

Abstract
The failure of pancreatic β‐cells to supply insulin in quantities sufficient to maintain euglycemia is a hallmark of type 2 diabetes. Perturbation of β‐cell cholesterol homeostasis, culminating in elevated intracellular cholesterol levels, impairs insulin secretion and has therefore been proposed as a mechanism contributing to β‐cell dysfunction. The manner in which this occurs, however, is unclear. Cholesterol is an essential lipid, as well as a major component of membrane rafts, and numerous proteins critical for the regulation of insulin secretion have been reported to associate with these domains. Although this suggests that alterations in membrane rafts could partially account for the reduction in insulin secretion observed when β‐cell cholesterol accumulates, this has not yet been demonstrated. In this review, we provide a brief overview of recent work implicating membrane rafts in some of the basic molecular mechanisms of insulin secretion, and discuss the insight it provides into the β‐cell dysfunction characteristic of type 2 diabetes. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2012.00200.x, 2012)
Keywords: Cholesterol, Pancreatic β‐cell, Type 2 diabetes
Introduction
The efficient regulation of circulating glucose levels is critical for proper health. This process requires insulin, a peptide hormone that lowers glycemia by triggering the uptake of glucose into cells. As the exclusive source of insulin, pancreatic β‐cells play an essential role in glucose homeostasis. They are responsible for the production, storage and release of insulin in a manner that is strictly coupled to demand. The molecular mechanisms and dynamics of β‐cell secretion have been widely described1,2. What is clear is that glucose‐stimulated insulin secretion (GSIS) is a complex and tightly regulated process, the disruption of which has severe consequences. Evidence of this can be seen in diabetes, a widespread metabolic disease resulting from the failure of β‐cells to meet the physiological requirements for insulin. In type 1 diabetes, this is caused by the autoimmune‐mediated destruction of β‐cells3. In type 2 diabetes, β‐cells fail to adequately compensate for the insulin resistance often ensuing in conditions such as obesity4. Early β‐cell dysfunction, characterized by impaired GSIS, is considered fundamental in this process5.
The deficits in GSIS observed in type 2 diabetes have been attributed to numerous factors. One receiving significant attention recently is altered β‐cell cholesterol homeostasis6. Cholesterol is an essential constituent of mammalian cell membranes, which influences their fluidity, permeability and curvature7. It is also a major component of membrane rafts – dynamic nanoscale assemblies of sterols, sphingolipids and proteins that can be stabilized to coalesce into platforms that facilitate various cellular processes8. These domains are now widely considered to participate in the regulation of GSIS, a conclusion based primarily on two observations. First, numerous proteins involved in either GSIS or the biogenesis of secretory granules (SGs), the intracellular storage organelles from which insulin is secreted, have been shown to partition with detergent‐resistant membranes (DRMs) in the light fractions of sucrose density gradients after solubilization at 4°C with Triton X‐100 or other non‐ionic detergents. Second, perturbation of membrane cholesterol often induced a repartitioning of these proteins that corresponded with alterations in their function, as well as in insulin secretion. These results have prompted speculation that changes in membrane rafts, triggered by altered cellular cholesterol homeostasis, contribute to the impaired GSIS in type 2 diabetes. Here we provide a brief overview of the work linking membrane rafts and GSIS, and the insight it provides into β‐cell dysfunction.
Cholesterol Homeostasis and β‐Cell Dysfunction
Recent studies in mice have emphasized the importance of β‐cell cholesterol homeostasis for insulin secretion. Disruption of cholesterol efflux through knockout of the cholesterol transporter adenosine triphosphate (ATP)‐binding cassette transporter A1 (ABCA1) selectively in β‐cells resulted in the accumulation of islet cholesterol, as well as glucose intolerance and impaired GSIS9. This was in agreement with the previous observation that knockout of liver X receptor β (LXRβ), a nuclear hormone receptor that upregulates ABCA1 expression in response to cholesterol, caused β‐cell dysfunction10. Mice lacking the cholesterol acceptor apolipoprotein E (APOE) also showed elevated islet cholesterol levels and reduced insulin secretion11,12. Unlike Abca1−/− mice, however, circulating cholesterol levels in Apoe−/− mice were elevated, suggesting a correlation between hypercholesterolemia and β‐cell dysfunction11. Knockout of the low density lipoprotein receptor (LDLR), which facilitates cholesterol uptake, also triggered hypercholesterolemia, although its impact on β‐cell function was inconsistent. One group observed elevated islet cholesterol levels, impaired glucose tolerance and reduced GSIS in these mice13, whereas another reported no significant alterations in islet cholesterol or β‐cell function12. Despite this discrepancy, the trend emerging from these and other studies is that cholesterol accumulation in β‐cells impairs insulin secretion. Work investigating the role of membrane rafts in GSIS has identified numerous stages at which this might occur.
