

Abstract
A mild, catalytic, atom economical synthesis of imidazo[1,2-a]pyridines has been developed: catalytic PicAuCl2 in the presence of an acid produces a range imidazo[1,2-a]pyridines in good yield. This strategy is mild and forseen to be of particular use for the installation of stereogenic centers adjacent to the imidazo[1,2-a]pyridine ring without loss of enantiomeric excess.
Keywords: gold; alkyne; pyridine N-oxide; imidazo[1,2-a]pyridine; redox; atom economic
The possibility of forming an α-oxo gold carbenoid species through an oxygen transfer reaction using an alkyne and a nucleophilic oxide (initially a sulfoxide)[1] was first exploited in 2007 and this concept has subsequently been expanded: specifically one area in which this redox chemistry has been developed is the accommodation of pyridine N-oxides[2] which serve as the oxidant in the formation of the α-oxo gold carbenoid species (Scheme 1).[3] This reaction combination has utilized both intra- and intermolecular attack of the N-oxide moiety on the alkyne however in cases where the N-oxide has been utilized in an intermolecular reaction a stochiometric amount of pyridine waste is produced.[4] As an alternative, we envisaged a process in which 2-aminopyridine N-oxide would serve as the oxidant and be incorporated in the molecular complexity of the product through trapping the carbenoid species.[5] Imidazo[1,2-a]pyridines have recently had significant interest in the pharmaceutical industry as they exhibit antibacterial,[6] antifungal,[7] antiviral,[8] and anti-inflammatory properties.[9] The imidazo[1,2-a]pyridine ring system can be formed in many ways: typically they are formed via condensation of aminopyridines and α-haloketones[10] and can be formed by a one-pot condensation of an aldehydes, isonitrile and a 2-aminopyridine though this approach can require harsh conditions so limiting the scope of functionalities tolerated in this reaction.[11] More recently a number of copper catalysed methodologies have been developed, namely a three-component reaction of a 2-aminopyridine with an aldehyde and an alkyne,[12] a dehydrogenative coupling of a ketone with a 2-aminopyridine conducted at 40°C,[13] an oxidative cyclization of a halo-alkyne with an amino-pyridine at 60°C,[14] and an oxidative reaction between a ketone and a 2-aminopyridine carried out at 120°C.[15] Two further methods reported recently are worthy of note, the iron(III)-catalysed reaction between a nitro-olefin and an amino-pyridine achieved at 80°C,[16] and the silver-mediated oxidative coupling between terminal alkynes and a 2-aminopyridine conducted at 110°C.[17]
Scheme 1.

Precedent using an intermolecular pyridine N-oxide and the new concept for imidazo[1,2-a]pyridine synthesis using a 2-aminopyridine N-oxide.
The concept reported here offers an atom economic redox[18] process to imidazo[1,2-a]pyridines by employing the pyridine component both as a nucleophile, directly attacking the gold carbenoid intermediate, and it is involved in the regioselective formation these N-bridgehead heterocycles (Scheme 1, equation 2). Interestingly this combination of reagents appears to offer more flexibility in the choice of components in the reaction.[19] We foresaw that this new methodology might allows access to a range N-bridgehead heterocycles with chiral substituents which would otherwise be extremely difficult to obtain using the classical routes. To test our initial hypothesis and delineate reaction parameters, the reaction of 2-aminopyrdine N-oxide and 1-chloro-3-ethynylbenzene was investigated.
Gratifyingly our initial efforts identified the use of 1.2 equiv of pyridine N-oxide and 10 mol% of PicAuCl2 in dichloromethane at room temperature as suitable conditions for the reaction, giving a 43% conversion to the desired imidazo[1,2-a]pyridine 2 (entry 1 in Table 1). Addition of 1 equivalent of MsOH improved the conversion, with 71% of starting alkyne being converted to the desired product. Alternative acids such as TFA, HNTf2, acetic acid and p-nitrobenzoic acid were then tested and as previously noted, faster reaction was seen on lowering of the pKa of the acid.[2]
Table 1.
Evaluation of reaction parameters.
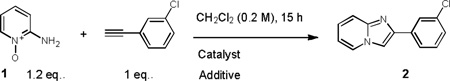 | |||
|---|---|---|---|
| Entry | Catalyst[a] | Additive | Conversion[b] |
| 1 | PicAuCl2 | None | 43 |
| 2 | PicAuCl2 | MsOH (1 eq.) | 71 |
| 3 | PicAuCl2 | TFA (1 eq.) | 70 |
| 4 | None | TFA (1 eq.) | none |
| 5 | PicAuCl2 | p-NO2PhCO2H (1 eq.) | none |
| 6 | PicAuCl2 | HNTf2 (1 eq.) | 72 |
| 7 | PicAuCl2 | TFA (0.5 eq.) | 54 |
| 8 | AuCl | TFA (1 eq.) | 33 |
| 9 | AuCl3 | TFA (1 eq.) | 53 |
| 10 | NHCAuNTf2 | TFA (1 eq.) | 36 |
| 11 | PPh3AuNTf2 | TFA (1 eq.) | 36 |
| 12 | PicAuCl2 | TFA (1 eq.)[c] | 89 (72)[d] |
10 mol % of catalyst was used.
