

Graphical abstract
Keywords: Lipopolysaccharide, Kdo, Ko, Oligosaccharide synthesis, Acinetobacter
Highlights
-
•
Synthesis of Acinetobacter LPS fragments with orthogonal protecting pattern.
-
•
First synthesis of Ko-glycosides substituted at position 5.
-
•
α-Selective glucosylation using benzylidene trifluoroacetimidate donor.
-
•
Regioselective phosphorylation at C-6 of a glucosyl substituent.
Abstract
The α-d-glucopyranosyl-(1→5)-substituted methyl glycosides of 3-deoxy-α-d-manno-oct-2-ulosonic acid (Kdo), 3-deoxy-α-d-lyxo-hept-2-ulosonic acid (Kdh), and d-glycero-α-d-talo-oct-2-ulosonic acid (Ko) were prepared using orthogonally protected glycosyl acceptor derivatives via glycosylation with a torsionally disarmed 4,6-O-benzylidene protected trifluoroacetimidate glucosyl donor followed by global deprotection. The related 6-O-phosphoryl-α-d-glucopyranosyl-(1→5)-substituted Kdo and Kdh derivatives were derived from a benzylidene-protected glucosyl intermediate using phosphoramidite and phosphoryl chloride-based phosphorylation steps, respectively. The deprotected disaccharides serve as ligands to study lectin binding of Acinetobacter lipopolysaccharide core oligosaccharides.
1. Introduction
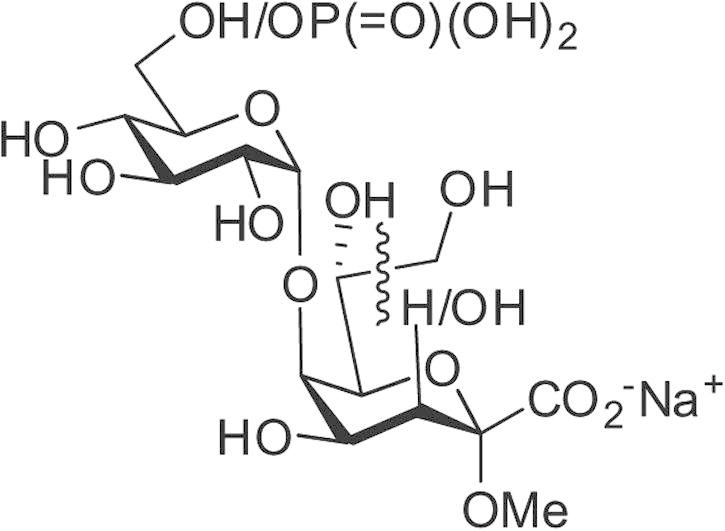
Bacteria of the genus Acinetobacter have increasingly been implicated in nosocomial infections which are difficult to eradicate due to resistance against major antimicrobial drugs.1 Lipopolysaccharide (LPS)—located in the outer leaflet of the bacterial cell membrane—is a major virulence factor contributing to bacterial evasion of adaptive and innate immune responses.2 A general but not exclusive architecture of LPS comprises a bisphosphorylated acylated diglucosamine backbone, termed lipid A, a core region and an antigenic polysaccharide which is the main target of specific antibodies allowing the distinction of O-serotypes and which is therefore called O-polysaccharide or O-antigen.3 The first sugar connecting the core region and the lipid A is—in general—3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) but it may be partially replaced by the isosteric, acid-stable 3-hydroxy-derivative d-glycero-d-talo-oct-2-ulosonic acid (Ko).4, 5, 6, 7 In LPS from Acinetobacter haemolyticus NCTC 10305 in particular, a large fraction (∼80%) contains Ko instead of Kdo (∼20%), providing the linkage to the lipid A. In addition, the core oligosaccharide contains several α- and β-configured glucosyl as well as two 3-deoxy-d-lyxo-hept-2-ulosaric acid (Dha) residues (Fig. 1).
Figure 1.

Structure of the deacylated inner core LPS fraction from Acinetobacter haemolyticus NCTC 10305.6
Previously, a 28 kDa murine serum protein has been described which binds to the inner core region of LPS from this Acinetobacter strain but also to oligosaccharides containing l-glycero-d-manno-heptosyl-Kdo units.8, 9, 10 This “serum factor” has only recently been recognized as mannose binding lectin-A (MBL-A). Most notably this MBL binds to isolated LPS oligosaccharides with an unusual high affinity for the individual binding site (with ELISA IC50 values in the mid to low nanomolar range).11 Extending our previous studies detailing the interaction of the core region with antibodies and lectins12, 13, 14, 15, we have set out to prepare a first series of fragments of the Acinetobacter haemolyticus NCTC 10305 inner core region in order to define the binding epitope at the molecular level. Since Smith degradation of the isolated LPS core resulted in ligands which were still bound by the lectin with similar affinity5, non-natural disaccharides containing 3-deoxy-d-lyxo-hept-2-ulosonic acid (Kdh) have been prepared in addition. The synthetic oligosaccharides serve as ligands in forthcoming binding and STD NMR studies with C-type lectins such as human lung surfactant protein D and mannose-binding lectins.
2. Results and discussion
2.1. Preparation of the disaccharides α-Glc-(1→5)-α-Kdo/Kdh
The glycosides were designed as methyl (Me) glycosides since the single 1H NMR signal of the Me group serves as a suitable reference for integration of STD effects in binding studies. Thus the previously reported Me α-Kdo glycoside 216 (easily available via the scalable reaction of the peracetate 1 with MeOH, catalyzed by Dowex (H+) ion-exchange resin) was employed for the preparation of the Kdo glycosyl acceptor derivative. The exocyclic 7,8-O positions of 2 were protected by a 1,1,3,3-tetraisopropyldisiloxane-1,3-diyl (TIPDS) group in order to utilize its arming effect and to provide orthogonality of fluoride-induced silyl deprotection.17 Thus, by using imidazole and TIPDSCl2 in DMF at −40 °C the bis-silyl ether derivative 3 was obtained in 68% yield (Scheme 1). In order to generate an option for eventual attachment of the lateral Kdo unit (Fig. 1) and to also activate the 5-OH group for subsequent glycosylation, a p-methoxybenzyl group (PMB) was installed at position 4. The diol derivative 3 was first reacted with dibutyltin oxide to give the intermediate stannylene derivative followed by treatment with PMB-chloride/tetrabutyl ammonium iodide/DMF in toluene. As previously reported, the activated stannylene acetal also induced formation of the corresponding PMB ester 5 and the 1.5-lactone 6 as by-products.18 Treatment of the mixture with sodium methoxide, however, eventually afforded the methyl ester derivative 4 in 70% yield.
Scheme 1.

Reagents and conditions: (a) TIPDSCl2, 1H-imidazole, DMF, −40 °C, 68%; (b) Bu2SnO, toluene, reflux, then DMF, PMBCl, Bu4NI, toluene, 60 °C; mixture of 4, 5 and 6; then (c) 0.1 M NaOMe, MeOH, rt, 70% for 4.
