the primary structural component of Lewy bodies, and toxic α-synuclein protofibrils form as intermediates in the fibrilization process [37]. Oxidized dopamine covalently binds α-synuclein, stabilizing these protofibril intermediates [38] (for a biophysical review of this subject, see [39]). In vitro, oligomeric α-synuclein can also disrupt SNARE complex formation [40]. Furthermore, α-synuclein protofibrils have been shown to permeabilize vesicle membranes, which would facilitate leakage of vesicular dopamine into the cytoplasm and ablate the vesicle electrochemical gradient [41]. These processes may synergistically interact in PD (Fig. 1d) and, in part, explain the vulnerability of dopamine neurons in PD.
Cytoplasmic Dopamine Toxicity and VMAT2
Despite the essential role of dopamine to life processes, it is neurotoxic if vesicular storage is disrupted. Under normal conditions, low levels of dopamine are present in the cytosol following synthesis from DOPA, plasmalemmal transport by the dopamine transporter (DAT), and vesicular leak [42]. Cytosolic dopamine is metabolized by enzymatic deamination or broken down by autoxidation, producing reactive, harmful products. Efficient transport of dopamine by VMAT2 prevents accumulation of these toxic byproducts. Thus, the relative expression of DAT to VMAT2 has been suggested as a determinant of neuronal vulnerability in PD [43, 44].
Enzymatic deamination of dopamine occurs at the mitochondria by monoamine oxidase (MAO), forming DOPAL, a toxic aldehyde intermediate, as well as H2O2 [45]. DOPAL is reactive, and it readily forms adducts to cytosolic proteins. Alternatively, it may autoxidize, generating reactive oxygen species [46]. The toxicity of DOPAL has been thoroughly studied in vitro [47] and has been shown to cause neurodegeneration in mice [48]. Relative DOPAL formation is elevated in the striata of postmortem PD brains [49]. Intraneuronally, DOPAL is preferentially detoxified by aldehyde dehydrogenase to form DOPAC [45].
While autoxidation of cytosolic dopamine occurs readily in the alkaline environment of the cytosol, dopamine is stable in the acidic environment of the vesicle. Autoxidation is particularly deleterious because it yields a reactive dopamine quinone, as well as superoxide, hydrogen peroxide, and a hydroxyl radical. Furthermore, the dopamine quinone itself is highly reactive and forms cysteinyl adducts [50], disrupting the function of target proteins and potentially DNA [51]. Another fate of cytosolic monoamines that is not necessarily toxic is the formation of neuromelanin, the dark pigment that gives the SNpc its color. Notably, neuromelanin is also present in the LC, where oxidation of dopamine and NE also occurs. Neuromelanin forms in the acidic lysosomal compartments, which contain VMAT2 and are capable of monoamine transport. Within the confines of these organelles, dopamine quinones may instead polymerize, forming neuromelanin, [52]. However, neuromelanin biosynthesis from autoxidized dopamine is prevented in cultured nigral rat neurons overexpressing VMAT2, suggesting that the ability to properly store dopamine in vesicles is critical in this process [53].
Numerous studies have shown that unregulated cytosolic dopamine is neurotoxic [50, 54–56]. Mice expressing DAT on non-dopaminergic striatal neurons that lack VMAT2 are able to take up dopamine but not store it in vesicles; these mice exhibit motor deficits and profound striatal neurodegeneration, accompanied by markers of increased dopamine oxidation [57]. In vitro experiments have also demonstrated that relative vulnerability of dopamine neurons in PD may be mediated by levels of cytosolic dopamine [58]. Additionally, transgenic mice with altered expression of VMAT2 (Fig. 1c) have illustrated how crucial vesicular storage of dopamine is to the integrity of the nigrostriatal system. Vesicular function is essential and VMAT2 knockout mice die soon after birth. Heterozygotes develop normally, but have increased sensitivity to amphetamine-induced locomotion, susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity [59–61], and levodopa induced death of SNpc neurons [58]. VMAT2 hypomorphic mice (~5 % wildtype expression; VMAT2LO) have emerged as a mouse model of PD [62–64, 65•]. These mice develop normally, but undergo progressive nigrostriatal degeneration, α-synuclein accumulation, as well as motor and nonmotor symptoms of PD [63, 64]. They also show markers of oxidative stress, including cysteinyl-catechol adducts. Notably, neurodegeneration occurs only when these mice express α-synuclein [63]. VMAT2LO mice exhibit levodopa-correctable motor deficits, including reduced stride-length and locomotor activity, as well as nonmotor symptoms of PD, including anosmia, gastric dysmotility, and depressive and anxiety-like behaviors. In contrast, PACAP38, a neuropeptide shown to increase VMAT2 expression, attenuates methamphetamine-induced neurotoxicity in mice, reduces markers of oxidative stress and neuroinflammation, and suggests that increase VMAT2 protects against oxidative stress [66].
In addition to VMAT2’s protective role against cytoplasmic dopamine toxicity, it also plays an important role in neuroprotection against exogenous toxicants that may damage dopamine neurons [16, 67]. The classic dopaminergic toxicant, MPTP, was serendipitously but tragically discovered to induce chronic parkinsonism in humans after intravenous administration [68]. After crossing the blood-brain barrier, MPTP is metabolized by glial monoamine oxidase to form the toxic metabolite, MPP+, which is then transported into neurons by the dopamine transporter [69–72]. MPP+ causes neurotoxicity by inhibiting mitochondrial respiration and triggering cell death. MPP+ is also a substrate for VMAT2 and can be sequestered away from sites of action. In fact, VMAT2 was initially cloned for its ability to confer resistance to MPP+ toxicity and is evolutionarily related to bacterial toxin extruding antiporters (TEXANS) [73–75]. In support of this, heterozygote VMAT2 knockout mice have heightened sensitivity to MPTP toxicity [61]. It is conceivable that VMAT2 expression may modulate the effects of other exogenous toxicants involved in PD pathogenesis [59, 76].
Pharmacologic Blockade of VMAT2
Carlsson and colleagues’ observation that reserpine induced a parkinsonian phenotype that was correctable with levodopa was a launching point for pharmacotherapy in PD [6, 77]. Subsequent experimentation revealed that the effects of reserpine were akin to sympathectomy, in that reserpinized subjects were still responsive to the inotropic and pressor effects of direct-acting sympathomimetics or adrenergic receptor agonists (directly acting sympathomimetics, eg, norepinephrine, epinephrine, neosynephrine), but abolished the effects of indirectly acting sympathomimetics including the phenethylamines, amphetamine, ephedrine, and tyramine [78, 79]. It was later realized that reserpine and the aforementioned indirectly acting sympathomimetics all function by inhibiting VMAT2 (Fig. 1b).
