

Abstract
Aims/Introduction: β‐cell function was evaluated by homeostasis model assessment of β‐cell function (HOMA‐B) index, proinsulin:insulin and proinsulin:C‐peptide ratios in adult, Japanese type 2 diabetes patients receiving liraglutide.
Materials and Methods: Data from two randomized, controlled clinical trials (A and B) including 664 Japanese type 2 diabetes patients (mean values: glycated hemoglobin [HbA1c] 8.61–9.32%; body mass index [BMI] 24.4–25.3 kg/m2) were analyzed. In two 24‐week trials, patients received liraglutide 0.9 mg (n = 268) or glibenclamide 2.5 mg (n = 132; trial A), or liraglutide 0.6, 0.9 mg (n = 176) or placebo (n = 88) added to previous sulfonylurea therapy (trial B).
Results: Liraglutide was associated with improved glycemic control vs sulfonylurea monotherapy or placebo. In liraglutide‐treated groups in trials A and B, area under the curve (AUC) insulin 0–3 h was improved (P < 0.001 for all) and the AUCinsulin 0–3 h:AUCglucose 0–3 h ratio was increased (estimated treatment difference [liraglutide–comparator] 0.058 [0.036, 0.079]). HOMA‐B significantly increased with liraglutide relative to comparator in trial B (P < 0.05), but not in trial A. The reduction in fasting proinsulin:insulin ratio was 50% greater than in comparator groups.
Conclusions: In Japanese type 2 diabetes patients, liraglutide was associated with effective glycemic control, restoration of prandial insulin response and indications of improved β‐cell function. This trial was registered with Clinicaltrials.gov (trial A: no. NCT00393718/JapicCTI‐060328 and trial B: no. NCT00395746/JapicCTI‐060324). (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2012.00193.x, 2012)
Keywords: Insulin‐secreting cells, Liraglutide, Type 2 diabetes
Introduction
Diabetes is increasing in Japan to levels that are comparable with those of other countries, and a recent publication reported that the number of individuals ‘strongly suspected of having diabetes’ in Japan was approximately 8.9 million in 20071. Type 2 diabetes is characterized by impaired β‐cell function and insulin resistance. The increasing rate of diabetes in Japan probably reflects a complex interplay between genetic and environmental factors, including an increasingly Westernized diet, a more sedentary lifestyle and the ‘thrifty’ genotype characteristic of many Japanese people2. Compared with other ethnic populations, Japanese patients with type 2 diabetes show markedly reduced basal and impaired early‐phase insulin secretion, but lower indices of insulin resistance3. Accordingly, body mass index (BMI), which has a positive correlation with insulin resistance, is generally lower in Japanese type 2 diabetes patients, with a mean BMI of 24 kg/m2, compared with 27–30 kg/m2 and >30 kg/m2 in European and US patients, respectively3. In summary, these observations suggest that β‐cell failure might play a relatively greater part than insulin resistance in the pathophysiology of type 2 diabetes in Japanese people. This might be a result of loss of β‐cell mass or function.
In Japan, sulfonylureas (SU) are widely used either as monotherapy or in combination with other oral antidiabetic drugs (OAD) to treat type 2 diabetes. In a cross‐sectional study of 17,000 Japanese type 2 diabetes patients, 72–78% on oral therapy were using SU4. This is consistent with the known etiology of the disease in this population, where the key feature appears to be insufficient insulin secretion3.
The A Diabetes Outcome Progression Trial (ADOPT) study showed that treatment efficacy of glyburide (also known as glibenclamide) waned with successive treatment years5. Whereas glyburide improved β‐cell function to almost normal levels within 6 months of initiation, the effect then decreased and β‐cell function declined to below baseline level. Inukai et al.6 reported that homeostasis model assessment of β‐cell function (HOMA‐B) gradually decreased over time after a transient improvement during 5‐year treatment with glibenclamide in Japanese type 2 diabetes patients. This paradoxical effect might result from increased β‐cell stress accelerating β‐cell apoptosis, as well as the natural decline in β‐cell function associated with disease progression7. An alternative explanation is that desensitization to SU occurs, in which case, the state of decreased β‐cell function might be reversible7.
Among the newer treatments for type 2 diabetes are the incretin‐based therapies that include the glucagon‐like peptide‐1 receptor (GLP‐1R) agonists and dipeptidyl peptidase‐4 inhibitors (DPP‐4i), which exert their actions through potentiation of incretin receptor signaling. GLP‐1R agonists control blood glucose through regulation of islet function, principally with the stimulation of insulin and inhibition of glucagon secretion8. Liraglutide (Novo Nordisk A/S, Bagsværd, Denmark) is a once‐daily, human GLP‐1R agonist. GLP‐1R agonists are glucose‐dependent insulin secretagogues, but their mechanism of action and target receptors on the β‐cell differ from SU. Response to physiological levels of GLP‐1 is reduced in type 2 diabetes patients; pharmacological levels of native GLP‐1 or GLP‐1 analog therapy can restore this response9. In clinical trials, in predominantly Caucasian populations, treatment with GLP‐1R agonists is associated with sustained improvements in glycemic control, weight reduction and low hypoglycemia risk10. In clinical trials in European and US populations, GLP‐1R agonists have shown favorable effects on several parameters of β‐cell function11. In animal models, exposure to GLP‐1 is associated with an increase in β‐cell mass12. In contrast to SU, it is therefore possible that GLP‐1R agonists might limit the progressive loss of β‐cell function.
