

Abstract
Objective
Early-onset epileptic encephalopathies have been associated with de novo mutations of numerous ion channel genes. We employed techniques of modern translational medicine to identify a disease-causing mutation, analyze its altered behavior, and screen for therapeutic compounds to treat the proband.
Methods
Three modern translational medicine tools were utilized: (1) high-throughput sequencing technology to identify a novel de novo mutation; (2) in vitro expression and electrophysiology assays to confirm the variant protein's dysfunction; and (3) screening of existing drug libraries to identify potential therapeutic compounds.
Results
A de novo GRIN2A missense mutation (c.2434C>A; p.L812M) increased the charge transfer mediated by N-methyl-D-aspartate receptors (NMDAs) containing the mutant GluN2A-L812M subunit. In vitro analysis with NMDA receptor blockers indicated that GLuN2A-L812M-containing NMDARs retained their sensitivity to the use-dependent channel blocker memantine; while screening of a previously reported GRIN2A mutation (N615K) with these compounds produced contrasting results. Consistent with these data, adjunct memantine therapy reduced our proband's seizure burden.
Interpretation
This case exemplifies the potential for personalized genomics and therapeutics to be utilized for the early diagnosis and treatment of infantile-onset neurological disease.
Introduction
Genomic research has identified an increasing number of human developmental and seizure disorders that are associated with mutations of subunits of the N-methyl-D-aspartate receptors (NMDARs).1–8 For example, mutations in the GluN2A subunit of the NMDARs have been associated with several childhood-onset epilepsy syndromes within the epilepsy-aphasia spectrum.6–8 These syndromes include benign epilepsy with centrotemporal spikes (BECTS), Landau-Kleffner syndrome (LKS), and epileptic encephalopathy with continuous-spike-and-waves-during-slow-wave-sleep (CSWSS).6–8 In contrast, other GluN2A mutations extend the range of phenotypes beyond disorders in the epilepsy-aphasia spectrum to include early-onset epileptic encephalopathy, which is characterized by severe infantile-onset epilepsy and lack of development.2,5
The NMDARs are ligand-gated ion channels that mediate a slow component of synaptic transmission in the central nervous system. The GluN1 and GluN2 subunits of NMDARs bind glutamate and glycine, respectively, leading to the opening of a cation-selective pore. This action results in a current flux with subsequent depolarization and increase intracellular Ca2+, which can lead to changes in synaptic strength.9 Currently, four GluN2 subtypes (A–D) have been identified. The four known GluN2 subtypes (A–D) have varying temporal and spatial expression patterns throughout the nervous system,10,11 and control key functional and pharmacological properties of synaptic NMDARs.12 The GluN2A mutations identified to date cause either haploinsufficiency of the GluN2A subunit or altered activity of the mutant-GluN2A-containing NMDAR complexes.1,2,6–8
Based on the observation of altered activity of mutant-GluN2A-containing NMDAR complexes, we hypothesized that understanding the alteration in activity and identification of a targeted therapy improves outcome. Hence, we report the clinical characteristics and genome-based therapy of a child with an early-onset epileptic encephalopathy due to a novel GluN2A missense mutation (NP_000824.1:p.L812M)13. When studied in heterologous expression systems, this GluN2A-L812M subunit had increased NMDAR activity that was inhibited by several channel blockers including memantine. A previously reported GRIN2A mutation was also evaluated in vitro using NMDA blockers with contrasting responses. Treatment of our proband with memantine improved his electroencephalogram (EEG) and reduced his seizure burden.
Materials and Methods
Patients and DNA samples
The NHGRI IRB reviewed and approved all clinical and laboratory protocols. Patients and family members who were enrolled in the clinical protocol gave written informed consent. Seizures were logged by parents and school teachers on a daily basis. Genomic DNA was extracted from peripheral whole blood using the Gentra Puregene Blood kit (Qiagen, Valencia, CA) per manufacturer's standards. The proband's examination and diagnostic evaluation included EEGs, electromyography/nerve conduction studies (EMG/NCV), magnetic resonance imaging (MRI) of the brain, buffy coat analysis, and skin and muscle biopsies.