Determined by correlation between HPLC-MS and crude 1H-NMR analysis using 3,5-dinitrobenzoic acid as internal standard.
The reaction mixture was heated at 40°C for 15h.
Isolated yield after column chromatography.
The acid appears to play a key role: this we believe increases the reaction rate by promoting the dissolution of the sparingly insoluble starting pyridine N-oxide. This fact was particularly highlighted in the case of p-nitrobenzoic acid (entry 5) where the formation of an insoluble salt resulted in no conversion to the product. The best conversion to product was achieved using MsOH or TFA as the acid (entries 2 and 3). On the other hand, no product was formed in the absence of gold catalysts (entry 4).[20] Interestingly slow formation of the desired compound was observed when 0.5 equivalent of TFA was used (entry 7), and therefore 1 equivalent of TFA was chosen for further studies. Dichloromethane proved to be the solvent of choice; toluene, chlorobenzene and 1,2-dichloroethane did not give any product and other solvents were screened but none produced a better conversion than dichloromethane.[21] The concentration of the reaction mixture appeared not to affect the reaction rate. Various alternative metal catalysts were screened during the optimisation of the catalyst: platinum and copper catalysts failed to produce any of the desired product[22] and gold(I) catalysts, such as AuCl and Ph3PAuNTf2 (entries 8 and 11), generally reacted sluggishly and produced many impurities. Finally, heating the reaction mixture to 40°C for 15 hours gave the desired product 2 in 72 % yield (89% conversion).
After establishing the optimal conditions we attempted to delineate the scope of the functionalities tolerated (Scheme 2).
Scheme 2.

Scope of reaction with substitution possible on all positions of the pyridine N-oxide and various terminal alkynes. The yields are determined after column chromatography or preparative HPLC.
Pleasingly, the reaction conditions were tolerant of various functional groups using alkyl-alkyne as well as aryl-alkyne substrates where electron rich and electron poor aryl alkynes give the desired imidazo[1,2-a]pyridine in good conversions. Predictably, the electron-poor substrates were the best, as the alkyne was more susceptible to nucleophilic attack from 2-aminopyridine N-oxide 1. Further heteroatom density was tolerated, with 3-ethynylthiophene affording the desired product 9 in good yield whereas we the reaction of 3-ethynylpyridine and 3-ethynylquinoline proceeded in lower yields presumably due to nitrogen coordination to the gold catalyst. We also noted that conversions were dependent on the sterics of the R group associated with the alkyne: 1° > 2° >> 3° carbon α to terminal alkyne; it should be noted that 1-ethynyl-2,4,6-trimethylbenzene and 3,3-dimethylbut-1-yne did not produce any product by crude 1H NMR analysis. Our preliminary data suggests that the pyridine reactant is tolerant of substitution in all positions with the 6-methylpyridine N-oxide giving a significantly poorer conversion to product when compared with the 3-methyl derivative. Our initial hypothesis that the efficient and mild reaction conditions would be applicable to chiral alkynes was validated by the formation of Boc-protected proline derivative 17 in good yield and as a single diastereoisomer. Unfortunately disubstituted alkynes did not produce any of the desired product.[23]
A mechanism for the formation of the imidazo[1,2-a]pyridine adducts is proposed in Scheme 3. Addition of the pyridine N-oxide to the alkyne is promoted by complexation to the gold catalyst. The resulting vinylgold intermediate can rearrange to form a gold-carbenoid intermediate.[24] This intermediate may form an α-chloro and α-mesylate ketones[25] that are known to be converted to imidazo[1,2-a]pyridine; however, neither of these α-functionalized ketones were detected in our investigations. In contrast, the mass consistent with the proposed pyridinium intermediate in Scheme 3 was identified by crude LCMS analysis of the reaction mixture. This species can be generated either by reaction of a gold carbenoid with a newly generated aminopyridine, or from direct rearrangement of a vinylgold precursor.[26] Finally, condensation of the amine and the generated ketone formed from the pyridinium intermediate furnishes the imidazo[1,2-a]pyridine.
Scheme 3.

Proposed mechanism for the formation of the imidazo[1,2-a]pyridine using gold catalysis.