It is of clinical interest to determine whether the beneficial effects of liraglutide on β‐cell function evident in other populations could also provide clinical benefits in Japanese patients, and if these benefits could be sustained for longer periods than are achievable with SU. The results of glycemic control parameters, such as glycated hemoglobin [HbA1c], fasting plasma glucose (FPG), postprandial glucose (PPG) and seven‐point self‐monitored plasma glucose, as well as safety data, have been reported for two clinical trials with Japanese type 2 diabetes patients receiving liraglutide, either as monotherapy in one trial or added on to SU therapy in the other trial, for 24 weeks13,14. Here, we report the short‐term effect of liraglutide on β‐cell function in these trials.
Materials and Methods
Adult Japanese type 2 diabetes patients were screened and enrolled in one of two double‐blind, multicenter, randomized, parallel‐group clinical trials (trial A or B) if they were ≥20 years of age, with HbA1c≥7.4 to <10.4% and BMI <3513,14. Patients were to be on diet and OAD (trial A: ±OAD monotherapy – biguanide, sulphonylamide, SU [≤50% approved dose in Japan], α‐glucosidase inhibitor, insulin secretagogue or insulin sensitizer, within approved Japanese dose ranges; trial B: SU monotherapy – glibenclamide 1.25–10 mg/day, gliclazide 40–160 mg/day or glimepiride 1–6 mg/day). Patients with clinical conditions likely to interfere with the conduct of the trial were excluded. Trials were carried out in accordance with the Declaration of Helsinki15, with informed consent of patients and approval of relevant ethics committees.
In trial A (24 weeks; n = 411), patients were randomized (2:1) to once‐daily liraglutide (0.9 mg) or once‐ or twice‐daily glibenclamide (1.25–2.5 mg). In trial B (24 weeks; n = 267), patients continued SU treatment (glibenclamide [1.25–10 mg], gliclazide [40–160 mg] or glimepiride [1–6 mg]), and were randomized to one of two daily doses of liraglutide (0.6 or 0.9 mg), or placebo. In trial A, a 4–6 week run‐in/screening period preceded a 2‐week dose‐escalation period followed by a 22‐week maintenance period. In trial B, a screening visit was followed by a start‐of‐treatment visit after 4 weeks, a 2‐week dose‐escalation period and a 22‐week treatment period.
Patients in trial A were stratified by pretreatment therapy (±OAD) and, in trial B, according to type of SU. In both trials, liraglutide was initiated with 0.3 mg during week 1 and increased weekly (in 0.3 mg increments) to the final dose to minimize gastrointestinal side‐effects. Randomization lists were prepared by the contract research organization responsible for the study, Transcosmos Inc. (Tokyo, Japan). This organization also ensured that liraglutide was unidentifiable from placebo, blinded trial products and randomized patients, and informed the investigator and sponsor of randomization numbers. Dynamic allocation was used to guarantee a balanced allocation within strata of pretrial treatment. Randomization codes were maintained in sealed conditions until broken according to schedule. Liraglutide was given by subcutaneous injection in the abdomen using a prefilled pen once daily in the morning or evening in the upper arm, abdomen or thigh. Injections were to be given at the same time every day.
The primary outcome measure in all trials was HbA1c at the end of the trial (expressed by National Glycohemoglobin Standardization Program values). Secondary end‐points included seven‐point self‐measured PPG profiles, FPG, glucose homeostasis‐related parameters (fasting insulin, proinsulin, C‐peptide, glucagon, postprandial insulin and glucagon). These end‐points have been reported elsewhere. Secondary end‐points reported here include measures of HOMA‐B index, proinsulin:insulin ratio and proinsulin:C‐peptide ratio.
A meal test (Japanese‐style breakfast) was also carried out at baseline and 24 weeks. For each individual patient, the content of the meal was identical at these time‐points. The meal test was standardized within each site, but differed across sites. Plasma glucose, insulin, glucagon, fasting proinsulin and C‐peptide were measured. Intact proinsulin concentrations were determined by enzyme‐linked immunosorbent assay, based on anti‐proinsulin monoclonal antibodies (IBL; Immuno‐Biological Laboratories, Hamburg, Germany). Human insulin does not cross‐react in this assay. Within a concentration range of 5–500 pmol/L, proinsulin Des 64–65 cross‐reacted with frequencies of 53–65%.
All analyses were carried out by a central laboratory (Mitsubishi Kagaku BCL Inc., Tokyo, Japan), except the seven‐point plasma glucose profile, which was measured before and approximately 2 h after each meal and at bedtime by self‐monitoring using standardized glucose meters (Glutest Ace; Glutest PRO, Sanwa‐Kagaku, Nagoya, Japan; Glucocard Diameter or Glucocard Diameter α; Arkray KDK Corp., Kyoto, Japan) before the start of treatment and at study end. β‐cell function was assessed using the HOMA‐B index, where HOMA‐B = 360 × fasting insulin/(FPG − 63) and units of insulin and glucose were μU/mL and mg/dL, respectively. For insulin and glucagon (meal test), the area under the curve (AUC) was calculated using the trapezoidal rule.
