

Abstract
High-risk human papillomaviruses (HPV) are sexually transmitted viruses causally associated with several cancers. During its natural life cycle, HPV16, the most common high-risk genotype, infects the epithelial basal cellsin a process facilitated through a recently identified receptor, the annexin A2 heterotetramer (A2t). During infection, HPV16 also interacts with Langerhans cells (LC), the antigen presenting cells of the epithelium, inducing immune suppression, which is mediated by the HPV16 L2 minor capsid protein. Despite the importance of these virus-immune cell interactions, the specific mechanisms of HPV16 entry into LC and HPV16-induced immune suppression remain undefined. An N-terminal peptide of HPV16 L2 (aa 108-126) has been shown to specifically interact with A2t. Here, we show that incubation of human LC with this peptide blocks binding of HPV16. Inhibiting this interaction with an A2t ligand or by siRNA downregulation of A2t, significantly decreases HPV16 internalization into LC in an L2-dependent manner. A2t is associated with suppression of LC maturation as demonstrated through attenuated secretion of Th1-associated cytokines and decreased surface expression of MHC II on LC exposed to A2t. Conversely, small molecule inhibition of A2t prevents HPV16-induced suppression of LC immune function as indicated by significantly increased secretion of inflammatory cytokines and surface expression of CD86 in HPV16 treated LC pre-exposed to A2t inhibitors. These results demonstrate that HPV16 suppresses LC maturation through an interaction with A2t, revealing a novel role for this protein.
Introduction
Cervical cancer is the second most common cancer among women worldwide with >500,000 new cases reported and >274,000 associated deaths each year (1, 2). Persistent high-risk human papillomavirus (HPV) infection is causally associated with several cancers, including cervical cancer (3–5). Over half of all cervical cancer cases are associated with HPV16, the most common of the cancer-causing high-risk genotypes (6). During the natural life cycle of HPV16, the virus infects the basal cells of the epithelium and interacts with Langerhans cells (LC), the resident antigen presenting cells (APC) within the epithelia (7), which are responsible for initiating immune responses against pathogens entering the epithelium (8). However, 15% of women with high-risk HPV infections do not produce an effective immune response against the virus (7), and therefore represent a critical population of women particularly susceptible for developing invasive cervical cancer. In contrast to the extensive body of research defining the mechanism of infection within epithelial cells, limited studies actually focus on identifying and characterizing the HPV16 internalization pathway in LC and how these pathways affect LC immune responses.
HPV16 is a non-enveloped double stranded DNA virus whose 55 nm-diameter capsid is composed of two proteins: the L1 (late protein 1) major capsid protein and the L2 (late protein 2) minor capsid protein (9), each of which has unique functions during the infectious process. Infectious HPV16 virion production depends on the differentiation of basal epithelial cells into mature keratinocytes as the expression of late genes is contingent on host RNA factors (10). Therefore the majority of the literature concerning receptors uses HPV pseudovirions (PsV) and/or virus-like particles (VLP) to report specific aspects of viral uptake. When expressed in vitro, the major capsid protein L1 self-assembles into L1 VLP, which possesses a 72-pentamer-icosahedral structure (11–13). If L1 is expressed at the same time as the minor capsid protein L2, they assemble into L1L2 VLP with up to 72 L2 proteins per particle (12, 14). The L2 minor capsid protein plays an important role in efficient encapsidation of DNA within HPV16 (15, 16), allowing for the production of HPV16 L1L2 PsV that incorporate DNA within the capsid.
In epithelial cells, HPV16 infection is initiated upon viral capsid binding through an initial interaction between L1 and heparan sulfate proteoglycans (HSPG) (17), as well as other interactions with α6β1/4 integrins, cyclophilin B, growth factors and growth factor receptors (GF and GFR), and various tetraspanins (18–21). The eventual uptake of HPV16 into epithelial cells has been shown to be clathrin-, caveolin-, cholesterol-, and dynamin-independent, which points to a non-canonical and possibly novel ligand-induced internalization pathway related to macropinocytosis (22). Similarly, it was previously demonstrated that HPV16 entry into LC occurred via a clathrin- and caveolin-independent pathway (23) implicating a related HPV16 entry pathway into both epithelial and Langerhans cells.
Many functions aside from DNA encapsidation have been identified for L2 in the context of HPV16 infection of epithelial cells [reviewed in (24)] including endosomal escape (25–28), cytoskeletal interaction and cytoplasmic trafficking (29), and chaperoning of packaged DNA to the host cell nucleus (30). Historically, the role of L2 in HPV16 internalization into epithelial cells was implicated by studies demonstrating the existence of L2-neutralizing epitopes (31–36). Additionally an L2 peptide (L2108-126) was shown to bind to the cell surface and blocked HPV16 pseudo-infection of multiple cell lines (37). We recently identified and characterized the role/mechanism of the annexin A2 heterotetramer (A2t) as an HPV16 L2-specific entry receptor (38). We demonstrated that A2t specifically binds to L2108-126, co-localizes with HPV16 at the cell surface, and mediates HPV16 VLP entry and HPV16 pseudo-infection in an L2-dependent manner (38). The role of A2t in HPV16 entry was confirmed by an independent group, and A2t was further shown to co-internalize with HPV16 and mediate intracellular trafficking (39).
Interestingly, the presence of L2 in the viral capsid has been shown to double the rate of HPV16 entry into LC, and is responsible for HPV16-induced suppression of LC immune function (40). To date, no specific L2 receptor on LC has been identified. Thus far, it has been demonstrated that LC internalize HPV16L1L2 VLP via a clathrin- and caveolae-independent mechanism (23), whereas LC internalize HPV6bL1 VLP through a caveolae dependent pathway (41) and HPV16L1 VLP through a clathrin dependent pathway (42). While these results may seem contradictory, it is likely due to the presence or absence of the L2 minor capsid protein within the VLP used for these studies. Collectively, these studies suggest that a specific L2 receptor and receptor-specific mechanism exist for HPV16 uptake into LC. (23, 40), and our previous report points to A2t as a potential candidate as it is the only identified L2-specific receptor (38). A2t is found at the cell surface as a heterotetramer consisting of two annexin A2 (anxA2) monomers and an S100A10 dimer (43–46), which are co-expressed by LC (47). Understanding HPV16-LC interactions and identifying HPV16 receptors on LC involved in internalization are critical to delineating the local immune events in the mucosa during an active HPV16 infection. Therefore, due to the previous identification of A2t as an L2-specific receptor on epithelial cells and the role of L2 in HPV16 internalization and immune escape in LC, we hypothesized that internalization of HPV16 into LC is mediated through A2t in an L2-dependent manner and that entry via A2t suppresses LC maturation. To explore this possibility, we examined the interactions between A2t and HPV16 in LC and the role of A2t in LC maturation. In the current study, we expanded the role for A2t as an HPV16 L2-specific receptor in epithelial cells to include LC, and additionally reveal a novel role for A2t as an immune modulator of LC maturation.
Materials and Methods
LC generation
Human peripheral blood monocytes (PBMC) from healthy donors were obtained by leukapheresis (48). LC were generated from human PBMC as previously described (49), and incubated in complete media (RPMI 1640 supplemented with 10% FBS, 1X Pen/Strep, 1X Non-essential amino acids, and 1X 2-mercaptoethanol) with the addition of 1000 U/mL (~180 ng/mL) GM-CSF, 1000 U/mL (~200 ng/mL) IL-4, and 10 ng/ml TGF-β for 7 days. All studies were approved by USC’s IRB and informed consent was obtained from donors.
Antibodies
The following antibodies were used in this study: mouse-anti-anxA2, mouse-anti-anxA2 light chain, mouse-CD86-FITC, mouse-HLA-DR, DP, DQ-FITC, isotype controls (BD Biosciences, San Jose, CA); H16.V5 mouse-anti-L1 (gift from Neil Christensen, Ph.D.); rabbit-anti-beta actin (Cell Signaling, Danvers, MA); rabbit-anti-pAKT (ser 473), rabbit-anti-Akt (Santa Cruz Biotechnology); rabbit-anti-GAPDH (Cell Signaling, Danvers, MA); Alexa Fluor 680 goat-anti-rabbit (Invitrogen, Carlsbad, CA) and IRDye 800 donkey-anti-mouse (Rockland, Gilbertsville, PA).
Virus-like particles and pseudovirions
HPV16L1 VLP and HPV16L1L2 VLP were produced as previously described (50). Western blot analyses confirmed the presence of L1 and L2 while an ELISA and transmission electron microscopy confirmed the presence of intact particles. An E-toxate kit (Sigma-Aldrich, Carlsbad, CA) was used to semi-quantitate endotoxin. The endotoxin level in the preparations was less than 0.06 endotoxin units/ml and this level does not activate LC (48). Baculovirus DNA used in VLP production procedure does not activate LC (48). VLP were validated in binding assays via pre-incubation with heparin, a component of heparan sulfate proteoglycans (HSPG) that binds positively charged residues of HPV16, or with H16.V5 neutralizing antibody (51), prior to cellular exposure, which inhibits intact capsids from binding with cell surface receptors, and minimal binding of less than 20% was observed. To produce VLP with a mutated L2108-126 region (aa substitution of GGDD for LVEE in the L2 capsid region aa 108-111) (37), site-directed mutagenesis was performed as previously described (38). HPV16 pseudovirions were produced by cotransfection of 293TT cells with plasmids encoding codon-optimized HPV16 L1 and L2 following published procedures (52). To produce pseudovirions with a mutated L2108-111 region (GGDD for LVEE), site-directed mutagenesis was performed following published procedures (38). L1 content was quantitated by Coomassie Blue staining next to BSA standards following SDS-PAGE.