In summary, we have developed a new, regioselective, atom economic synthesis of imidazo[1,2-a]pyridines using gold catalysis that has the potential to install stereogenic centres adjacent to the imidazo[1,2-a]pyridine ring without loss of stereochemical integrity. Many functional groups are tolerated in the reaction which can conducted under mild conditions either at room temperature or more optimally at reflux in dichloromethane: the reaction provides complex core targets quickly from the vast array of commercially available alkynes.
Experimental Section
Typical Procedure
TFA (0.016 ml, 0.22 mmol) was added to a solution of 2-aminopyridine N-oxide 1 (29.0 mg, 0.26 mmol) in CH2Cl2 (0.2 M). The reaction mixture was stirred for 10 min, then alkyne (0.027 ml, 0.22 mmol) and PicAuCl2 (8.7 mg, 0.022 mmol) were added. After stirring overnight at 40°C, Et3N (20µl) was added and the reaction mixture was concentrated under reduced pressure and purified by column chromatography to give pure imidazo[1,2-a]pyridine 2 (36.2 mg, 72%).
Supplementary Material
Acknowledgements
We are grateful to H. Douglas for analytical support. The authors thank the Novartis Education, Diversity and Inclusion (ED&I) office for a presidential postdoctoral fellowship (E.P.A.T.) and summer student funding (M. R.). F.D.T. thanks NIHGMS (RO1 GM073932) for financial support.
References
- 1.a) Shapiro ND, Toste FD. J. Am. Chem. Soc. 2007;129:4160–4161. doi: 10.1021/ja070789e. [DOI] [PubMed] [Google Scholar]; b) Witham CA, Mauleon P, Shapiro ND, Sherry BD, Toste FD. J. Am. Chem. Soc. 2007;129:5838–5839. doi: 10.1021/ja071231+. [DOI] [PubMed] [Google Scholar]
- 2.a) Ye L, Cui L, Zhang G, Zhang L. J. Am. Chem. Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ye L, He W, Zhang L. J. Am. Chem. Soc. 2010;132:8550–8551. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) He W, Li C, Zhang L. J. Am. Chem. Soc. 2011;133:8482–8485. doi: 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]
- 3.Xiao J, Li X. Angew. Chem., Int. Ed. 2011;50:7226–7236. doi: 10.1002/anie.201100148. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 4.a) Trost BM. Angew. Chem. Int. Ed. 1995;34:259–281. [Google Scholar]; b) Sheldon RA. Pure and Applied Chemistry. 2000;72:1233–1246. [Google Scholar]
- 5.For other approaches of “waste-free” carbene generation using gold: Johansson MJ, Gorin DJ, Toste FD. J. Am. Chem. Soc. 2005;127:18002. doi: 10.1021/ja0552500. Yeom H-S, Lee J-E, Shin S. Angew. Chem. Int. Ed. 2008;47:7040–7043. doi: 10.1002/anie.200802802. Yeom H-S, Lee Y, Jeong J, So E, Hwang S, Lee J-E, Lee SS, Shin S. Angew. Chem. Int. Ed. 2010;49:1611–1614. doi: 10.1002/anie.200906346. Jeong J, Yeom H-S, Kwon O, Shin S. Chem. Asian J. 2011;6:1977–1981. doi: 10.1002/asia.201100159. Murai M, Kitabata S, Okamoto K, Ohe K. Chem. Comm. 2012;48:7622–7624. doi: 10.1039/c2cc32628k. Yeom H-S, Lee Y, Lee J-E, Shin S. Org. Biomol. Chem. 2009;7:4744–4752. doi: 10.1039/b910757f. Mukherjee A, Dateer RB, Chaudhuri R, Bhunia S, Karad SN, Liu R-S. J. Am. Chem. Soc. 2011;133:15372–15375. doi: 10.1021/ja208150d.