Trial A was carried out between December 2006 and November 2008, and trial B between November 2006 and October 2007.
Statistical Analysis
Efficacy end‐point analyses included data from all patients who were randomized and received trial product with efficacy data. Safety analyses included all patients who received trial drugs. Primary and secondary end‐points were analyzed using an analysis of variance (anova) model, with treatment group and stratification factor as fixed effects and baseline value as a covariate. An ad hoc analysis, AUCinsulin 0–3 h:AUCglucose 0–3 h, was carried out using anova model with trial (A or B) and treatment group (liraglutide or comparator) as fixed effects and corresponding baseline values as covariate. In trial A, sample size calculation was based on 1.2% common standard deviation (SD) for both treatments, and 0.0% true difference in HbA1c at 80% power, a non‐inferiority margin for HbA1c of 0.4% at a significance level of 2.5%. The sample size calculation in trial B was based on a mean difference of 0.6% in HbA1c between 0.9 mg + SU and placebo + SU after 24 weeks, with a SD of 1.2 and 80% power.
Results
Baseline demographics, patient characteristics and patient disposition are shown in Table 1. No baseline differences were noted between the trials.
Table 1. Patient disposition and baseline characteristics by trial and by treatment group.
| Trial A | Trial B | ||||
|---|---|---|---|---|---|
| No. patients randomized | 411 | 267 | |||
| Glibenclamide monotherapy | Liraglutide monotherapy | Placebo + SU | Liraglutide + SU | ||
| 0.9 mg | 0.6 mg + SU | 0.9 mg + SU | |||
| Randomized to treatment | 139 | 272 | 89 | 89 | 89 |
| Not exposed | 7 | 4 | 1 | 1 | 1 |
| Completed | 120 | 246 | 74 | 83 | 84 |
| Included in the efficacy analysis | 132 | 268 | 88 | 88 | 88 |
| Age, years (mean [SD]) | 58.5 (10.4) | 58.2 (10.4) | 58.6 (9.7) | 59.1 (10.3) | 61.3 (11.0) |
| Male/female, n | 86/46 | 183/85 | 57/31 | 53/35 | 59/29 |
| BMI, kg/m2 (mean [SD]) | 24.6 (3.8) | 24.9 (3.7) | 24.9 (4.0) | 25.3 (3.6) | 24.4 (3.4) |
| HbA1c, %* | 9.18 (0.97) | 9.32 (1.08) | 8.85 (0.99) | 9.00 (0.91) | 8.61 (0.78) |
| Duration of diabetes, years (mean [SD]) | 8.5 (6.8) | 8.1 (6.7) | 10.1 (7.3) | 9.3 (5.8) | 11.6 (7.7) |
*At baseline. The value for glycated hemoglobin (HbA1c; %) is estimated as a National Glycohemoglobin Standardization Program (NGSP) equivalent value (%) calculated by the formula HbA1c (%) = HbA1c according to the Japanese Diabetes Society (JDS) (%) + 0.4%, considering the relational expression of HbA1c (JDS) (%) measured by the previous Japanese standard substance and measurement methods and HbA1c (NGSP). BMI, body mass index; SD, standard deviation; SU, sulfonylureas.
All liraglutide doses reduced mean HbA1c relative to comparator. In trial A, HbA1c was reduced by 0.50% points with liraglutide relative to glibenclamide, whereas in trial B, mean HbA1c was 1.00 and 1.27% points lower than placebo in the 0.6 and 0.9 mg liraglutide treatment groups, respectively. Significant improvements in all other measured parameters of glycemia (FPG, PPG and self‐monitored plasma glucose) were also reported in each trial (data not shown).
No major hypoglycemic events were reported, and liraglutide was well tolerated across both trials. In trial A, the overall rate of hypoglycemia (episodes/subject‐year of exposure) was significantly lower in liraglutide‐ than glibenclamide‐treated patients (0.8 vs 5.5; P < 0.0001). In trial B, the number of all hypoglycemic episodes was higher in the 0.6 and 0.9 mg/day liraglutide + SU groups than in the placebo + SU monotherapy group (P = 0.0159 and P = 0.0085, respectively).
Insulin levels in the 3‐h post‐breakfast period (AUCinsulin 0–3 h) were higher with liraglutide than with the comparator in both trials (Figure 1a). AUCinsulin 0–3 h was significantly higher at week 24 (last observation carried forward [LOCF]) in liraglutide‐treated groups than in the glibenclamide group (P = 0.0165; trial A) or placebo + SU group (P < 0.0001; trial B; Figure 1a).
Figure 1.
