

Abstract
Kinetic resolution during the catalytic allyl-propargyl cross-coupling with chiral starting materials can be accomplished with a chiral Palladium catalyst. These reactions offer ready access to enantiomerically enriched enyne products from simple readily available starting materials.
Keywords: Allylation, Asymmetric Synthesis, Asymmetric Catalysis, Boron, Palladium
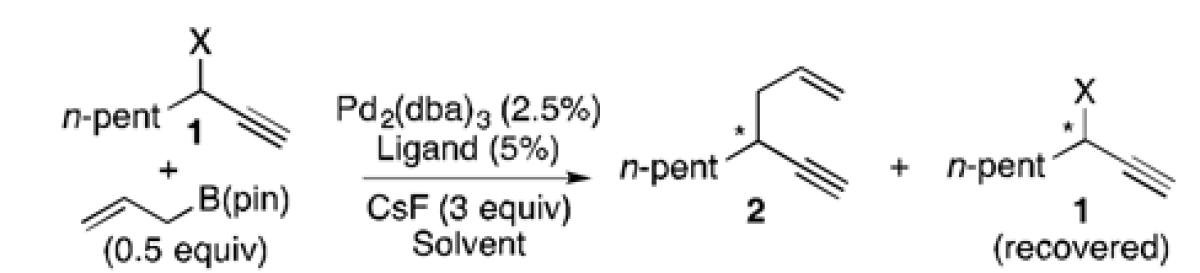
Palladium-catalyzed substitutions of propargylic electrophiles represent a powerful synthetic method for the construction of allene and alkyne containing compounds (Scheme 1, eq. 1).[1,2] These reactions typically proceed via oxidative addition of Pd(0) with propargyl electrophiles of type A to give interconverting allenyl (B) and propargyl palladium (C) intermediates, and yield allene (D) or alkyne (E) containing products upon subsequent reaction. While a number of methods have capitalized on these features resulting in catalytic carbon-carbon and carbon-heteroatom bond forming reactions,[2-4] there are few asymmetric variants.[3d, 5] This deficit is likely due to two issues: (1) unlike structurally similar (allyl)Pd intermediates that are configurationally dynamic,[6] both B and C are chiral and configurationally static, reflecting the configuration of the starting material after stereospecific SN2′ addition of Pd;[7] (2) in the absence of significant steric bias, bond formation generally occurs from the (allenyl)Pd complex B to furnish chiral allene products of type D,[8,9] which are known to be susceptible to racemization by Pd even under mild conditions.[10] To address these issues, asymmetric Pd-catalyzed propargyl substitutions have relied on stereospecific couplings with optically enriched propargyl electrophiles, and are limited in scope to allene products that are biased against racemization based on electronics or sterics.[3d, 5]
Scheme 1.

Palladium-Catalyzed Propargylic Substitutions
We recently reported a Pd-catalyzed cross-coupling of optically enriched propargyl acetates and allyl boron reagents to deliver 1,5-enynes (Scheme 1, eq. 2).[11] Mechanistically, it appeared that under the influence of a bidentate phosphine ligand (rac-BINAP), this transformation proceeds by 3,3′-reductive elimination from the allyl-allenyl-Pd complex F.[12,13] This allyl migration is highly regioselective for the configurationally-stable propargyl-substituted products of type E and allows for high stereospecificity for a range of sterically and electronically diverse electrophiles. Although this strategy provided practical access to synthetically useful intermediates, the need for enantiomerically pure starting material was a drawback. A potential solution revealed itself when, during the course of this study, we observed a small difference in reactivity of an optically enriched propargyl acetate during reactions catalyzed by complexes bearing either (R)- or (S)-BINAP. Reasoning that with a chiral catalyst that can more ably differentiate matched and mismatched substrates, a kinetic resolution might result and provide enantiomerically enriched 1,5-enynes from racemic starting material.[14] Although related Pd-catalyzed resolutions of allylic electrophiles have been reported,[15] such a strategy is unknown for propargyl systems.[16, 17]
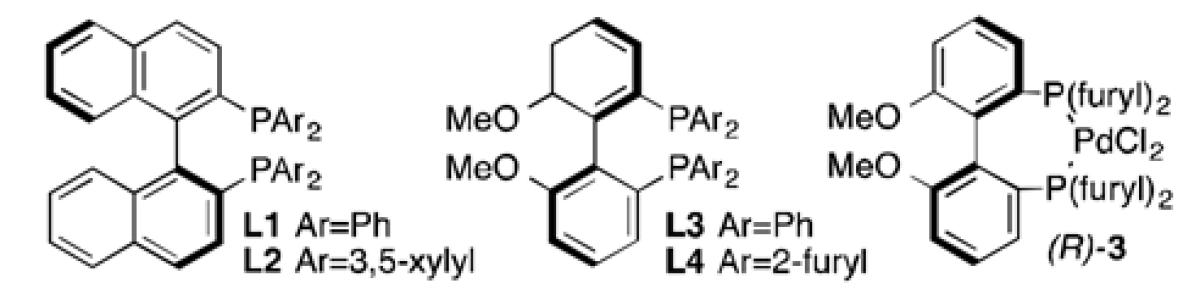
Initial experiments with (S)-BINAP (L1), a racemic mixture of substrate 1, and 0.5 equivalents of allylB(pin) in THF at 60 °C (Table 1, entry 1) offered moderate selectivity (s = 3.8).[18] The selectivity was relatively unchanged when the reaction was run at lower temperatures (entries 2 and 3) or in more polar solvents (entry 4), and decreased when run in the less polar solvent CH2Cl2 (entry 5). Use of a carbonate leaving group resulted in lower selectivity (entry 6, s = 1.3), while the pivaloyl ester showed a slight increase in selectivity (entry 7, s = 4.9).