Genotyping
Illumina (San Diego, CA) HumanOmni1-Quad genotyping arrays were run for the affected child, unaffected sister, and parents. Analyses, including homozygosity mapping, were carried out using Illumina GenomeStudio Software. SNP chip data were also used to verify exome sample IDs and parental identity for quality control purposes and to calculate sensitivity and specificity for genotype calling in exome sequence data.
Exome sequencing
Exome sequencing for this case was previously described13.
Neurophysiology and GRIN2A-L812M functional validation
For two-electrode voltage-clamp (TEVC) recordings, the cDNA for wild type human NMDA subunit GluN1-1a (hereafter GluN1) and GluN2A in pCI-neo were described previously14 (GenBank: NP_015566 and NP_000824). Site-directed mutagenesis was performed using the QuickChange protocol. Preparation of cRNA and the TEVC recordings from Xenopus laevis oocytes were performed as previously described15. The recording solution contained (in mmol/L) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, 0.01 EDTA (pH 7.4). The membrane potential was held at -40 mV in all oocyte experiments unless otherwise stated. EC50 values were obtained by fitting the concentration–response curve with
where N is the Hill slope.
Screening of FDA-approved NMDA receptor antagonists
FDA-approved drugs that might act as NMDA receptor open channel blockers were screened using TEVC recordings from oocytes coexpressing GluN1 with the wild-type or the mutant GluN2A. The concentration–effect curves were recorded at a holding potential of −40 mV and fitted with the equation:
where IC50 is the concentration of drug that produces a half-maximal effect, N is the Hill slope, and minimum is the degree of residual inhibition at a saturating concentration of drug.
Results
Clinical summary
The proband (UDP1130; Fig.1A) presented to the NIH Undiagnosed Diseases Program (UDP)16–18 at 6.5 years of age with a history of early-onset epileptic encephalopathy associated with profound cognitive impairment, absent motor development, and intractable seizures. An older sister was unaffected. His asymptomatic parents were non-consanguineous and of European and Hispanic descent. There was no family history of a similar condition.
Figure 1.
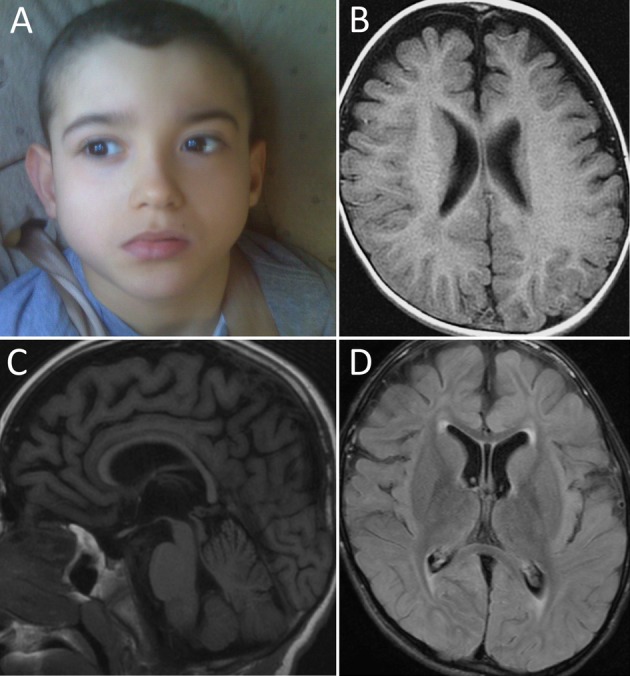
Proband and neuroimaging. (A) The proband was nondysmorphic. (B–D) MRI of the brain at 9 years of age (B-Axial T1, C-Sagittal T1, D-Axial FLAIR) showed progressive cerebral atrophy and a thin corpus callosum associated with subtle abnormalities of the white matter including hypomyelination of the terminal zones and the temporal lobes.