Recombinant protein expression and purification
Recombinant anxA2 and S100A10 was produced as previously described (38). Concentrations of all proteins and peptides, including the A2t complex, were determined using bicinchoninic acid assays (Pierce, Rockford, IL) compared to measured absorbance of albumin standards at 562nm. Purified A2t was produced by and purified by combining S100A10 and anxA2 at a molar ratio of 1:1 as previously described (38), and used in exogenous A2t activation assays.
HPV16 L1L2 VLP binding assay with L2108-126
LC were incubated with increasing concentrations of the HPV16 L2108-126 peptide (LVEETSFIDAGAPTSVPSI) (37), or a scrambled analog of the same peptide (IESPVSDTALGTPEIFVSA) with a maximum concentration of 50 μg/mL (0.5×106 cells) for 1 h at 4° C. Subsequently, the LC were incubated with 0.25μg of HPV16L1L2 VLP/treatment for 1h at 4° C and then incubated with an anti-L1 (H16.V5) Ab at a dilution of 1:25,000 for 30 min at 4° C. The cells were then incubated with biotinylated anti-mouse-IgG2b for 30 min at 4° C. Next, the HPV16L1L2 VLP/anti-L1/biotin treated cells were stained with streptavidin-FITC for 30 min. In control experiments, cells were left untreated or probed with either peptide/anti-L1/biotin-strepavidin-FITC (no VLP), VLP/anti-L1/biotin-strepavidin-FITC (no peptide), or peptide/Heparin-VLP/anti-L1/biotin-strepavidin-FITC (VLP incubated with 2.5μg Heparin for neutralization). Finally, HPV16L1L2 VLP binding to LC was assessed by flow cytometry with neutralization controls mentioned under VLP preparation. Data were normalized to the untreated groups.
L2108-126 peptide pulldown assay
LC were harvested, washed with PBS, and aliquoted into 1.5 ml microcentrifuge tubes in PBS. Then (6x)His-L2108-126 peptide was added to the LC, at a concentration of 50 μg/0.5 ×106 cells, and incubated for 1hr. In control experiments, LC were left untreated (no peptide added) but exposed to each condition thereafter. Following the incubation, the extracellular cross-linking agent, 3,3′-Dithiobis-(sulfosuccinimidylpropionate) (DTSSP) was added at a concentration of 1.5 mM to the LC and incubated for 2 hr to cross-link the peptide to the receptor. After the cross-linking reaction was quenched with 1 M Tris, LC were washed with PBS to remove excess unbound peptide, and resuspended in and incubated with a bursting solution [10 mM Hepes, 2 mM MgCl, 10 mM KCl2, 0.05% Tween-20, and Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL)] for 20 min. Next, the cells were centrifuged for 30 min at 13000 RCF. The supernatants were decanted and LC were resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 50 mM imidazole, 0.05% Tween-20, and Halt Protease Inhibitor Cocktail, pH 8.0). The cells were snap frozen, allowed to thaw, incubated on ice for 30 min, and sonicated for 10 sec. Subsequently, the lysates were centrifuged for 30 min at 10000 RCF. The lysate supernatants were decanted, mixed with 50% Ni-NTA agarose slurry (Qiagen, Valencia, CA) and incubated overnight. The following day an affinity column (Thermo Scientific) was assembled to elute the proteins from the Ni-NTA agarose slurry. Once the column was assembled, the lysate-Ni-NTA agarose slurry was washed twice with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 50 mM imidazole, and 0.05% Tween-20, pH 8.0). The proteins associated with the Ni-NTA agarose were eluted with imidazole containing elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, and 0.05% Tween-20, pH 8.0). All steps were performed at 4° C. The eluates then were separated via gel electrophoresis on 10% Bis-Tris gels using the NuPAGE Electrophoresis System (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions and transferred to nitrocellulose membranes for immunoblot analysis. The membranes were then probed for anxA2 and S100A10, and stained with secondary infrared Abs. Protein bands were visualized and quantified with the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE). Furthermore, the eluates were separated via gel electrophoresis on 10% Tris-HCL gels (Bio-Rad)under reducing conditions for silver stain analysis.
HPV16 VLP uptake assay with SLPI
HPV16L1 VLP and HPV16L1L2 VLP were labeled with carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE) (Invitrogen) as directed by the manufacturer’s instructions. After labeling the HPV16 VLP were column filtered with 2% agarose beads size standard 50–150 μm (Agarose Bead Technologies, Tampa, FL) with DPBS/0.5 M NaCl to remove excess free label. LC were harvested, washed with PBS, and aliquoted at a concentration of 106 cells/200 μl cold PBS into 1.5 ml amber tubes. Subsequently the cells were either left untreated or incubated with 30 μg/ml of rhu-SLPI (R&D Systems, Minneapolis, MN) for 1 h at 4° C (optimal concentration was determined through titration). Following the incubation the cells were washed with 500 μl cold PBS and spun down at 800 RCF for 5 min at 4° C. The supernatant was removed and the LC were resuspended in 400 μl of room temperature FACS buffer. Next, CFDA-SE labeled HPV16L1L2 VLP or HPV16L1 VLP (1 μg/1×106) were incubated with the LC at 37° C. In control experiments, LC were treated with H16.V5 (1:1500) neutralized CFDA-SE labeled VLP to ensure VLP integrity and lack of residual free CFDA-SE label. After 15 min, LC were harvested and fixed in 2% paraformaldehyde. Finally, HPV16 VLP internalization by LC was assessed via flow cytometry and data was normalized to untreated groups.
siRNA knockdown of anxA2 in LC and HPV16 VLP uptake assay
The S100A10 subunit of A2t is post-transcriptionally stabilized by anxA2 (53, 54), and knockdown of anxA2 has been shown to be sufficient for successful reduction in both subunits of A2t (38). A Human anti-anxA2 siRNA SMARTpool was synthesized (Thermo Scientific Rockford, IL) with the following sequences: #1 (AUACUAACUUUGAUGCUUGA); #2 (CGACGAGGACUCUCUCAUU); #3 (CUGUCAAAGCCUAUACUAA); #4 (AGACCAAAGGUGUGGAUGA). Control non-target siRNA (Thermo Scientific) was against no known protein. Anti-anxA2 or Control siRNA was transfected into LC using Lipofectamine 2000 (Invitrogen) as directed by the manufacturer’s instructions (20 pmol siRNA/2×105 cells). Protein from transfected cells was collected for 7 consecutive days and assessed via Western blot determining that minimal (approx. 50%) anxA2 levels were achieved 5 days post-transfection (data not shown). LC were then incubated for 5 days post-siRNA transfection before use in an HPV16 VLP uptake assay. HPV VLP were labeled with CFDA-SE as described above. CFDA-SE labeled, HPV16L1L2 VLP, or HPV16 L2 mutant VLP (37) (1 μg/2×105 cells) were incubated with the anti-anxA2 siRNA transfected, control siRNA transfected, or untreated LC at 37° C. After 45 min, LC were harvested and fixed in 2% paraformaldehyde. In control experiments LC were treated with H16.V5 neutralized VLP as described above. Finally, HPV16 VLP internalization by LC was assessed via flow cytometry and the data were normalized to the untreated groups. For anxA2 quantification, protein was collected from LC five days post-siRNA transfection with Mammalian Protein Extraction Reagent (Pierce), and reduced samples were run on 10% Bis-Tris gels using NuPAGE Electrophoresis System (Invitrogen) according to manufacturer’s instructions and transferred to nitrocellulose membranes for immunoblot analysis. The membranes were then probed for anxA2 and beta-actin, and stained with secondary infrared Abs. Protein bands were visualized and quantified with the Odyssey Imaging System.
LC activation assay with HPV16 wild type or L2 mutant PsV
106 LC were seeded in a 6-well plate and either left untreated, treated with 10 μg lipopolysaccharide (LPS) (Sigma-Aldrich), 10 μg HPV16 PsV, or 10 μg HPV16 L2 mutant PsV. The toll-like receptor 4 (TLR4) agonist LPS was chosen as a positive control as it has been shown to elicit strong immune responses in LC (55). The cells were then incubated at 37°C for 48 hr in 2 ml complete medium with periodic mixing for the first 1 hr. After 48 hr, the cells were harvested, washed, stained for surface MHC II (mouse-HLA-DR, DP, DQ-FITC) and CD86 or isotype controls, and analyzed by flow cytometry.
