

Abstract
Metabolic syndrome and its component phenotypes, hyperglycemia, hypertension, (abdominal) obesity and hypertriglyceridemia, are major risk factors for atherosclerosis. Recently, associations between exposure to endocrine‐disrupting chemicals (EDCs), mitochondrial dysfunction, metabolic syndrome and atherosclerosis have been established, suggesting a possible common mechanism underlying these phenomena. Extending a previously proposed mitochondria dysfunction theory of metabolic syndrome and using biophysical laws, such as metabolic scaling, Murray's law and fractal geometry of the vascular branching system, we propose that atherosclerosis could be explained as an ill‐adaptive change occurring in nutrient‐supplying arteries in response to the decreasing tissue energy demand caused by tissue mitochondrial dysfunction. Various aspects of this new hypothesis are discussed.
Keywords: Atherosclerosis, Endocrine‐disrupting chemicals, Mitochondrial dysfunction
Introduction
There is increasing awareness that endocrine‐disrupting chemicals (EDCs) are involved in the pathogenesis of metabolic syndrome, type 2 diabetes and related conditions1. As metabolic syndrome is a constellation of risk factors for atherosclerosis, namely hyperglycemia, hypertension, (abdominal) obesity, hypertriglyceridemia and other phenotypes frequently associated with atherosclerosis, exposure to EDCs is expected to cause atherosclerosis (Figure 1).
Figure 1.
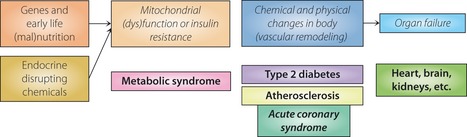
General outline of mitochondria‐based model. Genes, early life nutrition state and exposure to endocrine‐disrupting chemicals are causes of various disease states (shown in bold), such as metabolic syndrome. Mitochondrial dysfunction changes insulin sensitivity, and chemical and physical states of the body, including vascular remodeling or atherosclerosis. In contrast to the conventional view that emphasizes the narrowing of coronary arteries by atherosclerotic plaque or coronary microvascular dysfunction causes myocardial ischemia, followed by ischemic heart failure, our model emphasizes that organ failure is the end‐stage of mitochondrial dysfunction.
Persistent organic pollutants (POPs) are a most important group of EDCs, which persist in our environment, bioaccumulate in the body, and exert hazardous effects on humans and animals by disrupting the endocrine system. They are key chemicals designated among EDCs by the Stockholm Convention, a United Nations organization. Among the toxic effects of EDCs, mitochondrial toxicity is well established5. We suggest here that it could explain various aspects of atherosclerosis. Central to this thesis is that mitochondrial dysfunction would decrease energy (and heat) production of the body and lower its temperature, which in turn makes the body increase its mass to maintain an optimal core temperature7. This mechanism is believed to be necessary, because core temperature will no longer be optimal if there is no adaptive change in heat production per mass, according to body mass change. It is a reason why there is a natural law between the body mass and its unit mitochondrial function in the animal kingdom, the metabolic scaling law; the bigger the animal, the smaller the mass‐specific mitochondrial function is8.
A change in body mass would also require remodeling of vasculature in an adaptive response. The heart is special in that its high energy need is primarily derived almost exclusively from oxidative phosphorylation in mitochondria, with <5% of adenosine triphosphate (ATP) production coming from the glycolytic pathway10. Because of this dependence, increases of cardiac activity are matched with almost instantaneous parallel increases of oxygen availability. Changes in myocardial work and, thus, in energy demand, are accompanied by proportionate changes in coronary and, thus, myocardial blood flow. Decreased metabolism of an organ (because of decreased mitochondrial function), if it persists, could lead to the remodeling or narrowing in supplying arteries; atherosclerosis in coronary arteries in the case of the heart, and carotid arteries in the case of the brain.
We will show the evidence linking (EDCs induced) mitochondrial dysfunction and many biochemical features described in atherosclerosis, and also the recent conceptual advances made around biophysics, particularly the metabolic scaling, and fractal physiology could be applied as well.
Evidence Showing the Relationship Between Pops, Mitochondrial Dysfunction, Metabolic Syndrome and Atherosclerosis
Evidence supporting that exposure to EDCs including POPs leads to mitochondrial dysfunction, metabolic syndrome and atherosclerosis is increasing. For example;
Exposure to POPs induces mitochondrial dysfunction, such as mitochondrial DNA (mtDNA) depletion and damage to mtDNA and the genome5.
Mitochondrial dysfunction is associated with diabetes and metabolic syndrome14.
Serum levels of POPs are positively associated with diabetes, insulin resistance, metabolic syndrome and atherosclerosis3.
Exposure to POPs induces insulin resistance in cells and animals5.
Exposure to EDCs leads to (abdominal) obesity, enhanced inflammatory response and atherosclerosis5.
Mitochondrial dysfunction causes an increase in pro‐inflammatory signals and reactive oxygen species (ROS), and a decrease in endothelial nitric oxide (NO) availability21.
Exposure to dioxin leads to mtDNA depletion25, metabolic syndrome, hypercholesterolemia and atherosclerosis in animal studies6.
Some EDCs, such as phthalates and polychlorinated biphenyls (PCBs), are linked directly to human atherosclerosis26, and lipid extract from human atheroma has been shown to exert a toxic effect on cultured monocytes28.
However, these evidences are not included in current theories on atherosclerosis, such as the cholesterol hypothesis.
Weakness in the Current Explanations on the Pathogenesis of Atherosclerosis
Does Atheromatous Plaque Narrow the Arteries and Reduce Blood Flow?
Currently, atherosclerosis is considered to be a disease in which plaque inside the arteries limits the flow of oxygen‐rich blood to the organs and other parts of the body, which can lead to heart attack and stroke. Therefore, if the coronary artery is narrowed, the myocardial territory supplied by the diseased vessel will become ischemic, resulting in regional contractile dysfunction29. However, it is difficult to explain so‐called coronary microvascular dysfunction (CMVD), which is diagnosed clinically. In this disease, no apparent plaques are found in a coronary artery in an imaging study. In short, there is uncertainty around the concept that atherosclerotic artery obstructs blood flow, resulting in myocardial ischemia (obstruction‐ischemia theory). This chronic state should be differentiated from acute coronary syndrome, which develops when an atheroma in the coronary artery develops thrombosis and obstructs blood flow. These concepts are clearly shown in Figure 2.
Figure 2.
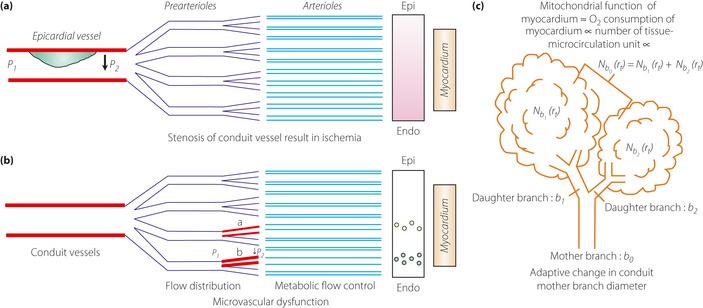
Comparison of current arterial obstruction‐ischemia concept of atherosclerosis, as shown in (a) and (b) (adapted from Lanza et al.29 with a permission from Wolters Kluwer Health) and (c) our concept. In the stenosis of (a) epicardial (Epi) artery, the territory supplied (gray area) results in regional contractile dysfunction (P1 and P2 indicate blood pressure proximal and distal to obstructive vessels), whereas in the case of (b) microvascular alterations, myocardial ischemia is considered localized only in small myocardial areas (small circles; a and b indicate dysfunctional microvessels). In these concepts, contractile abnormalities are results. (c) Our energy demand‐supply model (adapted from conceptual framework of Kamiya and Takahashi78 with a permission from The American Physiological Society); if the sum of tissue‐microcirculation units, as described here as Nb0 (rt), decrease, the diameters of daughter branches and the mother branch have to decrease. In other words, if metabolic demand of myocardium decreases (because of mitochondrial dysfunction), its blood supply would decrease, therefore, the diameter of supplying conduit vessel decreases adaptively. Endo, endocardial.
