

Abstract
Aims/Introduction: Excessive intake of sucrose can cause severe health issues, such as diabetes mellitus. In animal studies, consumption of a high‐sucrose diet (SUC) has been shown to cause obesity, insulin resistance and glucose intolerance. However, several in vivo experiments have been carried out using diets with much higher sucrose contents (50–70% of the total calories) than are typically ingested by humans. In the present study, we examined the effects of a moderate SUC on glucose metabolism and the underlying mechanism.
Materials and Methods: C57BL/6J mice received a SUC (38.5% sucrose), a high‐starch diet (ST) or a control diet for 5 weeks. We assessed glucose tolerance, incretin secretion and liver glucose metabolism.
Results: An oral glucose tolerance test (OGTT) showed that plasma glucose levels in the early phase were significantly higher in SUC‐fed mice than in ST‐fed or control mice, with no change in plasma insulin levels at any stage. SUC‐fed mice showed a significant improvement in insulin sensitivity. Glucagon‐like peptide‐1 (GLP‐1) secretion 15 min after oral glucose administration was significantly lower in SUC‐fed mice than in ST‐fed or control mice. Hepatic glucokinase (GCK) activity was significantly reduced in SUC‐fed mice. During the OGTT, the accumulation of glycogen in the liver was suppressed in SUC‐fed mice in a time‐dependent manner.
Conclusions: These results indicate that mice that consume a moderate SUC show glucose intolerance with a reduction in hepatic GCK activity and impairment in GLP‐1 secretion. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2012.00208.x, 2012)
Keywords: Glucagon‐like peptide‐1, Glucokinase, High‐sucrose diet
Introduction
The term ‘sugars’ is commonly used to describe mono‐ and disaccharides, which represent an important part of total caloric intake1. Glucose, fructose and sucrose are the most commonly consumed sugars. Approximately 10% of daily caloric intake can be attributed to fructose, and fructose consumption has dramatically increased2,3. Excessive ingestion of sucrose promotes the development of type 2 diabetes mellitus, which is associated with obesity and insulin resistance1. Animal studies have shown that diets extremely high in sucrose cause numerous metabolic abnormalities, such as obesity, insulin resistance, glucose intolerance and dyslipidemia4. The impact of a high‐sucrose diet (SUC) on insulin sensitivity remains controversial. Administration of a SUC (50–70% of the total calories) for 4–8 weeks induced insulin resistance in the liver and skeletal muscle5,6. However, in some experiments, mice fed a SUC (35–56% of the total calories) for 15–40 weeks developed glucose intolerance with enhanced insulin sensitivity7,8.
It is well recognized that the liver plays an important role in glucose homeostasis, in the rapid clearance of glucose in the postprandial state, and in the controlled production of glucose in the postabsorptive state9. The conversion of glucose into glycogen is a key pathway by which the liver removes glucose from the portal vein after a meal9. Hepatic glucokinase (GCK) plays a key role in glucose metabolism, as highlighted by the anomalies associated with Gck mutations10 and by the consequences of tissue‐specific knock‐out experiments11. Hepatic GCK, by which phosphorylation of glucose is a rate‐determining step in glucose uptake and glycogen synthesis12,13, is responsible for postprandial glucose disposal14. Insulin positively regulates Gck gene expression in the liver, and thereby stimulates hepatic glucose uptake and glycogen synthesis15. Expression of the hepatic Gck gene is reduced in diabetic animals with insulin deficiency and insulin resistance16,17.
The incretins glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP) play a major role in glucose homeostasis through stimulation of insulin secretion and suppressing glucagon secretion, thereby contributing to limiting postprandial glucose excursions18,19. Several nutrients, including triglycerides, fatty acids, proteins and carbohydrates, stimulate incretin secretion18,20–22. Among these nutrients, glucose is one of the most potent stimulators of incretin secretion in rodents and humans20–24. The two incretin hormones are responsible for approximately 50–70% of the postprandial insulin responses in healthy individuals25. Several studies have shown a significant reduction in GLP‐1 levels after mixed‐meal ingestion in type 2 diabetes patients25–27. However, whether chronic high‐carbohydrate ingestion can change incretin secretion remains unknown.
The aim of the present study was to examine the effects of a moderate SUC (38.5% of the total calories) on glucose metabolism and the effects of chronic high‐carbohydrate (corn starch or sucrose) ingestion on incretin secretion. The present findings show that consumption of a moderate SUC for 5 weeks results in glucose intolerance with a reduction in hepatic GCK expression and activity and impairment in GLP‐1 secretion.
Materials and Methods
Animals and Diets
Twelve‐week‐old male C57BL/6J mice were obtained from Japan SLC (Shizuoka, Japan) and housed in a temperature‐controlled room under a standard 12‐h light/dark cycle. All procedures were carried out according to a protocol approved by the Nagoya University Institutional Animal Care and Use Committee. Mice were fed a normal chow diet of CE‐2 (CLEA Japan, Osaka, Japan), containing 58.2% carbohydrates, 29.2% protein and 12.6% fat as energy content. After adaptation for 2 weeks, they were divided into three groups and fed a normal chow diet (NC), a high‐starch diet (ST) supplemented with 38.5% corn starch or a SUC containing 38.5% sucrose; the latter two diets were prepared by the addition of corn starch or sucrose, respectively, to CE‐2 (Table 1). Mice were fasted for 16 h or were re‐fed for 12 h after 24 h of starvation.