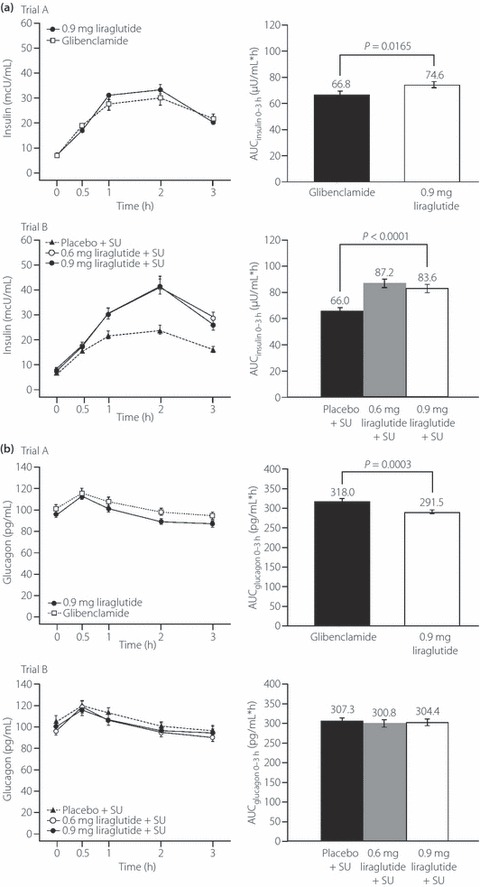
Insulin and glucagon profiles and area under the curve (AUC)0–3 h values for insulin and glucagon by trial (A and B) and by treatment group in the 3 h after the standard meal test at the end of the study period. (a) Insulin profiles (0–3 h) and comparison of AUCinsulin 0–3 h by trial and by treatment group. (b) Glucagon profiles (0–3 h) and comparison of AUCglucagon 0–3 h by trial and by treatment group. Data are last observation carried forward at week 24. Errors bars are standard error. SU, sulfonylureas.
The AUCinsulin 0–3 h:AUCglucose 0–3 h ratio was increased from baseline values (0.06–0.09) by more than 40% with liraglutide (0.14–0.16) relative to comparators (0.09–0.11) in both trials. The estimated mean (95% confidence interval [CI]) treatment difference for AUCinsulin 0–3 h:AUCglucose 0–3 h after administration of 0.9 mg liraglutide vs comparator was 0.038 (0.028, 0.048). After administration of 0.6 or 0.9 mg liraglutide, the treatment difference (liraglutide − comparator) was 0.038 (0.029, 0.048).
Fasting glucagon levels and AUCglucagon 0–3 h at week 24 (LOCF) in the liraglutide group were significantly lower than in the glibenclamide group (trial A), but not different to the SU monotherapy groups (trial B; Table 2 and Figure 1b).
Table 2. Analysis of the effect of liraglutide on glucose metabolism‐related parameters by trial and by treatment group.
| Trial A | Trial B | ||||
|---|---|---|---|---|---|
| Glibenclamide monotherapy | Liraglutide monotherapy | Placebo + SU | Liraglutide + SU | ||
| 0.9 mg | 0.6 mg | 0.9 mg | |||
| Fasting insulin, μU/mL | |||||
| End‐of‐study LS mean (SE) | 6.93 (0.36) | 7.16 (0.27) | 6.93 (0.38) | 7.29 (0.37) | 7.14 (0.38) |
| Liraglutide–comparator, mean (95% CI) | 0.24 (−0.53, 1.00) | 0.36 (−0.53, 1.26) | 0.21 (−0.68, 1.11) | ||
| P‐value for pairwise comparison | P = 0.5413 | NA | NA | ||
| Fasting glucagon, (pg/mL) | |||||
| End‐of‐study LS mean (SE) | 105.4 (2.6) | 96.5 (1.9) | 103.0 (3.5) | 98.7 (3.4) | 102.4 (3.5) |
| Liraglutide–comparator, mean (95% CI) | −8.9 (−14.5, −3.3) | −4.4 (−12.5, 3.8) | −0.6 (−8.8, 7.5) | ||
| P‐value for pairwise comparison | P = 0.002 | NA | NA | ||
| Fasting proinsulin, pmol/L | |||||
| End‐of‐study LS mean (SE) | 10.32 (0.54) | 6.04 (0.40) | 10.15 (0.92) | 9.15 (0.91) | 8.47 (0.93) |
| Liraglutide–comparator, mean (95% CI) | −4.27 (−5.44, −3.11) | −0.99 (−3.16, 1.18) | −1.67 (−3.84, 0.50) | ||
| P‐value for pairwise comparison | P < 0.0001 | NA | NA | ||
| Fasting C‐peptide, ng/mL | |||||
| End‐of‐study LS mean (SE) | 2.44 (0.07) | 2.55 (0.05) | 2.45 (0.08) | 2.76 (0.08) | 2.77 (0.08) |
| Liraglutide–comparator mean (95% CI) | 0.10 (−0.05, 0.25) | 0.31 (0.11, 0.50) | 0.32 (0.12, 0.51) | ||
| P‐value for pairwise comparison | P = 0.1740 | P = 0.0021 | P = 0.0016 | ||
CI, confidence interval; LS, least squares; NA, not available; SU, sulfonylureas.