Amphetamine (AMPH) and the substituted AMPH derivative methamphetamine (METH) are catecholamine releasers in the peripheral and central nervous systems. The rewarding effect of striatal dopamine release is largely responsible for the abuse potential of this class of drugs. When abused, AMPH and METH are capable of exerting profound dopaminergic toxicity [84]. AMPH and METH share the same mechanism of action and similar pharmacokinetic and pharmacodynamic properties [80, 81]. AMPH and METH mediate dopamine release by competing for vesicular uptake of dopamine by VMAT2. Furthermore, they are weak bases and may disrupt the electrochemical gradient of the vesicle by accepting free lumenal protons [81]. The increased level of cytosolic dopamine in turn facilitates non-exocytic dopamine release into the synaptic terminal by efflux via the dopamine transporter [81].
Administration of METH in animals causes striatal denervation, including loss of dopamine transporter expression, gliosis, and autophagic vacuolization [82–84]. In rats, this pathology is associated with cysteinyl dopamine adducts, suggesting that dopamine oxidation is indeed involved in METH toxicity [85]. This toxicity is relevant in humans; striatal dopamine transporter loss in METH-treated baboons is comparable with losses in human METH users who reported using similar doses of METH [86]. METH-induced striatal DAT loss in humans is not as severe as losses observed in PD, but it has been shown to persist in patients 3 years after quitting, suggesting long-term damage that may sensitize one to PD [87]. Recently, a retrospective statewide study in California found that patients admitted to the hospital for an AMPH or METH related incident were significantly more likely to develop PD than matched patient controls admitted for appendicitis (hazard ratio=1.75) [88]. In contrast, the same study showed that patients admitted to the hospital for cocaine-related incidents did not have an increased likelihood of developing PD. Because cocaine inhibits DAT and also increases dopamine levels in the synaptic cleft, these findings support the notion that PD risk may stem from cytosolic dopamine toxicity. In support of this notion, our lab has shown that VMAT2 expression in mice inversely correlates with their susceptibility to METH-induced dopaminergic neurotoxicity [66, 89].
Toxicological Blockade of VMAT2
Epidemiological evidence supporting an environmental connection to PD is compelling [90–99]. As we and others have recently reviewed, several classes of compounds have been associated with PD pathology, especially halogenated persistent organic pollutants (POPs) [16, 100, 101]. Halogenated POPs represent a broad class of environmental toxicants that include organochlorine insecticides, polychlorinated biphenyls (PCBs), and brominated flame retardants. Higher levels of many organochlorines and PCBs have been reported in postmortem brain tissue and serum of PD patients [102–106] and epidemiological studies have linked the compounds to PD [91]. Mechanistic studies into these compounds suggest that many exert selective toxicity to dopaminergic neurons, inhibit synaptosomal and vesicular uptake, and cause oxidative stress [107–125]. Taken together, these data suggest that many compounds epidemiologically linked to PD act by impairing vesicular function and increasing oxidative stress.
Genetic Variability of SLC18A2 in PD
Given the role of VMAT2 in regulating cytosolic dopamine toxicity, mutations affecting the function or expression of the transporter (Fig. 1c) might be expected to affect susceptibility to PD. Genetic mutations in the coding region of SLC18A2 are extremely rare, reflecting the essential nature of VMAT2 function in an evolutionary context. A recent report identified a crippling movement disorder in members of a consanguineous Saudi Arabian family carrying a recessive mutation in the coding region SLC18A2 resulting in a P387L substitution in the fifth luminal loop of VMAT2 [126•]. Affected individuals developed infantile-onset parkinsonism, severe cognitive impairment, mood disturbance, and autonomic dysfunction. Heterozygote parents were unaffected by the disorder, but were clinically depressed, consistent with a reduction in brain monoamines. The homozygous patients exhibited reductions in CSF and urinary monoamines, but increased monoamine metabolites. The investigators found that P387L mutation virtually abolished VMAT2 function in transfected cells. Treatment with the dopamine receptor agonist pramiprexole dramatically improved symptoms in all patients, though efficacy appeared to correlate inversely with age. Other coding mutations have been identified in humans, though they are very rare and have not been associated with neurological outcomes [127, 128].
While SLC18A2 has very little variability in the coding regions, the gene has a large and highly polymorphic promoter sequence (17.4 kb) [127, 129]. This high variability results in many low frequency haplotypes and renders genetic risk assessment difficult. Thus, studies of SLC18A2 haplotype and PD risk have been inconclusive [127, 130]. However, analyses of the functional consequences of promoter polymorphisms have provided useful information. Glatt and colleagues [131] screened the most prevalent promoter haplotypes in an American population and identified those that increased transcriptional activity of SLC18A2 in vitro. Collectively, gain-of-function haplotypes were found to be protective against risk of PD in women; the effect was particularly robust in women homozygous for gain-of-function haplotypes (odds ratio= 0.37). In a more recent study in Italy, investigators identified 2 SNPs within the promoter that conferred protection against PD, presumably by increasing VMAT2 expression [132•]. The variability of the SLC18A2 promoter suggests that genetic and epigenetic influences may contribute to variable VMAT2 expression and risk of PD within populations.
VMAT2 Imaging in PD
Positron emission tomography (PET) ligands of striatal dopamine terminal markers have been used to monitor the progression of PD and response to therapeutic treatments. VMAT2 has emerged as a reliable marker of striatal innervation in PD [76, 133, 134]. High-affinity radioligands for DAT and the fluorinated DOPA analog, 18F-DOPA, have been utilized for PET scans, but their binding substrates were found to be modulated by levodopa and dopamine agonists. For instance, changes in DAT binding by β-CIT in response to pramipexole (increased binding) and levodopa (reduced binding) had been mistakenly interpreted as changes in striatal innervation, when in fact DAT membrane expression is modified by these therapies [135, 136]. 18F-DOPA PET signal does not reliably correlate with striatal innervation because expression of AADC, for which 18F-DOPA is a substrate, increases in response to neuronal damage [137–140]. In contrast to DAT, VMAT2 expression is not modified by the PD therapeutics levodopa or selegiline [138, 141]. Bohnen and colleagues demonstrated that striatal C11-dihydrotetrabenazine binding correlated reliably to clinically rated (UPDRSIII) motor deficits in PD patients [133]. Another VMAT2 radioligand, F-18-AV-133 (a tetrabenazine derivative), is also now used to study nigrostriatal degeneration [142, 143]. While VMAT2 radioligand binding conveniently correlates to nigrostriatal denervation, it does not provide direct evidence of vesicular function or VMAT2 activity.
VMAT2 as a Peripheral Biomarker of PD
Studies examining VMAT2 in the human periphery may provide insight into how to measure central VMAT2 activity in PD. VMAT2 mediates the storage and release of norepinephrine in postganglionic neurons of the sympathetic nervous system. Sympathetic denervation commonly occurs in PD; orthostatic hypotension is a comorbid feature, affecting 30 % of PD patients [144, 145]. In vivo imaging of sympathetic denervation in PD patients is accomplished with radiolabeled ligands for both the norepinephrine transporter (expressed at the plasma membrane of sympathetic neurons) and VMAT2 [146–150]. Sympathetic denervation may occur prior to the onset of motor symptoms of PD, with noradrenergic denervation of the heart paralleling the dopaminergic denervation of the striatum [149, 151].