The proband was the full-term product of an uncomplicated pregnancy. He never visually tracked or made any purposeful movements. At 6 weeks of age, he had poor head control, axial hypotonia, and appendicular hypertonia. Parents noted frequent eye blinking movements and myoclonic jerks (not in clusters), which were sometimes associated with external stimuli. Neurological consultation at 7 months of age noted the above features and trace deep tendon reflexes. Brain MRI at the time reportedly showed enlarged frontal extra-axial spaces. At 2.5 years of age, the proband developed “near-constant flinging movements of his extremities”. His responses to stimuli included behavioral arrest, agitation, or laughter. Development was static at a neonatal level without further progression or regression.
The proband's epilepsy followed an intractable course. His first suspected seizure occurred at ∼2 months of age and was characterized as tonic extension of all extremities that lasted less than a minute. Between 2 and 18 months of age there were no documented seizures, but in retrospect, his parents feel that he was having frequent unrecognized episodes. His second definite seizure occurred at around 18 months of age and was described as having an initiating myoclonic jerk followed by tonic extension of all extremities, which evolved to slow rhythmic movements of the right arm and leg lasting for 5 min. The child was subsequently started on leviteracetam without efficacy. Prior to this time, the proband had frequent myoclonic jerks that were not treated. Seizures evolved to involve tonic flexion or extension of all extremities lasting minutes, with the myoclonic jerks continuing to occur alone or in association with these larger seizures. Over time, both types of episodes increased in frequency from monthly to a nearly daily frequency and were not responsive to numerous antiepileptic drug (AED) regimens (see Supplementary Materials).
The evolution of the probrand's EEG features paralleled his clinical course. An EEG at 2 months of age was reportedly normal. Another at 13 months of age identified a potential epileptic focus in the right cerebral hemisphere, as well as a single polyspike-like complex that correlated with a nocturnal episode of a whole body jerk with prominent trunk flexion. Therapy was not initiated because the treating physician was unsure that the episode was ictal. Over time, EEGs were reported to possess a diffusely slow and disorganized background with irregularly generalized or lateralized runs of spike and slow wave discharges (see Supplementary Materials). These EEGs were noted to only have very mild activation of the right-sided discharge during sleep and were without any features of electrical status epilepticus of sleep (ESES).
Upon initial examination at the NIH Clinical Center at 6.5 years of age, the patient did not visually track or exhibit any purposeful interaction with his environment. He was nondysmorphic and had axial hypotonia, appendicular hypertonia, and diffuse hyporeflexia. The child exhibited frequent random multifocal myoclonic movements of his extremities. Ophthalmological examination was normal. The patient was having near-daily seizures and was being treated with valproic acid. Over the next 2 years, rufinamide and lacosamide were added to his AED regimen with little improvement in seizure frequency.
Neuroimaging revealed widespread cerebral parenchymal volume loss, a thin corpus callosum, and abnormal myelination of the terminal zones and temporal lobes bilaterally. The cerebellum and brainstem were relatively spared (Fig.1B–D). These findings reflected progression compared to previous studies.
The proband's EEG was abnormal with slow activity of 3.5–4.5 hertz and 1–3 hertz delta and disorganized activity with intermittent frontally predominant irregular high amplitude 2.5-hertz spike-wave discharges that were greater on the right than left. The frequency of 2.5 hertz spike-wave discharges did not increase with sleep. More detailed information regarding the proband's phenotype and evaluation can be found in the Supplementary Materials.
Exome sequencing identifies a variant encoding the GluN2A p.L812M mutation
As previously reported,13 exome sequencing identified a novel de novo variant (NM_000833.3:c.2434C>A) in exon 13 of GRIN2A, which encodes the NMDAR subunit GluN2A (Fig.2A). This mutation resulted in a leucine to methionine substitution (NP_000824.1:p.L812M) in the linker region connecting the ligand-binding domain to the pore-forming transmembrane region of GluN2A (Fig.2B–C).19 This leucine is highly conserved across vertebrate species (Fig.2B).
Figure 2.