LC activation assay with recombinant proteins
106 LC were seeded in a 6-well plate and either left untreated, treated with 10 μg LPS (Sigma-Aldrich), 10 μg A2t, 10 μg anxA2, or 10 μg S100A10. The cells were then incubated at 37° C for 48 hr in 2 ml complete medium with periodic mixing for the first 1 hr. After 48 hr, supernatants were collected and cells were harvested, washed, stained for surface MHC II (mouse-HLA-DR, DP, DQ-FITC) or isotype controls, and analyzed by flow cytometry. Supernatants of selected groups were analyzed using the MILLIPLEX MAP Human Cytokine Kit (EMD Millipore).
LC signaling assay with recombinant A2t
LC were treated with as described above in the activation assay with recombinant proteins at 37° C for 15 min. Cellular extracts were prepared using the Mammalian Protein Extraction Reagent (Pierce). Normalized aliquots of cell lysates were electrophoresed on 10% NuPage Novex Bis-Tris gels (Invitrogen) under reducing conditions and transferred to nitrocellulose membranes. Immunoblotting was performed using Akt, pAkt, or GAPDH Abs followed by secondary infrared Abs. Protein bands were visualized and quantified with the Odyssey Imaging System.
LC activation assay with A2t inhibitor
106 LC were seeded in a 6-well plate and either left untreated, treated with 10 μg LPS (Sigma-Aldrich), or treated with increasing concentrations of a previously identified A2t inhibitor (A2ti: 2-[4-(2-Ethylphenyl)-5-o-tolyloxymethyl-4H-[1,2,4]triazol-3-ylsulfanyl]acetamide) (56) alone or A2ti for 1 hr prior to exposure to 10 μg HPV16 PsV. The half maximal inhibitory concentration (IC50) value of the A2ti was reported to be 24 μM (56). The cells were then incubated at 37°C for 48 hr in 2 ml complete medium with periodic mixing for the first 1 hr. After 48 hr, supernatants were collected and cells were harvested, washed, stained for surface MHC II and CD86 or isotype controls, and analyzed by flow cytometry. Supernatants were analyzed using the MILLIPLEX MAP Human Cytokine Kit (EMD Millipore).
Statistical analysis
All statistical analyses were performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA).
Results
The HPV16 L2108-126 peptide reduces binding of HPV16 L1L2 VLP to LC
We recently reported that the N-terminal L2108-126 peptide binds to the S100A10 subunit of A2t and is exposed on the capsid surface of HPV16 VLP and PsV, and through a competition assay showed that a scrambled version of the same peptide had no effect on L2108-126 binding to A2t in ten-fold excess (38). Interestingly, it was demonstrated that this A2t-binding peptide bound to A2t-expressing cervical epithelial cell lines (HeLa, SiHa, and CaSki) as much as four times more than A2t-negative cell lines (Alexander and Hep G2 cells), however no immune cells were tested (37). To determine whether this same A2t-binding L2 peptide facilitates attachment to LC, we incubated LC with either L2108-126, a scrambled version of the same peptide, or no peptide, and subsequently exposed the cells to HPV16L1L2 VLP. We then assessed the amount of bound HPV16 L1L2 VLP on the surface of LC with flow cytometry. When LC were pre-incubated with the L2108-126 peptide (50 μg/mL), there was a significant decrease (approx. 50% with p<0.05) in the amount of HPV16 L1L2 VLP bound to the LC surface compared to the scrambled peptide or untreated control (Fig. 1a), suggesting that the N-terminus of L2 facilitates HPV16 binding to LC, similar to epithelial cells. Titration experiments determined that the IC50 value of the peptide is 20μg/mL (data not shown).
Figure 1. The HPV16 L2108-126 peptide reduces binding of HPV16L1L2 VLP and binds to A2t on LC.
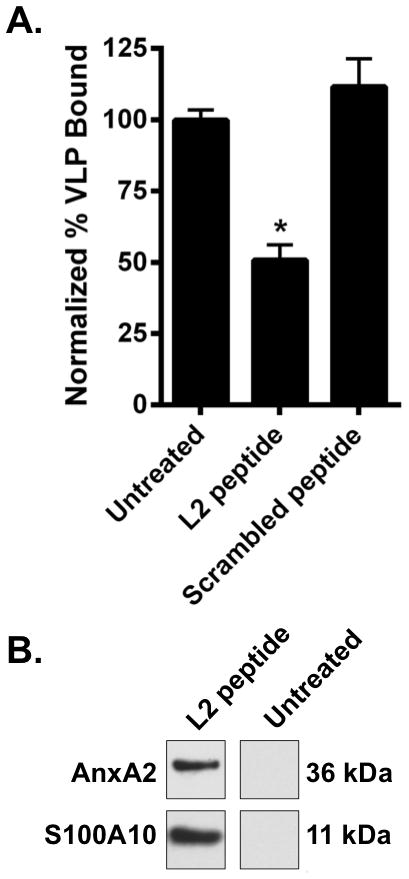
A. LC were left untreated or were treated with the L2108-126 or scrambled peptide and subsequently incubated with HPV16L1L2 VLP. After washing, HPV16L1L2 VLP remaining on the surface of LC were detected using an L1 specific conformational antibody (H16.V5). Binding was assessed by flow cytometry. These data are expressed as the mean of three separate experiments ± SEM (*P < 0.05 as determined by a two-tailed, unpaired t-test, as compared to untreated LC). B. LC were incubated with either no peptide or (6x)His-L2108-126 peptide and subsequently cross-linked with DTSSP. Cells were then lysed and mixed with a Ni-NTA agarose slurry overnight and eluted. Eluates were then electrophoresed, transferred to nitrocellulose and probed with either an anti-AnxA2 or an anti-S100A10 antibody. One representative experiment of three is shown.
HPV16 L2108-126 binds to LC cell surface A2t
Next, we wanted to determine if there was a direct interaction between the L2108-126 peptide and A2t on the LC cell surface to evaluate its potential as an L2-specific HPV16 internalization receptor on LC. Therefore, LC were either incubated with a (6x)His-L2108-126 peptide or left untreated, and subsequently exposed to the extracellular cross-linking agent DTSSP. After cross-linking L2108-126 to cell surface proteins, LC were lysed and the lysates were incubated in a Ni-NTA affinity column followed by elution with the Ni-binding competitor imidazole. Eluates were subsequently separated by gel electrophoresis for immuno-blot analysis. We found that both A2t subunits, anxA2 and S100A10, were present in the L2108-126 peptide pulldown eluate and not in the untreated control (Fig. 1b) demonstrating that A2t interacts with the L2108-126 peptide on the surface of LC. A reduced silver stain gel showed the annexin A2 subunit of A2t as a unique band (confirmed with mass spectrometry) in the L2108-126 peptide treated pulldown eluate compared to the untreated control (Supplemental Fig. 1)
SLPI reduces the uptake of HPV16L1L2 VLP by LC
We next sought to assess whether A2t plays a role in the internalization of HPV16 VLP into LC. SLPI is an anxA2 ligand that has been shown to inhibit HPV16 PsV infection of epithelial cells, which mimicked the effects of an anti-anxA2 antibody (38). Moreover, SLPI has been shown to block HIV-1 infection through its interaction with anxA2 (57), and HIV-1 is another virus that specifically targets LC during initial infection (58, 59). Therefore, LC were pretreated with SLPI prior to exposure to CFDA-SE-labeled HPV16 L1L2 VLP and internalization was analyzed via flow cytometry. Fluorescence of CFDA-SE occurs when acetate groups are cleaved by intracellular esterases, and consequently only VLP that have been internalized by cells are detected (60). LC exposed to SLPI showed a significant decrease in the internalization of HPV16 L1L2 VLP (Fig. 2). To examine if the decrease in uptake was dependent on the presence of the L2 protein, LC were pretreated with SLPI and similarly exposed to CFDA-SE-labeled HPV16 L1 VLP. Notably, no reduction in HPV16 L1 VLP internalization by LC treated with SLPI was observed (Fig. 2), indicating that SLPI inhibition of VLP uptake is L2 dependent. Similar to what we have previously observed on epithelial cells, L1 VLP enter LC less efficiently than L1L2 VLP (Fig. 2A) (38, 40). Notably, similar internalization was observed between baseline L1 VLP internalization and SLPI-blocked L1L2 VLP internalization, suggesting that when A2t is blocked, the L1L2 VLP may default to a pathway utilized by L1 VLP. To evaluate the effect of overall SLPI on internalization of either L1 or L1L2 VLP, the fluorescence of the SLPI-treated groups were normalized to the untreated groups independently for L1 and L1L2 VLP (Fig. 2B). To confirm that the CFDA-SE signals observed were not due to free CFDA-SE label, and to verify VLP integrity, HPV16 L1 and HPV16 L1L2 VLP were pre-incubated with H16.V5 (an anti-L1 Ab) for neutralization. Under these conditions, no significant internalization was observed (Fig. 2B).
Figure 2. SLPI reduces the uptake of HPV16L1L2 VLP by LC.