A novel approach pairing analysis of myocardial 18F-DA uptake and arterial 18F-DOPAC formation can provide an in vivo index of vesicular activity [152]. 18F-DA uptake is dependent on the presence of intact noradrenergic terminals, and MAO metabolism of 18F-DA to 18F-DOPAC is in competition with vesicular uptake [153]. Thus, one can determine both the extent of cardiac noradrenergic innervation (by PET imaging) and the relative uptake activity in vesicles (by measuring relative 18 F-DOPAC formation) with these combined methods. Goldstein and colleagues performed this analysis in patient cohorts including those with PD with orthostatic hypotension, pure autonomic failure (a Lewy body disease featuring orthostatic hypotension without parkinsonism), multiple systems atrophy (a non-Lewy body synucleinopathy that clinically resembles PD), and healthy controls. The authors concluded that myocardial vesicular function was impaired only in patients with Lewy body diseases (PD with orthostatic hypotension and in pure autonomic failure), consistent with the effects of pathogenic α-synuclein in disrupting vesicular function [39, 154].
VMAT2 is also present in platelets and may serve as a peripheral biomarker of monoaminergic vesicular function. Platelets express proteins involved in the storage and metabolism of serotonin, including the serotonin transporter (SERT), VMAT2, and monoamine oxidase. These blood cells have been used as models of serotonin neurons [155]. Profiles of imipramine binding to platelet SERT have been used as peripheral biomarkers of serotonergic neurotransmission in the brain [156–160]. Similarly, platelet VMAT2 levels may serve as a peripheral biomarker of central VMAT2 expression. Alterations in VMAT2 dihydrotetrabenazine binding in platelets have been linked to cases of depression, juvenile behavior disorders, and schizophrenia [161–169]. Platelet SLC18A2 mRNA levels may also be predictive of PD. In 39 PD patients vs 39 healthy control subjects, the relative quantity of SLC18A2 mRNA in platelets was decreased by 23 % in the PD patients [170]. Therefore, assessing platelet expression of VMAT2 may prove useful as a noninvasive biomarker of PD risk.
Conclusions
We have summarized ways in which alterations in vesicular storage of dopamine influences neuronal viability and susceptibility to neurodegeneration in PD. All disruptions of vesicular storage discussed have the overall effect of increasing cytosolic dopamine. In sporadic PD, it is conceivable that a combination of genetic and epigenetic factors decrease levels of functional VMAT2 while environmental exposure to compounds that alter vesicular function may have this same effect. Therefore, measuring vesicular uptake capacity in human patients may represent a useful measure of PD risk. Furthermore, interventions aimed at increasing vesicular function may be beneficial in the treatment of PD and may even be neuroprotective. These warrant a closer look in controlled trials.
Acknowledgments
P01 ES016731, T32 ES012870, T32 GM008602, and T32 DA015040.
Footnotes
Conflict of Interest Shawn P. Alter declares that he has no conflict of interest.
Gina M. Lenzi declares that she has no conflict of interest.
Alison I. Bernstein declares that she has no conflict of interest.
Gary W. Miller declares that he has no conflict of interest.
Compliance with Ethics Guidelines
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
This article is part of the Topical Collection on Movement Disorders
Contributor Information
Shawn P. Alter, Department of Environmental Health, Rollins School of Public Health, Emory University, Atlanta, GA, USA
Gina M. Lenzi, Department of Biochemistry, School of Medicine, Emory University, Atlanta, GA, USA
Alison I. Bernstein, Department of Environmental Health, Rollins School of Public Health, Emory University, Atlanta, GA, USA
Gary W. Miller, Email: gary.miller@emory.edu, Department of Neurology, School of Medicine, Emory University, Atlanta, GA, USA. Department of Pharmacology, School of Medicine, Emory University, Atlanta, GA, USA. Department of Environmental Health, Rollins School of Public Health, Claudia Nance Rollins Bldg, Room 8007, 1518 Clifton Road, NE, Atlanta, GA 30322, USA
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60(3):337–41. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- 2.Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR, et al. Neuropathology of immunohistochemically identified brainstem neurons in Parkinson’s disease. Ann Neurol. 1990;27(4):373–85. doi: 10.1002/ana.410270405. [DOI] [PubMed] [Google Scholar]
- 3.Richman A, Tyhurst JS. An extrapyramidal syndrome with reserpine. Can Med Assoc J. 1955;72(6):457–8. [Google Scholar]
- 4.Brodie BB, Shore PA, Silver SL. Potentiating action of chlorpromazine and reserpine. Nature. 1955;175(4469):1133–4. doi: 10.1038/1751133a0. [DOI] [PubMed] [Google Scholar]
- 5.Pletscher A, Shore PA, Brodie BB. Serotonin release as a possible mechanism of reserpine action. Science. 1955;122(3165):374–5. doi: 10.1126/science.122.3165.374. [DOI] [PubMed] [Google Scholar]
- 6.Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180(4596):1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- 7.Ehringer H. Hornykiewicz O [Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system] Klin Wochenschr. 1960;38:1236–9. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- 8.Birkmayer W. Hornykiewicz O [The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinsonakinesia] Wien Klin Wochenschr. 1961;73:787–8. [PubMed] [Google Scholar]
- 9.Coughenour HD, Spaulding RS, Thompson CM. The synaptic vesicle proteome: a comparative study in membrane protein identification. Proteomics. 2004;4(10):3141–55. doi: 10.1002/pmic.200300817. [DOI] [PubMed] [Google Scholar]
- 10.Blondeau F, Ritter B, Allaire PD, Wasiak S, Girard M, Hussain NK, et al. Tandem MS analysis of brain clathrin-coated vesicles reveals their critical involvement in synaptic vesicle recycling. Proc Natl Acad Sci U S A. 2004;101(11):3833–8. doi: 10.1073/pnas.0308186101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morciano M, Burre J, Corvey C, Karas M, Zimmermann H, Volknandt W. Immunoisolation of 2 synaptic vesicle pools from synaptosomes: a proteomics analysis. J Neurochem. 2005;95(6):1732–45. doi: 10.1111/j.1471-4159.2005.03506.x. [DOI] [PubMed] [Google Scholar]
- 12.Burre J, Beckhaus T, Schagger H, Corvey C, Hofmann S, Karas M, et al. Analysis of the synaptic vesicle proteome using three gel-based protein separation techniques. Proteomics. 2006;6(23):6250–62. doi: 10.1002/pmic.200600357. [DOI] [PubMed] [Google Scholar]
- 13.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127(4):831–46. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 14.Liu YJ, Edwards RH. The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu Rev Neurosci. 1997;20:125–56. doi: 10.1146/annurev.neuro.20.1.125. [DOI] [PubMed] [Google Scholar]