Table 1.
Optimization of Kinetic Resolution
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Entry | Ligand | X | solvent | t[h] | T[°C] | Conv [%][a] | s [b] |
| 1 | (S) -L1 | OAc | THF | 14 | 60 | 52 | 3.8 |
| 2 | (S) -L1 | OAc | THF | 14 | 40 | 33 | 3.7 |
| 3 | (S) -L1 | OAc | THF | 14 | 25 | 3 | 3.8 |
| 4 | (S) -L1 | OAc | dioxane | 14 | 60 | 50 | 4.2 |
| 5 | (S) -L1 | OAc | DCM | 14 | 60 | 50 | 1.9 |
| 6 | (S) -L1 | OBoc | THF | 5 | 60 | 52 | 1.3 |
| 7 | (S) -L1 | OPiv | THF | 14 | 60 | 67 | 4.9 |
| 8 | (S) -L1 | OTBS | THF | 14 | 60 | 0 | nd |
| 9 | (R) -L2 | OAc | THF | 14 | 60 | 52 | 1.9 |
| 10 | (R) -L3 | OAc | THF | 14 | 60 | 56 | 6.6 |
| 11 | (R) -L4 | OAc | THF | 14 | 60 | 50 | 18.6 |
| 12[c] | (R) -3 | OAc | THF | 8 | 60 | 45 | 14.9 |
|
| |||||||
| |||||||
Conv=[ee1/(ee1+ee2)], enantiomeric excess determined by GC analysis on a chiral stationary phase.
s=kfast/kslow=1n[(1-Conv/100)(1-ee1/100)]/1n[1-Conv/100)(l+ee1/100)].
0.75 mol % (R)-3 used m place of Pd2(dba)3/ligand.
The nature of the chiral ligand was found to have a significant effect on selectivity: with larger 3,5-xylyl groups on L2 selectivity was diminished compared to L1, while a small improvement was seen with the structurally similar MeO-BIPHEP (L3, s = 6.6). An evaluation of other members of the BIPHEP family revealed a marked jump in selectivity when MeO(furyl)BIPHEP (L4) was employed (entry 11, s = 18.6). Use of the pre-formed L4-Pd(II)Cl2 complex [(R)-3] allowed for cleaner and more efficient reaction even with significantly lower catalyst loadings (entry 12).[19]
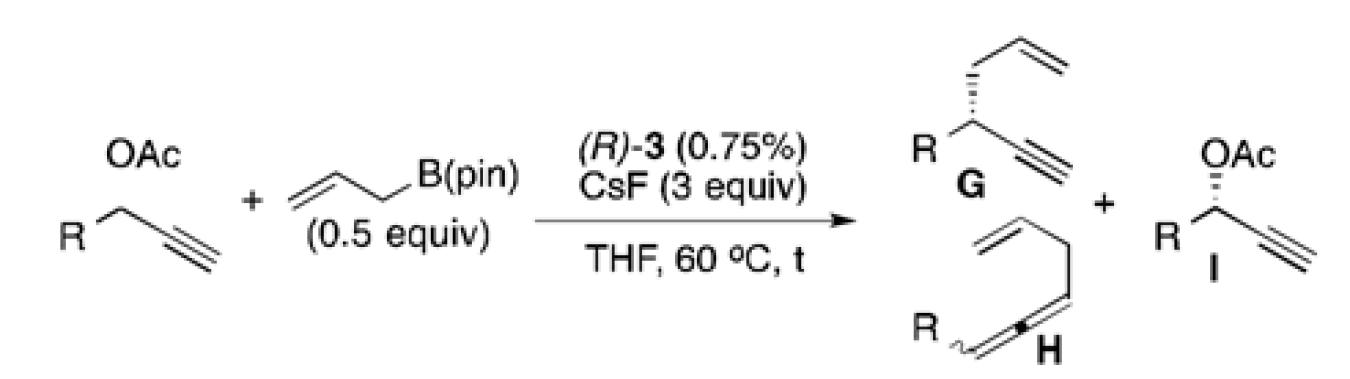
Having developed an active and selective catalyst system, the resolution of additional propargyl acetates was explored (Table 2). In addition to the n-alkyl example, the reaction tolerated aliphatic substrates with protected alcohols, giving similar selectivity for the benzyl protected substrate (entry 2) and an increased selectivity with the bulky TBDPS protected alcohol (entry 3). Pendant aromatic groups were tolerated; however, the selectivity appeared to be slightly sensitive to the proximity of the aromatic ring (entry 5). Diminished selectivity was also observed with an unsaturated R-group (entry 6). Use of the (S)-enantiomer of 3 (entry 8) yields the expected reversal of selectivity for both product and recovered substrate. Of note, aromatic propargyl acetates exhibited significantly enhanced reactivity (entries 9-14), and required only 1-2 hours for the resolution to proceed to completion. Although s values for the simple phenyl and p-tolyl substrate (entries 9 and 10) were similar to those of the aliphatic substrates, the reaction did appear sensitive to electronic perturbations to the aromatic ring (entries 11-14).