- 6.Rival Y, Grassy G, Michel G. Chem. Pharm. Bull. 1992;40:1170–1176. doi: 10.1248/cpb.40.1170. [DOI] [PubMed] [Google Scholar]
- 7.Fisher MH, Lusi A. J. Med. Chem. 1972;15:982–985. doi: 10.1021/jm00279a026. [DOI] [PubMed] [Google Scholar]
- 8.Hamdouchi C, de Blas J, del Prado M, Gruber J, Heinz BA, Vance L. J. Med. Chem. 1999;42:50–59. doi: 10.1021/jm9810405. [DOI] [PubMed] [Google Scholar]
- 9.Rupert KC, Henry JR, Dodd JH, Wadsworth SA, Cavender DE, Olini GC, Fahmy B, Siekierka JJ. Bioorg. Med. Chem. Lett. 2003;13:347–350. doi: 10.1016/s0960-894x(02)01020-x. [DOI] [PubMed] [Google Scholar]
- 10.a) Stasyuk AJ, Banasiewicz M, Cyranski MK, Gryko DT. J. Org. Chem. 2012;77:5552–5558. doi: 10.1021/jo300643w. [DOI] [PubMed] [Google Scholar]; b) Gueiffier A, Lhassani M, Elhakmaoui A, Snoeck R, Andrei G, Chavignon O, Teulade JC, Kerbal A, Essassi EM, Debouzy JC, Witvrouw M, Blache Y, Balzarini J, De Clercq E, Chapat JP. J. Med. Chem. 1996;39:2856–2859. doi: 10.1021/jm9507901. [DOI] [PubMed] [Google Scholar]; c) Gueiffier A, Mavel S, Lhassani M, Elhakmaoui A, Snoeck R, Andrei G, Chavignon O, Teulade JC, Witvrouw M, Balzarini J, De Clercq E, Chapat JP. J. Med. Chem. 1998;41:5108–5112. doi: 10.1021/jm981051y. [DOI] [PubMed] [Google Scholar]; d) Hand ES, Paudler WW. J. Org. Chem. 1978;43:2900–2906. [Google Scholar]
- 11.a) DiMauro EF, Kennedy JM. J. Org. Chem. 2007;72:1013–1016. doi: 10.1021/jo0622072. [DOI] [PubMed] [Google Scholar]; b) Rousseau AL, Matlab P, Parkinson CJ. Tetrahedron Lett. 2007;48:4079–4082. [Google Scholar]; c) Groebke K, Weber L, Mehlin F. Synlett. 1998:661–663. [Google Scholar]; d) Adib M, Sheikhi E, Rezaei N. Tetrahedron Lett. 2011;52:3191–3194. [Google Scholar]
- 12.a) Guchhait SK, Chandgude AL, Priyadarshani G. J. Org. Chem. 2012;77:4438–4444. doi: 10.1021/jo3003024. [DOI] [PubMed] [Google Scholar]; b) Chernyak N, Gevorgyan V. Angew. Chem. Int. Ed. 2010;49:2743–2746. doi: 10.1002/anie.200907291. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mishra S, Ghosh R. Synthesis. 2011:3463–3470. [Google Scholar]; d) Reddy BVS, Reddy PS, Reddy YJ, Yadav JS. Tetrahedron Lett. 2011;52:5789–5793. [Google Scholar]; e) Liu P, Fang LS, Lei X, Lin GQ. Tetrahedron Lett. 2010;51:4605–4608. [Google Scholar]
- 13.Cai Z-J, Wang S-Y, Ji S-J. Adv. Synth. Catal. 2013;355:2686–2692. [Google Scholar]
- 14.Goa Y, Yin M, Wu W, Huang H, Jiang H. Adv. Synth. Catal. 2013;355:2263–2273. [Google Scholar]
- 15.Bagdi AK, Rahman M, Soutra S, Majee A, Hajra A. Adv. Synth. Catal. 2013;355:1741–1747. [Google Scholar]
- 16.Santra S, Bagdi AK, Majee A, Hajra A. Adv. Synth. Catal. 2013;355:1065–1070. [Google Scholar]
- 17.He C, Hoa J, Xu H, Mo Y, Liu H, Han J, Lei A. Chem. Comm. 2012;48:11073–11075. doi: 10.1039/c2cc35927h. [DOI] [PubMed] [Google Scholar]
- 18.Burns NZ, Baran PS, Hoffman RW. Angew. Chem. Int. Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 19.Scifinder identified 74218 commercially available alkynes compared with a figure just less than 6200 for commercially available α-bromo or α-chloroketones. Date of the search: 14th of October 2013
- 20.a) Chen D-F, Han Z-Y, He YP, Yu J, Ging L-Z. Angew. Chem. Int. Ed. 2012;51:12307–12310. doi: 10.1002/anie.201205062. [DOI] [PubMed] [Google Scholar]; b) Graf K, Rühl CL, Rudolph M, Rominger F, K. Hashmi AS. Angew. Chem. Int. Ed. 2013 doi: 10.1002/anie.201304813. [DOI] [PubMed] [Google Scholar]
- 21.Formation of the desired product was also observed with EtOAc, THF and nitromethane in moderate yield.
- 22.Other catalyst were tested such as PtCl2, CuOTf2, CuOTf without success
- 23.Diphenyl acetylene and diethylacetylene were used without success.
- 24.Benitez D, Shapiro ND, Tkatchouk E, Wang Y, Goddard WA, Toste FD. Nature Chem. 2009;1:482–486. doi: 10.1038/nchem.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He W, Xie L, Xu Y, Xiang J, Zhang L. Org. Biomol. Chem. 2012;10:3168–3171. doi: 10.1039/c2ob25235j. [DOI] [PubMed] [Google Scholar]
- 26.Noey EL, Luo Y, Zhang L, Houk KN. J. Am. Chem. Soc. 2012;134:1078–1084. doi: 10.1021/ja208860x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.