Table 1. Composition of experimental diets.
| NC | ST | SUC | |
|---|---|---|---|
| Protein | 29.2 | 18.0 | 18.0 |
| Fat | 12.6 | 7.7 | 7.7 |
| Carbohydrates | 58.2 | 74.3 | 74.3 |
| (Sucrose) | – | – | (38.5) |
Data are expressed as % of total energy. NC, a normal chow diet; ST, a high‐starch diet; SUC, a high‐sucrose diet.
Plasma Biochemical Analyses
Blood glucose levels were measured with ANTSENSE II (Bayer Medical, Leverkusen, Germany). Plasma levels of insulin were determined by ELISA kit (Morinaga, Tokyo, Japan). Plasma triglycerides and free fatty acid levels were determined using the Triglyceride E test and NEFA C test (Wako Pure Chemical, Osaka, Japan), respectively. Plasma levels of total GIP and GLP‐1 were determined using the GIP (TOTAL) ELISA kit (Linco Research, St. Charles, MO, USA) and an electrochemiluminescent sandwich immunoassay (Meso Scale Discovery, Gaithersburg, MD, USA), respectively.
Glucose Tolerance Test, Insulin Tolerance Test and Pyruvate Tolerance Test
Oral and intravenous glucose tolerance tests (OGTT and IVGTT, respectively) were carried out after 5 weeks of feeding with the NC, SUC or ST. After 16 h of food deprivation, glucose was given either orally at a dose of 2 g/kg (OGTT) or intravenously at a dose of 2 g/kg (IVGTT). After administration, blood was collected at 0, 10, 15 and 60 min for the measurement of glucose and insulin. For the insulin tolerance test (ITT), mice were deprived of food for 6 h before the test. Insulin was injected intraperitoneally at a dose of 0.6 U/kg. Blood was collected 0, 30, 60, 90 and 120 min after insulin injection. For the pyruvate tolerance test, the mice were deprived of food for 16 h and then injected intraperitoneally with pyruvate dissolved in saline (2 g/kg). Blood was collected 0, 15, 30, 60 and 90 min after the injection of pyruvate.
Isolation of Tissue Ribonucleic Acid and Quantitative Real‐Time Reverse Transcription Polymerase Chain Reaction
Extraction of total ribonucleic acid (RNA), synthesis of complementary deoxyribonucleic acid (DNA) and quantitative real‐time reverse transcription‐polymerase chain reaction (RT–PCR) were carried out as previously described28. The primer sequences are shown in Table S1. The messenger RNA (mRNA) levels were normalized with respect to those of 36B4.
Measurement of Hepatic Glycogen and Triglyceride Content
For the determination of hepatic glycogen content, 100 mg of liver sample was digested in 0.5 mL of 1 mmol/L potassium hydroxide for 30 min in a 70°C water bath. The samples were briefly centrifuged, and 100 μL of the supernatant was removed and neutralized with 17 μL of 17.4 mol/L acetic acid. Glycogen was enzymatically cleaved to glucose by addition of 500 μL of 0.3 mol/L acetate buffer with 0.5% amyloglucosidase at 37°C. Glucose concentration was then measured using a Glucose C II test (Wako Pure Chemical)29. For the determination of hepatic triglyceride content, 100–200 mg of liver sample was homogenized for 10 min in 4 mL isopropanol with a Polytron disrupter. Triglyceride content was measured using a Triglyceride E test (Wako Pure Chemical)30.
Measurement of GCK Activity
GCK activity was measured as previously described31. Briefly, liver was homogenized with the sample buffer, and a part of the homogenate was centrifuged for 90 min at 40,000 g at 4°C. Thereafter, the supernatant was harvested. The final glucose concentration in the assay mixture (200 mmol/L HEPES, 50 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L NAD+, 5 mmol/L adenosine triphosphate, 100 μg/mL bovine serum albumin and 1 unit/mL glucose 6‐phosphate dehydrogenase) was adjusted to 0.5 or 50 mmol/L. The assay mixture (990 μL) was incubated for 5 min at 37°C, and the reaction was then started by the addition of 10 μL of the supernatant. The reaction velocity was measured as the rate of increase in absorbance monitored at 340 nm 2–3 min after initiation of the reaction. One unit was the amount of enzyme that catalyzed the phosphorylation of 1 μmol of glucose per minute. GCK activity was calculated using subtraction.
Immunoblotting Analysis
After 16 h of food deprivation, 5 units of human regular insulin (Eli Lilly, Indianapolis, IN, USA) were injected intravenously. The liver and skeletal muscle tissues samples were rapidly extracted 3 min after the injection. Immunoblotting analysis was then carried out as previously described28, by using an antitotal AKT antibody (at a dilution of 1:1000; Cell Signaling Technology, Beverly, MA, USA), an antiphospho‐AKT (Ser 473) antibody (at a dilution of 1:1000; Cell Signaling Technology), an antiglycogen synthase kinase‐3β (GSK‐3β) antibody (at a dilution of 1:1000; BD Biosciences, Sparks, MD, USA), or an antiphospho‐GSK‐3β (Ser 9) antibody (at a dilution of 1:1000; Cell Signaling Technology). The intensity of the signals was quantified using NIH image software (National Institutes of Health, Bethesda, MD, USA). Phosphorylation was expressed as a percentage of the amount of phospho‐AKT or phospho‐GSK‐3β relative to the total amount of AKT or GSK‐3β, respectively.