Previously, an α-(1→5)-linked glucosyl residue had been coupled to Kdo using an acetylated 2-O-benzyl thioglycoside donor under promotion with DMTST in 85% yield.19 The use of the known20 perbenzylated N-phenyl trifluoroacetimidate donor (NPTFA) 7α/β for the TMSOTf-catalyzed glycosylation of 4, however, resulted in decreased anomeric selectivity and a temperature dependent outcome.21, 22 Glycosylation of acceptor 4 promoted by 10% TMSOTf at −5 °C in CH2Cl2 afforded the α-(1→5)-linked disaccharide 8α as the major anomer (α/β ratio 2.4:1) accompanied by formation of the alcohols 9α and 9β (resulting from cleavage of the acid-labile PMB group). By lowering the temperature to −20 °C and reducing the amount of promoter (5% TMSOTf) the PMB cleavage could be largely suppressed. However, under these conditions, the undesired 8β was formed preferentially (α/β ratio 1:1.4). The change of the solvent to CH2Cl2/Et2O (9:1) resulted in a sluggish reaction with slightly enhanced α/β ratio (1.7:1), albeit in poor yield. Presumably, the formation of an intermediate α-anomeric triflate at a lower temperature leads to an increased contribution of an SN2-type glycosylation pathway.23 Alternatively, the torsionally disarmed 4,6-O-benzylidene trifluoroacetimidate donor 10 was prepared from its hemiacetal precursor24 as a separable anomeric mixture.25 The glycosylation of acceptor 4 with donor 10 in CH2Cl2 at −5 °C provided the disaccharide 11 in 80% yield as the α-anomer only, irrespective of the anomeric configuration of the donor (Scheme 2). Due to milder reaction conditions (lower temperature and promoter concentration) PMB cleavage could be completely suppressed without affecting the stereochemical outcome. The PMB group of the α-linked disaccharide derivative 8α was selectively removed by treatment with trifluoroacetic acid (TFA) affording 9α, followed by cleavage of the silyl ether group in order to generate the triol derivative 12. Treatment of 9α with tetrabutylammonium fluoride (TBAF) produced compound 12 in near quantitative yield, which was then fully deprotected by catalytic hydrogenation followed by alkaline hydrolysis of the methyl ester group to furnish disaccharide 13 as sodium salt in 90% yield. Global deprotection of the benzylidene protected disaccharide 11 provided disaccharide 13 in an excellent overall yield of 65% (based on acceptor 4). TFA-treatment of 11 simultaneously cleaved the benzylidene and the PMB group, respectively, to give triol 14 in 88% yield, followed by TIPDS removal and ensuing full deprotection of the resulting derivative 15 to give disaccharide 13.
Scheme 2.

Reagents and conditions: (a) TMSOTf, CH2Cl2, molecular sieves 4 Å, −5 °C, 34% for 8α, 14% for 8β, 15% for 9α, 7% for 9β, 80% for 11; (b) 99% TFA, CH2Cl2, 0 °C, 83% for 9α, 88% for 14; (c) TBAF, THF, rt, 98% for 12, 97% for 15; (d) H2 (1 atm), 10% Pd–C, MeOH, then 0.01 M aq NaOH, rt, 90% for 12→13, 95% for 15→13, 79% for 16→17; (e) NaIO4–SiO2, CH2Cl2, −10 °C, then NaBH4, MeOH, 0 °C, 34%.
In addition, the side-chain shortened analog was prepared by oxidative cleavage of the exocyclic diol of 12 with sodium metaperiodate generating the heptulosonic glycoside 16. The intermediate aldehyde formed upon oxidation could be analyzed by NMR, but proved to be rather labile upon attempted purification on silica gel. The ensuing borohydride reduction was accompanied by concomitant ester reduction. The reaction was therefore not allowed to run until completion, but was stopped when ester reduction was first detected on TLC. Global deprotection of 16 eventually afforded disaccharide 17 in 79% yield.
2.2. Preparation of the disaccharides α-Glc6P-(1→5)-α-Kdo/Kdh
Selective cleavage of the PMB-group of the benzylidene-protected disaccharide derivative 11 via DDQ-promoted oxidation afforded the glycosyl acceptor 18 (suitable for attachment of the lateral Kdo unit) in 83% yield (Scheme 3). The orthogonal protecting group pattern also allows for the selective introduction of the 6-phosphoester group as well as for additional glucosyl extension at position 4 of the glucosyl unit upon reductive opening of the benzylidene group (cf. Fig. 1). Reductive opening of the benzylidene toward the 6-OH derivative 19, however, met with difficulties when applying various combinations of Lewis acids and hydride sources (CoCl2/BH3.THF, TMSCl/NaCNBH3, TMSOTf/BH3, Bu2BOTf/BH3·THF). Lack of reactivity, loss of the PMB group or cleavage of the benzylidene group with formation of additional by-products were observed under these conditions. A modest conversion of compound 11 into the 4-O-benzyl ether 19 could eventually be accomplished in the presence of PhBCl2/Et3SiH in 43% yield. Thus, it was envisaged to introduce the 6-O-phosphate group at the 4,6-diol intermediate, with the additional option to utilize the remaining 4-hydroxyl group as an acceptor site for future extension by glucosyl residues. Hence, the benzylidene group of 11 was selectively removed—without cleavage of the PMB group—using p-toluenesulfonic acid in dry MeOH to afford the diol 20 in 91% yield. Short reaction times at 40 °C gave better and reproducible yields compared to prolonged reaction times at ambient temperature. Phosphorylation of the diol 20 was first approached using the phosphoramidite chemistry. Thus, treatment of 20 with dibenzyl N,N-diisopropylphosphoramidite/1H-tetrazole in the presence of ground molecular sieves (4 Å) in CH2Cl2 followed by oxidation with mCPBA gave the 6-O-phosphotriester derivative 21 in 67% yield and the corresponding 4-O-regioisomer 22 (10%). The phosphoramidite protocol also proved to be appropriate for the phosphorylation of the triol 14 and furnished the 6-O-phosphotriester derivative 25 in 63% yield. The assignment of the phosphate substitution at O-6 was based on the downfield shift of the 1H NMR signals of the H-6 protons and the 13C–31P coupling interaction leading to splitting of 13C NMR signals of C-6 and C-5, respectively. To increase the selectivity in the phosphorylation step, a more reactive phosphoryl halide was used at a low temperature. Indeed, reaction of the diol 20 with diphenyl phosphoryl chloride at 0 °C in the presence of 4-N,N-dimethylaminopyridine afforded the corresponding 6-O-phosphotriester derivative 23 in 96% yield.
Scheme 3.

Reagents and conditions: (a) DDQ, CH2Cl2/MeOH (3:1), rt, 83%; (b) PhBCl2, Et3SiH, CH2Cl2, −70 °C, 43%; (c) pTosOH, MeOH, 40 °C, 91%; (d) (BnO)2PN(iPr)2, 1H-tetrazole, CH2Cl2, −5 °C/0 °C, molecular sieves 4 Å, then mCPBA, 67% for 21, 10% for 22, 63% for 25; (e) (PhO)2P( O)Cl, DMAP, CH2Cl2, molecular sieves 4 Å, 0 °C, 96%; (f) Ac2O, pyr., DMAP, 0 °C, 96%; (g) TFA, CH2Cl2, 0 °C, 88%; (h) 1 M TBAF, THF, rt, 92%; (i) H2 (1 atm), 10% Pd–C, MeOH, then 0.01 M aq NaOH, rt, 98%.
TFA-treatment of the dibenzylphosphate derivative 21 proved to be selective for the removal of the PMB group and gave the diol derivative 25 in 88% yield. Subsequent reaction of 25 with TBAF afforded the tetraol derivative 26 (92%), which was subjected to hydrogenation on 10% Pd–C followed by alkaline hydrolysis to furnish the target disaccharide phosphate 27 as sodium salt in 98% yield. Deprotection of the silyl group of diphenylphosphate 23, however, required a modified protocol, since treatment with TBAF resulted in fluoride-induced nucleophilic displacement of the phenoxy groups on the phosphoester.26 The 4-OH group of 23 was thus acetylated to give compound 24 in order to prevent intramolecular phosphate migration or cyclization, respectively. The silyl ether groups of 24 were then removed by the action of triethylamine trihydrogen fluoride (TREAT), which afforded 28 in 82% yield with only minor (∼10%) phosphate cleavage (Scheme 4). Compound 28 was used for the preparation of the side-chain-shortened analog 29 by periodate oxidation (36%) followed by successive hydrogenolysis on Pd-C and PtO2, and saponification to afford the heptulosonic glycoside 30 in 52% yield. The poor yield was due to formation of an unidentified side product, which had to be separated on a HILIC column. Alternatively, dibenzylphosphate 26 was subjected to the oxidative degradation protocol affording 31 (47%), which reacted cleanly to target compound 30 upon hydrogenolysis with Pd–C and subsequent saponification.
Scheme 4.