- 15.Edwards RH. The neurotransmitter cycle and quantal size. Neuron. 2007;55(6):835–58. doi: 10.1016/j.neuron.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Chaudhry FA, Edwards RH, Fonnum F. Vesicular neurotransmitter transporters as targets for endogenous and exogenous toxic substances. Annu Rev Pharmacol Toxicol. 2008;48(1):277–301. doi: 10.1146/annurev.pharmtox.46.120604.141146. [DOI] [PubMed] [Google Scholar]
- 17.Hannah MJ, Schmidt AA, Huttner WB. Synaptic vesicle biogenesis. Annu Rev Cell Dev Biol. 1999;15:733–98. doi: 10.1146/annurev.cellbio.15.1.733. [DOI] [PubMed] [Google Scholar]
- 18.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–47. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 19.Segovia M, Ales E, Montes MA, Bonifas I, Jemal I, Lindau M, et al. Push-and-pull regulation of the fusion pore by synaptotagmin-7. Proc Natl Acad Sci U S A. 2010;107(44):19032–7. doi: 10.1073/pnas.1014070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17(15):5858–67. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat Neurosci. 1999;2(6):503–7. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 22.Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28(1):221–31. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- 23.Südhof TC. The synaptic vesiclecycle revisited. Neuron. 2000;28(2):317–20. doi: 10.1016/s0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 24.Rizo J, Sudhof TC. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices—guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–308. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- 25.Maximov A, Sudhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48(4):547–54. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Pang ZP, Sun J, Rizo J, Maximov A, Sudhof TC. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J. 2006;25(10):2039–50. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barker LA, Dowdall MJ, Whittaker VP. Choline metabolism in the cerebral cortex of guinea pigs. Stable-bound acetylcholine. Biochem J. 1972;130(4):1063–75. doi: 10.1042/bj1301063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceccarelli B, Hurlbut WP. Vesicle hypothesis of the release of quanta of acetylcholine. Physiol Rev. 1980;60(2):396–441. doi: 10.1152/physrev.1980.60.2.396. [DOI] [PubMed] [Google Scholar]
- 29.Paisan-Ruiz C, Nath P, Washecka N, Gibbs JR, Singleton AB. Comprehensive analysis of LRRK2 in publicly available Parkinson’s disease cases and neurologically normal controls. Hum Mutat. 2008;29(4):485–90. doi: 10.1002/humu.20668. [DOI] [PubMed] [Google Scholar]
- 30.Belluzzi E, Greggio E, Piccoli G. Presynaptic dysfunction in Parkinson’s disease: a focus on LRRK2. Biochem Soc Trans. 2012;40(5):1111–6. doi: 10.1042/BST20120124. [DOI] [PubMed] [Google Scholar]
- 31.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 32.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18(2):106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 33.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. Alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 34.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273(16):9443–9. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 35•.Scott D, Roy S. Alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32(30):10129–35. doi: 10.1523/jneurosci.0535-12.2012. Demonstrated that α-synuclein expression inversely correlates with synaptic vesicular trafficking and recycling pool size in hippocampal neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, et al. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123(5):653–69. doi: 10.1007/s00401-012-0963-y. Demonstrated that α-synuclein overexpression inhibits vesicular trafficking in DA terminals, and causes parkinsonian pathology and motor effects in rats. [DOI] [PubMed] [Google Scholar]
- 37.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 38.Conway KA, Rochet J-C, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294(5545):1346. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 39.Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, et al. Interactions among alpha-synuclein, dopamine, and biomembranes—some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23(1–2):23–33. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- 40.Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, et al. Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, et al. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40(26):7812–9. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- 42.Eisenhofer G, Kopin IJ, Goldstein DS. Leaky catecholamine stores: undue waste or a stress response coping mechanism? Ann NY Acad Sci. 2004;1018(1):224–30. doi: 10.1196/annals.1296.027. [DOI] [PubMed] [Google Scholar]
- 43.Uhl GR. Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson’s disease. Ann Neurol. 1998;43(5):555–60. doi: 10.1002/ana.410430503. [DOI] [PubMed] [Google Scholar]
- 44.Miller GW, Gainetdinov RR, Levey AI, Caron MG. Dopamine transporters and neuronal injury. Trends Pharmacol Sci. 1999;20(10):424–9. doi: 10.1016/s0165-6147(99)01379-6. [DOI] [PubMed] [Google Scholar]
- 45.Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56(3):331–49. doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- 46.Burke WJ, Li SW, Chung HD, Ruggiero DA, Kristal BS, Johnson EM, et al. Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role in neurodegenerative diseases. NeuroToxicol. 2004;25(1–2):101–15. doi: 10.1016/S0161-813X(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 47.Rees JN, Florang VR, Eckert LL, Doorn JA. Protein reactivity of 3,4-Dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem Res Toxicol. 2009;22(7):1256–63. doi: 10.1021/tx9000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One. 2012;7(2):e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldstein DS, Sullivan P, Holmes C, Kopin IJ, Basile MJ, Mash DC. Catechols in post-mortem brain of patients with Parkinson disease. Eur J Neurol. 2011;18(5):703–10. doi: 10.1111/j.1468-1331.2010.03246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Graham DG, Tiffany SM, Bell WR, Gutknecht WF. Autoxidation versus covalent binding of quinones as mechanism of toxicity of dopamine, 6-Hydroxydopamine, and related compounds toward C1300-Neuroblastoma cells in vitro. Mol Pharmacol. 1978;14(4):644–53. [PubMed] [Google Scholar]
- 51.Zahid M, Saeed M, Yang L, Beseler C, Rogan E, Cavalieri EL. Formation of dopamine quinone-DNA adducts and their potential role in the etiology of Parkinson’s disease. IUBMB Life. 2011;63(12):1087–93. doi: 10.1002/iub.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res. 1999;1(3):181–95. doi: 10.1007/bf03033289. [DOI] [PubMed] [Google Scholar]