Response to Injury Hypothesis
Among the hypotheses on the pathogenesis of atherosclerosis31, the most widely held view is the ‘response to injury’ hypothesis of Ross32, or its modifications. In this hypothesis, atherosclerosis is considered to be a result of a decades‐long inflammatory response process to endothelial injury, which stimulates migration and proliferation of smooth‐muscle cells that become intermixed with the area of inflammation. Various, possibly different, forms of insult might develop between the lining endothelium and the underlying cells of the artery wall. Ross explained that in hyperlipidemic individuals, injury inducing agents seem to be due principally to the lipids and lipoproteins associated with hyperlipidemia, whereas they might also result from molecules yet to be identified with cigarette smoking, hypertension, diabetes, or possibly even some infectious agents. EDCs fit in with the characteristics of the injury causing agent(s) Ross described very well.
Need for a New Hypothesis, as Atherosclerotic Processes are not Explained Well and the Cholesterol Hypothesis is only a part
‘Injurious Agent’ is Unknown
Ross32 developed his theory from the histological similarity observed between the atherosclerotic lesions and the intimal response to injury, which has been widely accepted33. Frink suspected that the ‘injurious agent’ (any agent or any process, singly or in combination) enters the wall and injures or stimulates resident vascular smooth muscle cells (VSMCs)34. It is part of our hypothesis that EDCs can injure the vascular wall (thus induce inflammation) and initiate fatty streaks, as shown in an animal experiment20, as well as the whole body. Indeed, a recent study found human atherosclerotic plaque lipid extract contains substances that impair the anti‐oxidant defense capacity of monocytes28. Those cells exposed to lipid extract showed a significant increase in the ROS level with a simultaneous rise of glutathione oxidation. A significant decrease in the intracellular anti‐oxidant enzyme activity of catalase, glutathione peroxidases and thioredoxin reductases was observed, along with an increase in intracellular superoxide dismutase activity, suggesting endogenous H2O2 overproduction. Those kinds of responses are very similar to the responses the cells show when they are exposed to EDCs, such as dioxin, bisphenol A or phthlates7. Here, we argue the response to injury hypothesis could be a better theory if it was modified to accept EDCs as injury agents.
Endothelial Dysfunction Occurring Long Before the Appearance of Atherosclerosis is Usually not Considered
The pathophysiological changes of atherosclerosis occur long before the occurrence of structural changes, such as intimal hyperplasia. Offspring born to nutritionally deprived mothers are predisposed to metabolic syndrome, and manifest endothelial dysfunction very early in life. Diminished levels of bioavailable NO are regarded as one of the hallmarks of endothelial dysfunction, and occur through several different mechanisms, such as reduced endothelial NO synthase (eNOS) expression levels, reduced eNOS enzymatic activity and reduced NO bioavailability24. Endothelial dysfunction is also associated with an increase in ROS production in the vasculature. This abnormality is difficult to explain with current theories, but could be explained easily with a mitochondria‐based model. We have presented evidence that the thrifty phenotype could be explained in terms of functional changes in mitochondrial unit function37.
Vascular Remodeling is not Explained
Atheroma development is compensated for by gradual dilation of the artery, or ‘remodeling’, so that the lumen remains unaltered up to a point. In 1987, Glagov et al.38 studied left main coronary arteries obtained at autopsy to evaluate whether atherosclerotic human coronary arteries enlarge in relation to plaque development. They found the internal elastic lamina area (representing the whole coronary artery, including the atherosclerotic area) correlates directly with the area of the atherosclerotic lesion, suggesting that coronary arteries enlarge as the lesion area increases. The lumen area does not decrease until a certain level is reached (lesion area/internal elastic lamina area ×100 over 40%; Figure 3a).
Figure 3.
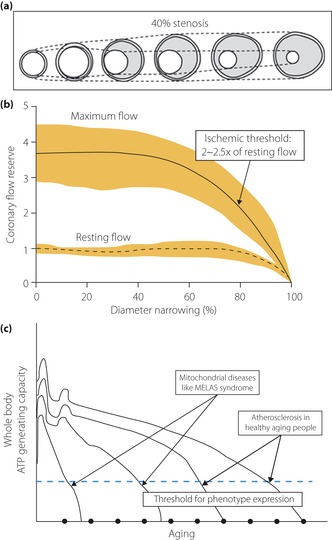
(a) The sequence of adaptive change or remodeling of atherosclerotic arteries. The artery enlarges initially in association with plaque accumulation to maintain the lumen area. When the lumen reaches more than 40% stenosis, the artery is believed to no longer enlarge at a rate sufficient to prevent narrowing of the lumen (adapted from Glagov et al.38 with permission from Massachusetts Medical Society). (b) However, resting coronary blood flow is maintained until arterial lumen is narrowed to approximately 80% (adapted from Gould40 with permission from American College of Cardiology Foundation). Coronary blood flow reserve (<2–2.5 times baseline value) is believed to be more important than the degree of narrowing itself for the manifestation of ischemic symptom. Most importantly, the figure shows the relentless decline of coronary artery narrowing along the aging. (c) Adenosine triphosphate (ATP) generating capacity of body decline along with aging. If it declines below a certain threshold, clinical phenotype(s) will manifest (adapted and modified from Shoffner and Wallace102). In our hypothesis, ischemic heart disease occurs when coronary blood flow reaches this threshold as a result of the decreasing bioenergetic function of the myocardium, in parallel with the decline of whole‐body metabolism.
Over the years, intensive studies on arterial remodeling have shown that despite comparable diffuse disease and arteriographic stenosis, functional reserve of the diseased segment might only be mildly reduced with remodeling. Readers are referred to a recent review by Kern and Samady39 for further details on this very complex issue.
Why should gradual dilation of the artery or remodeling occur, if narrowing of the artery is the primary process? Regional myocardial blood flow studies carried out with several new techniques, including positron emission tomography, suggested alternative explanations consistent with the mitochondria‐based model. This technology provided evidence that functional, rather than structural, disturbances might reflect more accurately the risks of developing coronary artery disease (Figure 3b)40. Furthermore, new technology showed the dissociation between the anatomical and functional measures of coronary stenosis severity as a result of diffuse atherosclerosis, and the extent of arterial remodeling. These observations are clearly against the concept that narrowing of the artery by atherosclerosis leads to functional derangement of downstream tissue; that is, myocardium. These findings are well matched with the age‐dependent decline of whole‐body mitochondrial function, as shown in Figure 3c.
Revascularization Therapy Does not Improve Prognosis
In a controlled trial, surgery to open a complete blockage in one of two carotid arteries did not prevent subsequent strokes41. Another clinical trial with a stent to open blocked arteries (by atherosclerosis) deep in the brain was also proven ineffective42. However, cerebral revascularization, either direct or indirect, was proven to be quite effective and sometimes recovered the lost brain function induced by ischemia in a disease called moyamoya disease, which is a chronic steno‐occlusive vasculopathy involving the distal supraclinoid internal carotid arteries43. Why is bypass surgery ineffective in atherosclerotic brain disease, while it is effective in moyamoya disease?
For coronary heart disease, many studies have also failed to show a benefit of opening a narrowed coronary artery, except in acute coronary syndrome with significant functional reserve44. For example, a randomized trial involving 2,287 patients with coronary artery disease at 50 USA and Canadian centers showed that percutaneous coronary intervention did not reduce the risk of death, myocardial infarction or other major cardiovascular events. After an average follow up of 4.5 years, the two groups were remarkably similar45.
Simple opening of stenotic coronary arteries will not affect the natural history of atherosclerotic heart disease, which will take a downhill course relentlessly, which is similar to the decline of the whole‐body bioenergetic function of the ageing process (Figure 4). Therefore, the preferred treatment for patients with stable coronary artery disease is the best available medical therapy.
Figure 4.
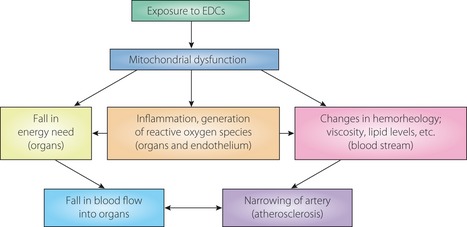
Various aspects of atherosclerosis are explained by the endocrine‐disrupting chemical (EDC)‐induced mitochondrial dysfunction. Atherosclerosis is ill‐adaptive changes (remodeling) occurring in arteries to reduce the blood supply as a response to decreased nutrient demand of organs, and blood supply and demand is in a balanced state. Exposure to EDCs, which are regarded as injurious agents, cause mitochondrial dysfunction, inflammatory response and increase reactive oxygen species production in the endothelium and tissue. Blood itself will be changed in its property; rheology, viscosity, and lipid components and levels.