Reagents and conditions: (a) TREAT, CH2Cl2, rt, 82%; (b) NaIO4–SiO2, CH2Cl2, 0 °C → rt, then NaBH4, MeOH, 0 °C, 36% for 29, 47% for 31; (c) H2 (1 atm), 10% Pd–C, MeOH, then H2 (1 atm), PtO2, MeOH, then 0.01 M aq NaOH, rt, 52%; (d) H2 (1 atm), 10% Pd–C, MeOH, then 0.01 M aq NaOH, rt, 87%.
2.3. Preparation of the disaccharide α-Glc-(1→5)-α-Ko
The previously reported intermediate 32 was employed for the preparation of a suitably protected methyl glycoside of d-glycero-α-d-talo-oct-2-ulopyranosylonic acid (Ko).27 The epimeric 3-O-acetate 32 was subjected to base-induced anomeric methylation followed by de-O-acetylation to produce 33. Ensuing Dess–Martin periodinane oxidation and reduction with ammonia–borane complex gave the alcohol 34 in 65% overall yield (Scheme 5). Reaction of 34 with benzyl bromide/NaH in DMF afforded the 3-O-benzyl derivative 35 in 94% yield. The reaction had to be performed in high dilution at 0 °C in order to prevent formation of the corresponding benzyl ester derivative. Next, the 4,5-O-isopropylidene ketal was selectively cleaved by the action of p-toluenesulfonic acid in wet acetone under equilibrating conditions.
Scheme 5.

Reagents and conditions: (a) NaH, MeI, DMF, 0°C, then NaOMe, rt, 86%; (b) Dess–Martin reagent, CH2Cl2, rt, then NH3·BH3, MeOH, 0 °C, 75%; (c) NaH, BnBr, DMF, 0 °C, 94%; (d) pTosOH, wet acetone, rt, 71%; (e) Bu2SnO, toluene, reflux, then DMF, PMBCl, Bu4NI, toluene, 60 °C, 81%; (f) TMSOTf, CH2Cl2, molecular sieves 4 Å, −30 °C, 42%; (g) pTosOH, MeOH, 0 °C → rt, 70%; (h) H2 (1 atm), 10% Pd–C, MeOH, then 0.01 M aq NaOH, rt, 92%.
The use of a defined amount of water was critical in order to prevent additional loss of the 7,8-O-acetonide. This way diol 36 was obtained in 71% yield ready for further processing into suitable glycosyl acceptor derivatives. Similar to the corresponding Kdo acceptor 4, the 4-O-PMB group was installed via the respective stannylidene acetal intermediate and ensuing treatment with PMBCl/Bu4NI/DMF in toluene affording the orthogonally protected acceptor 37 in 81% yield. Coupling of 37 with the benzylidene-protected NPTFA donor 10 promoted by TMSOTf in CH2Cl2 at −5 °C afforded the α-linked disaccharide 38 in 42% yield. The reduced yield of the coupling step was due to the concomitant cleavage of the acetonide during the glycosylation reaction. Deprotection of 38 was achieved by treatment with p-toluenesulfonic acid hydrate in methanol for 24 h which furnished the tetraol 39 in 70% yield. Hydrogenation of 39 with 10% Pd–C in methanol and final purification on Bio-Gel PD10 afforded the glucosyl-Ko disaccharide 40 as sodium salt in 92% yield. 13C NMR data of the deprotected target disaccharides 13, 17, 27, 30, and 40 were fully assigned and confirmed the respective structures (Table 1).
Table 1.
13C NMR chemical shifts (δ) of disaccharide derivatives 13, 17, 27, 30, and 40
| Atom position | Compound | ||||
|---|---|---|---|---|---|
| 13 | 17 | 27 | 30 | 40 | |
| OCH3 | 51.42 | 51.31 | 51.42 | 51.31 | 51.64 |
| Kdo/Kdh/Ko | |||||
| 1 | 176.03 | 175.93 | 176.06 | 175.95 | 174.16 |
| 2 | 101.29 | 100.98 | 101.30 | 100.99 | 103.26 |
| 3 | 35.47 | 35.36 | 35.22 | 35.16 | 72.76 |
| 4 | 66.60 | 66.45 | 66.42 | 66.31 | 66.95 |
| 5 | 75.95 | 76.73 | 75.48 | 76.46 | 77.19 |
| 6 | 72.35 | 73.90 | 72.35 | 73.95 | 71.75 |
| 7 | 69.24 | 61.67 | 69.01 | 61.63 | 68.96 |
| 8 | 63.94 | — | 64.07 | — | 63.87 |
| Glc | |||||
| 1 | 100.71 | 100.67 | 100.61 | 100.60 | 100.98 |
| 2 | 72.80 | 72.74 | 72.99 | 72.79 | 72.49 |
| 3 | 73.70 | 73.60 | 73.23 | 73.30 | 73.88 |
| 4 | 70.05 | 70.03 | 69.50 | 69.61 | 69.86 |
| 5 | 72.51 | 72.45 | 72.25 | 71.84 | 72.82 |
| JC,P 7.5 Hz | JC,P 7.7 Hz | ||||
| 6 | 60.77 | 60.73 | 62.93 | 63.73 | 60.77 |
| JC,P 4.1 Hz | JC,P 4.5 Hz | ||||
2.4. Conclusions and outlook
A benzylidene-protected glucosyl NPTFA donor proved as an efficient and α-selective glycosyl donor for the glycosylation of OH-5 of orthogonally protected Kdo and Ko glycosides and allowed for the regioselective introduction of the 6-O-phosphoryl group. Oxidative cleavage of the exocyclic side chain of Kdo provided the Kdh containing fragments. Global deprotection gave the disaccharide ligands related to the inner core region of Acinetobacter LPS which are suited to perform various binding assays to elucidate structural details of the interaction with MBL which are serum components and important in innate first-line immune reactions such as complement activation. The synthesis of larger fragments is currently in progress.
3. Experimental
3.1. General
All purchased chemicals were used without further purification unless stated otherwise. Solvents were dried over activated 3 Å (acetone) or 4 Å (CH2Cl2, DMF, pyridine, toluene) molecular sieves. THF was distilled on 4 Å molecular sieves shortly before use. Dry MeOH (secco solv) was purchased from Merck. Cation exchange resin DOWEX 50 H+ was regenerated by consecutive washing with HCl (3 M), water, and dry MeOH. Aqueous solutions of salts were saturated unless stated otherwise. Concentration of organic solutions was performed under reduced pressure <40 °C. Optical rotations were measured with a Perkin–Elmer 243 B polarimeter. values are given in units of 10−1 deg cm2 g−1. Thin layer chromatography was performed on Merck precoated plates: generally on 5 × 10 cm, layer thickness 0.25 mm, silica gel 60F254; alternatively on HPTLC plates with 2.5 cm concentration zone (Merck). Spots were detected by dipping reagent (anisaldehyde–H2SO4). For column chromatography silica gel (0.040–0.063 mm) was used. HP-column chromatography was performed on pre-packed columns (YMC-Pack SIL-06, 0.005 mm, 250 × 10 mm and 250 × 20 mm). Size exclusion chromatography was performed on Bio-Gel® P-2 Gel extra fine <45 μm (wet) (Bio-Rad, 45–90 μm) or on pre-packed PD-10 columns (GE Healthcare, Sephadex™ G-25 M). NMR spectra were recorded with a Bruker Avance III 600 instrument (600.22 MHz for 1H, 150.93 MHz for 13C and 242.97 MHz for 31P) using standard Bruker NMR software. 1H spectra were referenced to 7.26 (CDCl3), 5.32 (CD2Cl2), 3.31 (MeOD), and 0.00 (D2O, external calibration to 2,2-dimethyl-2-silapentane-5-sulfonic acid) ppm unless stated otherwise. 13C spectra were referenced to 77.00 (CDCl3), 53.84 (CD2Cl2), 49.00 (MeOD), and 67.40 (D2O, external calibration to 1,4-dioxane) ppm. 31P spectra in D2O were referenced to external ortho-phosphoric acid (0.00 ppm). ESI-MS data were obtained on a Waters Micromass Q-TOF Ultima Global instrument.