- 53.Sulzer D, Bogulavsky J, Larsen KE, Behr G, Karatekin E, Kleinman MH, et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci U S A. 2000;97(22):11869–74. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci U S A. 1996;93(5):1956–61. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benshachar D, Zuk R, Glinka Y. Dopamine neurotoxicity - inhibition of mitochondrial respiration. J Neurochem. 1995;64(2):718–23. doi: 10.1046/j.1471-4159.1995.64020718.x. [DOI] [PubMed] [Google Scholar]
- 56.Asanuma M, Miyazaki I, Ogawa N. Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox Res. 2003;5(3):165–76. doi: 10.1007/BF03033137. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Ding Y, Cagniard B, Van Laar AD, Mortimer A, Chi W, et al. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J Neurosci. 2008;28(2):425–33. doi: 10.1523/JNEUROSCI.3602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, et al. Interplay between cytosolic dopamine, calcium, and α-Synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62(2):218–29. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, et al. VMAT2 knockout mice: heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc Natl Acad Sci U S A. 1997;94(18):9938–43. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, et al. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19(6):1285–96. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 61.Gainetdinov RR, Fumagalli F, Wang YM, Jones SR, Levey AI, Miller GW, et al. Increased MPTP neurotoxicity in vesicular monoamine transporter 2 heterozygote knockout mice. J Neurochem. 1998;70(5):1973–8. doi: 10.1046/j.1471-4159.1998.70051973.x. [DOI] [PubMed] [Google Scholar]
- 62.Colebrooke RE, Humby T, Lynch PJ, McGowan DP, Xia J, Emson PC. Age-related decline in striatal dopamine content and motor performance occurs in the absence of nigral cell loss in a genetic mouse model of Parkinson’s disease. Eur J Neurosci. 2006;24(9):2622–30. doi: 10.1111/j.1460-9568.2006.05143.x. [DOI] [PubMed] [Google Scholar]
- 63.Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27(30):8138–48. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM, et al. Nonmotor symptoms of Parkinson’s Disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci. 2009;29(25):8103–13. doi: 10.1523/jneurosci.1495-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65•.Ulusoy A, Björklund T, Buck K, Kirik D. Dysregulated dopamine storage increases the vulnerability to α-synuclein in nigral neurons. Neurobiol Dis. 2012;47(3):367–77. doi: 10.1016/j.nbd.2012.05.012. Utilized lentiviralα-synuclein delivery to demonstrate synergistic toxicity of α-synuclein and cytosolic dopamine in VMAT2 hypomorphic mice. [DOI] [PubMed] [Google Scholar]
- 66.Guillot TS, Richardson JR, Wang MZ, Li YJ, Taylor TN, Ciliax BJ, et al. PACAP38 increases vesicular monoamine transporter 2 (VMAT2) expression and attenuates methamphetamine toxicity. Neuropeptides. 2008;42(4):423–34. doi: 10.1016/j.npep.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guillot T, Miller G. Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol. 2009;39(2):149–70. doi: 10.1007/s12035-009-8059-y. [DOI] [PubMed] [Google Scholar]
- 68.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):979–80. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 69.Glover V, Gibb C, Sandler M. Monoamine oxidase B(MAO-B) is the major catalyst for 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) oxidation in human brain and other tissues. Neurosci Lett. 1986;64(2):216–20. doi: 10.1016/0304-3940(86)90103-5. [DOI] [PubMed] [Google Scholar]
- 70.Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6 -tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci U S A. 1985;82(7):2173–7. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langston JW, Irwin I, Langston EB, Forno LS. 1-Methyl-4-phenylpyridinium ion (MPP+): identification of a metabolite of MPTP, a toxin selective to the substantia nigra. Neurosci Lett. 1984;48(1):87–92. doi: 10.1016/0304-3940(84)90293-3. [DOI] [PubMed] [Google Scholar]
- 72.Ransom BR, Kunis DM, Irwin I, Langston JW. Astrocytes convert the parkinsonism inducing neurotoxin, MPTP, to its active metabolite, MPP+ Neurosci Lett. 1987;75(3):323–8. doi: 10.1016/0304-3940(87)90543-x. [DOI] [PubMed] [Google Scholar]
- 73.Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, et al. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70(4):539–51. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- 74.Erickson JD, Eiden LE. Functional identification and molecular cloning of a human brain vesicle monoamine transporter. J Neurochem. 1993;61(6):2314–7. doi: 10.1111/j.1471-4159.1993.tb07476.x. [DOI] [PubMed] [Google Scholar]
- 75.Erickson JD, Eiden LE, Hoffman BJ. Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1992;89(22):10993–7. doi: 10.1073/pnas.89.22.10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miller GW, Erickson JD, Perez JT, Penland SN, Mash DC, Rye DB, et al. Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson’s disease. Exp Neurol. 1999;156(1):138–48. doi: 10.1006/exnr.1998.7008. [DOI] [PubMed] [Google Scholar]
- 77.Smith Y, et al. Parkinson’s disease therapeutics: new developments and challenges since the introduction of levodopa. Neuropsychopharmacology. 2012;37(1):213–46. doi: 10.1038/npp.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bertler A, Carlsson A, Nilsson J, Rosengren E. Effect of reserpine on the metabolism of catecholamies. Acta Physiologica Scand. 1957;42:22–3. [Google Scholar]
- 79.Burn JH, Rand MJ. The action of sympathomimetic amines in animals treated with reserpine. J Physiol. 1958;144(2):314–36. doi: 10.1113/jphysiol.1958.sp006104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Melega WP, Williams AE, Schmitz DA, DiStefano EW, Cho AK. Pharmacokinetic and pharmacodynamic analysis of the actions of D-amphetamine and D-methamphetamine on the dopamine terminal. J Pharmacol Exp Ther. 1995;274(1):90–6. [PubMed] [Google Scholar]