A. LC were left untreated (gray line) or treated with SLPI (30 μg/ml), then incubated with CFDA-SE labeled HPV16L1 or HPV16L1L2 VLP (black lines) for 15 min and internalization was assessed via flow cytometry. B. LC were incubated with SLPI (30 μg/ml), then incubated with CFDA-SE labeled HPV16L1, HPV16L1L2, or H16.V5 neutralized HPV16L1 or HPV16L1L2 VLP for 15 min. Uptake of CFDA-SE labeled VLP by LC was assessed by flow cytometry and normalized to untreated LC. The mean percentage of uptake ± SEM of three separate experiments is presented (*P < 0.05 by a two-tailed, unpaired t-test, as compared to the untreated LC).
siRNA-mediated knockdown of anxA2 in LC reduces HPV16 L1L2 VLP internalization
To further establish the role of A2t in HPV16 uptake into LC, we examined the effect of A2t siRNA knockdown in LC on HPV16 internalization. S100A10 is post-transcriptionally stabilized by annexin A2 (anxA2) and therefore it is sufficient to only target annexin A2 for knockdown of A2t (38). There was a significant reduction in anxA2 protein in LC treated with anxA2 siRNA compared to both mock-transfected untreated and control siRNA-treated LC (Fig. 3A). To specifically determine the effect of anxA2 knockdown on HPV16 L2-mediated uptake in LC, untreated and siRNA-transfected LC were exposed to CFDA-SE labeled-HPV16 L1L2 VLP or HPV16 VLP with a mutation in the A2t-binding region of L2 [a previously described substitution of GGDD for LVEE of L2 108-111 (37)] (Fig. 3B). This mutation in L2 significantly reduces HPV16 VLP binding to A2t and HPV16 PsV infectivity of epithelial cells (38). Knockdown of anxA2 resulted in a significant reduction in the uptake of HPV16 L1L2 VLP into LC compared to the untreated control. Importantly, no decrease in HPV16 L2 mutant VLP uptake was detected, demonstrating N-terminal L2 specificity for uptake through A2t. LC treated with non-target control siRNA showed no reduction in the expression of anxA2 protein (Fig. 3A), resulting in no change in HPV16 L1L2 or HPV16 L2 mutant VLP internalization (Fig. 3B). Similar to the experiments described above for SLPI-blocking, L2 mutant VLP enter slower than L1L2 VLP, therefore uptake was normalized to untreated controls independently. Collectively, the results from Figures 1–3 strongly indicate that A2t interacts with the L2 protein and is involved in the binding and internalization of HPV16 by LC.
Figure 3. Downregulation of anxA2 reduces uptake of HPV16L1L2 VLP.
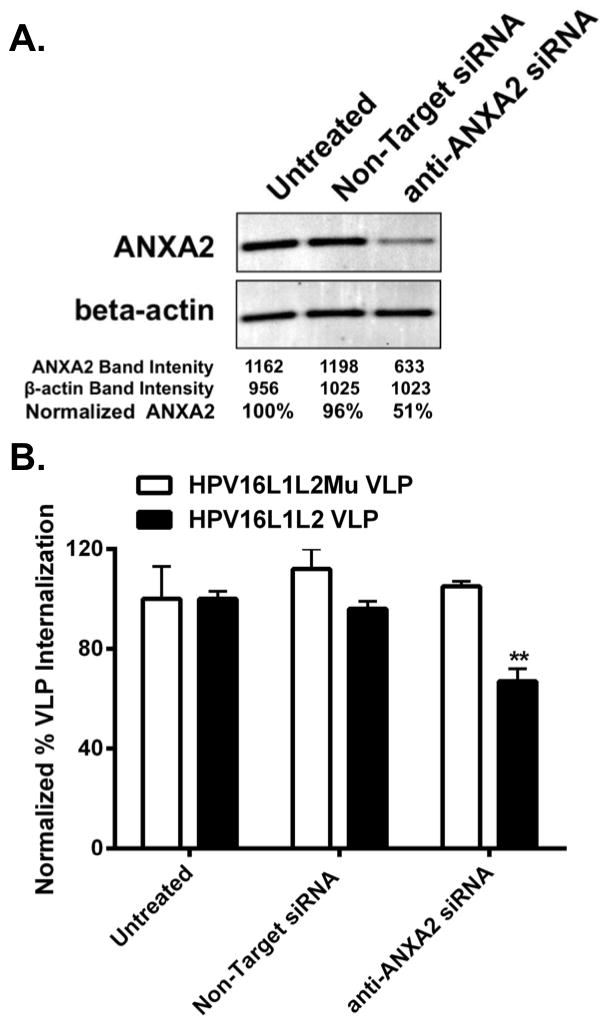
A. LC were transfected without siRNA (untreated), with control siRNA or an anti-anxA2 siRNA SMARTpool. The cells were incubated for 5 days before analysis of anxA2 protein expression by Western blot. Beta-actin served as the loading control. One representative experiment of four is shown. B. LC were transfected without siRNA (untreated), with Non-Target siRNA or with an anti-anxA2 siRNA SMARTpool. The cells were incubated for 5 days and exposed to CFDA-SE labeled HPV16 L1L2 VLP, or HPV16 L2 mutant VLP for 45 min. Uptake was assessed by flow cytometry normalized to untreated LC. These data are representative examples of three experiments performed in triplicate shown as the mean ± SD (**P < 0.01 as determined by a two-tailed, unpaired t-test, as compared to untreated LC)
Specific mutations in HPV16 L2108-111 increase HPV16 immunogenicity in LC
We have shown that LC exposed to HPV16 L1L2 VLP are not activated, implicating an HPV immune escape mechanism targeting LC (48, 49), and we further established that L2 is responsible for the suppression of LC immune function when LC are exposed to HPV16 VLP (40). Therefore, we next sought to determine the role of the A2t-binding region of L2 (L2108-126) in LC immune responses. To determine the effects of L2108-126 on the level of phenotypic activation of LC, the expression of cell surface MHC II and CD86 on LC after exposure to either wild type HPV16 PsV or HPV16 L2 mutant PsV was assessed. The MHC II profile of LC exposed to HPV16 L2 mutant PsV more closely resembled that of activated LC treated with LPS (Fig. 4A middle and right), whereas the MHC II profile of LC exposed to wild type HPV16 PsV closely mirrored that of untreated LC (Fig. 4A left). Furthermore, there was a statistically significant increase in MHC II and CD86 expression of LC treated with HPV16 L2 mutant PsV compared to wild type HPV16 PsV (Fig. 4B–C). These results show that altering the capsid’s interaction with A2t through mutation of the A2t binding region of L2 results in the phenotypic maturation of LC. While the L2 mutated PsV did not activate as strongly as LPS, the statistically significant increase in immunogenicity highlights the importance of L2 in LC immune responses, and specifically, the A2t-interacting region of L2. Though indirect, these results indicate that there is a role for A2t in HPV16-induced suppression of LC immune function.
Figure 4. Expression of MHC II on LC treated with wildtype HPV16 or L2 mutant HPV16 PsV.
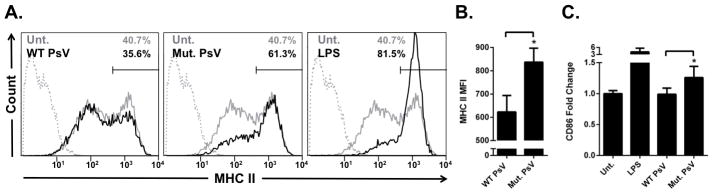
A. LC were either left untreated (grey lines), treated with LPS, HPV16 wild-type (WT) PsV, or HPV16 L2 mutant PsV (black lines). After 48 hr, the cell surface expression of MHC II was and analyzed by flow cytometry. The HPV16 L2 mutant PsV and LPS caused phenotypic activation of LC compared to untreated cells while there was a slight decrease in MHC II expression in WT HPV16PsV treated LC. Isotype controls are shown as grey dotted lines B. The mean fluorescent intensity (MFI) of MHC II-FITC stained cells was 624 for WT PsV and 837 for HPV16 L2 mutant PsV with p = 0.04 as determined by a two-tailed, unpaired t-test, for the means of 3 independent experiments. C. LC were either left untreated, treated with LPS, HPV16 WT PsV, or HPV16 L2 mutant PsV. After 48 hr, the cell surface expression of CD86 was and analyzed by flow cytometry. The HPV16 L2 mutant PsV caused a significantly greater expression of CD86 compared to WT HPV16 VLP (p<0.05 as determined by a two-tailed, unpaired t-test, for the means of 3 independent experiments).