Statistical Analysis
Results are expressed as mean ± SEM of values obtained from several experiments, and statistical significance was evaluated using analysis of variance (anova) with Bonferroni post‐hoc tests. P < 0.05 was considered statistically significant.
Results
Effects of Carbohydrates on Metabolic Parameters
To examine the effects of a diet containing high levels of not only sucrose, but also carbohydrates, we divided mice into three groups: NC, ST and SUC. After 5 weeks on the respective diets, no differences in bodyweight or energy intake were observed among the groups. Furthermore, no significant differences in the levels of plasma glucose, triglycerides or free fatty acids in either a fasted or a fed state were observed, although insulin levels in the fed state were higher in ST‐fed mice than in the other groups (Table 2).
Table 2. Bodyweight, insulin and metabolic parameters in fasted and fed state.
| Fasted | Fed | |||||
|---|---|---|---|---|---|---|
| NC | ST | SUC | NC | ST | SUC | |
| Bodyweight (g) | 26.7 ± 0.3 | 27.6 ± 0.8 | 27.5 ± 0.3 | |||
| Energy intake (kJ/day) | 48.0 ± 1.6 | 46.6 ± 0.4 | 47.5 ± 0.8 | |||
| Glucose (mmol/L) | 5.76 ± 0.21 | 5.69 ± 0.05 | 5.74 ± 0.29 | 11.4 ± 0.8 | 11.8 ± 0.5 | 12.5 ± 0.6 |
| Insulin (pmol/L) | 70.6 ± 14.8 | 63.4 ± 14.0 | 43.6 ± 7.8 | 145.6 ± 13.8 | 204.4 ± 11.1* | 138.9 ± 15.7 |
| Triglycerides (mmol/L) | 1.05 ± 0.08 | 1.23 ± 0.13 | 1.21 ± 0.07 | 1.28 ± 0.15 | 1.13 ± 0.09 | 1.17 ± 0.17 |
| Free fatty acids (mmol/L) | 1.54 ± 0.09 | 1.59 ± 0.11 | 1.45 ± 0.13 | 0.63 ± 0.09 | 0.61 ± 0.07 | 0.56 ± 0.08 |
Data are expressed as means ± SEM of values. *P < 0.05 compared with normal chow diet (NC)‐fed mice and high‐sucrose diet (SUC)‐fed mice, n = 4 or more per group. ST, a high‐starch diet.
Glucose Tolerance Test and Insulin Tolerance Test
The OGTT showed no differences in blood glucose levels at baseline or 60 min among the groups (Figure 1a). Plasma glucose levels in SUC‐fed mice were significantly higher 10 and 15 min after oral glucose administration than those in NC‐ and ST‐fed mice (Figure 1a), whereas insulin levels were similar among the groups at all times (Figure 1b). To investigate the ability of pancreatic islets to secrete insulin, we carried out an IVGTT. An IVGTT shows glucose‐stimulated insulin secretion in vivo, because glucose does not pass through the portal vein after an intravenous glucose injection32. The IVGTT showed no differences among the groups in glucose concentrations after intravenous administration of glucose at the indicated periods (Figure 1c). Insulin levels did not differ among the groups at any time (Figure 1d). The ITT showed that blood glucose levels after insulin injection were significantly lower in the SUC‐fed mice than in the NC‐ and ST‐fed mice (Figure 1e). Insulin‐induced phosphorylation of AKT in the skeletal muscle was enhanced in SUC‐fed mice (Figure 1f). These results show that the ability to secrete insulin is conserved, and that peripheral insulin sensitivity is enhanced in SUC‐fed mice.
Figure 1.
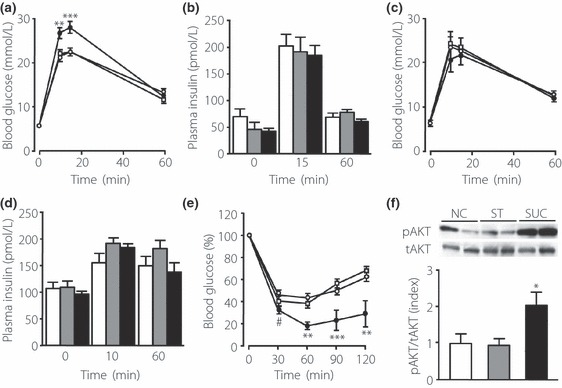
Characterization of glucose tolerance and insulin sensitivity in mice fed a high‐starch diet (ST) and a high‐sucrose diet (SUC), after 5 weeks of feeding. (a) Blood glucose levels during an oral glucose tolerance test (OGTT) in mice fed a normal chow diet (NC; open circle, n = 11), a ST (open square, n = 10) or a SUC (filled circle, n = 12). Glucose was given orally at a dose of 2 g/kg. (b) Plasma insulin levels during the OGTT in mice fed a NC (white bars), a ST (gray bars) or a SUC (black bars). (c) Blood glucose levels during an intravenous glucose tolerance test (IVGTT). Glucose (2 g/kg) was given intravenously to mice fed a NC (open circle, n = 8), a ST (open square, n = 9) or a SUC (filled circle, n = 5). (d) Plasma insulin levels during the IVGTT in mice fed a NC (white bars), a ST (gray bars) or a SUC (black bars). (e) Blood glucose levels during the insulin tolerance test in mice fed a NC (open circle, n = 10), a ST (open square, n = 12) or a SUC (filled circle, n = 7). Insulin was injected intraperitoneally at a dose of 0.6 U/kg. (f) Representative immunoblots and quantification of AKT phosphorylation by insulin in the skeletal muscle. AKT phosphorylation is expressed as the ratio of phospho‐AKT (pAKT) relative to the total amount of AKT. White, gray and black bars indicate data from mice fed a NC (n = 5), a ST (n = 5) or a SUC (n = 5), respectively. Values are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 compared with the NC‐ and ST‐fed mice at the indicated time‐points. #P < 0.05 compared with the NC‐fed mice at the indicated time points. tAKT, total AKT.