- 81.Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69(4):628–49. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181(1):151–60. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- 83.Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22(20):8951–60. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yamamoto BK, Moszczynska A, Gudelsky GA. Amphetamine toxicities. Ann NY Acad Sci. 2010;1187(1):101–21. doi: 10.1111/j.1749-6632.2009.05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19(4):1484–91. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB, et al. Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable with those recreationally abused by humans: evidence from C-11 WIN-35,428 positron emission tomography studies and direct in vitro determinations. J Neurosci. 1998;18(1):419–27. doi: 10.1523/JNEUROSCI.18-01-00419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with C-11 WIN-35,428. J Neurosci. 1998;18(20):8417–22. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Callaghan RC, Cunningham JK, Sykes J, Kish SJ. Increased risk of Parkinson’s disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol Depend. 2012;120(1–3):35–40. doi: 10.1016/j.drugalcdep.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 89.Guillot TS, Shepherd KR, Richardson JR, Wang MZ, Li YJ, Emson PC, et al. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J Neurochem. 2008;106(5):2205–17. doi: 10.1111/j.1471-4159.2008.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ascherio A, Chen H, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, et al. Pesticide exposure and risk for Parkinson’s disease. Ann Neurol. 2006;60:197–203. doi: 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- 91.Elbaz A, Clavel J, Rathouz PJ, Moisan F, Galanaud J-P, Delemotte B, et al. Professional exposure to pesticides and Parkinson disease. Ann Neurol. 2009;66:494–504. doi: 10.1002/ana.21717. [DOI] [PubMed] [Google Scholar]
- 92.Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson’s disease: a meta-analysis. Environ Res. 2001;86:122–7. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- 93.Priyadarshi A, Khuder SA, Schaub EA, Shrivastava S. A meta-analysis of Parkinson’s disease and exposure to pesticides. Neurotoxicology. 2000;21:435–40. [PubMed] [Google Scholar]
- 94.Semchuk KM, Love EJ, Lee RG. Parkinson’s disease and exposure to rural environmental factors: a population based case-control study. Can J Neurol Sci. 1991;18:279–86. doi: 10.1017/s0317167100031826. [DOI] [PubMed] [Google Scholar]
- 95.Semchuk KM, Love EJ, Lee RG. Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology. 1992;42:1328–35. doi: 10.1212/wnl.42.7.1328. [DOI] [PubMed] [Google Scholar]
- 96.Steenland K, Hein MJ, Cassinelli RT, Prince MM, Nilsen NB, Whelan EA, et al. Polychlorinated biphenyls and neurodegenerative disease mortality in an occupational cohort. Epidemiology. 2006;17:8–13. doi: 10.1097/01.ede.0000190707.51536.2b. [DOI] [PubMed] [Google Scholar]
- 97.Tanner CM. Parkinson’s disease: environmental etiologic factors. Curr Opin Neurol. 1990:3. [Google Scholar]
- 98.Ritz B, Costello S. Geographic model and biomarker-derived measures of pesticide exposure and Parkinson’s disease. Ann N Y Acad Sci. 2006;1076:378–87. doi: 10.1196/annals.1371.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gatto NM, Cockburn M, Bronstein J, Manthripragada AD, Ritz B. Well-water consumption and Parkinson’s disease in rural California. Environ Health Perspect. 2009;117(12):1912–8. doi: 10.1289/ehp.0900852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caudle WM, Guillot TS, Lazo CR, Miller GW. Industrial toxicants and Parkinson’s disease. Neurotoxicology. 2012;33:178–88. doi: 10.1016/j.neuro.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hatcher JM, Miller GW, Pennell KD. Parkinson’s disease and pesticides: a toxicological perspective. Trends Pharmacol Sci. 2008;29:322–9. doi: 10.1016/j.tips.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Corrigan F, Wienburg C, Daniel S, Mann D. Organochlorine insecticides in substantia nigra in Parkinson’s disease. J Toxicol Environ Health Part A. 2000;59:229–34. doi: 10.1080/009841000156907. [DOI] [PubMed] [Google Scholar]
- 103.Corrigan FM, Murray L, Wyatt CL, Shore RF. Diorthosubstituted polychlorinated biphenyls in caudate nucleus in Parkinson’s disease. Exp Neurol. 1998;150:339–42. doi: 10.1006/exnr.1998.6776. [DOI] [PubMed] [Google Scholar]
- 104.Fleming L, Mann JB, Bean J, Briggle T, Sanchez-Ramos JR. Parkinson’s disease and brain levels of organochlorine pesticides. Ann Neurol. 1994;36:100–3. doi: 10.1002/ana.410360119. [DOI] [PubMed] [Google Scholar]
- 105.Hatcher-Martin JM, Gearing M, Steenland K, Levey AI, Miller GW, Pennell KD. Association between polychlorinated biphenyls and Parkinson’s disease neuropathology. Neurotoxicology. 2012;33:1298–304. doi: 10.1016/j.neuro.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Weisskopf MG, Knekt P, O’Reilly EJ, Lyytinen J, Reunanen A, Laden F, et al. Persistent organochlorine pesticides in serum and risk of Parkinson disease. Neurology. 2010;74:1055–61. doi: 10.1212/WNL.0b013e3181d76a93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miller GW, Kirby M, Levey AI, Bloomquist J. Heptachlor alters expression and function of dopamine transporters. Neurotoxicology. 1999;20:631–7. [PubMed] [Google Scholar]
- 108.Bemis JC, Seegal RF. PCB-induced inhibition of the vesicular monoamine transporter predicts reductions in synaptosomal dopamine content. Toxicol Sci. 2004;80:288–95. doi: 10.1093/toxsci/kfh153. [DOI] [PubMed] [Google Scholar]
- 109.Caudle WM, Richardson JR, Delea KC, Guillot TS, Wang M, Pennell KD, et al. Polychlorinated biphenyl-induced reduction of dopamine transporter expression as a precursor to Parkinson’s disease-associated dopamine toxicity. Toxicol Sci. 2006;92:490–9. doi: 10.1093/toxsci/kfl018. [DOI] [PubMed] [Google Scholar]
- 110.Caudle WM, Richardson JR, Wang M, Miller GW. Perinatal heptachlor exposure increases expression of presynaptic dopaminergic markers in mouse striatum. Neurotoxicology. 2005;26:721–8. doi: 10.1016/j.neuro.2004.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fonnum F, Mariussen E, Reistad T. Molecular mechanisms involved in the toxic effects of polychlorinated biphenyls (PCBs) and brominated flame retardants (BFRs) J Toxicol Environ Health Part A. 2006;69(1–2):21–35. doi: 10.1080/15287390500259020. [DOI] [PubMed] [Google Scholar]
- 112.Hatcher JM, Di Monte DA, Richardson JR, Guillot TS, McCormack AL, Jones DP, et al. Dieldrin exposure induces oxidative damage in the mouse nigrostriatal dopamine system. Exp Neurol. 2007;204:619–30. doi: 10.1016/j.expneurol.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kitazawa M, Anantharam V, Kanthasamy A. Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptotic cell death in dopaminergic cells. Free Radical Biol Med. 2001;31:1473–85. doi: 10.1016/s0891-5849(01)00726-2. [DOI] [PubMed] [Google Scholar]
- 114.Lee DW, Opanashuk LA. Polychlorinated biphenyl mixture aroclor 1254-induced oxidative stress plays a role in dopaminergic cell injury. Neurotoxicology. 2004;25:925–39. doi: 10.1016/j.neuro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 115.Richardson JR, Caudle WM, Wang M, Dean ED, Pennell KD, Miller GW. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson’s disease. FASEB J. 2006;20:1695–7. doi: 10.1096/fj.06-5864fje. [DOI] [PubMed] [Google Scholar]
- 116.Richardson JR, Miller GW. Acute exposure to aroclor 1016 or 1260 differentially affects dopamine transporter and vesicular monoamine transporter 2 levels. Toxicol Lett. 2004;148:29–40. doi: 10.1016/j.toxlet.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 117.Sanchez-Ramos J, Facca A, Basit A, Song S. Toxicity of dieldrin for dopaminergic neurons in mesencephalic cultures. Exp Neurol. 1998;150:263–71. doi: 10.1006/exnr.1997.6770. [DOI] [PubMed] [Google Scholar]