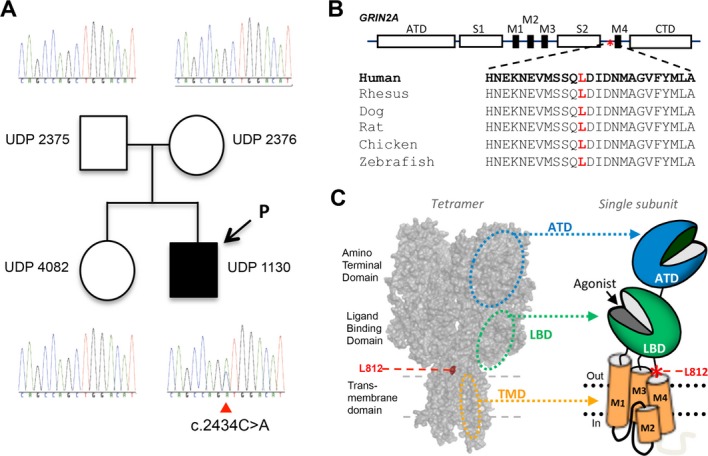
Genetic and protein changes of GRIN2A and GluN2A. (A) Family pedigree and genotypes reveal a de novo mutation (affected proband is indicated by arrow; parentage was confirmed by SNP array; data not shown). (B) Schematic representation of GluN2A subunit (asterisk notes the position of the L812M mutation). The L812 residue is highly conserved across vertebrate species. (C) A homology model of GluN2A subunit28 built from the GluA2 crystallographic data29 and shown as space fill. The red asterisk in the cartoon depicting the domain arrangement of an individual subunit shows the position of L812M in the linker region between the ligand-binding domain (S2, LBD) and the transmembrane domain (M4).
Functional analysis of mutant NMDA receptors
The pharmacological properties of this mutant receptor, GluN2A-L812M, were compared with wild-type GluN2A receptors using the Xenopus leavis oocyte expression system and TEVC recordings. We determined the concentrations of glutamate and glycine that can produce a half-maximal current response (EC50). Receptors containing GluN2A-L812M showed an eightfold reduction in EC50 (i.e., increased potency) for glutamate (0.42 μmol/L vs. 3.4 μmol/L of wild type) as well as glycine (0.14 μmol/L vs. 1.1 μmol/L of wild type), consistent with a previous study describing function of this residue in recombinant receptors at the single channel (Fig.3; Table1) (Yuan et al. in press). These data support the idea that GluN2A-L812M-containing NMDARs are overactive due to the increased activation at low concentrations of agonists. The resulting enhanced excitatory drive is consistent with the production of an epileptic phenotype. More extensive mechanistic studies of GluN2A-L812M-containing NMDARs are described elsewhere13.
Figure 3.
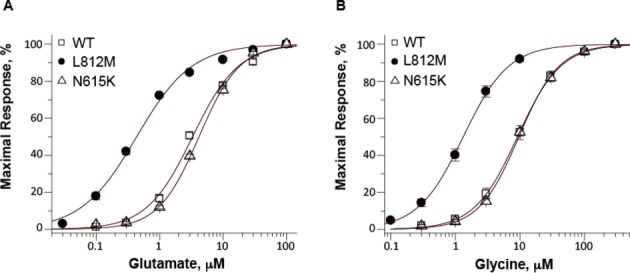
Functional analysis of GluN1/GluN2A receptors, GluN1/GluN2A-N615K receptors, and GluN1/GluN2A-L812M receptors. (A) The composite glutamate (in the presence of 100 μmol/L glycine) and (B) glycine (in the presence of 100 μmol/L glutamate) concentration-response curves indicate an increased agonist potency in GluN1/GluN2A-L812M compared to wild-type and GluN1/GluN2A-N615K NMDA receptors. Fitted EC50 values are given in μmol/L in the symbol legend.
Table 1.