Effects of Carbohydrates on Incretin Secretion
No differences in plasma GLP‐1 levels in the fasting state were observed among the groups (Figure 2b). Oral glucose administration induced GLP‐1 secretion, and peak plasma GLP‐1 levels were observed at 15 min (Figure 2a). An unexpected finding is that plasma GLP‐1 levels 15 min after oral glucose administration were significantly lower in the SUC‐fed mice than in the NC‐ and ST‐fed mice (Figure 2b). Plasma GIP levels at baseline were similar among the groups (Figure 2d). Peak GIP levels were observed 15 min after oral glucose administration (Figure 2c), and GIP levels at 15 min were not suppressed in the SUC‐fed mice (Figure 2d).
Figure 2.
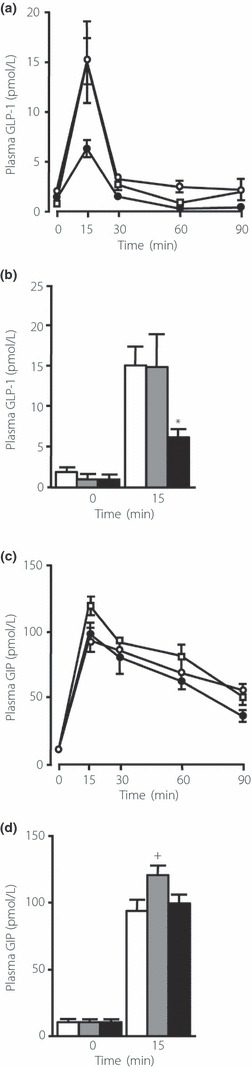
Effects of carbohydrates on incretin secretion. (a) Time course of total glucagon‐like peptide‐1 (GLP‐1) levels after oral glucose administration in mice fed a normal chow diet (NC; open circle), a high‐starch diet (ST; open square), or a high‐sucrose diet (SUC; filled circle). (b) The plasma levels of total GLP‐1 at baseline and 15 min after oral glucose loading in mice fed a NC (white bars, n = 15), a ST (gray bars, n = 11–15), or a SUC (black bars, n = 16). (c) Time course of total glucose‐dependent insulinotropic polypeptide (GIP) levels after oral glucose administration in mice fed a NC (open circle), a ST (open square) or a SUC (filled circle). (d) The plasma levels of total GIP at baseline and 15 min after oral glucose loading in mice fed a NC (white bars, n = 13–14), a ST (gray bars, n = 11–18) or a SUC (black bars, n = 14–17). Values are mean ± SEM. *P < 0.05 compared with the NC‐ and ST‐fed mice. +P < 0.05 compared with the NC‐ and SUC‐fed mice.
Hepatic Glucose Metabolism in SUC‐Fed Mice
To investigate the effects of carbohydrates on hepatic glucose metabolism, we carried out a pyruvate tolerance test. As shown in Figure 3a, glucose levels throughout the pyruvate tolerance test were significantly lower in the SUC‐fed mice than in the NC‐ and ST‐fed mice. Glucose‐6‐phosphatase (G6pc) mRNA expression in the fasted state was comparable among the groups, whereas phosphoenolpyruvate carboxykinase (Pepck) mRNA expression increased in the SUC‐fed mice (Figure 3b). The expression of G6pc and Pepck mRNA in the re‐fed state was comparable among the groups (Figure 3b). Thereafter, we assessed glycogen content in the liver. Storage of glycogen in the liver did not differ among the groups in the fed state (NC 60.7 ± 2.6 mg/g liver; ST 55.3 ± 3.2 mg/g liver; SUC 61.3 ± 2.5 mg/g liver), and after 16 h of fasting, glycogen content was undetectable in all groups. However, accumulation of glycogen throughout the OGTT was significantly suppressed in the SUC‐fed mice 60 and 120 min after oral glucose administration (Figure 3c). The expression levels of Gck mRNA were lower in the fasting SUC‐fed mice than in the NC‐fed mice (Figure 3d). Total GCK activity during fasting was reduced by approximately 50% in the SUC group compared with the other groups, showing decreased GCK expression in the SUC‐fed mice (Figure 3e).
Figure 3.