Fasting insulin was similar between liraglutide‐ and comparator‐treated patients (Table 2) in both trials. End‐of‐study fasting C‐peptide levels were significantly higher in liraglutide + SU‐treated patients than in those on placebo + SU in trial B (P = 0.0017), but there was no significant difference between liraglutide‐ and glibenclamide‐treated patients in trial A. The estimated mean of fasting proinsulin at trial end was significantly lower in liraglutide‐ than glibenclamide‐treated patients in trial A (P < 0.0001), and was not significantly different for patients on liraglutide + SU and placebo + SU in trial B (Table 2).
In trial B, the estimated treatment difference ([liraglutide + SU] − [placebo + SU]) in HOMA‐B index was significant for both doses of liraglutide (P = 0.047 and P = 0.0012 for 0.6 and 0.9 mg, respectively). No significant between‐treatment difference at the end of trial in HOMA‐B was observed in trial A (glibenclamide 34.9%; liraglutide 39.0%; P = 0.0997; Table 3).
Table 3. Analysis of the effect of liraglutide on β‐cell function: related parameters by trial and by treatment group.
| Trial A | Trial B | ||||
|---|---|---|---|---|---|
| Glibenclamide monotherapy | Liraglutide monotherapy | Placebo + SU | Liraglutide + SU | ||
| 0.9 mg | 0.6 mg | 0.9 mg | |||
| β‐cell function, HOMA‐B (%) | |||||
| End‐of‐study LS mean (SE) | 34.88 (2.31) | 39.04 (1.75) | 30.86 (5.26) | 43.35 (5.13) | 51.53 (5.30) |
| Treatment difference mean (95% CI) | 4.15 (−0.80, 9.10) | – | 12.49 (0.17, 24.81) | 20.67 (8.22, 33.13) | |
| P‐value for pairwise comparison | P = 0.0997 | – | P = 0.0470 | P = 0.0012 | |
| P‐value for overall test | – | P = 0.0050 | |||
| Proinsulin:insulin ratio, (pmol/L)/(μU/mL) | |||||
| End‐of‐study LS mean (SE) | 1.79 (0.08) | 0.99 (0.06) | 1.86 (0.13) | 1.29 (0.12) | 1.17 (0.13) |
| Treatment difference mean (95% CI) | −0.81 (−0.98, −0.63) | – | −0.57 (−0.86, −0.27) | −0.69 (−0.99, −0.40) | |
| P‐value for pairwise comparison | P < 0.0001 | – | P = 0.0002 | P < 0.0001 | |
| P‐value for overall test | – | P < 0.0001 | |||
| Proinsulin:C‐peptide ratio, (pmol/L)/(ng/mL) | |||||
| End‐of‐study LS mean (SE) | 4.17 (0.19) | 2.31 (0.14) | 4.10 (0.26) | 3.10 (0.26) | 2.71 (0.26) |
| Treatment difference mean (95% CI) | −1.85 (−2.26, −1.45) | – | −0.99 (−1.60, −0.38) | −1.38 (−1.20, −0.77) | |
| P‐value for pairwise comparison | P < 0.0001 | – | P = 0.0016 | P < 0.0001 | |
| P‐value for overall test | – | P < 0.0001 | |||
CI, confidence interval; HOMA‐B, homeostasis model assessment of β‐cell function; LS, least squares; SE, standard error; SU, sulfonylureas.
Decreases in the proinsulin:insulin ratio from baseline (baseline of 1.79–2.15 across all groups) for liraglutide‐treated groups (decrease of 0.65–0.87 across all groups) were greater than in SU‐treated groups (decrease of 0.15–0.26), resulting in lower values for the liraglutide‐treated group than for the comparator or placebo‐treated group at week 24. Reduction in the proinsulin:C‐peptide ratio from baseline (baseline of 4.09–4.76 across groups) was also greater in liraglutide‐treated groups (decrease of 1.24–1.61) than in SU‐treated patients (decrease of 0.06–0.55).
Discussion
The present report shows that 24 weeks’ treatment with liraglutide provides a significant improvement in β‐cell function in Japanese type 2 diabetes patients. Additionally, liraglutide was associated with significantly greater improvements in key parameters of glycemia, namely HbA1c, FPG, PPG and seven‐point self‐monitored plasma glucose, than comparators, and these results have been reported elsewhere13,14. As previously described, liraglutide resulted in weight loss or no weight gain in Japanese patients. The overall improvement of glycemic control with 0.9 mg liraglutide seen in Japanese populations was not different to that observed with 1.2 mg liraglutide in non‐Japanese populations13,14,16.