Alternative Explanation for Atherosclerosis: Linking Pops Exposure, Mitochondrial Damage and Biochemical Changes Observed During Atherosclerosis
Toxin‐Induced Mitochondrial Dysfunction Could Explain Vascular Inflammation
Persistent organic pollutants, such as dioxins, damage mitochondria and induce insulin resistance, metabolic syndrome and atherosclerosis. It is part of our hypothesis that these toxins can injure the vascular wall (thus induce inflammation) and initiate fatty streaks, as shown in an animal experiment20.
The importance of the role of mitochondria in this process has become clearer in recent years. For example, Manfredi and Rovere‐Querini23 suggested that mitochondria could be the primary organelle involved in initiating inflammation. The toxic effects of dioxin and other pollutants on mitochondrion are well established. For example, Chen et al.11 showed that one of most important POPs, dioxin, damages mitochondria and induces cells to release free radicals. Once mitochondria are damaged, they release additional ROS, which causes further damage in a vicious cycle. The mitochondria theory of aging is closely linked to the free radical theory of aging, with long history21, although this issue is far from settled. West and Bergman48 discussed the mechanism of aging from a systems biology framework, which is basically an extension of metabolic scaling law9. We want to emphasize that those points are incorporated into our mitochondria‐based model37.
Endothelial cells with damaged mitochondria release damage‐associated molecular pattern molecules (including high‐mobility group protein B1, heat shock proteins and S100 proteins), which recruit inflammatory cells such as macrophages. Thus, POPs could very likely induce inflammation in the endothelium, as seen in fatty streaks (a feature of atherosclerosis).
Other mitochondrial proteins activate formyl peptide receptor‐1 on neutrophils, resulting in the production of a collagenase that sustains leukocyte migration in peripheral tissues. Furthermore, intravenous injection of mitochondrial proteins into mice activates circulating neutrophils, with random extravasation at peripheral organs, such as the liver and lungs50. These results show that mitochondrion is a critical instigator of inflammation during the atherosclerotic process.
C1C12 myoblasts that are mtDNA‐depleted secrete increased amounts of interleukin‐6 and other inflammation mediators, as well as being resistant to insulin stimulation13. Another mtDNA‐depleted cell line became insulin‐resistant19. These findings clearly suggested that mitochondrial dysfunction is at a crossroads between insulin resistance and the inflammatory response, at least at the cell level.
Mitochondrial Dysfunction can Explain Increased Endothelial Cell ROS Production in Atherosclerosis
Endothelial cells depend heavily on the glycolytic pathway for ATP production and use very little oxidative phosphorylation. However, they contain mitochondrion, which is critically involved in maintaining the fine regulatory balance for calcium handling, ROS production and NO metabolism, which play important roles in controlling microcirculation51.
Recently, Davidson52 made a detailed review on this subject, and suggested how mitochondrial function in the endothelium could be linked to cardiovascular disease. He emphasized the experiment by Zhang et al.53, which showed that thioredoxin (Trx2) overexpression not only prevented mitochondrial oxidative stress in hypertensive mice and preserved endothelial function, but even when Trx2 overexpression was restricted to the endothelial mitochondria, it was still able to decrease oxidative stress, improve aortic endothelia cell function and prevent atherosclerotic lesions. Davidson54 showed evidence that the endothelium is a direct target of all major risk factors for heart disease, including diabetes, hyperlipidemia and hyperglycemia, oxidized low‐density lipoprotein, inflammation, smoking, aging, and hypertension, and that the common element was ROS. However, he also acknowledged that targeting mitochondrial ROS with various agents failed to prevent the progression of atherosclerosis in humans.
One might ask if current animal models of atherosclerosis really reproduce human atherosclerotic disease, because current animal models of atherosclerosis usually do not use POPs. If POPs exposure is a major cause of atherosclerosis in humans, and if ROS overproduction is secondary to mitochondrial damage incurred by POPs exposure, one could expect that targeting mitochondrial ROS with various agents in humans might not be effective.
Disturbed NO Bioavailability of Endothelial Cells can be Explained by Mitochondrial Dysfunction
Another important functional mediator of endothelial dysfunction in atherosclerosis is NO. Disturbances in NO bioavailability have been linked to endothelial dysfunction, leading to increased susceptibility to atherosclerotic lesions, hypertension, hypercholesterolemia, diabetes mellitus, thrombosis and stroke24. The most compelling evidence for this conclusion is provided by transgenic mice with homozygous inactivation of the eNOS gene. These mice could not produce NO in the endothelium, and showed features of metabolic syndrome, including obesity, insulin resistance, hyperlipidemia and hypertension55; eNOS is known to play an important role in regulation of mitochondrial function and its biogenesis57.
This role has not received much attention, however, because it is regarded as a phenomenon accompanying endothelial dysfunction caused by poor NO availability22. However, the realization of the ubiquitous presence of eNOS, previously thought to play a role only in the endothelial part of the vasculature, might change this view. For example, cardiac myocytes and adipocytes have eNOS, which play important roles in glucose and fatty acid metabolism, intracellular signaling, and control of mitochondrial function and its biogenesis in those cells58. Furthermore, mitochondrial dysfunction might modulate endothelial NO and superoxide generation60. More importantly to our discussion, Koh et al.61 reported that eNOS in adipocytes plays a major role in adiponectin synthesis, which has been proposed as a central player in the pathogenesis of metabolic syndrome62.
Although adiponectin and its receptors play important roles in the pathogenesis of insulin resistance, Koh et al.61 clearly showed they play a secondary role to mitochondrial dysfunction. One has to appreciate a close relationship exists between eNOS function (thus NO availability and other endocrine mediators including adiponectin) and mitochondrial function.
Proliferation of VSMCs Filled with Lipids Could be Another Manifestation of Response to Endothelial (Mitochondrial) Injury
The main cell type involved in narrowing of the arteries (type 2 lesions in particular) is VSMCs filled with lipids. One of the major functions of the endothelium is controlling VSMC contraction or a tonic role54. Endothelium also plays a trophic role, a negative influence on smooth muscle proliferation, and controlling macrophage and leukocyte traffic through the endothelial layer. NO plays very important roles here, as discussed previously. VSMC proliferation might be another epiphenomenon of NO deficiency24, thus mitochondrial dysfunction. Furthermore, there is evidence that mitochondrial dysfunction stimulates VSMC migration directly64. Note that the degree of mitochondrial dysfunction in the endothelium is somehow linked to VSMC proliferation and to narrowing of the vascular lumen, such that less blood flow is matched to the decreased metabolic demand of the myocardium it supplies. We believe that this explanation is informative; atherosclerosis could be both an inflammatory response to a toxic agent(s) and an adaptive response to the decreased tissue energy demand of its effect on mitochondrial function. Further evidence supporting this concept of adaptive response is provided later.
Atherosclerosis in Fractal Pathophysiology; an ILL‐Adaptive Response of the Vascular System to Mitochondrial Dysfunction – Supply and Demand of Energy
Glagov et al.38 pointed out that the atherosclerosis process itself does not cause cardiovascular events, because blood flow is preserved through outward remodeling of the arterial wall. However, acute, occlusive luminal thrombosis does occur in a small percentage of lesions, termed ‘culprit lesions’, leading to ischemia or death of distal tissues. Acute coronary syndrome is a clear example. We will not discuss this rather late‐stage phenomenon65, where antiplatelet therapy and opening of the narrowed artery were proven effective44.
We will be focusing on the entire stages of the atherosclerosis process and remodeling or narrowing of the artery itself. In our model, atherosclerosis is an ill‐adaptive response of the supplying coronary artery to the decreased metabolic demand (in the case of coronary artery disease) of the myocardium (as a result of mitochondrial dysfunction; Figure 4).
Microvascular Rarefaction, or the Reduced Number or Combined Length of Small Vessels in a Given Volume of Tissue: Basic Abnormality
Microcirculation, including the smallest arteries, arterioles, capillaries and venules, plays a critical role in the exchange of gases, nutrients and metabolites between blood and tissues. It comprises a basic unit of physiology, and in the pathology in atherosclerosis. Abnormalities of the microvascular system are common among patients with conventional cardiovascular risk factors, including hypertension, diabetes, obesity and dyslipidemia. By quantifying the microcirculatory system, either by counting capillaries in histological sections or by a functional analysis of capillary blood flow, Levy et al.67 showed that: (i) hypertensive patients have microvascular rarefaction (a reduced number or combined length of small vessels in a given volume of tissue); (ii) these microvascular changes occur very early in these conditions; and (iii) they are also hallmarks of the long‐term complications of hypertension and diabetes (a result of those disease states).