3.2. Methyl [methyl 3-deoxy-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (3)
A cooled mixture of 1H-imidazole (0.22 g, 3.228 mmol) and TIPDSCl2 (0.46 mL, 1.412 mmol) in dry DMF (7.4 mL) was added dropwise to a solution of 2 (0.36 g, 1.345 mmol) in dry DMF (8.6 mL) at −40 °C. After 2 h at −40 °C another portion of 1H-imidazole (22 mg, 0.323 mmol) and TIPDSCl2 (46 μL, 0.141 mmol) in dry DMF (0.74 mL) was slowly added and after 30 min excessive reagent was scavenged by addition of dry MeOH (2 mL). The mixture was allowed to warm up to ambient temperature, solid NaHCO3 (0.4 g) was added, and the suspension was concentrated. The residue was partitioned between EtOAc and aq NaHCO3, the aqueous phase was extracted with EtOAc (2 × 10 mL) and the combined organic layers were dried over MgSO4, filtered, and concentrated. The residual oil was purified by column chromatography (toluene/EtOAc 4:1 → 1:1) affording 3 (0.46 g, 68%) as a colorless amorphous solid: +36.4 (c 0.85, CHCl3); Rf 0.38 (toluene/EtOAc 1:1); 1H NMR (CDCl3): δ 4.30–4.26 (m, 1H, H-7), 4.18 (dd, 1H, J8a,8b 12.1, J8a,7 1.7 Hz, H-8a), 4.06–4.04 (m, 1H, H-5), 4.03–3.98 (m, 1H, H-4), 3.84 (dd, 1H, J8b,7 7.0 Hz, H-8b), 3.78 (s, 3H, CO2CH3), 3.50 (dd, J6,7 8.0, J6,5 1.0 Hz, H-6), 3.22 (s, 3 H, OCH3), 2.15 (dd, 1H, J3eq,3ax 13.0, J3eq,4 5.1 Hz, H-3eq), 1.86 (dd, 1H, J3ax,4 11.5 Hz, H-3ax), 1.12–0.93 (m, 28 H, TIPDS); 13C NMR (CDCl3): δ 168.53 (s, C-1), 99.36 (s, C-2), 74.08 (d, C-7), 71.35 (d, C-6), 66.81 (t, C-8), 66.67 (d, C-5), 66.17 (d, C-4), 52.52 (q, CO2CH3), 51.17 (q, OCH3), 35.10 (t, C-3), 17.40, 17.35, 17.33, 17.23, 17.21 [q, 8C, Si-CH-(CH3)2], 13.31, 12.77, 12.48 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 531.2409; calcd for C22H44O9Si2Na+: 531.2416.
3.3. Methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (4)
A mixture of 3 (91 mg, 0.179 mmol) and dibutyltin oxide (49 mg, 0.197 mmol) in dry toluene (4.0 mL) was heated to reflux on a Dean–Stark apparatus for 2 h. To the cooled solution dry DMF (166 μL, 2.146 mmol), 4-methoxybenzyl chloride (73 μL, 0.537 mmol) and tetrabutylammonium iodide (73 mg, 0.197 mmol) were added successively. After 16 h at 60 °C the solution was allowed to cool to ambient temperature. The solution was diluted with EtOAc and consecutively washed with HCl (1 M), aq NaHCO3, aq Na2S2O3 (50 g/L) and brine. The organic phase was dried (MgSO4), filtered, and concentrated. The residual oil was taken up in dry MeOH (4.0 mL) and treated with 0.1 M NaOMe in dry MeOH (0.179 mmol, 1.8 mL) at 0 °C. After 1 h at ambient temperature the solution was made neutral by adding DOWEX 50 H+ resin, the suspension was filtered, and the filtrate was concentrated. Column chromatography of the residue (toluene/EtOAc 19:1 → 9:1) provided 4 (79 mg, 70%) as a colorless oil: +24.5 (c 1.07, CHCl3); Rf 0.66 (toluene/EtOAc 3:1); 1H NMR (CDCl3): δ 7.27–7.24 (m, 2H, Ar), 6.89–6.86 (m, 2H, Ar), 4.56 (d, 1H, J 11.5 Hz, CHHPh), 4.50 (d, 1H, J 11.4 Hz, CHHPh), 4.36–4.32 (m, 1H, H-7), 4.25 (dd, 1H, J8a,8b 11.9, J8a,7 1.6 Hz, H-8a), 4.14–4.12 (m, 1H, H-5), 3.84 (ddd, 1H, J4,3ax 11.5, J4,3eq 5.2, J4,5 2.9, H-4), 3.80–3.75 (m, 7H, H-8b, PhOCH3, CO2CH3), 3.38 (dd, 1H, J6,7 8.6, J6,5 1.0 Hz, H-6), 3.20 (s, 3H, OCH3), 2.21 (bs, 1H, OH), 2.18 (dd, 1H, J3eq,3ax 12.8, H-3eq), 2.01 (dd, 1H, H-3ax), 1.18–0.91 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.60 (s, C O), 159.38, 130.03 (s, 2C, Ar), 129.28, 113.96 (d, 4C, Ar), 99.36 (s, C-2), 72.97 (d, 2C, C-4, C-7), 71.88 (d, C-6), 69.99 (t, CH2Ph), 67.16 (t, C-8), 63.77 (d, C-5), 55.26 (q, PhOCH3), 52.50 (q, CO2CH3), 50.99 (q, OCH3), 32.09 (t, C-3), 17.47, 17.41, 17.37, 17.27 [q, 8C, Si-CH-(CH3)2], 13.29, 12.82, 12.54, 12.49 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 651.2995; calcd for C30H52O10Si2Na+: 651.2991.
Data for 5: colorless oil; +18.2 (c 0.72, CHCl3), Rf 0.76 (toluene/EtOAc 3:1); 1H NMR (CDCl3): δ 7.32–7.29 (m, 2H, Ar), 7.26–7.22 (m, 2H, Ar), 6.89–6.85 (m, 4H, Ar), 5.15 (s, 2H, CH2Ph), 4.53 (d, 1H, J 11.6 Hz, CHHPh), 4.49 (d, 1H, J 11.4 Hz, CHHPh), 4.33 (ddd, 1H, J7,6 8.6, J7,8b 7.2, J7,8a 1.5 Hz, H-7), 4.24 (dd, 1H, J8a,8b 11.8 Hz, H-8a), 4.13–4.11 (m, 1H, H-5), 3.84–3.79 (m, 7H, H-4, 2 × PhOCH3), 3.76 (dd, 1H, H-8b), 3.37 (d, 1H, H-6), 3.13 (s, 3H, OCH3), 2.21 (d, 1H, JOH,5 2.6 Hz, OH), 2.15 (dd, 1H, J3eq,3ax 12.9, J3eq,4 4.9 Hz, H-3eq), 1.97 (dd, 1H, J3ax,4 11.7 Hz, H-3ax), 1.10–0.90 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 167.77 (s, C O), 159.74, 159.37 (s, 2C, Ar), 130.26 (d, 2C, Ar), 130.03 (s, Ar), 129.29 (d, 2C, Ar), 127.57 (s, Ar), 113.94 (d, 4C, Ar), 99.18 (s, C-2), 73.06 (d, 2C, C-4, C-7), 71.84 (d, C-6), 69.98 (t, CH2Ph), 67.10 (t, C-8), 66.98 (t, CH2Ph), 63.75 (d, C-5), 55.26 (q, 2C, 2 × PhOCH3), 50.94 (q, OCH3), 31.99 (t, C-3), 17.47, 17.40, 17.38, 17.27 [q, 8C, Si-CH-(CH3)2], 13.29, 12.81, 12.54, 12.51 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 757.3414; calcd for C37H58O11Si2Na+: 757.3410.