Glucose Uptake and Metabolism
Glycemia is normally maintained at concentrations of ∼5 mmol/L, and increases above this value trigger glucose uptake in β‐cells. In rodents, this is facilitated primarily by glucose transporter (GLUT)2, whereas in humans GLUT1 is the predominant glucose transporter14. Although there is little direct evidence for the association of these transporters with membrane rafts in β‐cells, GLUT1 partitioned with DRMs in various other cell types. In addition, cholesterol depletion of liver‐derived clone 9 cells with methyl‐β‐cyclodextrin (MβCD), which extracts cholesterol from the plasma membrane, disrupted the raft partitioning of GLUT1 and enhanced glucose transport15. Interestingly, Ldlr−/− islets with elevated cholesterol showed reduced glucose uptake13, as did cholesterol‐loaded primary β‐cells16. However, the possibility that this represented a membrane raft‐dependent effect on glucose transporter activity was not investigated.
After its uptake, glucose is phosphorylated to generate glucose‐6‐phosphate. This critical rate‐limiting step in glucose metabolism is catalyzed by glucokinase (GCK), the primary glucose sensor of β‐cells17. A pool of GCK associates with SGs, and glucose induces its dissociation and activation18. This association is mediated by an interaction with neuronal nitric oxide synthase (nNOS)19, and recent work suggests a role for membrane rafts in this process. Hao et al.11 have reported that MβCD reduced nNOS dimerization, promoted the translocation and activation of GCK, and enhanced GSIS. Cholesterol overloading had the converse effect.
How might membrane rafts regulate nNOS dimerization? nNOS has been previously shown to interact with islet cell autoantigen 512 (ICA512), a transmembrane SG protein also known as insulinoma‐associated protein 2 (IA‐2) or protein tyrosine phosphatase, receptor type, N (PTPRN)20. Hao et al. speculated that ICA512 associates with membrane rafts, and this facilitates nNOS dimerization and the retention of GCK on SGs (Figure 1a). Accordingly, elevated SG membrane cholesterol levels would reduce GCK translocation and impair GSIS11. Both ICA512 and nNOS partition with DRMs in insulinoma cells (Dirkx R and Solimena M, unpublished data, 2007–2009), and recent structural studies have indicated that the luminal/extracellular domain of ICA512 dimerizes21. However, it is not yet clear if membrane rafts influence ICA512 dimerization or its interaction with nNOS. Therefore, additional work is required, especially in light of reports that GCK does not translocate from granules in response to glucose and that NG‐nitro‐l‐arginine methyl ester (l‐NAME), an inhibitor of nNOS that enhanced its dimerization, amplified insulin secretion22,23. Apart from GCK activity, cholesterol overloading of β‐cells directly disrupted mitochondrial metabolism, and this together with reduced glucose uptake likely accounted for the near complete inability of glucose to elicit increases in cellular ATP levels in these cells16.
Figure 1.
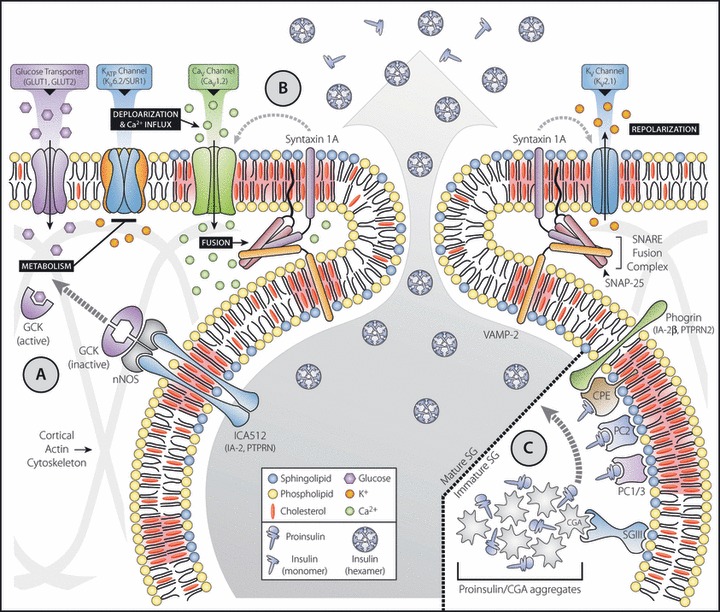
Insulin secretion – a membrane raft perspective. In pancreatic β‐cells, glucose uptake and metabolism triggers membrane depolarization. This initiates Ca2+ influx, leading to soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE)‐mediated secretory granule (SG) fusion and insulin secretion. Sphingolipid‐ and cholesterol‐enriched membrane rafts (highlighted in red) have been proposed to regulate various aspects of glucose‐stimulated insulin secretion including (a) glucose metabolism, and the secretion‐dependent translocation and activation of a SG‐associated pool of glucokinase (GCK); (b) KATP, Cav and Kv channel activity, SNARE‐mediated SG fusion and its spatial coupling to Ca2+ entry, fusion pore formation, and the syntaxin‐mediated regulation of channel gating; and (c) targeting and retention of prohormones, their processing enzymes, and granins in the cholesterol‐enriched membranes of newly forming SG. CaV, voltage‐gated Ca2+; CGA, chromogranin A; CPE, carboxypeptidase E; GLUT, glucose transporter; KATP, adenosine triphosphate‐sensitive K+; Kir, inwardly rectifying K+; KV, voltage‐gated K+; SUR, sulphonylurea receptor; nNOS, neuronal nitric oxide synthase; IA‐2, insulinoma‐associated protein 2; IA‐2β, insulinoma‐associated protein 2β; ICA512, islet cell autoantigen 512; PC1/3, prohormone convertase 1/3; PC2, prohormone convertase 2; PTPRN, protein tyrosine phosphatase, receptor type, N; PTPRN2, protein tyrosine phosphatase, receptor type, N polypeptide 2; SGIII, secretogranin III; SNAP‐25, synaptosomal‐associated protein 25; VAMP‐2, vesicle‐associated membrane protein 2.