It is appreciated that counting the capillary density, however, does not reflect the functional state of capillaries. Some microvessels are not perfused under the resting condition. They could be recruited during hyperemia. An estimate of the available flow reserve has been used as a useful index of microvascular function. Capillary recruitment (capacity) is often assessed in the skin by video microscopy, and functional blood flow reserve (capacity) is often measured for coronary circulation and expressed as the ratio of maximal to basal blood flow. Capillary recruitment capacity (thus functional reserve) is significantly reduced in the skin of hypertensive patients, as compared with normotensive individuals, and capillary recruitment is inversely correlated with blood pressure. This relationship extends across the normotensive and the hypertensive range, confirming impaired microcirculation in patients with hypertension68.
Coronary flow reserve capacity is significantly lower in obese humans than in non‐obese humans, and capillary recruitment is reduced compared with lean control subjects. Levy et al.67 also noted that microvascular abnormalities that lead to impaired tissue perfusion appear to represent a generalized condition that affects multiple tissues and organs. They cited the following studies: (i) coronary flow reserve in hypertension is correlated with the media: lumen ratios of small arteries in biopsies of subcutaneous fat69; (ii) dilatation of venules in the retina independently predict progression of cerebral small‐vessel disease70; and (iii) reduced coronary flow reserve predicts the occurrence of retinopathy in diabetes71. We will take these observations to the thrifty phenotype hypothesis, wherein a similar discussion is in progress.
Fetal Malnutrition and Metabolic Syndrome in Later Life: Rarefaction of Tissue‐Microcirculation System Complex
The role of the intrauterine environment, particularly maternal nutrition, in influencing fetal growth and cardiovascular health in offspring in later life is known as the thrifty phenotype hypothesis. In a recent review on this subject, Clough and Norman72 showed that suboptimal maternal nutrition and fetal growth result in reduced microvascular perfusion and functional reserve capacity, which are strongly associated with later development of obesity, type 2 diabetes and hypertension. They highlighted the fact that these conditions are linked to microvascular rarefaction and remodeling that together limit capillary recruitment, reduce exchange capacity, increased diffusion distances for metabolic substrates, and increased local and overall peripheral resistance72. These changes in small vessel structure and function are detectable very early, long before the onset of overt cardiovascular and metabolic disease67.
Previously, we showed the tissue mitochondrial density is decreased in offspring of a malnutrition‐afflicted dame37. If mitochondrial biogenesis of the offspring is not optimal as a result of malnutrition during development, resulting in decreased tissue mitochondrial density, the tissue‐microcirculation system complex would also develop rather poorly.
Murray's Law, Metabolic Scaling Law and the Fractal Nature of the Vascular System
Murray's law states that the cube of the radius of a parent vessel should equal the sum of the cubes of the radii of the daughter vessels75. In Murray's optimum system, flow and vessel radius are functionally related: an optimum radius is found for a given flow. For a given metabolic coefficient, the volume of the vascular system in an organ or organism will depend on the flow required of it: vasculature optimum for high flows will have larger vessels than that for low flows, the cubes of the vessel radii being proportional to the flow required.
The overall metabolic rate in animals is generally accepted to show negative allometry, scaling mass to a power ~0.75, known as Kleiber's law, and thus the mass‐specific metabolic rate is proportional to a power of approximately −0.25. This means that larger‐bodied species have lower mass‐specific metabolic rates than those of smaller‐bodied species. This scaling relationship is applicable to different sized animals within a species and goes down to the mitochondrial function unit8.
In 1997, West et al.49 proposed a model (known now as West–Brown–Enquist or WBE model) and claimed this provided a complete analysis of scaling relationships for mammalian circulatory systems. They also claimed it predicts structural and functional properties of the vertebrate cardiovascular system, among many other distribution systems with fractal characteristics. The fractal nature of the branching systems in our body, including vascular and bronchial trees, is well appreciated49. The optimization of the energy dissipated in this network system to a minimum is empirically established. Recently, Huo and Kassab77 derived scaling laws within an organ of a given species, and validated the volume‐diameter and flow‐length scaling laws using the minimum energy hypothesis, without using the controversial assumption of space‐filling terminal branches49, which has been debated79. It should be emphasized that this debate is on the ‘origin’ of the law, not the law itself.
Branching systems including vascular and bronchial trees increase their branch density toward terminals according to a power function with the exponent called the fractal dimension. From a stochastic model based on this feature, Kamiya and Takahashi78 formulated the fractal‐based integrals to calculate such morphological parameters as aggregated branch length, surface area and content volume for any given range of radius. Then they derived the branch number and cross‐sectional area by virtue of the logarithmic sectioning of the axis of the radius, and of the branch radius–length relationship also given by a power function of the radius with an exponent. Using those derivatives, they quantified various hydrodynamic parameters of vascular and bronchial trees as fluid conduit systems, including the individual branch flow rate, mean flow velocity, wall shear rate and stress, internal pressure, and circumferential tension. The validity of these expressions was then verified by comparing the outcomes with actual data measured in vivo in the vascular beds. Analyses of mammalian skeletal muscles have shown that the structure of the capillary‐tissue arrangement (Krogh cylinder) is most efficiently tailored for oxygen delivery to tissues during heavy muscular exercise, and that the optimum radius of the cylinder is scale‐independently constant. Most importantly, they concluded the organ size proportional to body mass was mainly attributable to the accumulated number of basic units.
Basic Unit of Structure and Function: Tissue‐Microcirculation System Unit
One could appreciate such units in work by Harel et al.,80 who used functional magnetic resonance imaging (MRI) to map the functional units down to the level of cortical columns and lamina of the brain. They advanced a theory in which a cortical column, which is an ensemble of neurons involved in a particular neuronal computation, is spatially correlated with a specific vascular unit; that is, a cluster of an emerging principal vein surrounded by a set of diving arteries. They suspected that such a correlation between functional (neuronal) and structural (vascular) units existed as a fundamental intrinsic cortical unit, as shown in Figure 5. This theory argues that the circular nature of a regular organization of vessels within the cortex and a relatively uniform distribution of these principal veins spaced every 0.75–1 mm. From postmortem observations of humans, they have suggested such vascular organization might manifest as a ‘vascular unit’, which is associated with a known ‘functional unit’ in the cortex81.
Figure 5.
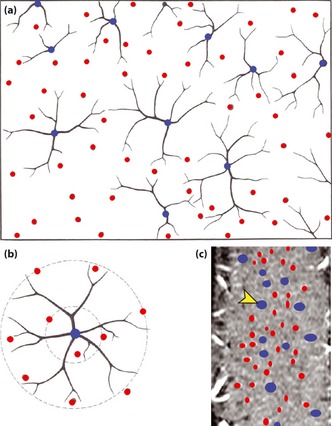
Vascular unit model proposed by Harel et al.80 (reproduced with permission from Frontiers in Neuroenergetics). The (a) circular nature of the vascular organization is shown in a cross‐section showing the venous units and (b) their arterial rings; a principle cortical vein (blue) is surrounded by several penetrating arteries (red). (c) Vessel classification maps in a tangential section in cat visual cortex, derived from the magnetic resonance imaging data.
Glenny83 suggested that the geometry of the airway and vascular trees might be considered a demonstration of emergent adaptive behavior of fractal geometry. The shapes of the bronchial and vascular trees are considered adaptive, because specific geometries are advantageous to the organism, reinforced through natural selection. As fractal geometry is conserved in branching trees throughout the plant and animal kingdoms, it is thought to confer an evolutionary advantage of efficient distribution of nutrients, suggesting that these fractal structures are important to the organism. We believe that optimal organization of the tissue‐microvascular system complex with tight regulatory control of vascular tree geometries is a necessity in the evolutionary process. This idea is similar to the concept of symmorphosis, which postulates a quantitative match of design and functional parameters within a defined system84.