Table 2.
Substrate Scope of Kinetic Resolution
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Entry | R | t[h] | run | er G[a] | G:H [b] | er I[a] | Conv [%][c] |
s AVG [d] |
| 1 |
|
8 |
a
b |
88:12 89:11 |
>20:1 >20:1 |
77:23 82:18 |
41 45 |
13.5 |
| 2 |
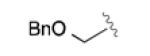
|
8 |
a
b |
87:13 88:12 |
>20:1 >20:1 |
72:28 74:26 |
37 39 |
11.1 |
| 3 |
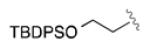
|
12 |
a
b |
n.d. n.d. |
>20:1 >20:1 |
74:26 75:25 |
36 36 |
26.3 |
| 4 |
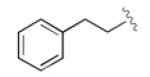
|
8 |
a
b |
n.d. n.d. |
>20:1 >20:1 |
79:21 84:16 |
45 49 |
10.4 |
| 5 |
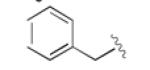
|
8 |
a
b |
85:15 83:17 |
>20:1 >20:1 |
61:39 63:37 |
24 29 |
6.5 |
| 6 [e] |
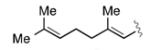
|
1.5 |
a
b |
83:17 82:18 |
90:10 90:10 |
70:30 77:23 |
42 46 |
7.4 |
| 7 |
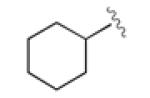
|
14 |
a
b |
90:10 90:10 |
>20:1 >20:1 |
62:38 64:36 |
23 26 |
11.6 |
| 8 [f] |
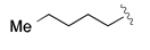
|
8 |
a
b |
14:86 12:88 |
>20:1 >20:1 |
13:87 15:85 |
50 48 |
14.5 |
| 9 [e] | Ph | 2 |
a
b |
86:14 85:15 |
92:8 92:8 |
78:22 88:12 |
44 52 |
11.5 |
| 10 [e] | p-tolyl | 2 |
a
b |
85:15 88:12 |
>20:1 >20:1 |
78:22 72:28 |
45 37 |
10.3 |
| 11 [e] | p-MeO | 1 |
a
b |
80:20 82:18 |
91:9 90:10 |
81:19 81:19 |
48 47 |
7.8 |
| 12 [e] | p-Cl | 2 |
a
b |
82:18 82:18 |
92:8 92:8 |
73:27 72:28 |
41 41 |
7.1 |
| 13 [e] |
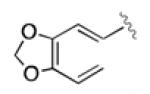
|
2 |
a
b |
84:16 84:16 |
91:9 91:9 |
75:25 82:18 |
42 48 |
9.4 |
| 14 [e] |
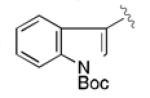
|
1.5 |
a
b |
76:24 68:32 |
90:10 91:9 |
79:21 88:12 |
53 67 |
5.0 |
Enantiomer ratios were determined by GC or SFC analysis on chiral stationary phase.
Determined by 1H NMR analysis.
Conv=[eeI/eeG+eeI)]
s=kfast/kslow=1n[(l-Conv/100)(l-eeI/100)]/ln[(l-Conv/100)(l+eeI/100)].
Run with 0.5 mol % (R)-3.
Run with (S)- 3.
The kinetic resolution by allyl-propargyl coupling also proved reliable on large scale, as the resolution of one gram of 1 shows a nearly identical s value and regioselectivity (Scheme 2, eq 3) as the example in Table 2. The ability to recover both enriched product as well as enriched starting material in good to high yield demonstrates the potential for efficient use of this resolution in total synthesis applications. In line with our previous study, the reaction was also tolerant of β-substituted allyl boron reagents: the selectivity and reactivity are nearly unchanged when methallylB(pin) is used to give the disubstituted 1,5-enyne 4 (Scheme 2, eq 4).
Scheme 2.

Further Synthetic Utility
To determine whether the selectivity of the resolution was derived during the oxidative addition or reductive elimination, kinetic isotope effects were studied (Scheme 3). A competition study between substrates 1 and d-1 showed a kH/kD of 0.71 based on recovered starting material.[20] The magnitude of this secondary inverse isotope effect is similar to what is expected for an sp to sp2 hybridization change,[21, 22] and suggests that oxidative addition is the rate- and stereochemistry-determining step of the reaction. In contrast, a normal secondary isotope effect might be expected if the later proposed 3,3′-reductive elimination were rate determining.