Summary of agonist potency data.
| Parameters/constructs | WT | L812M | N615K |
|---|---|---|---|
| Glutamate – EC50, μmol/L (n) | 3.4 ± 0.3 (8) | 0.42 ± 0.02* (17) | 4.2 ± 0.2 (15) |
| Glycine – EC50, μmol/L (n) | 1.1 ± 0.2 (14) | 0.14 ± 0.01* (10) | 1.0 ± 0.1 (14) |
P < 0.05 compared to WT; one way ANOVA, Tukey post hoc.
We compared the above results with those for another GRIN2A mutation (p.N615K), previously associated with early-onset epileptic encephalopathy in a young girl.2 The GluN2A-N615K mutation resides near the apex of the pore-lining reentrant loop in the receptor subunit, a site known to strongly influence Mg2+ block.2,20 Interestingly, GluN2A-N615K had EC50 values that were unchanged when compared to wild-type receptors (Fig.3; Table1), but the mutation had dramatic effects on Mg2+ inhibition of NMDAR activity.2 Although, the L812M mutation caused a significant but incomplete reduction in Mg2+ sensitivity, the GluN2A-N615K mutation was reported to eliminate Mg2+ block at all physiologically relevant concentrations.2
Screening of FDA-approved NMDA-antagonists
Since conventional anticonvulsant medications did not provide adequate control of the proband's seizures, a number of FDA-approved NMDAR antagonists were evaluated for their ability to inhibit receptors containing GluN2A-L812M (Table2). Of these, memantine had been shown to exert anticonvulsant effects in a wide range of animal models of epilepsy,21 and had been used in children without toxicity.22,23 In vitro analysis revealed that memantine inhibited GluN2A-L812M-containing NMDARs with an IC50 of 12 μmol/L compared with 4.6 μmol/L for the wild type (at a holding potential of −40 mV) indicating that this compound can effectively reduce GluN2A-L812M-related NMDAR hyperactivity (Fig.4A; Table2). We also evaluated the inhibition of FDA-approved NMDAR channel blockers on currents arising from GluN2A-N615K-containing NMDARs. Interestingly, both dextromethorphan and its metabolite dextrorphan24 showed enhanced potency on N615K-containing NMDARs in comparison to wild-type NMDARs (twofold and 5.6-fold, respectively; Table2). These values are in stark contrast to the potency of these blockers at L812M (Table2), which is lower than that observed in WT receptors.
Table 2.
Screening FDA-approved NMDAR antagonists.
| Name | Class | WT | L812M | N615K |
|---|---|---|---|---|
| Memantine | AD | 4.6 ± 0.5 (15, 89%) | 12 ± 0.8 (10, 87%) | 43 ± 2.8 (16, 69%) |
| Amantadine | Antiviral | 110 ± 11 (10, 94%) | 113 ± 5.1 (8, 88%) | 458 ± 25 (8, 76%) |
| Dextromethorphan | Antitussive | 18 ± 2.4 (18, 88%) | 33 ± 4.8 (10, 91%) | 9.0 ± 1.3 (21, 92%) |
| Dextrorphan | 1.9 ± 0.3 (18, 92%) | 6.1 ± 1.3 (8, 85%) | 0.34 ± 0.06 (11, 96%) |
IC50, μmol/L (n, percent maximal inhibition at 100 μmol/L for memantine, at 1000 μmol/L for amantadine, at 300 μmol/L for dextromethorphan, and at 30 μmol/L for dextrorphan).
Figure 4.

Screening of NMDAR antagonists and personalized therapy. (A) Composite concentration-response curves for memantine inhibition of GluN1/GluN2A-L812M and wild-type current responses to 100 μmol/L glutamate and glycine determined by TEVC recordings from Xenopus oocytes. Memantine inhibits the wild-type and the mutant receptors with IC50 values of 4.6 μmol/L and 12 μmol/L, respectively. (B) Adjunct-AED treatment with memantine reduced seizure frequency. Seizures per week were compared against AED treatment. Lacosamide and rufinamide were fully weaned at weeks 50 and 58, respectively. Memantine was titrated up to its full dosage over weeks 50–55; valproate dosing remained unchanged. The average number of episodes and associated standard deviations over the full course of the study are represented by the labeled dashed lines.25 (C–D) Routine EEG segments before (C) and after (D) memantine treatment. Both demonstrate a poorly organized, diffusely slow background. However, the right frontal-predominant spike-wave discharges recorded in the prememantine EEG were not present in the postmemantine EEG.