β‐Cell Channel Function and Secretory Granule Exocytosis
The enhanced intracellular ATP levels resulting from glucose metabolism inactivate ATP‐sensitive K+ (KATP) channels, causing membrane depolarization. This triggers Ca2+ influx through voltage‐gated Ca2+ (Cav) channels and subsequent soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE)‐mediated fusion of SGs. Depolarization also activates voltage‐gated K+ (Kv) channels, which act to re‐establish the resting membrane potential and thus suppress insulin secretion.
The inwardly rectifying K+ (Kir) channel 6.2 and its associated sulphonylurea receptor (SUR)1 constitute the main KATP channel responsible for glucose‐induced depolarization of β‐cells. While Kir6.2 associated with membrane rafts in cardiomyocytes24, Kir6.2 and SUR1 were excluded from raft fractions in HIT‐T15 insulinoma cells25. However, Kv2.1 and Cav1.2, and the SNARE proteins syntaxin 1A, synaptosomal‐associated protein 25 (SNAP‐25) and vesicle‐associated membrane protein 2 (VAMP‐2) were all partially associated with membrane rafts (Figure 1b)25. MβCD, which caused their redistribution out of raft fractions, increased GSIS, a result which capacitance measurements indicated was due, in part, to enhanced refilling of the readily releasable SG pool. Kv channel current amplitude was reduced in depleted cells, suggesting that membrane raft disruption promoted insulin secretion by hindering repolarization25. In this context, it is noteworthy that SNARE proteins, in addition to their fundamental role in granule fusion, have also been reported to regulate KATP, Kv and Cav channels26. Thus, membrane rafts appear to contribute to the modulation of β‐cell excitability, either by directly regulating channel activity or by spatially coordinating SNARE‐coupled channel gating.
Upregulated GSIS and enhanced depolarization were also observed in MβCD‐treated INS‐1 cells, as was increased cortical F‐actin remodeling27. Cholesterol overloading, in contrast, impaired secretion, depolarization and remodeling. These results were attributed to altered membrane phosphatidylinositol 4,5‐bisphosphate (PIP2) levels, as MβCD increased PIP2 hydrolysis whereas cholesterol loading appeared to enhance membrane PIP2 levels and disrupt glucose‐dependent hydrolysis27. PIP2 is an important regulator of insulin secretion. Its accumulation impaired insulin secretion by reducing the sensitivity of KATP channels to ATP and stabilizing the cortical F‐actin cytoskeleton28,29. This work suggests that the effects of cholesterol accumulation on GSIS are coupled in part through PIP2. Do membrane rafts play a role in this process? Possibly, as Triton X‐100 insoluble raft fractions were reportedly enriched in PIP230. However, it is still unclear how this might influence glucose‐dependent PIP2 hydrolysis or the regulation of KATP channels comprised of Kir6.2 and SUR1, both of which appear to be excluded from membrane rafts in β‐cells.
In contrast to the observations that cholesterol depletion enhances GSIS, further work by Xia et al.31 indicated that the inhibition of cholesterol biosynthesis in MIN6 insulinoma cells, which also disrupted the raft partitioning of Cav1.2, Kv2.1 and SNARE proteins, impaired GSIS. In this case, Cav, Kv and KATP channel activity were all reduced. Similar results were reported in β‐cells after cholesterol overloading, suggesting that these channels are sensitive to any deviations from optimal cholesterol levels, regardless of direction16. As reducing Kv and KATP channel activity promotes secretion, the component of impaired GSIS resulting from altered channel function was attributed in these studies to the observed deficit in Cav channel activity.
Alterations of the membrane lipid environment also disrupted insulin secretion by dispersing Ca2+ influx independent of changes in channel activity or SG release competence32. This spatial uncoupling of Ca2+ influx and SGs has been proposed to hinder fusion pore formation, thus reducing the frequency of full fusion events and the efficiency of insulin release33. A similar effect could partly explain the decreased GSIS resulting from cholesterol depletion. However, a direct disruption of exocytosis was also demonstrated in cholesterol‐depleted insulinoma cells31,34. In addition, reduced SG exocytosis, detected in the absence of altered channel function, was a primary deficit in the β‐cells of Abca1−/−islets35. This indicated that altered membrane cholesterol levels likely impaired SNARE function.