Data for 6: colorless amorphous solid; −13.8 (c 0.82, CHCl3); Rf 0.59 (toluene/EtOAc 9:1); 1H NMR (CDCl3): δ 7.26–7.23 (m, 2H, Ar), 6.89–6.86 (m, 2H, Ar), 5.00–4.99 (m, 1H, H-5), 4.54 (d, 1H, J 11.9 Hz, CHHPh), 4.49 (d, 1H, J 11.8 Hz, CHHPh), 4.16 (dd, 1H, J8a,8b 12.1, J8a,7 1.7 Hz, H-8a), 3.93 (ddd, 1H, J7,6 9.5, J7,8b 7.6 Hz, H-7), 3.89 (app. td, 1H, J4,3ax 8.8, J4,3eq = J4,5 2.2 Hz, H-4), 3.80 (s, 3H, PhOCH3), 3.69 (dd, 1H, H-8b), 3.58 (s, 3H, OCH3), 3.57 (d, 1H, H-6), 2.50 (dd, 1H, J3ax,3eq 14.7 Hz, H-3ax), 2.00–1.97 (m, 1H, H-3eq), 1.12–0.91 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.24 (s, C O), 159.54 (s, Ar), 129.36 (d, 2C, Ar), 128.92 (s, Ar), 114.03 (d, 2C, Ar), 94.91 (s, C-2), 73.37 (d, C-6), 72.71, 72.68 (d, 2C, C-5, C-7), 71.93 (d, C-4), 70.05 (t, CH2Ph), 66.64 (t, C-8), 55.25 (q, PhOCH3), 52.71 (q, OCH3), 38.72 (t, C-3), 17.40, 17.35, 17.32, 17.27, 17.20, 17.18 [q, 8C, Si-CH-(CH3)2], 13.20, 12.64, 12.45, 12.39 [d, 4C, Si-CH-(CH3)2].
3.4. 2,3,4,6-Tetra-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (8α), 2,3,4,6-tetra-O-benzyl-β-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (8β), 2,3,4,6-tetra-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (9α), and 2,3,4,6-tetra-O-benzyl-β-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (9β)
A solution of glycosyl acceptor 4 (50 mg, 0.080 mmol) in dry CH2Cl2 (0.6 mL) containing 4 Å molecular sieves (75 mg) was stirred for 20 min followed by addition of glycosyl donor 722 (85 mg, 0.120 mmol) in dry CH2Cl2 (0.6 mL) under Ar. TMSOTf (1.4 μL, 0.008 mmol) in dry CH2Cl2 (50 μL) was added dropwise to the cold (−5 °C) mixture and after 5 min the reaction was quenched by adding a solution of Et3N (23 μL, 0.160 mmol) in dry CH2Cl2 (0.3 mL). Filtration of the suspension over Celite®, concentration of the filtrate, and purification of the residue by HP-column chromatography (n-hexane/EtOAc 9:1 → 2:1) provided target compound 8α (31 mg, 34%) together with 8β (13 mg, 14%) and disaccharide alcohols 9α (14 mg, 15%) and 9β (6 mg, 7%) as colorless syrups.
Data for 8α: +68.4 (c 0.53, CHCl3); Rf 0.62 (n-hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 7.36–7.20 (m, 20H, Ar), 7.10–7.07 (m, 2H, Ar), 6.85–6.81 (m, 2H, Ar), 5.28 (d, 1H, J1′,2′ 3.7 Hz, H-1′), 4.94 (d, 1H, J 12.0 Hz, CHHPh), 4.86 (d, 1H, J 11.7 Hz, CHHPh), 4.83 (d, 1H, J 11.1 Hz, CHHPh), 4.75 (d, 1H, J 10.7 Hz, CHHPh), 4.68 (d, 1H, J 12.0 Hz, CHHPh), 4.54 (d, 1H, J 11.5 Hz, CHHPh), 4.51 (d, 1H, J 12.3 Hz, CHHPh), 4.50–4.45 (m, 2H, H-7, CHHPh), 4.41 (d, 1H, J 11.0 Hz, CHHPh), 4.27 (d, 1H, J5,4 2.2 Hz, H-5), 4.24–4.19 (m, 2H, H-5′, CHHPh), 4.06 (app. t, 1H, J3′,2′ = J3′,4′ 9.5 Hz, H-3′), 4.01 (dd, 1H, J8a,8b 13.1, J8a,7 1.9 Hz, H-8a), 3.86–3.80 (m, 4H, H-4, CO2CH3), 3.79 (s, 3H, PhOCH3), 3.72 (app. t, 1H, J4′,5′ 9.4 Hz, H-4′), 3.64–3.60 (m, 3H, H-2′, H-6, H-8b), 3.40 (dd, 1H, J6′a,6′b 10.7, J6′a,5′ 2.1 Hz, H-6′a), 3.29 (s, 3H, OCH3), 3.12 (dd, 1H, J6′b,5′ 1.9 Hz, H-6′b), 2.21 (dd, 1H, J3eq,3ax 12.5, J3eq,4 4.4 Hz, H-3eq), 2.16 (app. t, 1H, J3ax,4 12.2 Hz, H-3ax), 1.06–0.87 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.80 (s, C O), 159.07, 138.86, 138.78, 138.57, 138.11, 130.52 (s, 6C, Ar), 129.01, 128.25, 128.23, 128.21, 128.09, 127.98, 127.85, 127.82, 127.50, 127.48, 127.36, 127.00, 126.77, 113.70 (d, 24C, Ar), 99.13 (s, C-2), 98.66 (d, C-1′), 81.82 (d, C-3′), 80.19 (d, C-2′), 77.95 (d, C-4′), 75.10 (t, CH2Ph), 74.78 (t, CH2Ph), 73.62 (d, C-4), 73.29 (t, CH2Ph), 72.53 (t, CH2Ph), 71.82 (d, C-5), 71.75 (d, C-7), 71.39 (d, C-6), 70.23 (d, C-5′), 69.94 (t, CH2Ph), 67.79 (t, C-6′), 63.00 (t, C-8), 55.22 (q, PhOCH3), 52.34 (q, CO2CH3), 51.12 (q, OCH3), 33.21 (t, C-3), 17.60, 17.54, 17.50, 17.49, 17.29, 17.17, 17.09 [q, 8C, Si-CH-(CH3)2], 14.18, 13.55, 12.99, 12.75 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 1168.5848; calcd for C64H86O15Si2NH4+: 1168.5844.
Data for 8β: +14.4 (c 0.94, CHCl3); Rf 0.60 (n-hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 7.37–7.34 (m, 2H, Ar), 7.29–7.22 (m, 16H, Ar), 7.18–7.13 (m, 4H, Ar), 6.83–6.80 (m, 2H, Ar), 5.31 (d, 1H, J1′,2′ 7.8 Hz, H-1′), 5.08 (d, 1H, J 11.6 Hz, CHHPh), 4.89 (d, 1H, J 10.9 Hz, CHHPh), 4.81 (d, 1H, J 11.0 Hz, CHHPh), 4.78–4.76 (m, 1H, H-5), 4.74–4.71 (m, 2H, 2 × CHHPh), 4.62 (d, 1H, J 11.8 Hz, CHHPh), 4.52–4.45 (m, 4H, 1 × CHHPh, 3 × CHHPh), 4.35–4.31 (m, 1H, H-7), 4.27 (dd, 1H, J8a,8b 12.0, J8a,7 2.0 Hz, H-8a), 3.94 (ddd, 1H, J4,3ax 11.7, J4,3eq 4.6, J4,5 1.8 Hz, H-4), 3.81 (dd, 1H, J8b,7 6.4 Hz, H-8b), 3.78 (s, 3H, PhOCH3), 3.73–3.69 (m, 1H, H-6′a), 3.65–3.55 (m, 2H, H-3′, H-6′b), 3.49 (s, 3H, CO2CH3), 3.46–3.39 (m, 3H, H-4′, H-5′, H-6), 3.34 (dd, 1H, J2′,3′ 9.1 Hz, H-2′), 3.20 (s, 3H, OCH3), 2.26 (app. t, 1H, J3ax,3eq 12.7 Hz, H-3ax), 2.20 (dd, 1H, H-3eq), 1.11–0.87 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.52 (s, C O), 159.00, 138.95, 138.67, 138.22, 138.19, 130.32 (s, 6C, Ar), 128.66, 128.35, 128.29, 128.25, 128.15, 128.01, 127.96, 127.89, 127.68, 127.56, 127.51, 127.45, 127.19, 113.79 (d, 24C, Ar), 100.49 (d, C-1′), 99.36 (s, C-2), 84.73 (d, C-3′), 82.31 (d, C-2′), 78.36 (d, C-4′), 75.81 (d, C-4), 75.75 (t, CH2Ph), 74.94 (t, CH2Ph), 74.57 (d, C-6), 74.09 (t, CH2Ph), 73.28 (d, C-5′), 73.24 (t, CH2Ph), 72.69 (d, C-7), 69.86, 69.83 (t, 2C, CH2Ph, C6′), 66.57 (t, C-8), 64.98 (d, C-5), 55.24 (q, PhOCH3), 52.19 (q, CO2CH3), 50.94 (q, OCH3), 33.30 (t, C-3), 17.81, 17.79, 17.57, 17.48, 17.42, 17.34, 17.28 [q, 8C, Si-CH-(CH3)2], 13.38, 12.79, 12.74 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 1168.5854; calcd for C64H86O15Si2NH4+: 1168.5844.