- 118.Seegal R, Brosch K, Bush B. Polychlorinated biphenyls produces regional alterations of dopamine metabolism in rat brain. Toxicol Lett. 1986;30:197–202. doi: 10.1016/0378-4274(86)90103-7. [DOI] [PubMed] [Google Scholar]
- 119.Seegal R, Bush B, Shain W. Lightly chlorinated ortho-substituted PCB congeners decrease dopamine in nonhuman primate brain and in tissue culture. Toxicol Appl Pharmacol. 1990;106:136–44. doi: 10.1016/0041-008x(90)90113-9. [DOI] [PubMed] [Google Scholar]
- 120.Seegal RF. The neurotoxicological consequences of developmental exposure to PCBs. Toxicol Sci. 2000;57:1–3. doi: 10.1093/toxsci/57.1.1. [DOI] [PubMed] [Google Scholar]
- 121.Seegal RF, Okoniewski R, Brosch K, Bemis JC. Polychlorinated biphenyls alter extraneuronal but not tissue dopamine concentrations in adult rat striatum: an in vivo microdialysis study. Environ Health Perspect. 2002;110:1113–7. doi: 10.1289/ehp.021101113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bradner JM, Suragh TA, Wilson WW, Lazo CR, Stout KA, Kim HM, et al. Exposure to the polybrominated diphenyl ether mixture DE-71 damages the nigrostriatal dopamine system: role of dopamine handling in neurotoxicity. Exp Neurol. 2013;241:138–47. doi: 10.1016/j.expneurol.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mariussen E, Morch Andersen J, Fonnum F. The effect of polychlorinated biphenyls on the uptake of dopamine and other neurotransmitters into rat brain synaptic vesicles. Toxicol Appl Pharmaco. 1999;161(3):274–82. doi: 10.1006/taap.1999.8806. [DOI] [PubMed] [Google Scholar]
- 124.Mariussen E, Fonnum F. The effect of brominated flame retardants on neurotransmitter uptake into rat brain synaptosomes and vesicles. Neurochem Int. 2003;43(4–5):533–42. doi: 10.1016/s0197-0186(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 125.Mariussen E. Neurotoxic effects of perfluoroalkylated compounds: mechanisms of action and environmental relevance. Arch Toxicol. 2012;86(9):1349–67. doi: 10.1007/s00204-012-0822-6. [DOI] [PubMed] [Google Scholar]
- 126•.Rilstone JJ, Alkhater RA, Minassian BA. Brain dopamine-serotonin vesicular transport disease and its treatment. N Engl J Med. 2013 doi: 10.1056/NEJMoa1207281. A case report highlighting a recessive mutation in a coding region of the VMAT2 gene causing infantile-onset parkinsonism in a consanguineous pedigree. [DOI] [PubMed] [Google Scholar]
- 127.Glatt CE, DeYoung JA, Delgado S, Service SK, Giacomini KM, Edwards RH, et al. Screening a large reference sample to identify very low frequency sequence variants: comparisons between 2 genes. Nat Genet. 2001;27(4):435–8. doi: 10.1038/86948. [DOI] [PubMed] [Google Scholar]
- 128.Burman J, Tran CH, Glatt C, Freimer NB, Edwards RH. The effect of rare human sequence variants on the function of vesicular monoamine transporter 2. Pharmacogenetics. 2004;14(9):587–94. doi: 10.1097/00008571-200409000-00003. [DOI] [PubMed] [Google Scholar]
- 129.Lin ZC, Zhao Y, Chung CY, Zhou YH, Xiong NA, Glatt CE, et al. High regulatability favors genetic selection in SLC18A2, a vesicular monoamine transporter essential for life. FASEB J. 2010;24(7):2191–200. doi: 10.1096/fj.09-140368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genetics. 2012;8(3):e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Glatt CE, Wahner AD, White DJ, Ruiz-Linares A, Ritz B. Gain-of-function haplotypes in the vesicular monoamine transporter promoter are protective for Parkinson disease in women. Hum Mol Genet. 2006;15(2):299–305. doi: 10.1093/hmg/ddi445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132•.Brighina L, Riva C, Bertola F, Saracchi E, Fermi S, Goldwurm S, et al. Analysis of vesicular monoamine transporter 2 polymorphisms in Parkinson’s disease. Neurobiol Aging. 2013 doi: 10.1016/j.neurobiolaging.2012.12.020. Identified 2 SNPs in the VMAT2 promoter that are associated with risk of PD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bohnen NI, Albin RL, Koeppe RA, Wernette KA, Kilbourn MR, Minoshima S, et al. Positron emission tomography of monoaminergic vesicular binding in aging and Parkinson disease. J Cerebral Blood Flow Metab. 2006;26(9):1198–212. doi: 10.1038/sj.jcbfm.9600276. [DOI] [PubMed] [Google Scholar]
- 134.Frey KA, Koeppe RA, Kilbourn MR, VanderBorght TM, Albin RL, Gilman S, et al. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Ann Neurol. 1996;40(6):873–84. doi: 10.1002/ana.410400609. [DOI] [PubMed] [Google Scholar]
- 135.Albin RL, Nichols TE, Frey KA. Brain imaging to assess the effects of dopamine agonists on progression of Parkinson disease. JAMA. 2002;288(3):311–2. author reply 2–3. [PubMed] [Google Scholar]
- 136.Hauser RA, Koller WC, Hubble JP, Malapira T, Busenbark K, Olanow CW. Time course of loss of clinical benefit following withdrawal of levodopa/carbidopa and bromocriptine in early Parkinson’s disease. Mov Disord. 2000;15(3):485–9. [PubMed] [Google Scholar]
- 137.Kish SJ, Zhong XH, Hornykiewicz O, Haycock JW. Striatal 3,4-dihydroxyphenylalanine decarboxylase in aging: disparity between postmortem and positron emission tomography studies? Ann Neurol. 1995;38(2):260–4. doi: 10.1002/ana.410380220. [DOI] [PubMed] [Google Scholar]
- 138.Tedroff J, Ekesbo A, Rydin E, Langstrom B, Hagberg G. Regulation of dopaminergic activity in early Parkinson’s disease. Ann Neurol. 1999;46(3):359–65. [PubMed] [Google Scholar]
- 139.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain. 1999;122(8):1437–48. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 140.Lee CS, Samii A, Sossi V, Ruth TJ, Schulzer M, Holden JE, et al. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson’s disease. Ann Neurol. 2000;47(4):493–503. doi: 10.1002/1531-8249(200004)47:4<493::aid-ana13>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 141.Kilbourn MR, Frey KA, Vander Borght T, Sherman PS. Effects of dopaminergic drug treatments on in vivo radioligand binding to brain vesicular monoamine transporters. Nucl Med Biol. 1996;23(4):467–71. doi: 10.1016/0969-8051(96)00023-6. [DOI] [PubMed] [Google Scholar]
- 142.Okamura N, Villemagne VL, Drago J, Pejoska S, Dhamija RK, Mulligan RS, et al. In vivo Measurement of vesicular monoamine transporter type 2 density in Parkinson Disease with F-18-AV-133. J Nucl Med. 2010;51(2):223–8. doi: 10.2967/jnumed.109.070094. [DOI] [PubMed] [Google Scholar]
- 143.Chao KT, Tsao HH, Weng YH, Hsiao IT, Hsieh CJ, Wey SP, et al. Quantitative analysis of binding sites for 9-fluoropropyl-(+)-dihydrotetrabenazine (18F AV-133) in a MPTP-lesioned PD mouse model. Synapse. 2012;66(9):823–31. doi: 10.1002/syn.21572. [DOI] [PubMed] [Google Scholar]