A tight association between the ‘vascular unit’ and the ‘functional unit’ can be also seen in muscles. Weibel and Hoppeler79 showed that aerobic capacity is determined by two properties – body size and athletic prowess. They showed that the maximum oxygen consumption rate (VO2max) and the morphometric characteristics of muscle are parallel. If VO2max is plotted against the whole‐body muscle mitochondrial volume (V[mt]), the two variables appear to be tightly associated (Figure 6). They also observed that mitochondria can only perform at a high rate of oxidative phosphorylation if they receive an adequate supply of O2 from capillary blood.
Figure 6.
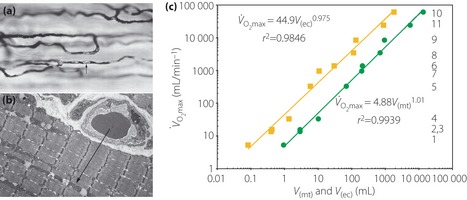
(a) Light micrograph of capillary network in muscle, with an arrow pointing to an erythrocyte in stained plasma and (b) an electron micrograph shows the path for oxygen from capillary erythrocyte to mitochondria in the muscle cell, suggesting tight linkage between mitochondrial function and blood supply at the subcellular level. (c) Maximum oxygen consumption rate (VO2max) plotted as function of total muscle mitochondrial volume (V[mt]; squares) and capillary erythrocyte volume (V[ec]; circles) in 11 species of animals show clearly the tight relations between mitochondrial function and its blood supply in quantitative terms (reproduced from Weibel and Hoppeler79 with permission from The Company of Biologists Ltd).
Based on a model developed by Bassingthwaighte and Goresky85, Beard86 developed a model of oxygen transport and cellular (bio)energetics that could explain observations of in vivo cardiac energy metabolism. He assumed that within the capillary, interstitial space and cellular (myocyte) space, the concentrations of oxygen and other metabolites vary primarily along the length of the capillary (the advective transport is modeled in the capillary region): the interstitial and cellular spaces are assumed to be stagnant (non‐flowing). The cellular region is further subdivided into cytoplasmic and mitochondrial compartments. He showed a ‘one‐dimensionally distributed blood–tissue exchange model integrating the three‐regions’ that is able to reproduce experimental observations on ATP, adenosine disphosphate, creatine phosphate and inorganic phosphate over a range of workloads and during coronary hypoperfusion (Figure 7). This model also explained metabolite levels observed at low to moderate workloads, and the changes in metabolite levels and tissue oxygenation detected during graded hypoperfusion.
Figure 7.
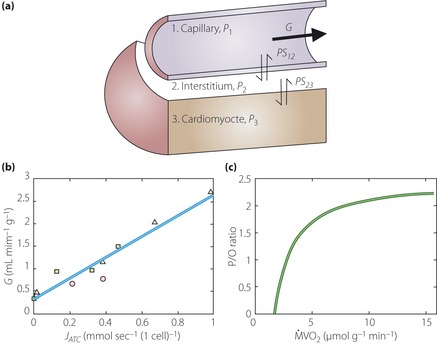
(a) Three‐region one dimensionally distributed blood–tissue exchange model for oxygen transport by Beard86 (reproduced with a permission). The three regions correspond to capillary, interstitium and cell (myocyte), and oxygen is transported by passive permeation between these regions. (b) Plot of coronary flow as a function of the predicted rate of adenosine triphosphate (ATP) consumption (JATC). The model‐predicted rate of oxygen consumption (MVO2) agrees well with the reported experimentally measured estimates of MVO2. (c) The predicted ratio between mitochondrial ATP production and rate of oxygen atom consumption (P/O ratio) is plotted for JATC values, and shows the oxygen consumption rate offsets the rate of proton leak across the inner mitochondrial membrane, which was also consistent with data. G, coronary flow.
A comparison of the model predictions with the data of Katz et al.87 provides independent validation of the model of Beard. Therefore, this three region one‐dimensionally distributed blood–tissue exchange model of Beard could be used to describe the integrated vascular and functional unit mathematically.
Previously, we applied the theory developed by West et al.9 in explaining the metabolic scaling law to the development of obesity and/or insulin resistance7. We reasoned that if mass‐specific mitochondrial function decreases, body mass must increase to compensate for a decreasing body core temperature. Earlier in the present review, we presented evidence showing that exposure to EDCs leads to mitochondrial dysfunction. The tissue‐microcirculation system unit would become dysfunctional, as it is in tight correlation with unit mitochondrial function of tissue. When the function of the tissue‐microcirculation system unit decreases, the blood flow into organs with less of these units will decrease, demanding remodeling of supplying arteries. This is a central tenet of our hypothesis. We concur with Harel et al.80, that the tools and knowledge being developed today could be applied to metabolic syndrome and then to atherosclerosis. We also believe that the synthesis of our hypothesis could benefit further by incorporating advances made in the fractal physiology88.
Localization of Atheroma in Arteries and the Underlying Physiological Principle: More Evidence for Mitochondrial Dysfunction as a Cause of Atherosclerosis
Decreased Blood Flow
With a color‐coded and duplex Doppler sonography system, Bai et al.89 found lower blood flow velocity and volume, higher resistance index in the carotid arteries, and more enlarged common carotid artery diameter in ischemic stroke patients than in controls. They also showed that common carotid end‐diastolic velocity could predict future development of ischemic stroke either alone or in combination with intima‐media thickness, a measure of atherosclerosis90.
Transcranial Doppler sonography shows an excellent correlation with MRI in measuring brain blood flow and flow velocity, suggesting measuring blood flow with Doppler sonography technique is a good alternative to more valid methods using MRI91. Blood flow decrease was also found to be quantitatively associated with future development of stroke in that study. Thus, we could safely conclude that blood flow into the brain is decreased in atherosclerosis and precedes the development of stroke.
Boundary‐Layer Separation of Blood Flow
In 1966, Fox and Hugh92 tried to explain the focal nature of atheroma lesions, and their peculiar predilection for the curved segments and the mouths of branched vessels based on a physiological principle. They pointed out that this peculiar localization of atheroma could not be explained by a ‘disorder of lipid metabolism’. They observed that in a fluid system, well‐defined static zones can form despite quite high velocities nearby as a result of a process known as ‘boundary‐layer separation’. From the striking similarity between the regions where this occurs and the sites of predilection of atheromas, they imagined that static zones occur in the arterial system, which might allow the interaction of platelets and fibrin to form a mesh in which lipid particles become trapped, and that this becomes organized to form a plaque of atheroma as envisaged by Duguid93.
They found the striking similarity between the areas at which atheroma is commonly found, and those where the occurrence of boundary‐layer separation is predicted. They suspected a causal relationship between the two processes; the stagnant or low‐velocity areas as a result of the separation of the boundary layer permit intravascular aggregation, leading to a variety of clotting; organization of the solid material or the development of a thrombus would follow, resulting in the formation of an atheromatous plaque.
Observing 12 adult human carotid bifurcations obtained at autopsy, Zarins et al.94 showed pathological evidence that intimal thickening is greatest and consistently eccentric in the carotid sinus. Wall shear stress along the inner wall was higher; and along the outer wall opposite the flow divider apex, where the intima was thickest, wall shear stress was lower. Regions of moderate to high shear stress, where flow remained unidirectional and axially aligned, were relatively spared of intimal thickening. Intimal thickening and atherosclerotic plaques develop largely in regions of relatively low wall shear stress, flow separation and departure from axially aligned, unidirectional flow94. Using quantitative model flow studies, this group had confirmed early plaque deposition occurs in regions characterized by flow separation, stasis, increased particle residence time, low mean shear stress and oscillation in shear stress direction.
Increased Blood Viscosity and Hypercholesterolemia
One of the major predictions of the theory based on boundary‐layer separation is that alteration in blood viscosity might be a contributing factor to atheroma, which was shown by Ercan et al.95 They showed that plasma viscosity is positively correlated with total serum cholesterol, the atherogenic index, total cholesterol (total‐C)/high‐density lipoprotein cholesterol (HDL‐C) ratio and low‐density lipoprotein cholesterol (LDL‐C)/HDL‐C ratio in hypercholesterolemic patients with peripheral arterial obstructive disease (POAD). Furthermore, plasma viscosity was higher in hypercholesterolemic patients with POAD than in normocholesterolemic patients with POAD.
Total serum cholesterol is a well‐known risk factor for atherosclerosis. Note that a positive linear correlation of plasma viscosity with total‐C and apolipoprotein (apo) A‐II and apoB was observed in the World Health Organization sponsored multinational monitoring of trends and determinants in cardiovascular disease (MONICA) project96. That study also showed that blood viscosity is negatively related to HDL‐C concentration, which reduces the risk of coronary heart disease.