Exogenous A2t induces suppression of LC immune function
While the results above suggest a role for A2t in suppression of LC maturation by HPV16, we next sought to characterize the precise contribution of A2t in LC activation in the absence of HPV16. Exogenous A2t was found to activate murine macrophages (61); and HPV16 was also shown to cause increased translocation and accumulation of A2t to the outer leaflet, which we hypothesized can be mimicked without HPV16 through the addition of excess exogenous A2t. Therefore we studied the effect of externally provided purified A2t on LC activation. To test this, LC were left untreated or treated with purified A2t or its individual subunits anxA2 and S100A10 to determine if the subunits or the heterotetramer form are associated with LC immune responses. The expression of cell surface MHC II and CD86 on LC after treatment with purified proteins was then assessed via flow cytometry. There was a significant reduction in the surface expression of MHC II on LC treated with A2t compared to untreated, anxA2 treated, or S100A10 treated LC (Fig. 5A). Since CD86 expression on immature LC is low, we were not able to detect any decrease in protein expression below baseline levels (data not shown). However, MHC II is normally expressed by immature LC and indeed a decrease in extracellular MHC II was observed in A2t treated LC. These results indicate a suppressive role for A2t in LC immune responses, and that the tetramer form of the receptor is required to induce a suppressed LC phenotype. Furthermore, we analyzed cytokine and chemokine secretion by LC that were exposed to purified A2t, and found that LC exposed to A2t showed a significant and pronounced decrease in the release of many pro-inflammatory cytokines and chemokines (Table 1) indicative of a Th1 cell-mediated immune response (40). Specifically, there was a statistically significant decrease in the secretion of MCP-1, MIP-1α, IP10, and RANTES. There was no significant change observed in the secretion of MIP-1β or TNF-α, and the secreted levels of IL-6 and IL-12 were below measurable levels for untreated and A2t treated LC. While there was a statistically significant increase in the secretion of IL-8 in A2t treated LC compared to untreated LC, the levels were much lower than LC treated with LPS.
Figure 5. Exogenous A2t suppresses LC immune function.
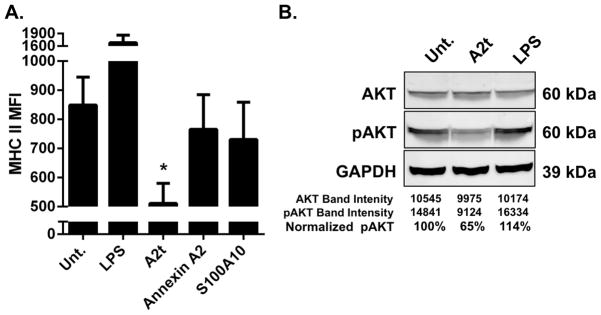
A. LC exposed to A2t show reduced surface expression of MHC II compared to untreated LC and LC exposed to annexin A2 or S100A10. LC were left untreated (Unt.) or incubated with LPS, A2t, Annexin A2, or S100A10 and subsequently analyzed via flow cytometry for the change in expression of MHC II. The mean of three experiments ± SD is presented (*P < 0.05 as determined by a two-tailed, unpaired t-test, as compared to untreated LC). B. A2t induces an immune suppressive signal transduction cascade in LC. LC were left untreated (Unt.) or incubated with LPS or A2t for 15 min. Cellular lysates were isolated and subjected to Immuno-blot analysis demonstrating a reduction in pAKT in the A2t treated cells. One representative example of three is shown.
Table I. LC exposed to A2t show reduced secretion of Th1-associated cytokines compared to controls.
LC were left untreated or incubated with LPS or A2t. Supernatants were collected at 48 hours and analyzed in triplicate for the presence of cytokines and chemokines.
| Untreated | LPS | A2t | |
|---|---|---|---|
| IL-6 | <LL | 2469 ± 86 | <LL |
| IL-8 | 718 ± 36 | 3927 ± 543 | 1239 ± 54** |
| IL-12 | <LL | 6 ± 0 | <LL |
| TNF-α | 8 ± 1 | 3721 ± 124 | 12 ± 3 |
| MCP1 | 862 ± 40 | 6189 ± 298 | 81 ± 6*** |
| MIP-1α | 58 ± 3 | 2612 ± 431 | 34 ± 2*** |
| MIP-1β | 56 ± 7 | 4900 ± 384 | 49 ± 3 |
| IP10 | 189 ± 2 | 761 ± 10 | 14 ± 1*** |
| RANTES | 125 ± 9 | 2541 ± | 92 ± 8** |
These data are a representative example of an experiment performed three times expressed as the mean concentration ± SD (**P<0.01, ***P < 0.001 as determined by a two-tailed, unpaired t-test, as compared to untreated LC).
A2t induces an immune suppressive signal cascade in LC
We have previously reported that HPV16 L2 induces an immune suppressive signal transduction cascade within LC that is hallmarked by a reduction in p-Akt in LC treated with HPV16 L2-containing VLP (40). To determine if A2t is associated with this suppressive signal transduction cascade, LC were treated with purified A2t prior to analysis of Akt and p-Akt levels via an immuno-blot assay. Cellular levels of p-Akt were reduced to 64% with p=0.008 (95% CI, 51–77%) in LC exposed to A2t compared to untreated (95% CI, 78–122%) and LPS-treated (95% CI, 94–136%) controls (representative example shown in Fig. 5B). These results indicate that exogenous A2t causes the same suppressive signaling events that are associated with HPV16 L2, further implicating a role for A2t in the suppression of LC immune function.
Small molecule inhibition of A2t prevents HPV16-induced immune suppression of LC
Recently, small molecule inhibitors of A2t (A2ti) have been identified that can disrupt the A2t tetramer by blocking the binding between anxA2 and S100A10 (56). As shown above, the role for A2t in the suppression of LC immune function is dependent on the tetramer form, and not on the individual subunits. In this manner, A2ti has the potential to disrupt the immune-suppressive properties of A2t in LC without affecting other cellular roles for either anxA2 or S100A10. Hence the effect of A2ti on LC activation with and without the addition of HPV16 L1L2 PsV was examined. Increasing concentrations of A2ti led to an increase in the surface expression of MHC II and the activation-associated surface marker CD86 on LC (Fig. 6A). At all concentrations of A2ti tested, there was not a significant increase in MHC II expression compared to LC treated with HPV16 alone, but there was a significant increase in MHC II expression on LC treated with 25 μM and 50 μM of A2ti with the addition of HPV16. Additionally, at 10 μM and 25 μM of A2ti, which are well below and near the reported IC50 value of 24 μM respectively, there was not a statistically significant increase in CD86 in the A2ti-treated groups compared to those treated with HPV16 alone. However, when HPV16 PsV were added to these groups (i.e. LC pre-treated with 10 μM or 25 μM A2ti), there was a significant increase in the surface expression of CD86 showing that the L2-containing PsV were no longer able to suppress the maturation of LC. At 50 μM of A2ti, LC were phenotypically activated as indicated by a significant increase in CD86 without HPV16 PsV, but were even further activated in the presence of HPV16 PsV, indicating a significant increase in the LC activating potential of HPV16 when A2t is disrupted.
Figure 6. Inhibition of A2t prevents HPV16-induced immune suppression of LC.

A. Inhibition of A2t increases immunogenicity of HPV16 PsV. LC were left untreated (Unt.), or treated with LPS, HPV16 PsV, or treated with increasing concentrations of A2t inhibitor (A2ti) alone or A2ti prior to exposure to HPV16 PsV and subsequently analyzed via flow cytometry for the change in expression of MHC II and CD86. B. LC exposed to A2ti and HPV16 PsV show increased secretion of Th1-associated chemokines compared to LC treated with HPV16 PsV alone. LC were left untreated (Unt.) or treated with LPS, HPV16 PsV, A2ti (25 μM), or A2ti plus HPV16 PsV. Supernatants were collected at 48 hours and analyzed in triplicate for the presence of cytokines and chemokines. These data are a representative example of an experiment performed three times expressed as the mean concentration ± SD (**P < 0.01, ***P < 0.001 as determined by a two-tailed, unpaired t-test, as compared to HPV16 PsV only treated LC).
In addition to cellular phenotype, the secretion of inflammatory cytokines and chemokines by LC pre-treated with A2ti was examined with and without the addition of HPV16 PsV. It was found that LC treated with 25 μM A2ti prior to exposure to HPV16 PsV showed a significant increase in the secretion of IL-8, MIP-1α, MIP-1β, and RANTES compared to both HPV16 only and A2ti only treated LC. There was a subtle yet non-significant increase observed in the secretion of TNF- α in LC treated with A2ti prior to exposure to HPV16 PsV compared to HPV16 only and A2ti only treated LC, and no change seen in the secretion of IL-10. The secreted levels of IL-6 and IL-12 were found to be below measurable levels while MCP-1 levels were at saturated levels for all treatment groups (all results can be seen in Sup. Table 1). Taken together, these data demonstrate that targeted small-molecule disruption of A2t reverses HPV16-induced LC-targeted immune suppression, and strongly suggest that HPV16 entry via A2t leads to the suppression of LC maturation.
Discussion
The HPV16 life cycle is strictly intraepithelial, and as a result HPV16 antigens should be processed and presented by LC, the professional APC that reside in the parabasal and lower suprabasal layers of squamous epithelium (62). Various studies have found that HPV L1 VLP and HPV L1L2 VLP can bind to and stimulate activation of human dendritic cells (DC) (49, 63–66), providing evidence that the capsids of HPV can induce the maturation of APC. However, HPV16 has evolved over time using a mechanism in which internalization of capsids of HPV16 into LC results in suppressive signaling and defective activation (48, 49). In this sense, LC that are normally targeted by HPV16 during a natural infection, may be uniquely manipulated by HPV compared to other immune cells or DC subsets through A2t, though future research is needed to determine if A2t plays a role in immune responses of other DC types.