Targeted therapy with memantine altered EEG appearance, decreased seizure frequency and allowed for tapering of conventional AEDs
On the basis of these findings, memantine was added as an adjunct medication to the proband's AED regimen at ∼9 years of age. Prior to this, the proband was being treated with lacosamide, rufinamide, and valproic acid without efficacy (mg/kg per day: lacosamide 11.25; rufinamide 45; valproic acid 25). Memantine dosage was titrated over several weeks and remained at a dosage of ∼0.5 mg/kg per day without side effects. Valproic acid dosing remained constant during memantine treatment, while lacosamide and rufinamide were removed during this period (Fig.4B).
Seizures on the child's previous AED regimen were frequent and logged by the parents; monitoring was continued after memantine initiation. Seizures consisted of two general types: one type involved tonic flexion of all extremities lasting several seconds to minutes, the other type consisted of brief unprovoked myoclonic jerks. Over a 54-week period prior to memantine treatment, the proband averaged 11.1 episodes per week (SEM = 0.5). Once the full memantine dosage was achieved, there was a decrease in average seizure frequency to 3.3 per week (SEM = 0.3). This decrease occurred within several weeks of reaching the full dose of memantine and has continued for over a year (averaged over the entire 116 week recorded period the seizure frequency was 7.0 per week [SEM = 0.5; Fig.4B])25 and almost all seizure activity occurred during sleep. Furthermore, the proband's myoclonic jerks ceased upon memantine treatment. His cognitive ability remained unchanged.
In parallel with the decrease in seizure frequency, there was improvement in the interictal EEG recordings. In comparison to his EEG before initiation of memantine (see Clinical summary), his postmemantine EEG still possessed a slow and disorganized background, but there were no asymmetries or epileptiform discharges during both wakefulness and sleep (Fig.4D). Cognitive ability has remained unchanged.
Discussion
The diagnosis and treatment of our patient's early-onset epileptic encephalopathy highlights three tools currently available for translational medicine: (1) the identification of a pathogenic mutation using high-throughput sequencing technology; (2) confirmation of the variant protein's dysfunction using in vitro expression and electrophysiology assays; and (3) identification of a potential therapeutic compound through screening of existing drug libraries. Specifically, we identified a de novo GRIN2A mutation that characterized the increased activity of NMDARs with which it was incorporated, and showed that memantine inhibited this hyperactivity in vitro. Completing this illustration of personalized medicine, we showed that initiation of memantine as an adjunct antiepileptic medication reduced seizure frequency and allowed for a reduction in the number of antiepileptic medications.25
GRIN2A-associated early-onset epileptic encephalopathy is a rare disorder, with only a few patients reported to date.2,5 Mutant NMDARs associated with this disorder underwent electrophysiological analysis to provide insight into their pathological mechanisms. Our proband's GRIN2A mutation (L812M) possessed increased intrinsic activity in response to agonists and decreased response to negative modulators, which translated into a charge transfer that was enhanced by over an order of magnitude13. In contrast, another child's early-onset epileptic encephalopathy was the result of a different GRIN2A mutation (N615K) acting via a different mechanism, the relief of Mg2+ blockade.2 Both of these cases of early-onset epileptic encephalopathy were the result of greatly increased current flow through mutant-GluN2A-containing NMDARs, leading to excessive excitatory drive, thereby inducing seizure activity and/or excitotoxicity.26 In our proband, this likely led to neuronal loss, motor dysfunction, cognitive impairment, and the other neurological features. Whether other cases of GRIN2A-associated early-onset epileptic encephalopathy (e.g., Pro522Arg mutation5) share these features requires further investigation.