Mitochondrial dysfunction might be the common biological reason behind all these phenomena and the relationships between them. Although there is no direct evidence linking mitochondrial dysfunction and increased blood viscosity in quantitative terms, there is indirect evidence. Analyzing longitudinal data of 12,881 initially non‐diabetic adults, who participated in the Atherosclerosis Risk in Communities Study (1987–1998) for blood viscosity, Tamariz et al.97 found that blood viscosity is independently associated with insulin resistance and several features of metabolic syndrome. Other studies have reported a similar association98. These findings suggest elevated blood viscosity and hypercholesterolemia might be different manifestations of systemic mitochondrial dysfunction. Blood flow through arteries must be lower in patients if there is systemic mitochondrial dysfunction, as the metabolic demand will be decreased. Combined with hypercholesterolemia and elevated blood viscosity, atheroma (deposition of platelets and lipid in mesh) would develop in the predilection sites, where the blood flow is stagnant and shear stress is lowest, by a process of boundary‐layer separation.
Supply and Demand Mismatch: Development of Angina Pectoris and Stroke
Angina pectoris is considered a manifestation of coronary artery disease caused by atherosclerosis. A mismatch between supply and demand is given again as an explanation for this condition; for example. by Lanza et al.29, which is shown in Figure 3. We suppose that blood supply to the myocardium is well adapted to the decreased metabolic needs, until lumen diameter is narrowed by approximately 80% or functional reserve is approximately 2–2.5 times the basal state, as shown in Figure 4. When myocardial energy demand is decreased for a long time, energy‐supplying vasculatures are expected to narrow, in an (ill‐)adaptive response. If a sudden increase in energy demand occurs, as in the case of sudden exercise, the blood supply through narrowed vessels simply cannot keep up as a result of decreased functional reserve, precipitating ischemia or angina pain.
In contrast, if myocardial metabolism is decreased well below the minimal demand to maintain cardiac function, heart failure will ensue. In this interpretation, CMVD could be regarded as a state of mitochondrial dysfunction of the myocardium, not a cause. High prevalence of small‐vessel ischemic disease, lacunar infarctions and other brain abnormalities in patients undergoing coronary artery bypass graft supports this notion99.
Conclusion
Extending our earlier mitochondrial dysfunction theory of metabolic syndrome, and the proposal for the necessity of research on POPs100, we extended this hypothesis further that mitochondrial dysfunction induced by environmental toxins, such as dioxins, could be a cause of metabolic syndrome and also of atherosclerosis. We explained atherosclerosis as an adaptive change occurring in supplying arteries in response to the decreasing tissue energy demand. EDCs, such as dioxins, could induce mitochondrial dysfunction, which in turn would lead to decreased tissue metabolism, which in turn induce most of the biochemical features observed in atherosclerosis.
With systems bioenergetics and fractal physiology in mind, we showed that physiological laws, such as the metabolic scaling law, Murray's law and the fractal dimension of the vascular branching system, are useful in explaining the phenotypes of metabolic syndrome and also the pathogenesis of atherosclerosis.
These biophysical laws and biomathematics provided strong support for a concept that mitochondrial dysfunction at the level of the tissue‐microvascular system complex could cause atherosclerosis in the supplying arteries. These facts suggest that mitochondrial function of the tissue‐microcirculation unit could be used in the synthesis of fractal physiology. In this synthesis, therefore, EDCs‐induced mitochondrial dysfunction will be seen as an environmental ‘cause’ of metabolic syndrome and atherosclerosis. There are mathematical tools and knowledge, such as fractal physiology, which could be developed further to describe metabolic syndrome and atherosclerosis.
In a recent essay on physics and medicine, West wrote that ‘traditional medicine has typically focused on specific events at a localized level of organization and often in isolation with a narrow focus and timeframe’101. There might be no better example than coronary heart disease and atherosclerosis. He stressed the ‘need to understand quantitatively, from underlying principles, the mechanisms that determine the baseline scale of life, and thereby develop corresponding metrics to define the average, idealized, healthy human being based on fundamental measurable parameters associated with different levels–e.g., cells, mitochondria, proteins, repair mechanisms, metabolism, environment, specific diseases, individual life history, genomic variation, and diet’.
We conclude that such an idealized model could be synthesized based on the tissue‐microcirculation unit or mitochondrion and biophysical laws.
Acknowledgements
Kyong Soo Park and Young Min Cho, both at Seoul National University College of Medicine, are gratefully acknowledged for their critical comments to the paper. We also acknowledge that their previous works contributed significantly in developing this article. This article was supported in part by RIC 2012‐02‐02 and the National Research Foundation of Korea (No. ROA‐2008‐000‐20127‐0). We do not have any conflicts of interest related to the content of this paper.
(J Diabetes Invest, doi: 10.1111/jdi.12048, 2013)
References
- 1.Carpenter DO. Environmental contaminants as risk factors for developing diabetes. Rev Environ Health 2008; 23: 59–74 [DOI] [PubMed] [Google Scholar]
- 2.Everett CJ, Frithsen I, Player M. Relationship of polychlorinated biphenyls with type 2 diabetes and hypertension. J Environ Monit 2011; 13: 241–251 [DOI] [PubMed] [Google Scholar]
- 3.Lee DH, Lee IK, Porta M, et al Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non‐diabetic adults: results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 2007; 50: 841–851 [DOI] [PubMed] [Google Scholar]
- 4.Lee D‐H, Lee I‐K, Song K, et al A strong dose‐response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999–2002. Diabetes Care 2006; 29: 1638–1644 [DOI] [PubMed] [Google Scholar]
- 5.Lim S, Ahn SY, Song IC, et al Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS ONE 2009; 4: e5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruzzin J, Petersen R, Meugnier E, et al Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ Health Perspect 2010; 118: 465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HK, Cho YM, Kwak SH, et al Mitochondrial dysfunction and metabolic syndrome‐looking for environmental factors. Biochim Biophys Acta 2010; 1800: 282–289 [DOI] [PubMed] [Google Scholar]
- 8.Savage VM, Allen AP, Brown JH, et al Scaling of number, size, and metabolic rate of cells with body size in mammals. Proc Natl Acad Sci USA 2007; 104: 4718–4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West GB, Woodruff WH, Brown JH. Allometric scaling of metabolic rate from molecules and mitochondria to cells and mammals. Proc Natl Acad Sci USA 2002; 99(Suppl 1): 2473–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopaschuk GD, Ussher JR, Folmes CD, et al Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010; 90: 207–258 [DOI] [PubMed] [Google Scholar]
- 11.Chen S‐C, Liao T‐L, Wei Y‐H, et al Endocrine disruptor, dioxin (TCDD)‐induced mitochondrial dysfunction and apoptosis in human trophoblast‐like JAR cells. Mol Hum Reprod 2010; 16: 361–372 [DOI] [PubMed] [Google Scholar]
- 12.Ibrahim MM, Fjære E, Lock E‐J, et al Chronic consumption of farmed salmon containing persistent organic pollutants causes insulin resistance and obesity in mice. PLoS ONE 2011; 6: e25170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park S‐Y, Choi JH, Ryu HS, et al C1q tumor necrosis factor alpha‐related protein isoform 5 is increased in mitochondrial DNA‐depleted myocytes and activates AMP‐activated protein kinase. J Biol Chem 2009; 284: 27780–27789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang C‐H, Su S‐L, Hsieh M‐C, et al Depleted leukocyte mitochondrial DNA copy number in metabolic syndrome. J Atheroscler Thromb 2011; 18: 867–873 [DOI] [PubMed] [Google Scholar]