Data for 9α: +34.3 (c 0.64, CHCl3); Rf 0.39 (n-hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 7.33–7.24 (m, 18H, Ar), 7.13–7.10 (m, 2H, Ar), 5.04 (d, 1H, J1′,2′ 3.4 Hz, H-1′), 4.93 (d, 1H, J 11.0 Hz, CHHPh), 4.82 (d, 1H, J 11.0 Hz, CHHPh), 4.79 (d, 1H, J 10.7 Hz, CHHPh), 4.75 (s, 2H, CH2Ph), 4.54 (d, 1H, J 12.2 Hz, CHHPh), 4.52 (m, 1H, H-7), 4.45 (d, 1H, J 12.2 Hz, CHHPh), 4.44 (d, 1H, J 10.5 Hz, CHHPh), 4.16 (dd, 1H, J8a,8b 12.9, J8a,7 2.9 Hz, H-8a), 4.09–3.99 (m, 5H, H-3′, H-5′, H-4, H-5, H-8b), 3.79–3.77 (m, 4H, H-6, CO2CH3), 3.61–3.56 (m, 2H, H-6′a, H-6′b), 3.54 (dd, 1H, J2′,3′ 9.6 Hz, H-2′), 3.48 (dd, 1H, J 9.9, J 8.9 Hz, H-4′), 3.44 (d, 1H, JOH,4 9.6 Hz, OH), 3.31 (s, 3H, OCH3), 2.13 (dd, 1H, J3eq,3ax 12.8, J3eq,4 4.4 Hz, H-3eq), 1.86 (app. t, 1H, J3ax,4 12.5 Hz, H-3ax), 1.06–0.82 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.57 (s, C O), 138.67, 138.30, 137.96, 137.69 (s, 4C, Ar), 128.39, 128.38, 128.35, 138.33, 127.99, 127.89, 127.87, 127.80, 127.73, 127.58, 127.53, 127.45 (d, 20C, Ar), 99.46 (d, C-1′), 99.03 (s, C-2), 81.42 (d, C-3′), 78.74 (d, C-2′), 78.60 (d, C-5), 77.87 (d, C-4′), 75.37 (t, CH2Ph), 75.08 (t, CH2Ph), 73.50 (t, CH2Ph), 73.08 (d, C-7), 72.56 (t, CH2Ph), 72.05 (d, C-6), 71.39 (d, C-5′), 68.67 (t, C-6′), 66.79 (d, C-4), 63.87 (t, C-8), 52.37 (q, CO2CH3), 51.19 (q, OCH3), 36.13 (t, C-3), 17.63, 17.54, 17.53, 17.50, 17.35, 17.22, 17.14 [q, 8C, Si-CH-(CH3)2], 13.98, 13.60, 12.96, 12.77 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 1048.5273; calcd for C56H78O14Si2NH4+: 1048.5268.
Data for 9β: +19.7 (c 0.85, CHCl3); Rf 0.27 (n-hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 7.33–7.23 (m, 18H, Ar), 7.17–7.13 (m, 2H, Ar), 5.00 (d, 1H, J 11.0 Hz, CHHPh), 4.90 (d, 1H, J 10.9 Hz, CHHPh), 4.83 (d, 1H, J 10.7 Hz, CHHPh), 4.78 (2d, 2H, J 10.7 Hz, CHHPh, CHHPh), 4.70 (d, 1H, J1′,2′ 7.9 Hz, H-1′), 4.62 (d, 1H, J 11.9 Hz, CHHPh), 4.57 (d, 1H, J 10.7 Hz, CHHPh), 4.41 (d, 1H, J 11.9 Hz, CHHPh), 4.38 (app. dt, 1H, J7,6 = J7,8a 8.7, J7,8b 2.5 Hz, H-7), 4.17–4.09 (m, 3H, H-5, H-8a, H-8b), 3.90 (app. ddt, 1H, J4,3ax = J4,OH 11.6, J4,3eq 4.8, J4,5 2.6 Hz, H-4), 3.81 (dd, 1H, J6′a,6′b 10.9, J6′a,5′ 3.2 Hz, H-6′a), 3.80 (app. t, 1H, J4′,3′ = J4′,5′ 9.4 Hz, H-4′), 3.78 (s, 3H, CO2CH3), 3.72–3.68 (m, 3H, H-3′, H-6′b, OH), 3.63 (d, 1H, H-6), 3.55 (dd, 1H, J2′,3′ 9.1 Hz, H-2′), 3.41–3.37 (m, 1H, H-5′), 3.26 (s, 3H, OCH3), 1.82 (dd, 1H, J3eq,3ax 12.7 Hz, H-3eq), 1.64 (app. t, 1H, H-3ax), 1.08–0.85 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.73 (s, C O), 138.15, 137.16 (s, 4C, Ar), 128.47, 128.44, 128.39, 128.34, 127.96, 127.94, 127.8, 127.71, 127.69, 127.67, 127.60 (d, 20C, Ar), 103.86 (d, C-1′), 99.21 (s, C-2), 85.57 (d, C-3′), 82.12 (d, C-2′), 78.06 (d, C-4′), 77.23 (d, C-5), 75.53 (t, CH2Ph), 75.50 (t, CH2Ph), 75.13 (d, C-5′), 74.90 (t, CH2Ph), 73.43 (t, CH2Ph), 72.73 (d, C-7), 71.12 (d, C-6), 68.75 (t, C-6′), 67.43 (d, C-4), 63.88 (t, C-8), 52.33 (q, CO2CH3), 51.20 (q, OCH3), 35.88 (t, C-3), 17.83, 17.74, 17.54, 17.47, 17.37, 17.34, 17.21, 17.15 [q, 8C, Si-CH-(CH3)2], 13.77, 13.67, 12.95, 12.84 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 1053.4818; calcd for C56H78O14Si2Na+: 1053.4822.
Alternatively compound 9α was obtained by acid treatment of 8α:
A solution of 8α (17.6 mg, 0.015 mmol) in CH2Cl2 (14.2 mL) was treated with 99% TFA (3.6 mL) at 0 °C for 10 min. The solution was concentrated and coevaporated with toluene. Subsequent column chromatography of the residue (n-hexane/EtOAc 5:1) yielded 9α (13.0 mg, 83%) as a colorless oil.