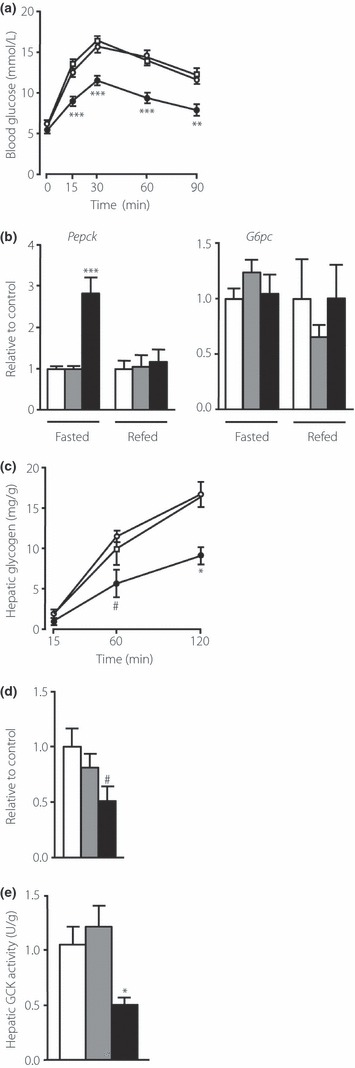
Effects of carbohydrates on hepatic glucose metabolism. (a) Blood glucose levels during the pyruvate tolerance test in mice fed normal chow diet (NC; open circle, n = 8), a high‐starch diet (ST; open square, n = 10) or a high‐sucrose diet (SUC; filled circle, n = 6). Pyruvate was given intravenously at a dose of 2 g/kg. (b) Messenger ribonucleic acid (mRNA) expression of glucose‐6‐phosphatase (G6pc) and phosphoenolpyruvate carboxykinase (Pepck) in the liver in fasted and re‐fed states. (c) Hepatic glycogen content during an oral glucose tolerance test in mice fed a NC (open circle, n = 3–5), a ST (open square, n = 3) or a SUC (filled circle, n = 3–5). (d) The mRNA expression of glucokinase (Gck) in the liver in a fasted state. In (b) and (d), mRNA abundance relative to 36B4 was determined using real‐time polymerase chain reaction (PCR) and expressed as a fold increase relative to the control. White, gray and black bars indicate data from mice fed a NC (n = 7–8), a ST (n = 6–7) or a SUC (n = 5–8), respectively. (e) Total GCK activity in the livers of mice fed a NC (white bars, n = 4), a ST (gray bars, n = 4) or a SUC (black bars, n = 4). Values are mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 compared with the NC‐ and ST‐fed mice at the indicated time‐points. #P < 0.05 compared with the NC‐fed mice at the indicated time‐points.
Lipid Metabolism in the Liver
Next we examined lipid metabolism. Hematoxylin–eosin staining showed no vacuolization in the liver in the SUC‐fed mice (data not shown). However, hepatic triglyceride content in the fed state was significantly higher in the SUC‐fed mice than in the NC‐ and ST‐fed mice (Figure 4a). In the fed state, gene expression of lipogenic enzymes, including fatty acid synthase, acetyl‐CoA carboxylase α (Acaca) and stearoyl‐CoA desaturase 1 (Scd1), was significantly higher in the livers of the SUC‐fed mice than in the livers of the NC‐fed mice; furthermore, the expression of Acaca and Scd1 mRNA was higher than in the NC‐ and ST‐fed mice (Figure 4b). Gene expression of lipolytic enzymes, including acyl‐CoA oxidase, medium‐chain acyl‐CoA dehydrogenase and carnitine palmitoyltransferase 1α, in the liver was comparable among the groups (Figure 4c). However, the ratio of insulin‐induced phosphorylation of AKT and GSK‐3β in the liver was similar among the groups (Figure 4d,e), showing that the insulin signal was conserved in the liver of the SUC‐fed mice.
Figure 4.
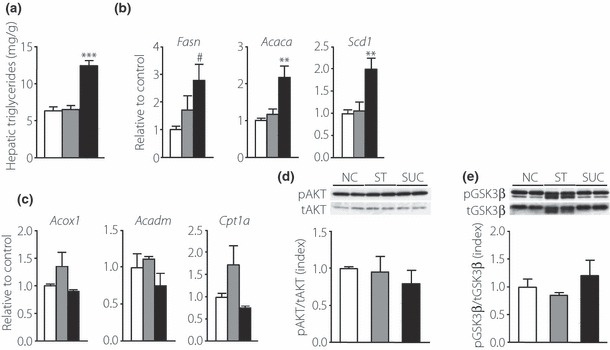
Effects of carbohydrates on hepatic lipid metabolism. (a) Hepatic triglyceride content in the livers of mice fed a normal chow diet (NC; white bars, n = 6), a high‐starch diet (ST; gray bars, n = 8) or a high‐sucrose diet (SUC; black bars, n = 9). (b) Messenger ribonucleic acid (mRNA) expression of the lipogenic enzymes fatty acid synthase (Fasn), acetyl‐CoA carboxylase α (Acaca) and stearoyl‐CoA desaturase 1 (Scd1) and (c) the lipolytic enzymes acyl‐CoA oxidase (Acox1), medium‐chain acyl‐CoA dehydrogenase (Acadm), and carnitine palmitoyltransferase 1α (Cpt1a) in the livers of mice fed a NC (white bars, n = 6), a ST (gray bars, n = 7–8) or a SUC (black bars, n = 5–7). The abundance of mRNA relative to 36B4 mRNA was determined using real‐time polymerase chain reaction, and is expressed as a fold increase relative to the control. (d, e) Insulin‐induced phosphorylation of AKT and glycogen synthase kinase‐3β (GSK‐3β) in the liver. Mice were injected intravenously with insulin. Liver was extracted 3 min after the injection, and immunoblotted with indicated antibodies. (d) Representative immunoblots and quantification of the phosphorylation of AKT by insulin. AKT phosphorylation is expressed as the ratio of phospho‐AKT (pAKT) relative to the total amount of AKT. (e) Representative immunoblots and quantification of the phosphorylation of GSK‐3β by insulin. GSK‐3β phosphorylation is expressed as the ratio of phospho‐GSK‐3β (pGSK‐3β) relative to the total amount of GSK‐3β. White, gray and black bars indicate data from mice fed a NC (n = 3), a ST (n = 3) or a SUC (n = 3), respectively. Values are mean ± SEM. **P < 0.01; ***P < 0.001 compared with the NC‐ and ST‐fed mice. #P < 0.05 compared with the NC‐fed mice. tAKT, total AKT; tGSK‐3β, total GSK‐3β.