Indirectly, these observations suggest that different degrees of SU insensitivity rather than β‐cell apoptosis are responsible for the failure of pretrial treatment in Japanese type 2 diabetes patients. Although SU and liraglutide have powerful insulin‐releasing effects on β‐cells, they exert their effect through separate, independent receptors17,18. GLP‐1R exist in the β‐cell plasma membrane, and receptor interaction leads to mobilization and exocytosis of insulin‐containing granules19. This action is strictly glucose‐dependent. In contrast, SU stimulate insulin secretion by closing β‐cell adenosine‐5′‐triphosphate‐sensitive potassium channels (KATP) through binding to SU receptor 1 (SUR1) and, according to recent evidence, by activating the cyclic adenosine monophosphate (cAMP) sensor exchange protein activated by cAMP – with the exception of gliclazide – also through direct binding17. These events are not glucose‐dependent. The differences in β‐cell function reported here should, however, be considered in respect to the different levels of glycemic control achieved with liraglutide vs comparators. Recently, it has been shown that GLP‐1R agonists can improve impaired glucose metabolism in diabetic pancreatic β‐cells, resulting in an increase in adenosine‐5′‐triphosphate (ATP) production20. As the closure of KATP channels by SU is ATP‐dependent21, it seems likely that the combination of a GLP‐1R agonist and an SU would be more effective at stimulating insulin secretion than a GLP‐1R agonist alone.
We have recently shown that active GLP‐1 levels after meal ingestion are extremely low in healthy Japanese subjects and Japanese patients with type 2 diabetes22. It seems likely, therefore, that supplementation of GLP‐1R agonists that are resistant to degradation by DPP‐4 would be effective in enhancing the GLP‐1 effect.
A significant improvement in β‐cell function with liraglutide was observed across the two trials. All indicators of β‐cell function were substantially ameliorated, showing that liraglutide positively affected the insulin response to glucose. A significant increase in postprandial insulin secretion (AUCinsulin 0–3 h) compared with comparators was shown. In support of this observation, the postprandial insulin:postprandial glucose level ratio increased with liraglutide by >40% across the two trials. Although insulin secretion by liraglutide from β‐cells is glucose‐dependent, insulin secretion diminishes despite the continued presence of liraglutide as glucose levels normalize23. This could be expected to counter postprandial hyperglycemia, a feature of type 2 diabetes, even at an early stage, in Japanese type 2 diabetes patients.
Liraglutide appears to have a positive impact on pancreatic glucoregulatory function, as shown by the trend to a reduction in fasting and postprandial glucagon levels. Trial A, in particular, showed significant glucagon reductions with liraglutide treatment compared with glibenclamide, and a similar trend was observed in trial B, despite not achieving significance. The absence of significance in trial B might be related to the long‐term use of SU, which are reported to increase prandial glucagon levels24. Therefore, in patients receiving combination therapy with liraglutide and an SU, the observable effect on glucagon levels would be attenuated as a result of the opposing effects each agent has on glucagon secretion.
In type 2 diabetes, normal suppression of glucagon after a meal is blunted, resulting in hyperglucagonemia and increased hepatic glucose production in many patients, thus exacerbating hyperglycemia25. Therefore, counter‐regulatory responses affecting glucagon secretion are impaired in these patient groups, and larger, more focused studies using appropriately matched patient groups will be required to unambiguously determine the effect of liraglutide on glucagon secretion.
Liraglutide promotes β‐cell preservation in animal studies, increasing β‐cell mass in rodents and inhibiting β‐cell apoptosis in vitro12,26. In our studies, liraglutide significantly increased HOMA‐B relative to both baseline and the comparators. In trial A, HOMA‐B measurements with 0.9 mg liraglutide and glibenclamide were significantly improved from baseline. These results were also in accordance with a previous monotherapy trial (LEAD‐3) carried out in a predominantly Caucasian population16. Although liraglutide did not outperform glibenclamide regarding improvements in HOMA‐B, it is encouraging to observe that it can be as equally effective as such a potent and widely used insulin secretagogue27. Consistent with this finding, Madsbad et al.28 reported a significant improvement from baseline in HOMA‐B with 0.75 mg liraglutide (23.6%), which was similar to that observed with glimepiride (1–4 mg, 24.6%) in 193 Caucasian type 2 diabetes patients. The magnitude of the liraglutide‐associated increase in HOMA‐B from baseline in the present study (in the order of 90–100%) was greater than that shown in other studies in mainly Caucasian study populations. In another study, treatment with liraglutide increased HOMA‐B relative to baseline by approximately 30%29,30.
During liraglutide treatment, the fasting proinsulin:insulin ratio was decreased. This might suggest improved processing of insulin in the β‐cell, possibly as a result of a more appropriate pattern of insulinotropic action reducing overall β‐cell stress. An elevated proinsulin:insulin ratio is a principal feature of type 2 diabetes and pre‐diabetes31, and shows β‐cell dysfunction32. Hyperproinsulinemia might be caused by increased demand on the β‐cells (during hyperglycemia and as a consequence of insulin resistance), increasing the release of incompletely processed granules containing proinsulin. Alternatively, it is suggested that the increased proinsulin concentration might be a result of an intrinsic β‐cell defect in type 2 diabetes32. The improvement in proinsulin:insulin ratio with liraglutide relative to SU therapy might reflect the different modes of action of the two agents. While both enhance insulin secretion from β‐cells, liraglutide’s effect appears to be glucose‐dependent, and hence predominantly a postprandial effect, whereas that of the SU appears more or less continuous, leading to increased β‐cell stress and, potentially, an increased rate of β‐cell apoptosis33.