3.5. 2,3-Di-O-benzyl-4,6-O-benzylidene-α,β-d-glucopyranosyl (N-phenyl)trifluoroacetimidate (10α/β)
2,3-Di-O-benzyl-4,6-O-benzylidene-α,β-d-glucopyranose28 (128 mg, 0.285 mmol) was dissolved in a mixture of dry CH2Cl2 (2.0 mL) and dry acetone (0.9 mL) followed by successive addition of K2CO3 (79 mg, 0.570 mmol) and 2,2,2-trifluoro-N-phenylacetimidoyl chloride (92 μL, 0.570 mmol). The heterogenous mixture was stirred at ambient temperature for 16 h, filtered through a pad of Celite®, and rinsed with CH2Cl2. After addition of one drop of Et3N the filtrate was concentrated and the crude residue was purified by chromatography (n-hexane/EtOAc 10:1) affording an anomeric mixture (α/β ∼ 1:8) of 10 (169 mg, 96%).
Data for 10β: colorless amorphous solid; +49.1 (c 2.57, CHCl3); Rf 0.75 (n-hexane/EtOAc 4:1); 1H NMR (CD2Cl2): δ 7.54–7.46 (m, 2H, Ar), 7.42–7.27 (m, 15H, Ar), 7.17–7.13 (m, 1H, Ar), 6.88–6.75 (m, 2H, Ar), 5.83 (bs, 1H, H-1), 5.60 (s, 1H, CHPh), 4.92 (d, 1H, J 11.4 Hz, CHHPh), 4.84 (d, 1H, J 11.0 Hz, CHHPh), 4.81 (2d, 2H, J 11.1 Hz, 2 × CHHPh), 4.38–4.29 (m, 1H, H-6a), 3.88–3.65 (m, 4H, H-2, H-3, H-4, H-6b), 3.45 (bs, 1H, H-5); 13C NMR (CD2Cl2): δ 143.78, 138.94, 138.41, 137.84 (s, 4C, Ar), 129.37, 129.20, 128.75, 128.67, 128.59, 128.49, 128.41, 128.27, 128.06, 126.49, 124.88, 119.63 (d, 20C, Ar), 101.69 (d, CHPh), 81.50, 81.13, 81.11 (d, 3C, C-2, C-3, C-4), 75.71, 75.21 (t, 2C, 2 × CH2Ph), 68.91 (t, C-6), 67.06 (d, C-5).
Data for 10α: colorless oil; +43.0 (c 1.92, CHCl3); Rf 0.61 (n-hexane/EtOAc, 4:1); 1H NMR (CD2Cl2): δ 7.53–7.49 (m, 2H, Ar), 4.42–7.27 (m, 15H, Ar), 7.15–7.11 (m, 1H, Ar), 6.80–6.74 (m, 2H, Ar), 6.45 (bs, 1H, H-1), 5.61 (s, 1H, CHPh), 4.94 (d, J 11.4 Hz, CHHPh), 4.85 (d, J 11.3 Hz, CHHPh), 4.84 (d, J 11.8 Hz, CHHPh), 4.76 (d, J 11.9 Hz, CHHPh), 4.36 (dd, 1H, J6a,6b 10.2, J6a,5 4.8 Hz, H-6a), 4.09 (app. t, 1H, J3,2 = J3,4 9.5 Hz, H-3), 4.03–3.96 (m, 1H, H-5), 3.81–3.70 (m, 3H, H-2, H-4, H-6b); 13C NMR (CD2Cl2): δ 144.06, 139.23, 138.46, 137.89 (s, 4C, Ar), 129.37, 129.20, 128.83, 128.63, 128.60, 128.30, 128.27, 128.12, 127.96, 126.52, 124.69, 119.75 (d, 20C, Ar), 101.82 (d, CHPh), 81.77 (d, C-4), 79.14 (d, C-2), 78.62 (d, C-3), 75.50, 74.29 (t, 2C, 2 × CH2Ph), 69.07 (t, C-6), 65.52 (d, C-5).
3.6. 2,3-Di-O-benzyl-4,6-O-benzylidene-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (11)
A solution of donor 10α/β (74 mg, 0.119 mmol) in dry CH2Cl2 (1.0 mL) was added to dry acceptor 4 (50 mg, 0.080 mmol) under Ar followed by addition of 4 Å molecular sieves. After stirring for 2.5 h at ambient temperature a solution of TMSOTf (0.4 μL, 0.002 mmol) in dry CH2Cl2 (0.1 mL) was added dropwise at −5 °C. The reaction was quenched after 15 min by addition of Et3N (23 μL, 0.159 mmol) in dry CH2Cl2 (0.7 mL). The mixture was allowed to warm to ambient temperature and was filtered through a pad of Celite®. The filtrate was concentrated and the residue was purified by chromatography (n-hexane/EtOAc 8:1→3:1, with 0.1% TEA) to give 11 (67 mg, 80%) as a coloress oil: +59.7 (c 0.53, CHCl3); Rf 0.63 (n-hexane/EtOAc 3:1), 1H NMR (CDCl3): δ 7.51–7.47 (m, 2H, Ar), 7.42–7.21 (m, 15H, Ar), 6.88–6.84 (m, 2H, Ar), 5.51 (s, 1H, CHPh), 5.28 (d, 1H, J1′,2′ 3.8 Hz, H-1′), 5.08 (d, 1H, J 11.8 Hz, CHHPh), 4.96 (d, 1H, J 11.3 Hz, CHHPh), 4.75 (d, 1H, J 11.0 Hz, CHHPh), 4.66 (d, 1H, J 11.5 Hz, CHHPh), 4.57 (s, 2H, CH2Ph), 4.42–4.39 (m, 1H, H-7), 4.35 (app. dt, 1H, J5′,4′ = J5′,6′b 9.9, J5′,6′a 5.1 Hz, H-5′), 4.25 (d, 1H, J5,4 2.3 Hz, H-5), 4.19 (app. t, 1H, J3′,2′ = J3′,4′ 9.5 Hz, H-3′), 3.96 (dd, 1H, J8a,8b 13.2, J8a,7 1.6 Hz, H-8a), 3.89 (dd, 1H, J6′a,6′b 10.1 Hz, H-6′a), 3.85 (ddd, 1H, J4,3ax 11.6, J4,3eq 4.7, H-4), 3.81 (s, 3H, CO2CH3), 3.77 (s, 3H, PhOCH3), 3.67 (dd, 1H, H-2′), 3.64–3.59 (m, 3H, H-4′, H-6, H-8b), 3.55 (app. t, 1H, J6′b,5′ 10.2 Hz, H-6′b), 3.28 (s, 3H, OCH3), 2.23 (dd, 1H, J3eq,3ax 12.5, H-3eq), 2.17 (app. t, 1H, H-3ax), 1.14–0.77 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.76 (s, C O), 159.08, 138.93, 138.78, 137.79, 130.34 (s, 5C, Ar), 129.01, 128.69, 128.23, 128.10, 128.05, 127.91, 127.43, 126.96, 126.08, 113.85 (d, 19C, Ar), 100.99 (d, CHPh), 99.24 (d, C-1′), 99.16 (s, C-2), 82.96 (d, C-4′), 79.28 (d, C-2′), 78.04 (d, C-3′), 74.70 (t, CH2Ph), 73.09 (d, C-4), 71.99 (t, CH2Ph), 71.65 (d, C-5), 71.54 (d, C-7), 71.23 (d, C-6), 70.11 (t, CH2Ph), 69.08 (t, C-6′), 62.79 (t, C-8), 62.37 (d, C-5′), 55.19 (q, PhOCH3), 52.34 (q, CO2CH3), 51.06 (q, OCH3), 33.20 (t, C-3), 17.55, 17.49, 17.47, 17.44, 17.27, 17.16, 17.07 [q, 8C, Si-CH-(CH3)2], 14.22, 13.44, 12.99, 12.79 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 1076.5210; calcd for C57H78O15Si2NH4+: 1076.5218.