The core SNARE fusion complex is comprised of the target (t)‐SNAREs syntaxin 1A, and SNAP‐25 on the plasma membrane and the vesicle (v)‐SNARE VAMP‐2 on vesicles2. As previously described in neurons36, cholesterol‐sensitive syntaxin 1 and SNAP‐25 clusters define sites where SGs preferentially dock and fuse37. In insulinoma cells, MβCD dispersed syntaxin 1 and induced a redistribution of SNAP‐25 to the cytosol34,37, both of which were accompanied by reductions in the number of docked SGs and GSIS. Interestingly, diffusion of syntaxin 1A from granule docking sites and reduced GSIS were also observed in INS‐1 cells cultured in high glucose, a condition that downregulated cholesterol biosynthesis and disrupted syntaxin 1A‐containing membrane rafts38.
These results suggest that membrane rafts promote SNARE clustering and fusion. However, studies demonstrating cholesterol‐dependent t‐SNARE clustering did not always detect SNARE proteins in membrane raft fractions36,37. In addition, syntaxin 1A clustering was detected in proteoliposomes that did not support the formation of raft‐like liquid ordered (Lo) domains39. When β‐cell SNARE proteins did partition with DRMs, a substantial pool was also detected in soluble fractions31. In neuroendocrine cells, cross‐linked syntaxin 1A and SNAP‐25 complexes partitioned in both raft and non‐raft fractions40. More recently, these proteins have been shown to form two conformationally distinct and spatially segregated t‐SNARE dimer intermediates, only one of which supports fusion. After cholesterol depletion, only the fusion competent form was detected41. In addition, cross‐linked complexes containing syntaxin 1A and the Sec1/Munc18‐like (SM) protein mammalian uncoordinated 18 (Munc‐18), an accessory protein essential for secretion that participates in multiple stages of SNARE assembly and fusion, were restricted to non‐raft fractions40. Although these observations suggest that SNARE assembly and fusion might proceed in non‐raft membrane regions, more work is required to understand the role of membrane rafts in the multiple, and often spatially and temporally distinct, stages of SNARE assembly and function.
Granule Biogenesis: Cholesterol and Secretory Granule Membrane Integrity
Proper SG biogenesis is an obvious prerequisite for secretion, and cholesterol, which is highly enriched in the granule membrane, is critical for their formation and integrity. In neuroendocrine cells, disruption of cholesterol biosynthesis with lovastatin, a 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A (HMG‐CoA) reductase inhibitor, blocked the formation of granules from the trans‐Golgi network (TGN) and caused swelling of the TGN cisterna42. In MIN6 cells, lovastatin decreased SG numbers while increasing their average size, and this corresponded with reduced insulin content and impaired regulated secretory response43.
SG malformation was also observed in two mouse models with deficient cholesterol production. Disruption of either lanthosterol‐5‐desaturase (SC5D) or 7‐dehydrocholesterol reductase (DHCR7), the enzymes responsible for the last two steps in cholesterol synthesis, decreased pancreatic exocrine and endocrine SG numbers, and increased granule size44. Studies in model membranes demonstrated that lanthosterol enhanced membrane flexibility while reducing intrinsic membrane curvature relative to cholesterol, suggesting that the altered SG biogenesis in Sc5d−/− mice resulted from the accumulation of lanthosterol in granule membranes44. Cholesterol accumulation in Abca1−/− islets was also suggested to disrupt SG biogenesis, a conclusion based on observations that β‐cell Golgi ultrastructure was altered and circulating proinsulin levels were enhanced35. However, although the insulin SGs of these mice were described to be ‘heterogeneous with respect to mean diameter’ compared with controls, no significant changes in SG numbers, size or distribution were observed.
Enlarged SGs and impaired GSIS downstream of depolarization were also observed in islets and insulinoma cells lacking the cholesterol transporter ATP‐binding cassette transporter G1 (ABCG1)45. In β‐cells, ABCG1 was reportedly expressed on SGs, and its loss led to reductions in SG cholesterol levels without altering total cellular or circulating cholesterol levels. Therefore, ABCG1 has been proposed to maintain SG integrity by facilitating the retention of cholesterol within the granule inner membrane leaflet, thus counteracting the carrier‐mediated diffusion of cholesterol from the SG outer membrane leaflet45. Granule membranes account for a large portion of total β‐cell cholesterol. In addition to reducing SG membrane integrity, the dispersal of this pool could alter raft‐dependent processes elsewhere – a possibility supported by the observation that MβCD extracted more cholesterol from cells lacking ABCG145. This apparent redistribution of cholesterol to the plasma membrane might explain the late‐stage deficit in secretion (i.e. downstream of Ca2+ influx) detected in these islets, although it is tempting to speculate that a corresponding reduction of SG membrane cholesterol might have also perturbed raft‐associated SG proteins, such as VAMP‐2. Whatever the case, this work suggests a unique mechanism of β‐cell dysfunction that could be of particular interest given the observation that ABCG1 expression is reduced in diabetic mice45.