Scheme 3.
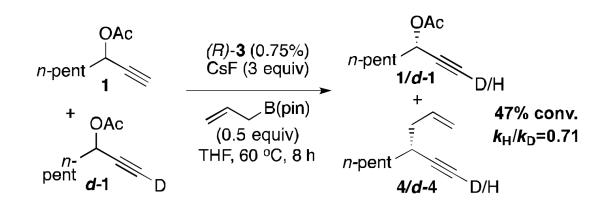
Kinetic Isotope Effect
To gain additional insight into the nature of possible stereodifferentiation during oxidative addition, density functional theory (DFT) calculations were carried out. The reaction coordinate was examined for the addition of (R)-3 to both the matched (R)- and mismatched (S)-enantiomers of methyl substituted propargyl acetate II (Scheme 5). In both cases, results show a strong stabilization upon formation of the Pd-alkyne complex (GS) from the free catalyst and substrate. This complex exhibits a slightly distorted square-planar geometry in which the alkyne is placed almost parallel to the P-Pd-P plane and the acetate group is directed away from the metal. This is in agreement with previous DFT calculations,[23] spectroscopic,[24] and crystallographic [25] data for Pd-alkyne complexes with phosphine ligands. Such a binding mode maximizes the orbital overlap required for back donation from the filled Pd d-orbitals into the C1≡C2 π* orbital. This back-bonding is strongly stabilizing and brings C1 and C2 in close proximity to the metal to cause a 30° distortion of the C1-H and C2-C3 bonds and form a structure that resembles a pallada(II)cyclopropene.[26]
From this structure, the calculated transition state shows bond formation between Pd and C3, with a slightly distorted trigonal bipyramidal geometry about the carbon characteristic of a SN2-type oxidative addition.[27] Natural Bond Order (NBO) analysis of the transition state confirms this feature, showing bonding character between the Pd and an sp2 hybridized C3 with concomitant loss of bonding character between C3 and the oxygen of the acetate. Such an addition pathway allows for direct formation of the η3-allenyl complex Int-a, which could isomerize to the more stable allenyl-Pd complex Int-b.
Calculated energy differences between both the ground and transition states for the diasteromeric matched and mismatched substrate/catalyst complexes are in good agreement with selectivities observed experimentally. Key to these observed energy differences are the interactions of the substituents at the propargylic carbon of the substrate and the ligand structure. In GSfast, the pentyl group is positioned into an open quadrant of the C2 symmetric ligand, away from the steric bulk of the pseudoaxial furyl ring (J), whereas in GSslow, the group is positioned into this blocked quadrant (K). This interaction is enhanced during oxidative addition (TSfast and TSslow) as C3 is brought closer to the catalyst, resulting in a 1.3 kcal/mol energy difference between the two transition states.
If stereodifferentiation is indeed derived during oxidative addition, the resolution should also be operative with nucleophiles other than allyl boronates. In this respect, it was considered that a kinetic resolution in a Pd-catalyzed reduction of propargyl electrophiles could allow access to optically enriched propargyl acetates utilizing a hydride as an inexpensive nucleophile. [28]
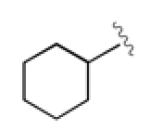
A survey of hydride sources revealed that tetramethylammonium triacetoxyborohydride (Me4NBH(OAc)3) was an ideal reagent for the desired transformation. This mild hydride source allowed use of catalyst 3 with little modification to reaction conditions (Table 3). The reaction required shorter reaction times, and showed s values similar to those with allylB(pin) as the nucleophile. Importantly, these reactions were clean and allowed facile isolation of the recovered enriched starting material in good to excellent yields. The reaction was especially efficient for aromatic (entries 1 and 2), and unsaturated (entry 5) substrates, and running these reactions to slightly higher conversion allowed for easy access to highly enriched substrate (entry 1c). The hydrogenolysis required longer reaction time for aliphatic substrates (entries 3 and 4), potentially suffering from slower oxidative additions or the formation of the non-conjugated allene byproduct. Synthetically useful selectivity could still be achieved for the more substituted cyclohexyl substrate (entry 4a).
Table 3.