- 144.Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RM. Prevalence of orthostatic hypotension in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2011;17(10):724–9. doi: 10.1016/j.parkreldis.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jain S, Goldstein DS. Cardiovascular dysautonomia in Parkinson disease: from pathophysiology to pathogenesis. Neurobiol Dis. 2012;46(3):572–80. doi: 10.1016/j.nbd.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Orimo S, Ozawa E, Nakade S, Sugimoto T, Mizusawa H. I-123-metaiodobenzylguanidine myocardial scintigraphy in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1999;67(2):189–94. doi: 10.1136/jnnp.67.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Li S-T, Dendi R, Holmes C, Goldstein DS. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann Neurol. 2002;52(2):220–3. doi: 10.1002/ana.10236. [DOI] [PubMed] [Google Scholar]
- 148.Orimo S, Suzuki M, Inaba A, Mizusawa H. 123I-MIBG myocardial scintigraphy for differentiating Parkinson’s disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2012;18(5):494–500. doi: 10.1016/j.parkreldis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 149.Tijero B, Gomez-Esteban JC, Llorens V, Lezcano E, Gonzalez-Fernandez MC, de Pancorbo MM, et al. Cardiac sympathetic denervation precedes nigrostriatal loss in the E46K mutation of the alpha-synuclein gene (SNCA) Clin Autonomic Res. 2010;20(4):267–9. doi: 10.1007/s10286-010-0068-4. [DOI] [PubMed] [Google Scholar]
- 150.Wong KK, Raffel DM, Koeppe RA, Frey KA, Bohnen NI, Gilman S. Pattern of cardiac sympathetic denervation in idiopathic Parkinson Disease studied with 11C Hydroxyephedrine PET. Radiology. 2012 doi: 10.1148/radiol.12112723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Goldstein DS, Holmes C, Sewell L, Park MY, Sharabi Y. Sympathetic noradrenergic before striatal dopaminergic denervation: relevance to Braak staging of synucleinopathy. Clin Autonomic Res. 2012;22(1):57–61. doi: 10.1007/s10286-011-0136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Goldstein DS, Holmes C, Kopin IJ, Sharabi Y. Intra-neuronal vesicular uptake of catecholamines is decreased in patients with Lewy body diseases. J Clin Invest. 2011;121(8):3320–30. doi: 10.1172/JCI.45803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Eisenhofer G, Hovevey-Sion D, Kopin IJ, Miletich R, Kirk KL, Finn R, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol ExpTherapeut. 1989;248(1):419–27. [PubMed] [Google Scholar]
- 154.Lotharius J, Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Hum Mol Genet. 2002;11(20):2395–407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- 155.Pletscher A. Platelets as models: use and limitations. Experientia. 1988;44(2):152–5. doi: 10.1007/bf01952200. [DOI] [PubMed] [Google Scholar]
- 156.Paul SM, Rehavi M, Skolnick P, Ballenger JC, Goodwin FK. Depressed patients have decreased binding of tritiated imipramine to platelet serotonin transporter. Arch Gen Psychiatry. 1981;38(12):1315–7. doi: 10.1001/archpsyc.1981.01780370017001. [DOI] [PubMed] [Google Scholar]
- 157.Weizman R, Carmi M, Tyano S, Rehavi M. Reduced 3H-imipramine binding but unaltered 3H-serotonin uptake in platelets of adolescent enuretics. Psychiatry Res. 1986;19(1):37–42. doi: 10.1016/0165-1781(86)90090-9. [DOI] [PubMed] [Google Scholar]
- 158.Weizman R, Carmi M, Tyano S, Apter A, Rehavi M. High affinity [3H]imipramine binding and serotonin uptake to platelets of adolescent females suffering from anorexia nervosa. Life Sci. 1986;38(13):1235–42. doi: 10.1016/0024-3205(86)90179-7. [DOI] [PubMed] [Google Scholar]
- 159.Weizman A, Carmi M, Hermesh H, Shahar A, Apter A, Tyano S, et al. High-affinity imipramine binding and serotonin uptake in platelets of eight adolescent and ten adult obsessive-compulsive patients. Am J Psychiatry. 1986;143(3):335–9. doi: 10.1176/ajp.143.3.335. [DOI] [PubMed] [Google Scholar]
- 160.Yubero-Lahoz S, Robledo P, Farre M, de la Torre R. Platelet SERT as a peripheral biomarker of serotonergic neurotransmission in the central nervous system. Curr Med Chem. 2013 doi: 10.2174/0929867311320110003. [DOI] [PubMed] [Google Scholar]
- 161.Zalsman G, Aslanov-Farbstein D, Rehavi M, Roz N, Vermeiren R, Laor N, et al. Platelet vesicular monoamine transporter 2 density in the disruptive behavior disorders. J Child Adolesc Psychopharmacol. 2011;21(4):341–4. doi: 10.1089/cap.2010.0148. [DOI] [PubMed] [Google Scholar]
- 162.Zalsman G, Rehavi M, Roz N, Laor N, Weizman A, Toren P. Altered affinity of the platelet vesicular monoamine transporter 2 to dihydrotetrabenazine in children with major depression. J Neural Transm. 2011;118(9):1383–7. doi: 10.1007/s00702-011-0643-4. [DOI] [PubMed] [Google Scholar]
- 163.Schwartz K, Iancu I, Stryjer R, Chelben J, Kotler M, Weizman A, et al. Reduced platelet vesicular monoamine transporter density in smoking schizophrenia patients. Eur Neuropsychopharmacol. 2005;15(5):557–61. doi: 10.1016/j.euroneuro.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 164.Schwartz K, Weizman A, Rehavi M. Decreased platelet vesicular monoamine transporter density in habitual smokers. Eur Neuropsychopharmacol. 2005;15(2):235–8. doi: 10.1016/j.euroneuro.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 165.Toren P, Rehavi M, Luski A, Roz N, Laor N, Lask M, et al. Decreased platelet vesicular monoamine transporter density in children and adolescents with attention deficit/hyperactivity disorder. Eur Neuropsychopharmacol. 2005;15(2):159–62. doi: 10.1016/j.euroneuro.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 166.Zucker M, Aviv A, Shelef A, Weizman A, Rehavi M. Elevated platelet vesicular monoamine transporter density in untreated patients diagnosed with major depression. Psychiatry Res. 2002;112(3):251–6. doi: 10.1016/s0165-1781(02)00223-8. [DOI] [PubMed] [Google Scholar]
- 167.Zucker M, Valevski A, Weizman A, Rehavi M. Increased platelet vesicular monoamine transporter density in adult schizophrenia patients. Eur Neuropsychopharmacol. 2002;12(4):343–7. doi: 10.1016/s0924-977x(02)00041-x. [DOI] [PubMed] [Google Scholar]
- 168.Zucker M, Weizman A, Harel D, Rehavi M. Changes in vesicular monoamine transporter (VMAT2) and synaptophysin in rat Substantia nigra and prefrontal cortex induced by psychotropic drugs. Neuropsychobiology. 2001;44(4):187–91. doi: 10.1159/000054941. [DOI] [PubMed] [Google Scholar]
- 169.Zucker M, Weizman A, Rehavi M. Characterization of high-affinity [3H]TBZOH binding to the human platelet vesicular monoamine transporter. Life Sci. 2001;69(19):2311–7. doi: 10.1016/s0024-3205(01)01301-7. [DOI] [PubMed] [Google Scholar]
- 170.Sala G, Brighina L, Saracchi E, Fermi S, Riva C, Carrozza V, et al. Vesicular monoamine transporter 2 mRNA levels are reduced in platelets from patients with Parkinson’s disease. J Neural Transm. 2010;117(9):1093–8. doi: 10.1007/s00702-010-0446-z. [DOI] [PubMed] [Google Scholar]