Granule Biogenesis: Protein Sorting to Secretory Granules
Biogenesis of SGs requires proper sorting of cargo proteins to immature secretory granules. Although the mechanisms governing this process are still debated46, membrane rafts appear to play a role (Figure 1c). Prohormone convertase 1/3 (PC1/3) and 2 (PC2), two cargo endoproteases responsible for the conversion of proinsulin to insulin during SG maturation, both partitioned with DRMs in neuroendocrine cells47,48. In addition, PC1/3 associated with the luminal leaflet of SGs in a cholesterol‐sensitive manner49, and deletion of its membrane‐binding domain led to missorting of PC1/3 and its constitutive release50. The membrane raft association of PC2, in contrast, appeared to be sphingolipid‐dependent. Inhibition of sphingolipid synthesis with fumonisin blocked its targeting to SGs48, as did truncation of its C‐terminal raft‐binding domain51.
The insulin processing enzyme carboxypeptidase E (CPE) was also reported to associate with membrane rafts in SG membranes purified from pituitary52. Cholesterol depletion dissociated CPE from SG membranes, and its sorting to SGs was disrupted in lovastatin‐treated AtT‐20 pituitary cells52. A similar missorting was observed in neuroendocrine cells when the C‐terminus of CPE, which mediated its partitioning with DRMs, was removed53. It should be noted, however, that CPE did not partition with raft fractions in MIN6 cells43.
Cholesterol appears essential for targeting the granin protein, secretogranin III (SGIII), to immature granules, as it bound to SG‐like liposomes and isolated SG membranes in a cholesterol‐dependent manner, and deletion of its N‐terminal membrane‐binding domain caused its constitutive secretion54. In INS‐1 cells, SGIII binds CPE55, and its interaction with chromogranin A (CGA), another granin that promotes prohormone aggregation in the acidic luminal milieu of SGs, targeted CGA to insulin SG membranes56. Therefore, SGIII has been described as a multi‐functional adaptor that facilitates the retention of prohormone aggregates in the cholesterol‐enriched membranes of nascent SGs, thus promoting their processing and maturation by raft‐associated convertases57. Does SGIII associate with membrane rafts? This does not appear to be the case, as it was soluble in Triton X‐100 and several other detergents typically used to designate raft association54. It has instead been proposed to reside in distinct cholesterol‐enriched membrane domains57. The transmembrane SG protein phogrin, additionally referred to as insulinoma‐associated protein 2β (IA‐2β) or protein tyrosine phosphatase, receptor type, N polypeptide 2 (PTPRN2), was also shown to interact with CPE, and this facilitated the targeting of both proteins to granules58. Phogrin and its paralog, ICA512, like SGIII were also primarily soluble in Triton X‐100. However, ICA512 partitioned to a greater extent with DRMs after solubilization in lubrol (Dirkx R and Solimena M, unpublished data, 2007), a finding similar to that reported previously for VAMP‐240. These results suggest that SG membranes, although highly enriched in cholesterol and sphingolipids, still appear to be organized in heterogeneous functional membrane raft domains with distinct protein profiles.
Conclusions
Cholesterol is essential for proper β‐cell function, and despite discrepancies regarding the effect of cholesterol depletion on some aspects of GSIS, there now appears to be a clear consensus that accumulation of cellular cholesterol impairs insulin secretion (Figure 2). In the end, however, the question remains – do membrane rafts regulate GSIS? Conceivably yes, as compelling evidence indicates that numerous proteins essential for granule biogenesis and insulin secretion associate with these domains in a cholesterol‐depletion sensitive fashion. However, at this time there is little evidence that physiological stimuli that trigger or augment insulin secretion (e.g. glucose, glucagon‐like peptide 1 etc.) do so by altering the partitioning of these proteins in or out of membrane rafts. In addition, although a recent study suggested that hyperglycemia‐induced reductions in cholesterol might impair GSIS in part by disrupting membrane rafts, it has not been clearly demonstrated that physiopathological conditions that induce cholesterol accumulation also alter membrane rafts. For these reasons, it is difficult to assess at this time if membrane raft perturbation is a key factor contributing to β‐cell dysfunction in type 2 diabetes. Thus, while it remains an appealing possibility, the hypothesis that impaired cellular cholesterol homeostasis disrupts insulin secretion by altering β‐cell membrane rafts remains to be proven.
Figure 2.