- 15.Lee HK, Song JH, Shin CS, et al Decreased mitochondrial DNA content in peripheral blood precedes the development of non‐insulin‐dependent diabetes mellitus. Diabetes Res Clin Pract 1998; 42: 161–167 [DOI] [PubMed] [Google Scholar]
- 16.Song J, Oh JY, Sung YA, et al Peripheral blood mitochondrial DNA content is related to insulin sensitivity in offspring of type 2 diabetic patients. Diabetes Care 2001; 24: 865–869 [DOI] [PubMed] [Google Scholar]
- 17.Lind PM, Lind L. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly. Atherosclerosis 2011; 218: 207–213 [DOI] [PubMed] [Google Scholar]
- 18.Ronn M, Lind L, Van Bavel B, et al Circulating levels of persistent organic pollutants associate in divergent ways to fat mass measured by DXA in humans. Chemosphere 2011; 85: 335–343 [DOI] [PubMed] [Google Scholar]
- 19.Park KS, Nam KJ, Kim JW, et al Depletion of mitochondrial DNA alters glucose metabolism in SK‐Hep1 cells. Am J Physiol Endocrinol Metab 2001; 280: E1007–E1014 [DOI] [PubMed] [Google Scholar]
- 20.Arsenescu V, Arsenescu RI, King V, et al Polychlorinated biphenyl‐77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect 2008; 116: 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev 1998; 78: 547–581 [DOI] [PubMed] [Google Scholar]
- 22.Le Gouill E, Jimenez M, Binnert C, et al Endothelial nitric oxide synthase (eNOS) knockout mice have defective mitochondrial beta‐oxidation. Diabetes 2007; 56: 2690–2696 [DOI] [PubMed] [Google Scholar]
- 23.Manfredi AA, Rovere‐Querini P. The mitochondrion–a Trojan horse that kicks off inflammation? N Engl J Med 2010; 362: 2132–2134 [DOI] [PubMed] [Google Scholar]
- 24.Napoli C, Ignarro LJ. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch Pharmacal Res 2009; 32: 1103–1108 [DOI] [PubMed] [Google Scholar]
- 25.Biswas G, Srinivasan S, Anandatheerthavarada HK, et al Dioxin‐mediated tumor progression through activation of mitochondria‐to‐nucleus stress signaling. Proc Natl Acad Sci USA 2008; 105: 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lind PM, Van Bavel B, Salihovic S, et al Circulating levels of persistent organic pollutants (POPs) and carotid atherosclerosis in the elderly. Environ Health Perspect 2012; 120: 38–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sergeev AV, Carpenter DO. Exposure to persistent organic pollutants increases hospitalization rates for myocardial infarction with comorbid hypertension. Prim Prev Insights 2010; 2: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szuchman‐Sapir A, Etzman M, Tamir S. Human atherosclerotic plaque lipid extract impairs the antioxidant defense capacity of monocytes. Biochem Biophys Res Commun 2012; 423: 884–888 [DOI] [PubMed] [Google Scholar]
- 29.Lanza GA, Crea F. Primary coronary microvascular dysfunction: clinical presentation, pathophysiology, and management. Circulation 2010; 121: 2317–2325 [DOI] [PubMed] [Google Scholar]
- 30.Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med 2012; 366: 54–63 [DOI] [PubMed] [Google Scholar]
- 31.Järvilehto M, Tuohimaa P. Vasa vasorum hypoxia: initiation of atherosclerosis. Med Hypotheses 2009; 73: 40–41 [DOI] [PubMed] [Google Scholar]
- 32.Ross R. Rous‐Whipple award lecture. atherosclerosis: a defense mechanism gone awry. Am J Pathol 1993; 143: 987–1002 [PMC free article] [PubMed] [Google Scholar]
- 33.Stary HC, Chandler AB, Dinsmore RE, et al A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995; 92: 1355–1374 [DOI] [PubMed] [Google Scholar]
- 34.Frink S, Grummer B, Pohlenz JF, et al Changes in distribution and numbers of CD4+ and CD8+ T‐lymphocytes in lymphoid tissues and intestinal mucosa in the early phase of experimentally induced early onset mucosal disease in cattle. J Vet Med B Infect Dis Vet Public Health 2002; 49: 476–483 [DOI] [PubMed] [Google Scholar]
- 35.Ambruosi B, Uranio MF, Sardanelli AM, et al In vitro acute exposure to DEHP affects oocyte meiotic maturation, energy and oxidative stress parameters in a large animal model. PLoS ONE 2011; 6: e27452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon MK, Kim MJ, Jung IK, et al Bisphenol A impairs mitochondrial function in the liver at doses below the no observed adverse effect level. J Korean Med Sci 2012; 27: 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HK, Park KS, Cho YM, et al Mitochondria‐based model for fetal origin of adult disease and insulin resistance. Ann N Y Acad Sci 2005; 1042: 1–18 [DOI] [PubMed] [Google Scholar]
- 38.Glagov S, Weisenberg E, Zarins CK, et al Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med 1987; 316: 1371–1375 [DOI] [PubMed] [Google Scholar]
- 39.Kern MJ, Samady H. Current concepts of integrated coronary physiology in the catheterization laboratory. J Am Coll Cardiol 2010; 55: 173–185 [DOI] [PubMed] [Google Scholar]
- 40.Gould KL. Does coronary flow trump coronary anatomy? JACC Cardiovasc Imaging 2009; 2: 1009–1023 [DOI] [PubMed] [Google Scholar]
- 41.Powers WJ, Clarke WR, Grubb RL, et al Extracranial‐intracranial bypass surgery for stroke prevention in hemodynamic cerebral ischemia: the Carotid Occlusion Surgery Study randomized trial. JAMA 2011; 306: 1983–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chimowitz MI, Lynn MJ, Derdeyn CP, et al Stenting versus aggressive medical therapy for intracranial arterial stenosis. N Engl J Med 2011; 365: 993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pandey P, Steinberg GK. Neurosurgical advances in the treatment of moyamoya disease. Stroke 2011; 42: 3304–3310 [DOI] [PubMed] [Google Scholar]
- 44.De Bruyne B, Pijls NH, Kalesan B, et al Fractional flow reserve‐guided PCI versus medical therapy in stable coronary disease. N Engl J Med 2012; 367: 991–1001 [DOI] [PubMed] [Google Scholar]
- 45.Boden WE, O'rourke RA, Teo KK, et al Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med 2007; 356: 1503–1516 [DOI] [PubMed] [Google Scholar]
- 46.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc 1972; 20: 145–147 [DOI] [PubMed] [Google Scholar]
- 47.Miquel J, Economos AC, Fleming J, et al Mitochondrial role in cell aging. Exp Gerontol 1980; 15: 575–591 [DOI] [PubMed] [Google Scholar]
- 48.West GB, Bergman A. Toward a systems biology framework for understanding aging and health span. J Gerontol A Biol Sci Med Sci 2009; 64: 205–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science 1997; 276: 122–126 [DOI] [PubMed] [Google Scholar]
- 50.Zhang Q, Raoof M, Chen Y, et al Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464: 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kusza K, Siemionow M. Is the knowledge on tissue microcirculation important for microsurgeon? Microsurgery 2011; 31: 572–579 [DOI] [PubMed] [Google Scholar]
- 52.Davidson SM. Endothelial mitochondria and heart disease. Cardiovasc Res 2010; 88: 58–66 [DOI] [PubMed] [Google Scholar]
- 53.Zhang H, Luo Y, Zhang W, et al Endothelial‐specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am J Pathol 2007; 170: 1108–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res 2007; 100: 1128–1141 [DOI] [PubMed] [Google Scholar]
- 55.Duplain H, Burcelin R, Sartori C, et al Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001; 104: 342–345 [DOI] [PubMed] [Google Scholar]
- 56.Huang PL, Huang Z, Mashimo H, et al Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995; 377: 239–242 [DOI] [PubMed] [Google Scholar]
- 57.Nisoli E, Falcone S, Tonello C, et al Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA 2004; 101: 16507–16512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Hu X, Selvakumar P, et al Role of the nitric oxide pathway in AMPK‐mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab 2004; 287: E834–E841 [DOI] [PubMed] [Google Scholar]
- 59.Tanaka T, Nakatani K, Morioka K, et al Nitric oxide stimulates glucose transport through insulin‐independent GLUT4 translocation in 3T3‐L1 adipocytes. Eur J Endocrinol 2003; 149: 61–67 [DOI] [PubMed] [Google Scholar]
- 60.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II‐mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 2008; 102: 488–496 [DOI] [PubMed] [Google Scholar]