Reduction of Propargyl Acetates
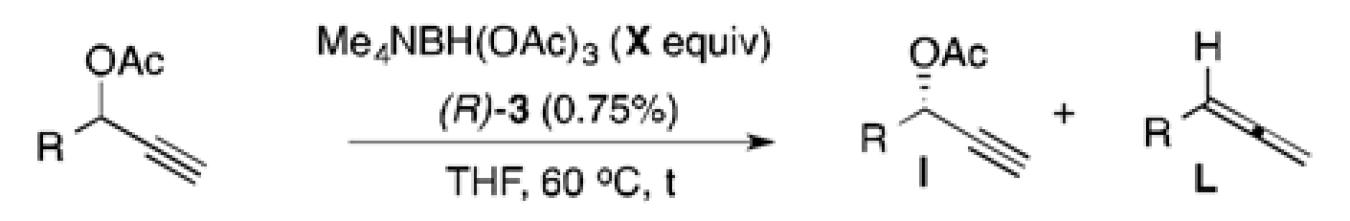
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Entry | R | t[h] | run | Equiv. X |
er I [a] | Conv [%][b] |
Yield I
[%][c] |
sAVG[d] |
| 1 |
|
0.75 |
a
b c[e] |
0.65 0.55 0.7 |
95:5 83:16 98:2 |
59 49 62 |
93 | 12.2 |
| 2 |
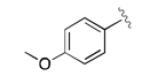
|
1 |
a
b |
0.5 0.5 |
80:20 76:24 |
49 45 |
89 | 7.8 |
| 3 |
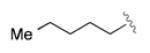
|
4.5 |
a
b |
0.6 0.6 |
85:15 78:22 |
54 44 |
71 | 9.4 |
| 4 [f] |
|
6.5 |
a
b |
0.75 0.75 |
92:8 79:21 |
55 42 |
88 | 14.9 |
| 5 |
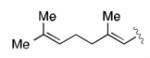
|
0.75 |
a
b |
0.5 0.5 |
80:20 77:23 |
49 46 |
90 | 7.1 |
Enantiomer ratios were determined by GC or SFC analysis on chiral stationary phase.
Determined by 1H NMR analysis
Yield based on theoretical 100% recovery at listed conversion, average of two or three runs
s=kfast/kslow=1n[(1-Conv/100)(1-eeI/100)]/1n[(1-Conv/100)(1+eeI/100)].
Run for 1 h
1.0% cat. loading.
In summary, a subtle difference in reactivity observed during the development of a stereospecific Pd-catalyzed cross-coupling of optically enriched propargyl acetates and allylmetal reagents has inspired the development of a kinetic resolution of racemic propargyl acetates to access enantioenriched 1,5-enyne products. This methodology obviates the need for optically enriched starting materials, and demonstrates previously unknown catalyst controlled selectivity in Pd-catalyzed reactions of propargyl electrophiles. Mechanistic studies of the resolution through laboratory and computational experimentation have evinced that oxidative addition could play the key stereodefining step. This understanding has allowed for the development of a hydrogenolysis to give entry to highly optically enriched propargyl acetates. In light of the number of developments in catalyst-controlled asymmetric manipulations of structurally similar allylic systems, we hope that the strategies described herein may find use in other contexts.
Experimental Section
General Procedure for Kinetic Resolution with allylboronic acid pinacol ester
An oven-dried vessle equipped with a magnetic stir bar was charged successively with catalyst 3, (0.75 mol%), THF [0.5M], the propargyl acetate 1 (1.0 equiv.), allylboronic acid pinacol ester (0.5 equiv), and cesium fluoride (3.0 equiv) in a dry-box under argon atmosphere. The vessel was sealed, removed from the dry-box, and heated to 60 °C while allowing to stir for 8 h. After this time, the reaction mixture was diluted with diethyl ether, filtered through a plug of silica gel and concentrated in vacuo.
General Procedure for Kinetic Resolution with Me4NBH(OAc)3
An oven-dried reaction vessel equipped with a magnetic stir bar was charged with catalyst 3, (0.75 mol%), tetramethylammonium triacetoxyborohydride (0.5 equiv), and THF [0.5M] in a dry-box under argon atmosphere. The vessel was sealed and allowed to stir for five minutes, over which time the solution turned a deep red color. The propargyl acetate, 1, (1.0 equiv) was added, and the vessel was sealed, removed from the dry-box, and heated to 60 °C while allowing to stir for 45 minutes. After this time, the reaction mixture was diluted with diethyl ether, filtered through a plug of silica gel and concentrated in vacuo.
Computational Details
All calculations were performed using Gaussian 09 with all geometry optimizations, energies and frequencies were calculated at the DFT level utilizing the B3LYP hybrid functional.[29, 30] The 6-31G** basis set was used for the elements C, H, P, and O in conjunction with the LANL2DZ relativistic pseudopotential for Pd. The two oxygens and carbonyl carbon of the acetate group were augmented with diffuse functions. All free energies were calculated at 333.15 K. The PCM model was used to estimate the effect of solvation (THF).[31] The frequency calculations for transition states demonstrated one imaginary frequency each, and were found to connected with the correct ground states through IRC calculations. NBO analysis was carried out with Gaussian NBO version 3.1.[32] The three-dimensional structures presented in Figure 1 were visualized utilizing CYLview.[33]
Figure 1.

DFT optimized structures, reaction coordinate, and simplified stereochemical model for oxidative addition (R)-3 into (R)- and (S)- substrate II. Energy values reported in kcal/mol relative to GSfast. Calculations performed at the B3LYP-PCM(THF)/LANL2DZ-6-31G**, some hydrogen atoms removed for clarity.