Taken together, the results from the present report suggest that treatment with liraglutide is at least as effective in Japanese patients with type 2 diabetes as in comparable populations. Despite the fact that the insulin secretory capacity of the β‐cells in Japanese type 2 diabetes patients might be substantially more impaired than in other populations, there appears to be sufficient β‐cell mass to preserve significant capacity for a glucose‐dependent insulin secretory response to liraglutide. This could indirectly show that the decreased insulin secretion seen in this population is more a consequence of impaired β‐cell function than lost β‐cell mass. The reported improvement in parameters of β‐cell function and glucoregulation provides scope for optimism that the therapeutic effects of liraglutide could be sustainable with long‐term therapy, possibly retarding the eventual decline in β‐cell secretory function that typifies SU therapy. This remains to be established in long‐term studies.
Acknowledgements
The two trials used for this analysis were part of the development program for liraglutide in Japan, and were supported by Novo Nordisk Pharma Limited, Japan. We are grateful to Tomoyuki Nishida from Novo Nordisk Pharma Limited for assistance with statistical analysis, and Penny Butcher and Steve Banner at Watermeadow Medical, UK, for assistance with writing, which was funded by Novo Nordisk. Professor Yutaka Seino has served as a consultant/advisor and on speakers’ bureaus for GlaxoSmithKline, Merck Global, Novartis, Novo Nordisk, Otsuka, Sanofi‐Aventis, Taisho and Takeda. Professor Kohei Kaku has received speaking honoraria/lecture fees from Astellas, AstraZeneca, Banyu, Daiichi‐Sankyo, Dainippon‐Sumitomo, Novartis, Novo Nordisk, Sanofi‐Aventis, Sanwa and Takeda, served as a consultant/advisor to Novo Nordisk, and received research funding from Astellas, Banyu, Daiichi‐Sankyo, Novo Nordisk, Sanofi‐Aventis and Takeda. Mads Frederik Rasmussen and Per Clauson are employees of and hold shares in Novo Nordisk.
References
- 1.Morimoto A, Nishimura R, Tajima N. Trends in the epidemiology of patients with diabetes in Japan. Jpn Med Assoc J 2010; 53: 36–40 [Google Scholar]
- 2.Kawamori R. Diabetes trends in Japan. Diabetes Metab Res Rev 2002; 18(Suppl. 3): S9–S13 [DOI] [PubMed] [Google Scholar]
- 3.Fukushima M, Usami M, Ikeda M, et al. Insulin secretion and insulin sensitivity at different stages of glucose tolerance: a cross‐sectional study of Japanese type 2 diabetes. Metab Clin Exp 2004; 53: 831–835 [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi M, Yamazaki K, Hirao K, et al. The status of diabetes control and antidiabetic drug therapy in Japan – a cross‐sectional survey of 17,000 patients with diabetes mellitus (JDDM 1). Diabetes Res Clin Pract 2006; 73: 198–204 [DOI] [PubMed] [Google Scholar]
- 5.Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin or glyburide monotherapy. N Engl J Med 2006; 355: 2427–2443 [DOI] [PubMed] [Google Scholar]
- 6.Inukai K, Watanabe M, Kurihara S, et al. Effects of long‐term monotherapy with glimepiride vs. glibenclamide on glycemic control and macrovascular events in Japanese type 2 diabetic patients. J New Rem & Clin 2010; 59: 622–629 [Google Scholar]
- 7.Karam JH, Sanz N, Salamon E, et al. Selective unresponsiveness of pancreatic beta‐cells to acute sulfonylurea stimulation during sulfonylurea therapy in NIDDM. Diabetes 1986; 35: 1314–1320 [DOI] [PubMed] [Google Scholar]
- 8.Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide 1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 2006; 368: 1696–1705 [DOI] [PubMed] [Google Scholar]
- 9.Chang AM, Jakobsen G, Sturis J, et al. The GLP‐1 derivative NN2211 restores beta‐cell sensitivity to glucose in type 2 diabetic patients after a single dose. Diabetes 2003; 52: 1786–1791 [DOI] [PubMed] [Google Scholar]
- 10.Peters A. Incretin‐based therapies: review of current clinical trials. Am J Med 2010; 123(Suppl. 3): S28–S37 [DOI] [PubMed] [Google Scholar]
- 11.Juhl CB, Hollingdal M, Sturis J, et al. Bedtime administration of NN2211, a long‐acting GLP‐1 derivative, substantially reduces fasting and postprandial glycemia in type 2 diabetes. Diabetes 2002; 51: 424–429 [DOI] [PubMed] [Google Scholar]
- 12.Rolin B, Larsen MO, Gotfredsen CF, et al. The long‐acting GLP‐1 derivative NN2211 ameliorates glycemia and increases beta‐cell mass in diabetic mice. Am J Physiol Endocrinol Metab 2002; 283: E745–E752 [DOI] [PubMed] [Google Scholar]
- 13.Seino Y, Rasmussen MF, Nishida T, et al. Efficacy and safety of the once‐daily human GLP‐1 analogue, liraglutide, vs glibenclamide monotherapy in Japanese patients with type 2 diabetes. Curr Med Res Opin 2010; 26: 1013–1022 [DOI] [PubMed] [Google Scholar]
- 14.Kaku K, Rasmussen MF, Clauson P, et al. Improved glycaemic control with minimal hypoglycaemia and no weight change with the once‐daily human glucagon‐like peptide‐1 analogue liraglutide as add‐on to sulphonylurea in Japanese patients with type 2 diabetes. Diabetes Obes Metab 2010; 12: 341–347 [DOI] [PubMed] [Google Scholar]
- 15.World Medical Association . Declaration of Helsinki – ethical principles for medical research involving human subjects. 2008. Available at: http://www.wma.net/en/30publications/10policies/b3/index.html (accessed August 25 2011).