The next step in our analysis involved screening these mutations with known inhibitors of NMDAR function to reduce activity. Interestingly, memantine was shown to antagonize L812M-associated hyperactivity, and consistent with this result, memantine treatment of our proband resulted in the substantial reduction in his seizure burden. Although this change may have had other causes (e.g., the placebo effect or the loss of the potentially seizure-provoking effects of rufinamide and lacosamide), our proband's persistent response (loss of lifelong myoclonic jerks and a reduction in seizure frequency) to over a year of treatment implies a memantine effect is the more likely possibility. Whether higher memantine dosages may further reduce his seizure burden needs to be determined; however, at this time, the pediatric safety guidelines of memantine dosing have limited our ability to test this hypothesis.
Our results show that other NMDAR antagonists (dextromethorphan and dextrorphan) possess enhanced potency at a different GluN2A mutation (N615K),2 and raise the possibility that treatment with either dextromethorphan or dextrorphan may have some efficacy in treating patients possessing the GluN2A-N615K or other mutations within the channel pore. The differential response of GluN2A-N615K and GluN2A-L812M-containing NMDARs to NMDAR antagonists is consistent with a previous report that dextromethorphan engages different structural determinants at its binding site in the pore than do other channel blockers.27 Together, these results indicate a need for specific electrophysiological evaluation of each mutant GRIN2A to evaluate its response to NMDAR antagonists.
These cases, with their infantile-onset and their dramatic effect on development, differ from GRIN2A-associated epilepsy-aphasia syndromes, such as BECTS, LKS or CSWSS.6–8 These disorders have periods of normal development prior to the development of epilepsy, and their associated encephalopathy is typically less severe. Interestingly, several families with these epilepsy–aphasia disorders were shown to possess GRIN2A missense mutations. Further electrophysiological characterization of these missense mutations may provide more insight into GRIN2A phenotype–genotype correlations, as well as additional therapeutic options for these disorders.
Finally, it is important to note that future evaluation of GRIN2A in patients with undiagnosed epileptic disorders, including early-onset epileptic encephalopathies, may expand the clinical phenotype associated with GRIN2A defects. High-throughput sequencing may be employed for genomic screening with subsequent electrophysiological studies and similar therapeutic screening strategies performed on mutated proteins. These strategies, especially in the context of early-onset encephalopathies, may dramatically improve a patient's overall development if instituted early. Eventually, patient-derived induced pluripotent stem cells could also serve as models for investigation (especially in GRIN2A-related cases due to haploinsufficiency6–8), and therapies identified in vitro can be applied in vivo, as described in this report.
Acknowledgments
We are grateful to Shannon McNeil, Chevalia Robinson, Jose Salas, and Cheryl Hipple for excellent administrative assistance; Mary Andriola, Eva Baker, Craig Blackstone, Joy Bryant, Barrington Burnett, Roxanne Fischer, Phuong Le, Kasper Hansen, Edwin Kolodny, Greg Pastores, Swati Sathe, David Sleat, Chigolum Umeonyido, Lissenka Vissers, Lynne Wolfe, and Jing Zhang for clinical and technical assistance and critical analysis. We especially thank the family of our patient for the loving care for their child and cooperation with our work. During this work T. M. P., D. A., T. M., K. F. F., D. R. S., C. F. B., and C. T. were supported by the National Institutes of Health (NIH) Undiagnosed Diseases Program; T. M., G. G., C. H., C. T., J. C. M., and W. A. G. were supported by the Intramural Research Program of the National Human Genome Research Institute of the National Institutes of Health. H. Y. was supported by NIH HSN268201300162P. S. T. was supported by NIH-NINDS NS036654. T. M. P. was also supported by the Cedars-Sinai Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Clinical data.
Ultrastructural analysis of (A) Buffy Coat and (B) Skin biopsies. Lysosomal storage is present in lymphocytes and skin cells (please note differences in scale).