Cholesterol homeostasis and glucose‐stimulated insulin secretion (GSIS). A summary of the reported effects of cholesterol perturbation in pancreatic islets, β‐cells or insulinoma cells. Arrows indicate increase (↑) or decrease (↓) in the described effect. Those effects associated with enhanced or reduced GSIS are listed in green or red, respectively. Instances of cholesterol‐sensitive repartitioning of raft‐associated proteins are highlighted in yellow. Superscripted numbers: references. Except as noted below, exocytosis is listed only when observed directly (i.e. changes in Ca2+‐dependent exocytotic response monitored by capacitance measurements taken independent of voltage‐gated Ca2+ [Cav] channel‐dependent Ca2+ influx). *The deficit in regulated secretion associated with the Sc5d−/−mice was reported from exocrine cells. **Although no net change in cholesterol was reported in Abcg1−/− islets, secretory granule (SG) cholesterol levels were reduced, whereas plasma membrane levels appeared to be elevated. ***Although not measured directly, impaired exocytosis likely accounted in part for the secretory deficit in Abcg1−/− islets, as glucose‐induced Ca2+ influx was unchanged compared with controls. Abca1, ATP‐binding cassette transporter A1; Abcg1, adenosine triphosphate‐binding cassette transporter G1; DRM, detergent resistant membrane; GCK, glucokinase; HMG‐CoA, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A; KATP, adenosine triphosphate‐sensitive K+ channel; KV, voltage‐gated K+ channel; Ldlr, low density lipoprotein receptor; MβCD, methyl‐β‐cyclodextrin; PIP2, phosphatidylinositol 4,5‐bisphosphate; Sc5d, lanthosterol‐5‐desaturase; SNAP‐25, synaptosomal‐associated protein 25; VAMP‐2, vesicle‐associated membrane protein 2.
Acknowledgements
We are grateful to Andreas Müller for the EM micrograph of a high pressure frozen rat β−cell, Erdinc Sezgin and Petra Schwille for discussion, and Kai Simons for critical reading of this manuscript. This work has been supported by funds from the Deutsche Forschungsgemeinschaft (grant SO 818/1‐1) and the German Ministry for Education and Research (BMBF) to the German Centre for Diabetes Research (DZD; http://www.dzd‐ev.de). We declare no conflicts of interest related to this manuscript.
References
- 1.Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003; 46: 1029–1045 [DOI] [PubMed] [Google Scholar]
- 2.Hou JC, Min L, Pessin JE. Insulin granule biogenesis, trafficking and exocytosis. Vitam Horm 2009; 80: 473–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.La Torre D, Lernmark A. Immunology of beta‐cell destruction. Adv Exp Med Biol 2010; 654: 537–583 [DOI] [PubMed] [Google Scholar]
- 4.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006; 116: 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kahn SE, Zraika S, Utzschneider KM, et al. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia 2009; 52: 1003–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunham LR, Kruit JK, Verchere CB, et al. Cholesterol in islet dysfunction and type 2 diabetes. J Clin Invest 2008; 118: 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maxfield FR, van Meer G. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol 2010; 22: 422–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lingwood D, Simons K. Lipid rafts as a membrane‐organizing principle. Science 2010; 327: 46–50 [DOI] [PubMed] [Google Scholar]
- 9.Brunham LR, Kruit JK, Pape TD, et al. Beta‐cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 2007; 13: 340–347 [DOI] [PubMed] [Google Scholar]
- 10.Gerin I, Dolinsky VW, Shackman JG, et al. LXRbeta is required for adipocyte growth, glucose homeostasis, and beta cell function. J Biol Chem 2005; 280: 23024–23031 [DOI] [PubMed] [Google Scholar]
- 11.Hao M, Head WS, Gunawardana SC, et al. Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic beta‐cell dysfunction. Diabetes 2007; 56: 2328–2338 [DOI] [PubMed] [Google Scholar]
- 12.Kruit JK, Kremer PHC, Dai L, et al. Cholesterol efflux via ATP‐binding cassette transporter A1 (ABCA1) and cholesterol uptake via the LDL receptor influences cholesterol‐induced impairment of beta cell function in mice. Diabetologia 2010; 53: 1110–1119 [DOI] [PubMed] [Google Scholar]
- 13.Bonfleur ML, Vanzela EC, Ribeiro RA, et al. Primary hypercholesterolaemia impairs glucose homeostasis and insulin secretion in low‐density lipoprotein receptor knockout mice independently of high‐fat diet and obesity. Biochim Biophys Acta 2010; 1801: 183–190 [DOI] [PubMed] [Google Scholar]