We have shown that internalization of HPV16 by human LC indicated the presence of a specific L2 receptor and internalization mechanism that results in the suppression of LC maturation (40). The highly evolutionarily conserved HPV16 L2108-126 epitope has been shown to be vital in the binding and infectivity of HPV16 in different cell types (37), and it binds specifically to the recently identified HPV16 uptake receptor A2t on epithelial cells (38). In the current study, we demonstrate that this conserved A2t binding region of L2 is associated with HPV16 binding to LC, and interacts with A2t on the LC surface. We further show that mutations in this region increase HPV16 immunogenicity, highlighting the importance of this region in the suppression of LC maturation. Through uptake assays we demonstrate that internalization of HPV16 L1L2 VLP by LC is facilitated by A2t, which can be inhibited by SLPI- or siRNA-mediated knockdown of A2t in LC. A2t was shown to suppress LC immune function, and small molecule disruption of A2t prevented HPV16-induced LC-targeted immune suppression. Collectively, these data demonstrate that A2t is involved in the binding and internalization of HPV16 into LC through an L2-dependent mechanism, and that this entry mechanism is associated with suppression of LC immune function.
It is conventional to use VLP and PsV to study HPV receptor binding and entry, especially with primary immune cells (23, 40–42, 48, 63, 67). Here, L1 and L1L2 VLP, as well as L2 mutated PsV, were used to examine the significance of the interaction between the L2 protein and A2t in HPV16 uptake in LC, where the primary difference was the absence or presence of the wild-type or mutated L2 protein. A noteworthy observation from the present assays is that HPV16 L1L2 VLP binding and internalization was never completely inhibited on LC with down-regulation or inhibition of A2t. Additionally, while A2t-mediated entry was shown to be dependent on L2, L1-only VLP still entered LC, and this type of entry has been shown to induce LC activation (49). This implies that there may be multiple and/or redundant uptake pathways for HPV16 that differentially activate LC. For these reasons, it is attractive to hypothesize a model where L1-mediated entry leads to LC activation, and conversely that L2/A2t-mediated entry suppresses LC immune responses. This model would further imply that inhibiting the L2/A2t-mediated pathway would not completely block entry, but would rather lead to HPV entry via an L1-mediated activating pathway thus preventing HPV16-induced immune suppression, which fully fits with the data presented herein. Future studies will aim to investigate such a model and determine all necessary cellular factors for each entry mechanism. Our current study identifies A2t as a novel HPV16 receptor on LC, but other studies are needed to determine if previously identified HPV receptors and cofactors of keratinocytes such as HSPG, integrins, cyclophilins, growth factors and growth factor receptors, and tetraspanins [reviewed in (68)] are also used for HPV16 internalization by LC.
Our data clearly show that A2t plays a role in suppressing the maturation of LC in experiments where purified A2t was added to LC in vitro, and this is the first study to identify a function for A2t in suppression of LC maturation. In contrast, exogenous A2t was previously shown to activate murine macrophages, which is in stark opposition to the suppressive role A2t plays on human LC (61). This may indicate a unique function for A2t in LC-mediated immune suppression that HPV16 has evolved to take advantage of. A recent report that confirmed A2t is an HPV16 receptor on epithelial cells provided evidence that initial HPV16 binding increases recruitment and translocation of A2t to the extracellular surface (39). Therefore, we hypothesized that the addition of A2t would bind to the outer leaflet of LC in culture simulating the reported increase in A2t on the cell surface on HPV16 treated cells. Though no mechanism has yet been delineated, evidence in the literature suggests that the early binding of HPV16 to α6 integrins prior to A2t binding and internalization can be linked to the local recruitment of A2t to the cell membrane. For example, it has been shown that binding and clustering of α6β1/4 integrins cause the recruitment of talin and facilitate the activation of focal adhesion kinase (FAK) (69), which plays an important role in HPV16 infection (70). Talin directly interacts with and activates phosphatidylinositol 4-phosphate, 5-kinase (PIP5K) (71, 72), which then catalyzes the focal production of phosphatidylinositol (4,5)-bisphosphate (PIP2) (73). Of note, it was demonstrated that PIP2 actively recruits A2t to specific regions of the cell membrane (74, 75). As mentioned above, early HPV16-integrin binding activates FAK, which in turn activates src-family kinases (SFK) (69, 76). Importantly, it was shown that SFK regulates the translocation of the A2t to the cell surface both in vitro and in vivo (45). Now, we can begin to see a hypothetical signal cascade in which the binding of HPV16 to the cell surface leads to the local recruitment and subsequent translocation of A2t to the cell surface to which HPV16 can then bind. This HPV16-induced local recruitment of A2t may lead to the suppression of LC maturation observed in our exogenous A2t experiments and perhaps initiates clathrin-, caveolin-, lipid raft-, flotillin-, cholesterol-, dynamin-independent endocytosis of HPV16.
Protein-protein interactions are key players in cellular processes, and are increasingly targets for small molecule discovery. The interaction between anxA2 and S100A10 has been well characterized by mutagenesis and crystallography (77, 78). In the current study, we show that LC derived from primary human PBMC express both proteins, and it has previously been reported that S100A10 is highly expressed by LC derived from umbilical blood CD34+ progenitor cells (47), and the S100 family of proteins is commonly used as an LC marker in vivo (79, 80), indicating similarities in the expression of A2t in LC from tissue in vivo and cellular progenitors in vitro. The 14 residue N-terminal region of two separate anxA2 molecules bind, primarily through hydrophobic interactions, to two binding pockets created by S100A10 dimerization. Recently, a group used a ligand-guided method and information about the topological arrangement of chemical features of the anxA2 N-terminus to successfully identify compounds that are able to compete with the binding of the anxA2 N-terminus to S100A10 (56). The most efficient of these A2t small molecule inhibitors (A2ti) was used to disrupt A2t on LC and was shown to effectively prevent HPV16-induced immune suppression.
Aside from HPV16, anxA2 has been shown to play a role in the binding and uptake of a variety of different viruses including human cytomegalovirus, respiratory syncytial virus, enterovirus 71, and was shown to be a cofactor for HIV-1 infection (58, 81–84). Like HPV16, HIV is another virus that specifically targets LC during infection (59), which may have implications in HIV-HPV co-infections through targeting of the same receptor and cell type. Though unknown for other viruses, it is unlikely that HPV16 binding to A2t is mediated indirectly by association with other cell surface binding proteins, because we have previously reported a strong direct interaction exists between the L2 protein and A2t in the absence of other cellular proteins (38). Our findings herein are the first to identify A2t as an HPV16 receptor on LC, and represent the first demonstration of specifically targeting A2t to overcome HPV16-induced immune suppression. Interestingly, our lab has recently demonstrated that LC exposed to capsids of HPV types 18, 31, 45, 11, and HPV5 similarly suppress LC activation (85), and future research will aim to determine if A2t is involved. The targeted inhibition of A2t in viral studies is both exciting and promising, and ongoing studies are currently underway to test the ability of A2ti to prevent HPV16 infection of epithelial cells, and in the future, the targeted inhibition of A2t may have broad anti-viral implications.
Supplementary Material
Acknowledgments
Support from the Karl H. and Ruth M. Balz Trust is gratefully acknowledged. The authors would like to thank Harold Kochounian for excellent technical assistance. We also would like to acknowledge the Beckman Center for Immune Monitoring Core Facility for technical assistance.