Supplementary Material
Acknowledgements
We thank Dr. Fredrik Haeffner for helpful discussions. MJA is grateful for American Chemical Society (DOC) and AstraZeneca Fellowships. This work was supported by a grant from the US National Institutes of Health (NIGMS 64451).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- 1.Detz RJ, Heimstra H, van Maarseveen JH. Eur. J. Org. Chem. 2009:6263. For a general review on catalytic propargylic substitutions, see: [Google Scholar]
- 2.(a) Tsuji J, Mandai T. Angew. Chem. Int. Ed. 1995;34:2589. For reviews on Pd-catalyzed reactions of propargylic systems see: [Google Scholar]; (b) Ma S, Zhang A. Pure Appl. Chem. 2001;73:337. [Google Scholar]; (c) Ma S. Eur. J. Org. Chem. 2004:1175. [Google Scholar]; (d) Guo L-N, Duan X-H, Liang Y-M. Acc. Chem. Res. 2011;44:111. doi: 10.1021/ar100109m. [DOI] [PubMed] [Google Scholar]
- 3.(a) Tsutsumi K, Ogoshi S, Kakiuchi K, Nishiguchi S, Kurosawa H. Inorg. Chim. Act. 1999;296:37. For a review on Pd-catalyzed cross-couplings of these systems, see: [Google Scholar]; (b) Condon-Gugenot S, Linstrumelle G. Tetrahedron, 2000;56:1851. [Google Scholar]; (c) Konno T, Tanikawa M, Ishihara T, Yamanaka H. Chem. Lett. 2000:1360. [Google Scholar]; (d) Molander GA, Sommers EM, Baker SR. J. Org. Chem. . 2006;71:1563. doi: 10.1021/jo052201x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yoshida M, Higuchi M, Shishido K. Tetrahedron, 2010;66:2765. [Google Scholar]; (f) S. Li J. Ye,, Ma S. Org. Lett. 2012;14:2312. doi: 10.1021/ol3007293. [DOI] [PubMed] [Google Scholar]
- 4.(a) Tsuji Y, Taniguchi M, Yasuda T, Kawamura T, Obora Y. Org. Lett. 2000;2:2635. doi: 10.1021/ol006160x. For some recent examples of Pd-catalyzed carbon-heteroatom bond formations in propargyl substitutions, see: [DOI] [PubMed] [Google Scholar]; (b) Nishioka N, Koizumi T. Tetrahedron Lett. 2011;52:3662. [Google Scholar]; (c) Kalek M, Stawinski J. Adv. Synth. Catal. 2011:353. [Google Scholar]; (d) Chen Z-S, Duan X-H, Wu L-Y, Ali S, Ji K-G, Zhou P-X, Liu X-Y, Liang Y-M. Chem. -Eur. J. 2011;17:6918. doi: 10.1002/chem.201100248. [DOI] [PubMed] [Google Scholar]
- 5.(a) Elsevier CJ, Stehouwer PM, Westmijze H, Vermeer PJ. J. Org. Chem., 1983;48:1103. [Google Scholar]; (b) Marshall JA, Wolf MA. J. Org. Chem. 1996;61:3238. [Google Scholar]; (c) Yoshida M, Gotou T, Ihara M. Tetrahedron Lett. 2004;45:5573. [Google Scholar]; (d) Yoshida M, Ueda H, Ihara M. Tetrahedron Lett. 2005;46:6705. [Google Scholar]
- 6.(a) Trost BM, Van Vranken DL. Chem. Rev. 1996;96:395. doi: 10.1021/cr9409804. For reviews on transition-metal catalyzed substitutions of allylic systems, see: [DOI] [PubMed] [Google Scholar]; (b) Lu Z, Ma S. Angew. Chem. Int. Ed. 2008;47:258. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- 7.(a) Elsevier CJ, Kleijn H, Boersma J, Vermeer P. Organometallics, 1986;5:716. [Google Scholar]; (b) Ogoshi S, Tsutsumi K, Kurosawa HJ. J. Organomet. Chem. 1995;493:C19. [Google Scholar]; (c) Baize MW, Blosser PW, Plantevin V, Schimpff DG, Gallucci JC, Wokcicki A. Organometallics, 1996;15:164. [Google Scholar]; (d) Tsutsumi K, Kawase T, Kakiuchi T, Ogoshi S, Okada Y, Kurosawa HJ. Bull. Chem. Soc. Jpn. 1999;72:2687. [Google Scholar]
- 8.(a) Keinan E, Peretz M. J. Org. Chem. 1983;48:5302. [Google Scholar]; (b) Elsevier CJ, Mooiweer HH, Kleijnm H, Vermeer P. Tetrahedron Lett. 1984;48:5571. [Google Scholar]; (c) Elsevier CJ, Vermeer PJ. J. Org. Chem. 1985;50:3042. [Google Scholar]; (d) Moriya T, Miyaura N, Suzuki A. Synlett, 1994:149. [Google Scholar]; (e) Dixneuf PH, Guyot T, Ness MD, Roberts SM. Chem. Comm. 1997:2083. [Google Scholar]; (f) Wang Y, Zhang W, Ma S. J. Am. Chem. Soc. 2013;135:11517. doi: 10.1021/ja406135t. [DOI] [PubMed] [Google Scholar]