- 14.De Vos A, Heimberg H, Quartier E, et al. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest 1995; 96: 2489–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes K, Ingram JC, Bennett MDM, et al. Methyl‐beta‐cyclodextrin stimulates glucose uptake in Clone 9 cells: a possible role for lipid rafts. Biochem J 2004; 2: 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee AK, Yeung‐Yam‐Wah V, Tse FW, et al. Cholesterol elevation impairs glucose‐stimulated Ca(2+) signaling in mouse pancreatic β‐cells. Endocrinology 2011; 152: 3351–3361 [DOI] [PubMed] [Google Scholar]
- 17.Zelent D, Najafi H, Odili S, et al. Glucokinase and glucose homeostasis: proven concepts and new ideas. Biochem Soc Trans 2005; 33(Pt 1): 306–310 [DOI] [PubMed] [Google Scholar]
- 18.Rizzo MA, Magnuson MA, Drain PF, et al. A functional link between glucokinase binding to insulin granules and conformational alterations in response to glucose and insulin. J Biol Chem 2002; 277: 34168–34175 [DOI] [PubMed] [Google Scholar]
- 19.Rizzo MA, Piston DW. Regulation of beta cell glucokinase by S‐nitrosylation and association with nitric oxide synthase. J Cell Biol 2003; 161: 243–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ort T, Maksimova E, Dirkx R, et al. The receptor tyrosine phosphatase‐like protein ICA512 binds the PDZ domains of beta2‐syntrophin and nNOS in pancreatic beta‐cells. Eur J Cell Biol 2000; 79: 621–630 [DOI] [PubMed] [Google Scholar]
- 21.Primo ME, Klinke S, Sica MP, et al. Structure of the mature ectodomain of the human receptor‐type protein‐tyrosine phosphatase IA‐2. J Biol Chem 2008; 283: 4674–4681 [DOI] [PubMed] [Google Scholar]
- 22.Arden C, Harbottle A, Baltrusch S, et al. Glucokinase is an integral component of the insulin granules in glucose‐responsive insulin secretory cells and does not translocate during glucose stimulation. Diabetes 2004; 53: 2346–2352 [DOI] [PubMed] [Google Scholar]
- 23.Lajoix A‐D, Pugnière M, Roquet F, et al. Changes in the dimeric state of neuronal nitric oxide synthase affect the kinetics of secretagogue‐induced insulin response. Diabetes 2004; 53: 1467–1474 [DOI] [PubMed] [Google Scholar]
- 24.Garg V, Jiao J, Hu K. Regulation of ATP‐sensitive K+ channels by caveolin‐enriched microdomains in cardiac myocytes. Cardiovasc Res 2009; 82: 51–58 [DOI] [PubMed] [Google Scholar]
- 25.Xia F, Gao X, Kwan E, et al. Disruption of pancreatic beta‐cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J Biol Chem 2004; 279: 24685–24691 [DOI] [PubMed] [Google Scholar]
- 26.Leung YM, Kwan EP, Ng B, et al. SNAREing voltage‐gated K+ and ATP‐sensitive K+ channels: tuning beta‐cell excitability with syntaxin‐1A and other exocytotic proteins. Endocr Rev 2007; 28: 653–663 [DOI] [PubMed] [Google Scholar]
- 27.Hao M, Bogan JS. Cholesterol regulates glucose‐stimulated insulin secretion through phosphatidylinositol 4,5‐bisphosphate. J Biol Chem 2009; 284: 29489–29498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin C‐W, Yan F, Shimamura S, et al. Membrane phosphoinositides control insulin secretion through their effects on ATP‐sensitive K+ channel activity. Diabetes 2005; 54: 2852–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomas A, Yermen B, Regazzi R, et al. Regulation of insulin secretion by phosphatidylinositol‐4,5‐bisphosphate. Traffic 2010; 11: 123–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pike LJ, Casey L. Localization and turnover of phosphatidylinositol 4,5‐bisphosphate in caveolin‐enriched membrane domains. J Biol Chem 1996; 271: 26453–26456 [DOI] [PubMed] [Google Scholar]
- 31.Xia F, Xie L, Mihic A, et al. Inhibition of cholesterol biosynthesis impairs insulin secretion and voltage‐gated calcium channel function in pancreatic beta‐cells. Endocrinology 2008; 149: 5136–5145 [DOI] [PubMed] [Google Scholar]
- 32.Hoppa MB, Collins S, Ramracheya R, et al. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metab 2009; 10: 455–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eliasson L, Abdulkader F, Braun M, et al. Novel aspects of the molecular mechanisms controlling insulin secretion. J Physiol (Lond.) 2008; 586: 3313–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vikman J, Jimenez‐Feltström J, Nyman P, et al. Insulin secretion is highly sensitive to desorption of plasma membrane cholesterol. FASEB J 2009; 23: 58–67 [DOI] [PubMed] [Google Scholar]
- 35.Kruit JK, Wijesekara N, Fox JEM, et al. Islet cholesterol accumulation due to loss of ABCA1 leads to impaired exocytosis of insulin granules. Diabetes 2011; 60: 3186–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang T, Bruns D, Wenzel D, et al. SNAREs are concentrated in cholesterol‐dependent clusters that define docking and fusion sites for exocytosis. EMBO J 2001; 20: 2202–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohara‐Imaizumi M, Nishiwaki C, Nakamichi Y, et al. Correlation of syntaxin‐1 and SNAP‐25 clusters with docking and fusion of insulin granules analysed by total internal reflection fluorescence microscopy. Diabetologia 2004; 47: 2200–2207 [DOI] [PubMed] [Google Scholar]