Footnotes
This study was supported by NIH grants R01 CA74397 and RC2 CA148298 to WM Kast who holds the Walter A. Richter Cancer Research Chair. Andrew Woodham and Lisa Yan are both TL1 Scholars and supported by SC CTSI (NIH/NCRR/NCATS) Grant # TL1TR000132. Andrew Woodham is supported in part by award number NIH grant 5P30CA014089-37 from the NCI through the Norris Comprehensive Cancer Center Heidelberger Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
References
- 1.Arbyn M, Castellsague X, de Sanjose S, Bruni L, Saraiya M, Bray F, Ferlay J. Worldwide burden of cervical cancer in 2008. Ann Oncol. 2011;22:2675–2686. doi: 10.1093/annonc/mdr015. [DOI] [PubMed] [Google Scholar]
- 2.Haedicke J, Iftner T. Human papillomaviruses and cancer. Radiother Oncol. 2013 doi: 10.1016/j.radonc.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Munoz N, Bosch FX, de Sanjos S, Herrero R, Castellsagu X, Shah KV, Snijders PJF, Meijer CJLM. Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer. New England Journal of Medicine. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 4.Walboomers JMM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJF, Peto J, Meijer CJLM, Muñoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. The Journal of Pathology. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 5.Harald zH. Human papillomaviruses in the pathogenesis of anogenital cancer. Virology. 1991;184:9–13. doi: 10.1016/0042-6822(91)90816-t. [DOI] [PubMed] [Google Scholar]
- 6.Bosch FMM, Munoz N, Sherman M, Jansen A, Peto J, Schiffman M, Moreno V, Kurman R, Shah K. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on servical cancer (IBSCC) Study Group. J Natl Cancer Inst. 1995;87:796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- 7.Stanley MAP, MR, Coleman N. HPV: from infection to cancer. Biochemical Society Transactions. 2007;35:1456–1460. doi: 10.1042/BST0351456. [DOI] [PubMed] [Google Scholar]
- 8.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol. 2008;8:935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 9.Modis Y, Trus BL, Harrison SC. Atomic model of the papillomavirus capsid. Embo J. 2002;21:4754–4762. doi: 10.1093/emboj/cdf494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cumming SC-IT, Milligan S, Graham SV. Human Papillomavirus type 16 late gene expression is regulated by cellular RNA processing factors in response to epithelial differentiation. Biochemical Society Transactions. 2008;36:522–524. doi: 10.1042/BST0360522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 12.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buck CB, Day PM, Trus BL. The papillomavirus major capsid protein L1. Virology. 2013 doi: 10.1016/j.virol.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. Arrangement of L2 within the papillomavirus capsid. J Virol. 2008;82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou JS, XY, Louis K, Frazer H. Interaction of human papillomavirus (HPV) type 16 capsid proteins with HPV DNA requires an intact L2 N-terminal sequence. J Virol. 1994;68:619–625. doi: 10.1128/jvi.68.2.619-625.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao KN, Sun XY, Frazer IH, Zhou J. DNA packaging by L1 and L2 capsid proteins of bovine papillomavirus type 1. Virology. 1998;243:482–491. doi: 10.1006/viro.1998.9091. [DOI] [PubMed] [Google Scholar]
- 17.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. Human papillomavirus infection requires cell surface heparan sulfate. J Virol. 2001;75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evander MF, IH, Payne E, Qi YM, Hengst K, et al. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J Virol. 1997;71:2449–2456. doi: 10.1128/jvi.71.3.2449-2456.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bienkowska-Haba MP, HD, Sapp M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathogens. 2009;5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Surviladze Z, Dziduszko A, Ozbun MA. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathogens. 2012;8:e1002519. doi: 10.1371/journal.ppat.1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spoden GFK, Husmann M, Boller K, Sapp M, et al. Clathrin- and caveolin-independent entry of human papillomavirus type 16--involvement of tetraspanin-enriched microdomains (TEMs) PLoS ONE. 2008;3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathogens. 2012;8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fausch SC, DSD, Kast WM. Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and Langerhans cells. Cancer Research. 2003;63:3478–3482. [PubMed] [Google Scholar]
- 24.Wang JW, Roden RB. L2, the minor capsid protein of papillomavirus. Virology. 2013 doi: 10.1016/j.virol.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kämper ND, PM, Nowak T, Selinka H, Florin L, et al. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol. 2006;80:759–768. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronnimann MP, Chapman JA, Park CK, Campos SK. A Transmembrane Domain and GxxxG Motifs Within L2 are Essential for Papillomavirus Infection. J Virol. 2012 doi: 10.1128/JVI.01539-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergant Marusic M, Ozbun MA, Campos SK, Myers MP, Banks L. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic. 2012;13:455–467. doi: 10.1111/j.1600-0854.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Day PM, Schiller JT. The role of furin in papillomavirus infection. Future Microbiol. 2009;4:1255–1262. doi: 10.2217/fmb.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang R, Yutzy WHt, Viscidi RP, Roden RB. Interaction of L2 with beta-actin directs intracellular transport of papillomavirus and infection. J Biol Chem. 2003;278:12546–12553. doi: 10.1074/jbc.M208691200. [DOI] [PubMed] [Google Scholar]
- 30.Day PM, Baker CC, Lowy DR, Schiller JT. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A. 2004;101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang R, Day PM, Yutzy WHt, Lin KY, Hung CF, Roden RB. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J Virol. 2003;77:3531–3541. doi: 10.1128/JVI.77.6.3531-3541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. Common neutralization epitope in minor capsid protein L2 of human papillomavirus types 16 and 6. J Virol. 1999;73:6188–6190. doi: 10.1128/jvi.73.7.6188-6190.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gambhira R, Karanam B, Jagu S, Roberts JN, Buck CB, Bossis I, Alphs H, Culp T, Christensen ND, Roden RB. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J Virol. 2007;81:13927–13931. doi: 10.1128/JVI.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slupetzky K, Gambhira R, Culp TD, Shafti-Keramat S, Schellenbacher C, Christensen ND, Roden RB, Kirnbauer R. A papillomavirus-like particle (VLP) vaccine displaying HPV16 L2 epitopes induces cross-neutralizing antibodies to HPV11. Vaccine. 2007;25:2001–2010. doi: 10.1016/j.vaccine.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo K, Ishii Y, Ochi H, Matsumoto T, Yoshikawa H, Kanda T. Neutralization of HPV16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the HPV16 minor capsid protein L2 surface region. Virology. 2007;358:266–272. doi: 10.1016/j.virol.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 36.Conway MJ, Cruz L, Alam S, Christensen ND, Meyers C. Cross-neutralization potential of native human papillomavirus N-terminal L2 epitopes. PLoS ONE. 2011;6:e16405. doi: 10.1371/journal.pone.0016405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawana Y, Kawana K, Yoshikawa H, Taketani Y, Yoshiike K, Kanda T. Human papillomavirus type 16 minor capsid protein l2 N-terminal region containing a common neutralization epitope binds to the cell surface and enters the cytoplasm. J Virol. 2001;75:2331–2336. doi: 10.1128/JVI.75.5.2331-2336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodham AW, Da Silva DM, Skeate JG, Raff AB, Ambroso MR, Brand HE, Isas JM, Langen R, Kast WM. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE. 2012;7:e43519. doi: 10.1371/journal.pone.0043519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dziduszko A, Ozbun MA. Annexin A2 and S100A10 Regulate Human Papillomavirus Type 16 Entry and Intracellular Trafficking in Human Keratinocytes. J Virol. 2013 doi: 10.1128/JVI.00519-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fahey LM, Raff AB, Da Silva DM, Kast WM. A major role for the minor capsid protein of human papillomavirus type 16 in immune escape. J Immunol. 2009;183:6151–6156. doi: 10.4049/jimmunol.0902145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan M, Peng J, Jabbar IA, Liu X, Filgueira L, Frazer IH, Thomas R. Despite differences between dendritic cells and Langerhans cells in the mechanism of papillomavirus-like particle antigen uptake, both cells cross-prime T cells. Virology. 2004;324:297–310. doi: 10.1016/j.virol.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 42.Bousarghin L, Hubert P, Franzen E, Jacobs N, Boniver J, Delvenne P. Human papillomavirus 16 virus-like particles use heparan sulfates to bind dendritic cells and colocalize with langerin in Langerhans cells. J Gen Virol. 2005;86:1297–1305. doi: 10.1099/vir.0.80559-0. [DOI] [PubMed] [Google Scholar]
- 43.Waisman DM. Annexin II tetramer: structure and function. Molecular and Cellular Biochemistry. 1995;149/150:301–322. doi: 10.1007/BF01076592. [DOI] [PubMed] [Google Scholar]
- 44.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 45.Deora AB, Kreitzer G, Jacovina AT, Hajjar KA. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J Biol Chem. 2004;279:43411–43418. doi: 10.1074/jbc.M408078200. [DOI] [PubMed] [Google Scholar]