- 9.(a) Marshall JA, Grant CM. J. Org. Chem. 1999;64:8214. doi: 10.1021/jo990938e. In mechanistically aligned chemistry, stereospecific addition of Pd into propargyl electrophiles, followed by subsequent transmetallation to In, Bi, Sn, Zn, and Yb forms new chiral metal-allenyl species. These nucleophiles can undergo stereospecific SN2′ additions to carbonyl compounds to give enantiomerically enriched propargyl alcohols, see: [DOI] [PubMed] [Google Scholar]; (b) Aurreocoechea JM, Arrare M, López B. Synlett, 2001:872. [Google Scholar]; (c) Arrate M, Durana A, Lorenzo P, de Lera ÁR, Álvarez R, Aurrecoechea JM. Chem. Eur. J. doi: 10.1002/chem.201301170. DOI:10.1002/chem.201301170. [DOI] [PubMed] [Google Scholar]
- 10.(a) Horváth A, Bächvall J. Chem. Comm. 2004:964. doi: 10.1039/b316482a. [DOI] [PubMed] [Google Scholar]; (b) Burks HE, Liu S, Morken JP. J. Am. Chem. Soc. 2007;129:8766. doi: 10.1021/ja070572k. [DOI] [PubMed] [Google Scholar]
- 11.Ardolino MJ, Morken JP. J. Am. Chem. Soc. 2012;134:8770. doi: 10.1021/ja302329f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Chem.- Eur. J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. For experimental and computational studies of the 3,3’-reductive elimination, see: [DOI] [PubMed] [Google Scholar]; (b) Cárdenas DJ, Echavarren AM. New J. Chem. 2004;28:338. [Google Scholar]; (c) Perez-Rodriguez M, Braga AAC, de Lera AR, Maseras F, Alvarez R, Espinet P. Organometallics, 2010;29:4983. [Google Scholar]
- 13.(a) Zhang P, Brozek LA, Morken JP. J. Am. Chem. Soc. 2010;132:10686. doi: 10.1021/ja105161f. For related allyl-allyl couplings thought to operate through a 3,3’-reductive elimination, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang P, Le H, Kyne RE, Morken JP. J. Am. Chem. Soc. 2011;133:9716. doi: 10.1021/ja2039248. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brozek LA, Ardolino MJ, Morken JP. J. Am. Chem. Soc. 2011;133:16778. doi: 10.1021/ja2075967. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Le H, Kyne RE, Brozek LA, Morken JP. Org. Lett. 2013;15:1432. doi: 10.1021/ol400088g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Kagan BH, Fiaud JC. Top. Stereochem. 1988;18:249. For some general reviews on kinetic resolutions, see: [Google Scholar]; (b) Vedejas E, Jure M. Angew. Chem. Int. Ed. 2005;44:3974. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 15.(a) Choi YK, Suh JH, Lee D, Lim IT, Jung JY, Kim MJ. J. Org. Chem. 1999;64:8423. doi: 10.1021/jo990956w. For selected examples of Pd-catalyzed resolutions of non-symmetric allylic systems, see: [DOI] [PubMed] [Google Scholar]; (b) Gais HJ, Bondarev O, Hetzer R. Tetrahedron Lett. 2005;46:6279. [Google Scholar]; (c) Mao B, Ji Y, Fañanás-Mastral M, Caroli G, Meetsma A. B. Feringa. Angew. Chem. Int. Ed. 2012;51:3168. doi: 10.1002/anie.201109075. [DOI] [PubMed] [Google Scholar]
- 16.(a) Tao B, Ruble JC, Hoic DA, Fu GC. J. Am. Chem. Soc. 1999;121:5091–5092. For nonenzymatic kinetic resolutions of propargyl alcohols, see: [Google Scholar]; (b) Tanaka K, Shoji T. Org. Lett. 2005;7:3561. doi: 10.1021/ol051335u. [DOI] [PubMed] [Google Scholar]; (c) Birman VB, Guo L. Org. Lett. 2006;8:4859. doi: 10.1021/ol061906y. [DOI] [PubMed] [Google Scholar]; (d) Li X, Jiang H, Uffman EW, Guo L, Zhang Y, Yang X, Birman VB. J. Org. Chem. 2012;77:1722. doi: 10.1021/jo202220x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Smith SW, Fu GC. J. Am. Chem. Soc. 2008;130:12645. doi: 10.1021/ja805165y. Fu and coworkers have reported a stereoconvergent Ni-catalyzed cross-coupling of racemic propargyl halides and carbonates with aryl zinc reagents, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Oelke AJ, Sun J, Fu GC. J. Am. Chem. Soc. 2012;134:2966. doi: 10.1021/ja300031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.s=(rate of fast reacting enantiomer)/(rate of slow reacting enantiomer); see ref 13a.