Discussion
In the present study, we observed hyperglycemia in the early phase after oral glucose administration. The liver is known to play an important role in the disposal of an oral glucose load in the early phase, whereas peripheral tissues play a role in the late phase after oral glucose loading33,34. These findings suggest that abnormal glucose metabolism in the liver caused glucose intolerance in the SUC‐fed mice.
The impaired hepatic glucose metabolism in type 2 diabetes involves both the fasting state, in which gluconeogenesis appears to be increased35,36, and the fed state, in which insulin suppression of glucose production is reduced37. Experimental studies have shown that consumption of sucrose enhances gluconeogenesis with impaired insulin sensitivity, resulting in impaired glucose metabolism in the liver38–40. We found that liver glycogen was completely depleted after a 16‐h fast, indicating that glucose production through glycogenolysis was minimized. Furthermore, we showed that glucose production from pyruvate was suppressed, which conflicts with the expression of G6pc and Pepck mRNA in a fasting state. The pyruvate tolerance test is known to measure the capacity of the liver to convert pyruvate to glucose, which would normally be inhibited by insulin. In the present study, hepatic lipogenesis was enhanced in SUC‐fed mice. Enhanced lipogenesis in the liver might increase fatty acid synthesis, and thereby reduce glucose production, from pyruvate. Therefore, the present results show that gluconeogenesis in SUC‐fed mice is suppressed in a fasted state.
A previous study showed that hepatic glucose uptake was reduced, and that hepatic glucose excursion was increased in patients with type 2 diabetes, because inadequate activation of hepatic GCK presumably decreased the uptake of extracellular glucose15. Clamp studies in animals with euglycemic hyperinsulinemia showed that impaired insulin action in the liver reduced glucose disposal in SUC‐fed rats, although a change in hepatic GCK activity was not examined5,6,41. Thus, we investigated GCK activity and glycogen storage in the liver, which represents glucose uptake. We showed that hepatic GCK expression and activity were attenuated in the SUC‐fed mice, and that glycogen storage was suppressed during the OGTT, suggesting that reduced glucose disposal through the reduction of GCK activity is responsible for glucose intolerance in mice fed a moderate SUC.
As mentioned previously, impaired insulin action is thought to modify GCK activity in the livers of SUC‐fed mice. An unexpected finding from the present study was that insulin‐induced phosphorylation of AKT and GSK‐3β did not change in the SUC‐fed mice, showing that the insulin signal is conserved in the liver. Thus, this finding suggests that a SUC reduces hepatic GCK expression and activity independent of insulin action. Further investigations are required to clarify the mechanism of reduced GCK expression in the liver of SUC‐fed mice.
We found elevated triglyceride content and lipogenic enzyme expression with conserved hepatic action of insulin in the SUC‐fed mice. Liver fat accumulation is proposed to link obesity and insulin resistance. Whether liver fat accumulation is a result or a cause of peripheral insulin resistance and glucose intolerance remains controversial42. Exposure of the liver to large quantities of fructose (and sucrose‐containing fructose) leads to rapid stimulation of lipogenesis and accumulation of triglycerides, which in turn contributes to reduced insulin sensitivity and hepatic insulin resistance/glucose intolerance43. Accordingly, our observations suggest that liver fat accumulation is not a result of insulin resistance.
Previous studies have shown that SUC feeding for a long period decreases insulin secretion41,44. In our experiments, no apparent decrease in insulin secretion was observed. However, we assumed that insulin secretion was relatively impaired in this model. In fact, we found that serum insulin levels were similar throughout the OGTT, although serum glucose levels in SUC‐fed mice were higher in the early phase after oral glucose administration, but not after intravenous glucose administration. We further showed impaired GLP‐1 secretion in response to oral glucose loading in SUC‐fed mice. These findings therefore suggest that insulin secretion is relatively impaired throughout the OGTT in SUC‐fed mice.
An animal study showed lower basal plasma GLP‐1 levels and diminished GLP‐1 response to oral glucose administration in mice on a high‐fat diet compared with those on a low‐fat diet45. High levels of non‐esterified fatty acids during fasting and after meals, accompanied by insulin resistance, have been speculated to inhibit nutrient‐mediated GLP‐1 secretion in obese individuals46. However, the effects of a high‐carbohydrate diet on incretin secretion have not yet been reported. In the present study, we found impaired GLP‐1 secretion, but not GIP secretion, in response to oral glucose loading in the SUC‐fed mice, but not in the ST‐fed mice; ours is the first study that determined the effects of chronic high‐carbohydrate ingestion on incretin secretion. Our observations are consistent with those of previous studies in humans that postprandial GLP‐1 levels were reduced in patients with type 2 diabetes compared with healthy subjects, but that GIP secretion remained relatively intact26,27,47. This finding suggests that excess ingestion of sucrose contributes to impaired GLP‐1 secretion in patients with type 2 diabetes. A reduction in insulin signaling in the peripheral tissues could not explain impaired GLP‐1 secretion in this model. Therefore, further investigations of the effects of a SUC on the function of intestinal L‐cells are required to clarify this mechanism.