- 46.Rescher U, Gerke V. S100A10/p11: family, friends and functions. Pflugers Arch. 2008;455:575–582. doi: 10.1007/s00424-007-0313-4. [DOI] [PubMed] [Google Scholar]
- 47.Rust R, Kluiver J, Visser L, Harms G, Blokzijl T, Kamps W, Poppema S, van den Berg A. Gene expression analysis of dendritic/Langerhans cells and Langerhans cell histiocytosis. The Journal of Pathology. 2006;209:474–483. doi: 10.1002/path.2003. [DOI] [PubMed] [Google Scholar]
- 48.Fausch SC, Da Silva DM, Rudolf MP, Kast WM. Human papillomavirus virus-like particles do not activate Langerhans cells: a possible immune escape mechanism used by human papillomaviruses. J Immunol. 2002;169:3242–3249. doi: 10.4049/jimmunol.169.6.3242. [DOI] [PubMed] [Google Scholar]
- 49.Fahey LM, Raff AB, Da Silva DM, Kast WM. Reversal of human papillomavirus-specific T cell immune suppression through TLR agonist treatment of Langerhans cells exposed to human papillomavirus type 16. J Immunol. 2009;182:2919–2928. doi: 10.4049/jimmunol.0803645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirnbauer RBF, Cheng N, Lowy DR, Schiller JT. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc Natl Acad Sci U S A. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dasgupta J, Bienkowska-Haba M, Ortega ME, Patel HD, Bodevin S, Spillmann D, Bishop B, Sapp M, Chen XS. Structural basis of oligosaccharide receptor recognition by human papillomavirus. J Biol Chem. 2011;286:2617–2624. doi: 10.1074/jbc.M110.160184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buck CB, Thompson CD. Production of papillomavirus-based gene transfer vectors. Curr Protoc Cell Biol. 2007;Chapter 26(Unit 26):21. doi: 10.1002/0471143030.cb2601s37. [DOI] [PubMed] [Google Scholar]
- 53.Puisieux A, Ji J, Ozturk M. Annexin II up-regulates cellular levels of p11 protein by a post-translational mechanisms. Biochem J. 1996;313(Pt 1):51–55. doi: 10.1042/bj3130051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He KL, Deora AB, Xiong H, Ling Q, Weksler BB, Niesvizky R, Hajjar KA. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J Biol Chem. 2008;283:19192–19200. doi: 10.1074/jbc.M800100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renn CN, Sanchez DJ, Ochoa MT, Legaspi AJ, Oh CK, Liu PT, Krutzik SR, Sieling PA, Cheng G, Modlin RL. TLR activation of Langerhans cell-like dendritic cells triggers an antiviral immune response. J Immunol. 2006;177:298–305. doi: 10.4049/jimmunol.177.1.298. [DOI] [PubMed] [Google Scholar]
- 56.Reddy TR, Li C, Fischer PM, Dekker LV. Three-Dimensional Pharmacophore Design and Biochemical Screening Identifies Substituted 1,2,4-Triazoles as Inhibitors of the Annexin A2-S100A10 Protein Interaction. Chem Med Chem. 2012;7:1435–1446. doi: 10.1002/cmdc.201200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma G, Greenwell-Wild T, Lei K, Jin W, Swisher J, Hardegen N, Wild CT, Wahl SM. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J Exp Med. 2004;200:1337–1346. doi: 10.1084/jem.20041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawamura T, Kurtz SE, Blauvelt A, Shimada S. The role of Langerhans cells in the sexual transmission of HIV. J Dermatol Sci. 2005;40:147–155. doi: 10.1016/j.jdermsci.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 59.de Jong MA, Geijtenbeek TB. Human immunodeficiency virus-1 acquisition in genital mucosa: Langerhans cells as key-players. J Intern Med. 2009;265:18–28. doi: 10.1111/j.1365-2796.2008.02046.x. [DOI] [PubMed] [Google Scholar]
- 60.Drobni P, Mistry N, McMillan N, Evander M. Carboxy-fluorescein diacetate, succinimidyl ester labeled papillomavirus virus-like particles fluoresce after internalization and interact with heparan sulfate for binding and entry. Virology. 2003;310:163–172. doi: 10.1016/s0042-6822(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 61.Swisher JF, Burton N, Bacot SM, Vogel SN, Feldman GM. Annexin A2 tetramer activates human and murine macrophages through TLR4. Blood. 2010;115:549–558. doi: 10.1182/blood-2009-06-226944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stanley MA. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev. 2012;25:215–222. doi: 10.1128/CMR.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lenz P, Lowy DR, Schiller JT. Papillomavirus virus-like particles induce cytokines characteristic of innate immune responses in plasmacytoid dendritic cells. Eur J Immunol. 2005;35:1548–1556. doi: 10.1002/eji.200425547. [DOI] [PubMed] [Google Scholar]
- 64.Lenz P, Day PM, Pang YY, Frye SA, Jensen PN, Lowy DR, Schiller JT. Papillomavirus-like particles induce acute activation of dendritic cells. J Immunol. 2001;166:5346–5355. doi: 10.4049/jimmunol.166.9.5346. [DOI] [PubMed] [Google Scholar]
- 65.Rudolf MP, Fausch SC, Da Silva DM, Kast WM. Human dendritic cells are activated by chimeric human papillomavirus type-16 virus-like particles and induce epitope-specific human T cell responses in vitro. J Immunol. 2001;166:5917–5924. doi: 10.4049/jimmunol.166.10.5917. [DOI] [PubMed] [Google Scholar]
- 66.Da Silva DM, Fausch SC, Verbeek JS, Kast WM. Uptake of human papillomavirus virus-like particles by dendritic cells is mediated by Fcgamma receptors and contributes to acquisition of T cell immunity. J Immunol. 2007;178:7587–7597. doi: 10.4049/jimmunol.178.12.7587. [DOI] [PubMed] [Google Scholar]
- 67.Renoux VM, Bisig B, Langers I, Dortu E, Clemenceau B, Thiry M, Deroanne C, Colige A, Boniver J, Delvenne P, Jacobs N. Human papillomavirus entry into NK cells requires CD16 expression and triggers cytotoxic activity and cytokine secretion. Eur J Immunol. 2011;41:3240–3252. doi: 10.1002/eji.201141693. [DOI] [PubMed] [Google Scholar]
- 68.Raff AB, Woodham AW, Raff LM, Skeate JG, Yan L, Da Silva DM, Schelhaas M, Kast WM. The evolving field of human papillomavirus receptor research: a review of binding and entry. J Virol. 2013;87:6062–6072. doi: 10.1128/JVI.00330-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mitra SKS, DD Integrin-regulated FAK-Src signaling in normal and cancer cells. Curren Opinion in Cell Biology. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 70.Abban CY, Meneses PI. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology. 2010;403:1–16. doi: 10.1016/j.virol.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ling KD, RL, Firestone AJ, Bunce MW, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 2002;420:89–93. doi: 10.1038/nature01082. [DOI] [PubMed] [Google Scholar]
- 72.Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- 73.van den Bout I, Divecha N. PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. Journal of Cell Science. 2009;122:3837–3850. doi: 10.1242/jcs.056127. [DOI] [PubMed] [Google Scholar]
- 74.Rescher URD, Ludwig C, Zobiack N, Gerke V. Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to actin assembly sites at cellular membranes. Journal of Cell Science. 2004;117:3473–3480. doi: 10.1242/jcs.01208. [DOI] [PubMed] [Google Scholar]
- 75.Hayes MJ, Shao D, Grieve A, Levine T, Bailly M, et al. Annexin A2 at the interface between F-actin and membranes enriched in phosphatidylinositol 4,5,-bisphosphate. Biochim Biophys Acta. 2009;1793:1086–1095. doi: 10.1016/j.bbamcr.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schlaepfer DDM, SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 77.Becker T, Weber K, Johnsson N. Protein-protein recognition via short amphiphilic helices; a mutational analysis of the binding site of annexin II for p11. Embo J. 1990;9:4207–4213. doi: 10.1002/j.1460-2075.1990.tb07868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rety S, Sopkova J, Renouard M, Osterloh D, Gerke V, Tabaries S, Russo-Marie F, Lewit-Bentley A. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nat Struct Biol. 1999;6:89–95. doi: 10.1038/4965. [DOI] [PubMed] [Google Scholar]
- 79.Miyagi J, Kinjo T, Tsuhako K, Higa M, Iwamasa T, Kamada Y, Hirayasu T. Extremely high Langerhans cell infiltration contributes to the favourable prognosis of HPV-infected squamous cell carcinoma and adenocarcinoma of the lung. Histopathology. 2001;38:355–367. doi: 10.1046/j.1365-2559.2001.01067.x. [DOI] [PubMed] [Google Scholar]
- 80.Muderspach L, Wilczynski S, Roman L, Bade L, Felix J, Small LA, Kast WM, Fascio G, Marty V, Weber J. A phase I trial of a human papillomavirus (HPV) peptide vaccine for women with high-grade cervical and vulvar intraepithelial neoplasia who are HPV 16 positive. Clin Cancer Res. 2000;6:3406–3416. [PubMed] [Google Scholar]
- 81.Wright JF, Kurosky A, Wasi S. An endothelial cell-surface form of annexin II binds human cytomegalovirus. Biochem Biophys Res Commun. 1994;198:983–989. doi: 10.1006/bbrc.1994.1140. [DOI] [PubMed] [Google Scholar]
- 82.Malhotra R, Ward M, Bright H, Priest R, Foster MR, Hurle M, Blair E, Bird M. Isolation and characterisation of potential respiratory syncytial virus receptor(s) on epithelial cells. Microbes Infect. 2003;5:123–133. doi: 10.1016/s1286-4579(02)00079-5. [DOI] [PubMed] [Google Scholar]
- 83.Yang SL, Chou YT, Wu CN, Ho MS. Annexin II Binds to Capsid Protein VP1 of Enterovirus 71 and Enhances Viral Infectivity. J Virol. 2011;85:11809–11820. doi: 10.1128/JVI.00297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buchbinder SP. HIV epidemiology and breakthroughs in prevention 30 years into the AIDS epidemic. Top Antivir Med. 2011;19:38–46. [PMC free article] [PubMed] [Google Scholar]
- 85.Da Silva DM, Movius CA, Raff AB, Brand HE, Skeate JG, Wong MK, Kast WM. Suppression of Langerhans cell activation is conserved amongst human papillomavirus alpha and beta genotypes, but not a mu genotype. Virology. 2014;452–453:279–286. doi: 10.1016/j.virol.2014.01.031. http://dx.doi.org/10.1016/j.virol.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.