References
- Reutlinger C, Helbig I, Gawelczyk B, et al. Deletions in 16p13 including GRIN2A in patients with intellectual disability, various dysmorphic features, and seizure disorders of the rolandic region. Epilepsia. 2010;51:1870–1873. doi: 10.1111/j.1528-1167.2010.02555.x. [DOI] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Araki Y, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabeux J, Kebir O, Gauthier J, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011;1:e55. doi: 10.1038/tp.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;20:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013;9:1073–1076. doi: 10.1038/ng.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke JR, Lal D, Reinthaler EM, et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet. 2013;9:1067–1072. doi: 10.1038/ng.2728. [DOI] [PubMed] [Google Scholar]
- Lesca G, Rudolf G, Bruneau N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. 2013;9:1061–1066. doi: 10.1038/ng.2726. [DOI] [PubMed] [Google Scholar]
- Lisman J, Raghavachari S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci STKE. 2006 doi: 10.1126/stke.3562006re11. :356. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, et al. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Akazawa C, Shigemoto R, Bessho Y, et al. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Zhang J, et al. Function analysis of a de novo GRIN2A missense mutation association with early-onset epileptic encephalopathy. Nat Commun. 2014;5:3251. doi: 10.1038/ncomms4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard M, Hansen KB, Andersen KT, et al. Molecular pharmacology of human NMDA receptors. Neurochem Int. 2012;61:601–609. doi: 10.1016/j.neuint.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Burgess MF, Zheng F, et al. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl WA, Tifft CJ. The NIH Undiagnosed Diseases Program: lessons learned. JAMA. 2011;305:1904–1905. doi: 10.1001/jama.2011.613. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Markello TC, Toro C, et al. The NIH Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson TM, Simeonov DR, Sincan M, et al. Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid 2-hydroxylase associated neurodegeneration. Eur J Hum Genet. 2012;20:476–479. doi: 10.1038/ejhg.2011.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder I, Borker P, Wollmuth LP. Specific sites within the ligand-binding domain and ion channel linkers modulate NMDA receptor gating. J Neurosci. 2010;35:11792–11804. doi: 10.1523/JNEUROSCI.5382-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Sakmann B. Adjacent asparagines in the NR2-subunit of the NMDA receptor channel control the voltage-dependent block by extracellular Mg2+ J Physiol. 1998;506(Pt 1):13–32. doi: 10.1111/j.1469-7793.1998.013bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemi M, Schachter SC. The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav. 2011;22:617–640. doi: 10.1016/j.yebeh.2011.07.024. [DOI] [PubMed] [Google Scholar]
- Chez MG, Burton Q, Dowling T, et al. Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J Child Neurol. 2007;22:574–579. doi: 10.1177/0883073807302611. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Posey DJ, Stigler KA, et al. A retrospective study of memantine in children and adolescents with pervasive developmental disorders. Psychopharmacology. 2007;191:141–147. doi: 10.1007/s00213-006-0518-9. [DOI] [PubMed] [Google Scholar]
- Küpfer A, Schmid B, Pfaff G. Pharmacogenetics of dextromethorphan O-demethylation in man. Xenobiotica. 1986;16:421–433. doi: 10.3109/00498258609050249. [DOI] [PubMed] [Google Scholar]
- Pujar S, Calvert S, Cortina-Borja M, et al. Statistical process control (SPC) – a simple objective method for monitoring seizure frequency and evaluating effectiveness of drug interventions in refractory childhood epilepsy. Epilepsy Res. 2010;91:205–213. doi: 10.1016/j.eplepsyres.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- LePage KT, Ishmael JE, Low CM, et al. Differential binding properties of [3H]dextrorphan and [3H]MK-801 in heterologously expressed NMDA receptors. Neuropharmacology. 2005;49:1–16. doi: 10.1016/j.neuropharm.2005.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acker TM, Yuan H, Hansen KB, et al. Mechanism for noncompetitive inhibition by novel GluN2C/D N-methyl-D-aspartate receptor subunit-selective modulators. Mol Pharmacol. 2011;80:782–795. doi: 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical data.
Ultrastructural analysis of (A) Buffy Coat and (B) Skin biopsies. Lysosomal storage is present in lymphocytes and skin cells (please note differences in scale).