In conclusion, we showed that a moderate SUC results in glucose intolerance with a reduction in hepatic GCK expression and activity, and impaired GLP‐1 secretion.
Supplementary Material
Table S1 Primers used for quantitative real‐time polymerase chain reaction
Supporting info item
Acknowledgements
We thank Ken‐ichi Miyamoto (Department of Molecular Nutrition, The University of Tokushima Graduate School) for helpful suggestions. We also thank Michiko Yamada and Mayumi Katagiri for technical assistance. This study was supported by a grant‐in‐aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (no. 22790855). The authors declare no conflicts of interest.
References
- 1.Laville M, Nazare JA. Diabetes, insulin resistance and sugars. Obes Rev 2009; 10(Suppl 1): 24–33 [DOI] [PubMed] [Google Scholar]
- 2.Bray GA, Nielsen SJ, Popkin BM. Consumption of high‐fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr 2004; 79: 537–543 [DOI] [PubMed] [Google Scholar]
- 3.Vos MB, Kimmons JE, Gillespie C, et al. Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med 2008; 10: 160 [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson RJ, Segal MS, Sautin Y, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr 2007; 86: 899–906 [DOI] [PubMed] [Google Scholar]
- 5.Pagliassotti MJ, Shahrokhi KA, Moscarello M. Involvement of liver and skeletal muscle in sucrose‐induced insulin resistance: dose‐response studies. Am J Physiol 1994; 266: R1637–R1644 [DOI] [PubMed] [Google Scholar]
- 6.Storlien LH, Kraegen EW, Jenkins AB, et al. Effects of sucrose vs starch diets on in vivo insulin action, thermogenesis, and obesity in rats. Am J Clin Nutr 1988; 47: 420–427 [DOI] [PubMed] [Google Scholar]
- 7.Shirakawa J, Fujii H, Ohnuma K, et al. Diet‐induced adipose tissue inflammation and liver steatosis are prevented by DPP‐4 inhibition in diabetic mice. Diabetes 2011; 60: 1246–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sumiyoshi M, Sakanaka M, Kimura Y. Chronic intake of high‐fat and high‐sucrose diets differentially affects glucose intolerance in mice. J Nutr 2006; 136: 582–587 [DOI] [PubMed] [Google Scholar]
- 9.Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J 2008; 414: 1–18 [DOI] [PubMed] [Google Scholar]
- 10.Vionnet N, Stoffel M, Takeda J, et al. Nonsense mutation in the glucokinase gene causes early‐onset non‐insulin‐dependent diabetes mellitus. Nature 1992; 356: 721–722 [DOI] [PubMed] [Google Scholar]
- 11.Postic C, Shiota M, Niswender KD, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell‐specific gene knock‐outs using Cre recombinase. J Biol Chem 1999; 274: 305–315 [DOI] [PubMed] [Google Scholar]
- 12.Pilkis SJ, Weber IT, Harrison RW, et al. Glucokinase: structural analysis of a protein involved in susceptibility to diabetes. J Biol Chem 1994; 269: 21925–21928 [PubMed] [Google Scholar]
- 13.Seoane J, Barbera A, Telemaque‐Potts S, et al. Glucokinase overexpression restores glucose utilization and storage in cultured hepatocytes from male Zucker diabetic fatty rats. J Biol Chem 1999; 274: 31833–31838 [DOI] [PubMed] [Google Scholar]
- 14.Okamoto Y, Ogawa W, Nishizawa A, et al. Restoration of glucokinase expression in the liver normalizes postprandial glucose disposal in mice with hepatic deficiency of PDK1. Diabetes 2007; 56: 1000–1009 [DOI] [PubMed] [Google Scholar]
- 15.Rizza RA. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes 2010; 59: 2697–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iynedjian PB, Gjinovci A, Renold AE. Stimulation by insulin of glucokinase gene transcription in liver of diabetic rats. J Biol Chem 1988; 263: 740–744 [PubMed] [Google Scholar]
- 17.Michael MD, Kulkarni RN, Postic C, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 2000; 6: 87–97 [PubMed] [Google Scholar]
- 18.Holst JJ. The physiology of glucagon‐like peptide 1. Physiol Rev 2007; 87: 1409–1439 [DOI] [PubMed] [Google Scholar]
- 19.Seino Y, Fukushima M, Yabe D. GIP and GLP‐1, two incretin hormones: similarities and differences. J Diabetes Invest 2010; 1: 8–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baggio LL, Drucker DJ. Biology of incretins: GLP‐1 and GIP. Gastroenterology 2007; 132: 2131–2157 [DOI] [PubMed] [Google Scholar]
- 21.Roberge JN, Brubaker PL. Secretion of proglucagon‐derived peptides in response to intestinal luminal nutrients. Endocrinology 1991; 128: 3169–3174 [DOI] [PubMed] [Google Scholar]
- 22.Tolhurst G, Reimann F, Gribble FM. Nutritional regulation of glucagon‐like peptide‐1 secretion. J Physiol 2009; 587: 27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott RM, Morgan LM, Tredger JA, et al. Glucagon‐like peptide‐1 (7‐36) amide and glucose‐dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post‐prandial and 24‐h secretion patterns. J Endocrinol 1993; 138: 159–166 [DOI] [PubMed] [Google Scholar]