- 61.Koh EH, Kim M, Ranjan KC, et al eNOS plays a major role in adiponectin synthesis in adipocytes. Am J Physiol Endocrinol Metab 2010; 298: E846–E853 [DOI] [PubMed] [Google Scholar]
- 62.Kadowaki T, Yamauchi T, Kubota N, et al Adiponectin and adiponectin receptors in obesity‐linked insulin resistance. Novartis Found Symp 2007; 286: 164–176 Discussion 176–182, 200–203. [DOI] [PubMed] [Google Scholar]
- 63.Yamauchi T, Kadowaki T. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes 2008; 32(Suppl 7): S13–S18 [DOI] [PubMed] [Google Scholar]
- 64.Ahn SY, Choi Y‐S, Koo H‐J, et al Mitochondrial dysfunction enhances the migration of vascular smooth muscles cells via suppression of Akt phosphorylation. Biochim Biophys Acta 2010; 1800: 275–281 [DOI] [PubMed] [Google Scholar]
- 65.Tabas I. Pulling down the plug on atherosclerosis: finding the culprit in your heart. Nat Med 2011; 17: 791–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arora RR, Rai F. Antiplatelet intervention in acute coronary syndrome. Am J Ther 2009; 16: e29–e40 [DOI] [PubMed] [Google Scholar]
- 67.Levy BI, Schiffrin EL, Mourad J‐J, et al Impaired tissue perfusion: a pathology common to hypertension, obesity, and diabetes mellitus. Circulation 2008; 118: 968–976 [DOI] [PubMed] [Google Scholar]
- 68.De Jongh RT, Serné EH, Ijzerman RG, et al Impaired microvascular function in obesity: implications for obesity‐associated microangiopathy, hypertension, and insulin resistance. Circulation 2004; 109: 2529–2535 [DOI] [PubMed] [Google Scholar]
- 69.Rizzoni D, Palombo C, Porteri E, et al Relationships between coronary flow vasodilator capacity and small artery remodelling in hypertensive patients. J Hypertens 2003; 21: 625–631 [DOI] [PubMed] [Google Scholar]
- 70.Ikram MK, De Jong FJ, Bos MJ, et al Retinal vessel diameters and risk of stroke: the Rotterdam Study. Neurology 2006; 66: 1339–1343 [DOI] [PubMed] [Google Scholar]
- 71.Nishino M, Hoshida S, Egami Y, et al Coronary flow reserve by contrast enhanced transesophageal coronary sinus Doppler measurements can evaluate diabetic microvascular dysfunction. Circ J 2006; 70: 1415–1420 [DOI] [PubMed] [Google Scholar]
- 72.Clough GF, Norman M. The microcirculation: a target for developmental priming. Microcirculation 2011; 18: 286–297 [DOI] [PubMed] [Google Scholar]
- 73.Antonios TFT, Rattray FM, Singer DRJ, et al Rarefaction of skin capillaries in normotensive offspring of individuals with essential hypertension. Heart 2003; 89: 175–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanabe Y, Kawasaki R, Wang JJ, et al Retinal arteriolar narrowing predicts 5‐year risk of hypertension in Japanese people: the Funagata Study. Microcirculation 2010; 17: 94–102 [DOI] [PubMed] [Google Scholar]
- 75.Sherman TF. On connecting large vessels to small. the meaning of Murray's law. J Gen Physiol 1981; 78: 431–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bassingthwaighte JB, Liebovitch LS, West BJ. Fractal Physiology. Oxford University Press, Oxford, UK, 1994 [Google Scholar]
- 77.Huo Y, Kassab GS. Intraspecific scaling laws of vascular trees. J R Soc Interface 2012; 9: 190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kamiya A, Takahashi T. Quantitative assessments of morphological and functional properties of biological trees based on their fractal nature. J Appl Physiol 2007; 102: 2315–2323 [DOI] [PubMed] [Google Scholar]
- 79.Weibel ER, Hoppeler H. Exercise‐induced maximal metabolic rate scales with muscle aerobic capacity. J Exp Biol 2005; 208: 1635–1644 [DOI] [PubMed] [Google Scholar]
- 80.Harel N, Bolan PJ, Turner R, et al Recent advances in high‐resolution MR application and its implications for neurovascular coupling research. Front Neuroenergetics 2010; 2: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Duvernoy HM, Delon S, Vannson JL. Cortical blood vessels of the human brain. Brain Res Bull 1981; 7: 519–579 [DOI] [PubMed] [Google Scholar]
- 82.Wolff Jr, Goerz C, Bär T, et al Common morphogenetic aspects of various organotypic microvascular patterns. Microvasc Res 1975; 10: 373–395 [DOI] [PubMed] [Google Scholar]
- 83.Glenny RW. Emergence of matched airway and vascular trees from fractal rules. J Appl Physiol 2011; 110: 1119–1129 [DOI] [PubMed] [Google Scholar]
- 84.Weibel ER, Taylor CR, Hoppeler H. The concept of symmorphosis: a testable hypothesis of structure–function relationship. Proc Natl Acad Sci USA 1991; 88: 10357–10361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bassingthwaighte JB, Goresky CA. Modeling in the analysis of solute and water exchange in the microvasculature In: Renkin EM, Michel CC, (eds). Handbook of Physiology. Section 2, The Cardiovascular System, Volume IV, The Microcirculation. American Physiological Society, Bethesda, MD, 1984; 549–626 [Google Scholar]
- 86.Beard DA. Modeling of oxygen transport and cellular energetics explains observations on in vivo cardiac energy metabolism. PLoS Comput Biol 2006; 2: e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Katz LA, Swain JA, Portman MA, et al Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol 1989; 256: H265–H274 [DOI] [PubMed] [Google Scholar]
- 88.West BJ. Fractal physiology and the fractional calculus: a perspective. Front Physiol 2010; 1: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bai C‐H, Chen J‐R, Chiu H‐C, et al Lower blood flow velocity, higher resistance index, and larger diameter of extracranial carotid arteries are associated with ischemic stroke independently of carotid atherosclerosis and cardiovascular risk factors. J Clin Ultrasound 2007; 35: 322–330 [DOI] [PubMed] [Google Scholar]
- 90.Chuang S‐Y, Bai C‐H, Chen J‐R, et al Common carotid end‐diastolic velocity and intima‐media thickness jointly predict ischemic stroke in Taiwan. Stroke 2011; 42: 1338–1344 [DOI] [PubMed] [Google Scholar]
- 91.Sorond FA, Hollenberg NK, Panych LP, et al Brain blood flow and velocity: correlations between magnetic resonance imaging and transcranial Doppler sonography. J Ultrasound Med 2010; 29: 1017–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fox JA, Hugh AE. Localization of atheroma: A theory based on boundary layer separation. Br Heart J 1966; 28: 388–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duguid JB. Mural thrombosis in arteries. Br Med Bull 1955; 11: 36–38 [DOI] [PubMed] [Google Scholar]
- 94.Zarins CK, Giddens DP, Bharadvaj BK, et al Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res 1983; 53: 502–514 [DOI] [PubMed] [Google Scholar]
- 95.Ercan M, Koksal C, Konukoglu D, et al Impaired plasma viscosity via increased cholesterol levels in peripheral occlusive arterial disease [correction of disase]. Clin Hemorheol Microcirc 2003; 29: 3–9 [PubMed] [Google Scholar]
- 96.Koenig W, Sund M, Ernst E, et al Association between rheology and components of lipoproteins in human blood. Results from the MONICA project. Circulation 1992; 85: 2197–2204 [DOI] [PubMed] [Google Scholar]
- 97.Tamariz LJ, Young JH, Pankow JS, et al Blood viscosity and hematocrit as risk factors for type 2 diabetes mellitus: The atherosclerosis risk in communities (ARIC) study. Am J Epidemiol 2008; 168: 1153–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith S, Julius S, Jamerson K, et al Hematocrit levels and physiologic factors in relationship to cardiovascular risk in Tecumseh, Michigan. J Hypertens 1994; 12: 455–462 [PubMed] [Google Scholar]
- 99.Selnes OA, Gottesman RF, Grega MA, et al Cognitive and neurologic outcomes after coronary‐artery bypass surgery. N Engl J Med 2012; 366: 250–257 [DOI] [PubMed] [Google Scholar]
- 100.Lee HK. Persistent organic pollutants and epidemic of diabetes and metabolic syndrome. J Diabetes Invest 2010; 1: 121–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.West GB. The importance of quantitative systemic thinking in medicine. Lancet 2012; 379: 1551–1559 [DOI] [PubMed] [Google Scholar]
- 102.Shoffner JM, Wallace DC, Scriver CR, et al Oxidative phosphorylation diseases: chapter 46 In: Sriver CR, Beaudet AL, Sly WS, Valle D (eds). Metabolic and Molecular Basis of Inherited Diseases, 7th edn Mcgraw‐Hill, New York: 1995; 1535–1609 [Google Scholar]