- 38.Somanath S, Barg S, Marshall C, et al. High extracellular glucose inhibits exocytosis through disruption of syntaxin 1A‐containing lipid rafts. Biochem Biophys Res Commun 2009; 389: 241–246 [DOI] [PubMed] [Google Scholar]
- 39.Murray DH, Tamm LK. Molecular mechanism of cholesterol‐ and polyphosphoinositide‐mediated syntaxin clustering. Biochemistry 2011; 50: 9014–9022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chamberlain LH, Burgoyne RD, Gould GW. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc Natl Acad Sci USA 2001; 98: 5619–5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rickman C, Medine CN, Dun AR, et al. t‐SNARE protein conformations patterned by the lipid microenvironment. J Biol Chem 2010; 285: 13535–13541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Thiele C, Huttner WB. Cholesterol is required for the formation of regulated and constitutive secretory vesicles from the trans‐Golgi network. Traffic 2000; 1: 952–962 [DOI] [PubMed] [Google Scholar]
- 43.Tsuchiya M, Hosaka M, Moriguchi T, et al. Cholesterol biosynthesis pathway intermediates and inhibitors regulate glucose‐stimulated insulin secretion and secretory granule formation in pancreatic beta‐cells. Endocrinology 2010; 151: 4705–4716 [DOI] [PubMed] [Google Scholar]
- 44.Gondré‐Lewis MC, Petrache HI, Wassif CA, et al. Abnormal sterols in cholesterol‐deficiency diseases cause secretory granule malformation and decreased membrane curvature. J Cell Sci 2006; 119(Pt 9): 1876–1885 [DOI] [PubMed] [Google Scholar]
- 45.Sturek JM, Castle JD, Trace AP, et al. An intracellular role for ABCG1‐mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J Clin Invest 2010; 120: 2575–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dikeakos JD, Reudelhuber TL. Sending proteins to dense core secretory granules: still a lot to sort out. J Cell Biol 2007; 177: 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blázquez M, Docherty K, Shennan KI. Association of prohormone convertase 3 with membrane lipid rafts. J Mol Endocrinol 2001; 27: 107–116 [DOI] [PubMed] [Google Scholar]
- 48.Blázquez M, Thiele C, Huttner WB, et al. Involvement of the membrane lipid bilayer in sorting prohormone convertase 2 into the regulated secretory pathway. Biochem J 2000; 349(Pt 3): 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnaoutova I, Smith AM, Coates LC, et al. The prohormone processing enzyme PC3 is a lipid raft‐associated transmembrane protein. Biochemistry 2003; 42: 10445–10455 [DOI] [PubMed] [Google Scholar]
- 50.Lou H, Smith AM, Coates LC, et al. The transmembrane domain of the prohormone convertase PC3: a key motif for targeting to the regulated secretory pathway. Mol Cell Endocrinol 2007; 267: 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Assadi M, Sharpe JC, Snell C, et al. The C‐terminus of prohormone convertase 2 is sufficient and necessary for Raft association and sorting to the regulated secretory pathway. Biochemistry 2004; 43: 7798–7807 [DOI] [PubMed] [Google Scholar]
- 52.Dhanvantari S, Arnaoutova I, Snell CR, et al. Carboxypeptidase E, a prohormone sorting receptor, is anchored to secretory granules via a C‐terminal transmembrane insertion. Biochemistry 2002; 41: 52–60 [DOI] [PubMed] [Google Scholar]
- 53.Zhang C‐F, Dhanvantari S, Lou H, et al. Sorting of carboxypeptidase E to the regulated secretory pathway requires interaction of its transmembrane domain with lipid rafts. Biochem J 2003; 369(Pt 3): 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hosaka M, Suda M, Sakai Y, et al. Secretogranin III binds to cholesterol in the secretory granule membrane as an adapter for chromogranin A. J Biol Chem 2004; 279: 3627–3634 [DOI] [PubMed] [Google Scholar]
- 55.Hosaka M, Watanabe T, Sakai Y, et al. Interaction between secretogranin III and carboxypeptidase E facilitates prohormone sorting within secretory granules. J Cell Sci 2005; 118(Pt 20): 4785–4795 [DOI] [PubMed] [Google Scholar]
- 56.Hosaka M, Watanabe T, Sakai Y, et al. Identification of a chromogranin A domain that mediates binding to secretogranin III and targeting to secretory granules in pituitary cells and pancreatic beta‐cells. Mol Biol Cell 2002; 13: 3388–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hosaka M, Watanabe T. Secretogranin III: a bridge between core hormone aggregates and the secretory granule membrane. Endocr J 2010; 57: 275–286 [DOI] [PubMed] [Google Scholar]
- 58.Saito N, Takeuchi T, Kawano A, et al. Luminal interaction of phogrin with carboxypeptidase E for effective targeting to secretory granules. Traffic 2011; 12: 499–506 [DOI] [PubMed] [Google Scholar]