- 24.Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient‐induced glucagon‐like peptide‐1 secretion. Endocrinology 1999; 140: 1687–1694 [DOI] [PubMed] [Google Scholar]
- 25.Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C‐peptide responses. J Clin Endocrinol Metab 1986; 63: 492–498 [DOI] [PubMed] [Google Scholar]
- 26.Toft‐Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon‐like peptide‐1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001; 86: 3717–3723 [DOI] [PubMed] [Google Scholar]
- 27.Vilsboll T, Krarup T, Deacon CF, et al. Reduced postprandial concentrations of intact biologically active glucagon‐like peptide 1 in type 2 diabetic patients. Diabetes 2001; 50: 609–613 [DOI] [PubMed] [Google Scholar]
- 28.Yamada T, Ozaki N, Kato Y, et al. Insulin downregulates angiopoietin‐like protein 4 mRNA in 3T3‐L1 adipocytes. Biochem Biophys Res Commun 2006; 347: 1138–1144 [DOI] [PubMed] [Google Scholar]
- 29.Sloop KW, Cao JX, Siesky AM, et al. Hepatic and glucagon‐like peptide‐1‐mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest 2004; 113: 1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanda H, Tateya S, Tamori Y, et al. MCP‐1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006; 116: 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hara H, Miwa I, Okuda J. Inhibition of rat liver glucokinase by alloxan and ninhydrin. Chem Pharm Bull (Tokyo) 1986; 34: 4731–4737 [DOI] [PubMed] [Google Scholar]
- 32.Nagase H, Inoue S, Tanaka K, et al. Hepatic glucose‐sensitive unit regulation of glucose‐induced insulin secretion in rats. Physiol Behav 1993; 53: 139–143 [DOI] [PubMed] [Google Scholar]
- 33.Ferrannini E, Bjorkman O, Reichard GA Jr, et al. The disposal of an oral glucose load in healthy subjects. A quantitative study. Diabetes 1985; 34: 580–588 [DOI] [PubMed] [Google Scholar]
- 34.Katz LD, Glickman MG, Rapoport S, et al. Splanchnic and peripheral disposal of oral glucose in man. Diabetes 1983; 32: 675–679 [DOI] [PubMed] [Google Scholar]
- 35.Consoli A, Nurjhan N, Capani F, et al. Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes 1989; 38: 550–557 [DOI] [PubMed] [Google Scholar]
- 36.Magnusson I, Rothman DL, Katz LD, et al. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest 1992; 90: 1323–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care 1992; 15: 318–368 [DOI] [PubMed] [Google Scholar]
- 38.Pagliassotti MJ, Prach PA. Quantity of sucrose alters the tissue pattern and time course of insulin resistance in young rats. Am J Physiol 1995; 269: R641–R646 [DOI] [PubMed] [Google Scholar]
- 39.Pagliassotti MJ, Prach PA. Increased net hepatic glucose output from gluconeogenic precursors after high‐sucrose diet feeding in male rats. Am J Physiol 1997; 272: R526–R531 [DOI] [PubMed] [Google Scholar]
- 40.Pagliassotti MJ, Prach PA, Koppenhafer TA, et al. Changes in insulin action, triglycerides, and lipid composition during sucrose feeding in rats. Am J Physiol 1996; 271: R1319–R1326 [DOI] [PubMed] [Google Scholar]
- 41.Chicco A, D’Alessandro ME, Karabatas L, et al. Muscle lipid metabolism and insulin secretion are altered in insulin‐resistant rats fed a high sucrose diet. J Nutr 2003; 133: 127–133 [DOI] [PubMed] [Google Scholar]
- 42.Raubenheimer PJ, Nyirenda MJ, Walker BR. A choline‐deficient diet exacerbates fatty liver but attenuates insulin resistance and glucose intolerance in mice fed a high‐fat diet. Diabetes 2006; 55: 15–20 [DOI] [PubMed] [Google Scholar]
- 43.Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2005; 2: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pighin D, Karabatas L, Rossi A, et al. Fish oil affects pancreatic fat storage, pyruvate dehydrogenase complex activity and insulin secretion in rats fed a sucrose‐rich diet. J Nutr 2003; 133: 4095–4101 [DOI] [PubMed] [Google Scholar]
- 45.Anini Y, Brubaker PL. Role of leptin in the regulation of glucagon‐like peptide‐1 secretion. Diabetes 2003; 52: 252–259 [DOI] [PubMed] [Google Scholar]
- 46.Ranganath L, Norris F, Morgan L, et al. Inhibition of carbohydrate‐mediated glucagon‐like peptide‐1 (7–36) amide secretion by circulating non‐esterified fatty acids. Clin Sci (Lond) 1999; 96: 335–342 [DOI] [PubMed] [Google Scholar]
- 47.Muscelli E, Mari A, Casolaro A, et al. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008; 57: 1340–1348 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers used for quantitative real‐time polymerase chain reaction
Supporting info item