- 16.Garber A, Henry R, Ratner R, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD‐3 Mono): a randomised, 52‐week, phase III, double‐blind, parallel‐treatment trial. Lancet 2009; 373: 473–481 [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Katoh M, Shibasaki T, et al. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 2009; 325: 607–610 [DOI] [PubMed] [Google Scholar]
- 18.Flamez D, Gilon P, Moens K, et al. Altered cAMP and Ca2+ signaling in mouse pancreatic islets with glucagon‐like peptide‐1 receptor null phenotype. Diabetes 1999; 48: 1979–1986 [DOI] [PubMed] [Google Scholar]
- 19.Seino Y, Fukushima M, Yabe D. GIP and GLP‐1, the two incretin hormones: similarities and differences. J Diabetes Invest 2010; 1: 8–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujimoto S, Mukai E, Inagaki N. Role of endogenous ROS production in impaired metabolism‐secretion coupling of diabetic pancreatic β cells. Prog Biophys Mol Biol 2011; 107: 304–310 [DOI] [PubMed] [Google Scholar]
- 21.Mukai E, Ishida H, Kato S, et al. Metabolic inhibition impairs ATP‐sensitive K+ channel block by sulfonylurea in pancreatic beta‐cells. Am J Physiol 1998; 274: E38–E44 [DOI] [PubMed] [Google Scholar]
- 22.Yabe D, Kuroe A, Lee S, et al. Little enhancement of meal‐induced glucagon‐like peptide 1 secretion in Japanese: comparison of type 2 diabetes patients and healthy controls. J Diabetes Invest 2010; 1: 56–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mari A, Degn K, Brock B, et al. Effects of the long‐acting human glucagon‐like peptide‐1 analog liraglutide on beta‐cell function in normal living conditions. Diabetes Care 2007; 30: 2032–2033 [DOI] [PubMed] [Google Scholar]
- 24.Ahrén B, Foley JE, Ferrannini E, et al. Changes in prandial glucagon levels after a 2‐year treatment with vildagliptin or glimepiride in patients with type 2 diabetes inadequately controlled with metformin monotherapy. Diabetes Care 2010; 33: 730–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunning BE, Gerich JE. The role of alpha‐cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev 2007; 28: 253–283 [DOI] [PubMed] [Google Scholar]
- 26.Bregenholt S, Moldrup A, Blume N, et al. The long‐acting glucagon‐like peptide‐1 analogue, liraglutide, inhibits β‐cell apoptosis in vitro. Biochem Biophys Res Commun 2005; 330: 577–584 [DOI] [PubMed] [Google Scholar]
- 27.Riddle MC. Sulfonylureas differ in effects on ischemic preconditioning – is it time to retire glyburide? J Clin Endocrinol Metab 2003; 88: 528–530 [DOI] [PubMed] [Google Scholar]
- 28.Madsbad S, Brock B, Perrild H, et al. Fourteen weeks of liraglutide therapy in subjects with type 2 diabetes significantly improves first phase insulin secretion and maximal beta‐cell secretory capacity. Diabetologia 2006; 49: 4–5 [Google Scholar]
- 29.Degn KB, Juhl CB, Sturis J, et al. One week’s treatment with the long‐acting glucagon‐like peptide 1 derivative liraglutide (NN2211) markedly improves 24‐h glycemia and alpha‐ and beta‐cell function and reduces endogenous glucose release in patients with type 2 diabetes. Diabetes 2004; 53: 1187–1194 [DOI] [PubMed] [Google Scholar]
- 30.Marre M, Shaw J, Brändle M, et al. Liraglutide, a once‐daily human GLP‐1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with Type 2 diabetes (LEAD‐1 SU). Diabet Med 2009; 26: 268–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Røder ME, Porte D, Schwartz RS, et al. Disproportionately elevated proinsulin levels reflect the degree of impaired B cell secretory capacity in patients with noninsulin‐dependent diabetes mellitus. J Clin Endocrinol Metab 1998; 83: 604–608 [DOI] [PubMed] [Google Scholar]
- 32.Porte D, Kahn SE. Beta‐cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes 2001; 50(Suppl. 1): S160–S163 [DOI] [PubMed] [Google Scholar]
- 33.Knop FK, Holst JJ, Vilsbøll T. Replacing SUs with incretin‐based therapies for type 2 diabetes mellitus: challenges and feasibility. IDrugs 2008; 11: 497–501 [PubMed] [Google Scholar]