- 19.The discrepancies in selectivity for entries 11 and 12 was attributed to a background reaction that occurred with the Pd2(dba)3 system that might have led to an artificially high calculated s value for entry 11. This reaction was not seen with 3, and could have been attributed to the presence of dba or non-ligated palladium species.
- 20.The 1H-NMR ratio of 1:d-1 recovered from the reaction was compared to a sample of the 1:d-1 mixture prior to reaction. See supporting information for details on these experiments.
- 21.(a) Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. p. 429. We were unable to find experimental proof that the sp to sp2 hybridization experiences the same type of inverse secondary isotope effect as the sp2 to sp3 hybridization, however this might be expected based on trends in bond strength and stretching frequencies. This assumption has been made previously for kinetic isotope effects in deuterium labeled alkynes: see: [Google Scholar]; (b) Impaktschi J, Mohsseni-Ala J, Ihlig S. Eur. J. Inorg. Chem. 2003:4313. [Google Scholar]; (c) Arndt M, Salih KSM, Fromm A, Goossen LJ, Menges F, Niedner-Schatterburg G. J. Am. Chem. Soc. 2011;133:7428. doi: 10.1021/ja111389r. [DOI] [PubMed] [Google Scholar]
- 22.(a) Simmons EM, Hartwig JF. Angew. Chem. Int. Ed. 2012;51:3066. doi: 10.1002/anie.201107334. Our kinetic isotope labeling study does not preclude the possibility that oxidative addition could be a reversible step preceding a later rate-determining step. This phenomenon is known for primary isotope effects, however secondary equilibrium isotope effects are typically very small, see. [DOI] [PubMed] [Google Scholar]; (b) ref. 20a. pp. 432–444.
- 23.(a) Cui Q, Musaev DG, Morokuma K. Organometallics, 1998;17:1383. [Google Scholar]; (b) Ahlquist M, Fabrizi G, Cacchi S, Norrby P. Chem. Comm. 2005:4196. doi: 10.1039/b507784b. [DOI] [PubMed] [Google Scholar]
- 24.Schager F, Bonrath W, Pörschke K, Kessler M, Krüger C, Seevogel K. Organometallics, 1997;16:4276. [Google Scholar]
- 25.(a) Huh HS, Lee YK, Lee SW. J. Mol. Struct. 2006:209. [Google Scholar]; (b) Caeiro J, Peña D, Cobas A, Pérez D, Guitián E. Adv. Synth. Catal. 2006;348:2466. [Google Scholar]
- 26.A conformational analysis was performed in which the C2-C3 bond of the optimized ground state was rotated 360° by varying the Pd-C2-C3-O dihedral angle. This analysis confirmed that the GS shown was the lowest energy conformation about C2-C3. See supporting information for more details.
- 27.(a) Ogoshi S, Fukunishi Y, Tsutsumi K, Kurosawa H. J. Chem. Soc. Chem. Comm. 1995:2485. A similar alkyne coordination/SN2 oxidative addition pathway has been proposed by Kurosawa for the addition of Pt into propargyl chlorides, see: [Google Scholar]; (b) Kurosawa H. Pure Appl. Chem. 1998;70:1105. [Google Scholar]; (c) Kurosawa H, Ogoshi S. Bull. Chem. Soc. Jpn. 1998;71:973. [Google Scholar]
- 28.(a) Tsuji J, Mandai T. Synthesis, 1996:1. review: [Google Scholar]; (b) Tsuji J, Sugigura T, Yuhara M, Minami I. J. Chem. Soc., Chem. Commun. 1986:922. [Google Scholar]; (c) Tsuji J, Sugiuara T, Minami I. Synthesis, 1987;2:603. [Google Scholar]; (d) Mandai T, Matsumoto T, Kawada M, Tsuji J. Tetrahedron Lett. 1993;13:2161. [Google Scholar]; (e) Mandai T, Matsumoto T, Kawada M, Tsuji J. J. Organomet. Chem. 1994;473:343. [Google Scholar]; (f) Darcel C, Bartsch S, Bruneau C, Dixneuf PH. Synlett, 1994:475. [Google Scholar]; (g) Radinov R, Hutchings SD. Tetrahedron Lett. 1999;40:8955. [Google Scholar]; (h) Ohmiya H, Yang M, Yamauchi Y, Ohtsuka Y, Sawamura M. Org. Lett. 2010;12:1796. doi: 10.1021/ol100405k. [DOI] [PubMed] [Google Scholar]
- 29.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Gaussian, Inc.; Wallingford CT; 2009. [Google Scholar]
- 30.(a) Becke AD. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 31.(a) Miertus S, Scrocco E, Tomassi J. Chem. Phys. 1981;55:117. [Google Scholar]; (b) Barone V, Cossi M, Tomassi J. Chem. Phys. 1997;107:3210. [Google Scholar]
- 32.Glendening ED, Reed AE, Carpenter JE, Weinhold F. NBO Version 3.1 [Google Scholar]
- 33.Legault CY. CYLview, 1.0b. Université de Sherbrooke. 2009 http://www.cylview.org
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.