3.7. 2,3,4,6-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl (methyl 3-deoxy-α-d-manno-oct-2-ulopyranosid)onate (12)
Compound 9α (16.0 mg, 0.016 mmol) was dissolved in dry THF (3.3 mL) and was treated with TBAF (1 M in THF, 23 μL, 0.023 mmol) at ambient temperature for 20 min. After addition of dry MeOH (1.7 mL) the solvent was removed in vacuo. Purification of the residue by chromatography (toluene/EtOAc 1:1) afforded 12 (12.0 mg, 98%) as a colorless oil; +36.0 (c 0.53, CHCl3); Rf 0.18 (toluene/EtOAc 1:1); 1H NMR (CDCl3): δ 7.39–7.24 (m, 18H, Ar), 7.18–7.15 (m, 2H, Ar), 4.96 (d, 1H, J 11.1 Hz, CHHPh), 4.93 (d, 1H, J 11.1 Hz, CHHPh), 4.89 (d, 1H, J 11.6 Hz, CHHPh), 4.83 (d, 1H, J 10.7 Hz, CHHPh), 4.73 (d, 1H, J1′,2′ 3.7 Hz, H-1′), 4.64 (d, 1H, J 11.8 Hz, CHHPh), 4.52 (d, 1H, J 12.3 Hz, CHHPh), 4.49 (d, 1H, J 10.7 Hz, CHHPh), 4.45 (d, 1H, J 12.1 Hz, CHHPh), 4.28 (d, 1H, J 5.6 Hz, OH), 4.07 (app. t, 1H, J3′,2′ = J3′,4′ 9.5 Hz, H-3′), 4.03–3.95 (m, 4H, H-5′, H-4, H-5, H-7), 3.86–3.81 (m, 1H, H-8a), 3.79 (s, 3H, CO2CH3), 3.68 (dd, 1H, J6,7 8.9, J6,5 1.6 Hz, H-6), 3.66–3.61 (m, 1H, H-8b), 3.60 (dd, 1H, J6′a,6′b 10.4, J6′a,5′ 2.2 Hz, H-6′a), 3.58 (dd, 1H, H-2′), 3.55 (dd, 1H, J6′b,5′ 5.6 Hz, H-6′b), 3.54 (d, 1H, J 12.5 Hz, OH), 3.51 (app. t, 1H, J4′,5′ 9.6 Hz, H-4′), 3.18 (s, 3H, OCH3), 2.16–2.11 (m, 1H, H-3eq), 2.08–2.00 (m, 1H, OH), 1.68 (app. t, 1H, J3ax,3eq = J3ax,4 12.3 Hz, H-3ax); 13C NMR (CDCl3): δ 168.20 (s, C O), 138.15, 137.55, 137.48, 136.60 (s, 4C, Ar), 128.85, 128.72, 128.61, 128.54, 128.53, 128.40, 128.03, 128.01, 127.89, 127.86, 127.83 (d, 20C, Ar), 100.65 (d, C-1′), 98.99 (s, C-2), 81.83 (d, C-3′), 80.06 (d, C-5), 78.83 (d, C-2′), 78.31 (d, C-4′), 75.77 (t, CH2Ph), 75.37 (t, CH2Ph), 74.96 (t, CH2Ph), 73.51 (t, CH2Ph), 71.98 (d, C-6), 71.75 (d, C-5′), 69.06 (d, C-7), 68.44 (t, C-6′), 65.85 (d, C-4), 64.32 (t, C-8), 52.46 (q, CO2CH3), 51.02 (q, OCH3), 36.12 (t, C-3); ESI-TOF HRMS: m/z = 811.3299; calcd for C44H52O13Na+: 811.3300.
3.8. α-d-Glucopyranosyl-(1→5)-sodium (methyl 3-deoxy-α-d-manno-2-oct-2-ulopyranosid)onate (13)
Compound 12 (11.2 mg, 0.014 mmol) was dissolved in dry MeOH (0.5 mL). The atmosphere was exchanged to argon by alternating evacuation and flushing with argon. Then, palladium on active charcoal (10%, 1 mg) was added to the flask followed by successive exchange of the atmosphere to argon and hydrogen using the same method described before. The mixture was stirred intensively for 3 h, diluted with MeOH, and passed through a 0.45 μm syringe filter. Concentration of the filtrate afforded debenzylated methyl ester which was saponified with aq NaOH (0.01 M, 2.0 mL) at ambient temperature for 1.5 h. The solution was neutralized by addition of freshly regenerated DOWEX 50 H+, the ion-exchange resin was filtered off, and the filtrate was lyophilized. Purification by SEC (Bio-Gel P2 gel, 5% aq EtOH) and freeze-drying of pooled fractions provided 13 (5.6 mg, 90%) as a colorless amorphous solid; +99.9 (c 0.56, D2O); 1H NMR (D2O): 5.11 (d, 1H, J1′,2′ 4.0 Hz, H-1′), 4.12–4.03 (m, 4H, H-4, H-5, H-7, H-5′), 3.89 (dd, 1H, J8a,8b 11.8, J8a,7 2.8 Hz, H-8a), 3.79–3.68 (m, 3H, H-3′, H-6′a, H-6′b), 3.62 (dd, 1H, J8b,7 6.1 Hz, H-8b), 3.56–3.54 (m, 1H, H-6), 3.49 (dd, 1H, J2′,3′ 10.0 Hz, H-2′), 3.41 (dd, 1H, J 10.0, J 9.0 Hz, H-4′), 3.11 (s, 3H, OCH3), 1.99–1.95 (m, 1H, H-3eq), 1.82 (app. t, 1H, J3ax,3eq = J3ax,4 12.5 Hz, H-3ax); 13C NMR data: see Table 1; ESI-TOF HRMS: m/z = 413.1296; calcd for C15H25O13−: 413.1301.
3.9. 2,3-Di-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (14)
Compound 11 (50.0 mg, 0.047 mmol) was treated with trifluoroacetic acid (99%, 1.0 mL) in CH2Cl2 (8.0 mL) at 0 °C for 10 min. The solution was concentrated, coevaporated with toluene, and the crude product was purified by chromatography (n-hexane/EtOAc 3:1, then 1:2) yielding 14 (40.2 mg, 88%) as a colorless oil; +79.6 (c 0.52, CHCl3); Rf 0.24 (n-hexane/EtOAc 1:2); 1H NMR (CDCl3): δ 7.36–7.26 (m, 10H, Ar), 5.09 (d, 1H, J1′,2′ 3.4 Hz, H-1′), 4.91 (d, 1H, J 11.6 Hz, CHHPh), 4.79 (d, 1H, J 12.4 Hz, CHHPh), 4.75 (d, 1H, J 12.5 Hz, CHHPh), 4.71 (d, 1H, J 11.5 Hz, CHHPh), 4.44 (app. td, 1H, J7,6 8.3, J7,8a = J7,8b 2.3 Hz, H-7), 4.14 (dd, 1H, J8a,8b 12.8 Hz, H-8a), 4.10–4.05 (m, 2H, H-4, H-5), 4.03–3.97 (m, 2H, H-5′, H-8b), 3.85 (app. t, 1H, J3′,2′ = J3′,4′ 8.6 Hz, H-3′), 3.81–3.76 (m, 5H, CO2CH3, H-6, H-6′a), 3.73 (dd, 1H, J6′b,6′a 11.7, J6′b,5′ 5.6 Hz, H-6′b), 3.53 (dd, 1H, H-2′), 3.49 (app. t, 1H, J4′,5′ 8.7 Hz, H-4′), 3.31 (s, 3H, OCH3), 2.17 (m, 1H, H-3eq), 1.90 (app. t, 1H, J3ax,3eq = J3ax,4 12.2 Hz, H-3ax), 1.07–0.81 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.62 (s, C O), 138.38, 138.02 (s, 2C, Ar), 128.58, 128.41, 127.96, 127.91, 127.67, 127.42 (d, 10C, Ar), 99.07 (s, C-2), 98.90 (d, C-1′), 80.20 (d, C-3′), 78.39 (d, C-2′), 77.62 (d, C-5), 74.73 (t, CH2Ph), 73.12 (d, C-5′), 72.91 (d, C-7), 72.55 (t, CH2Ph), 71.79 (d, C-6), 69.91 (d, C-4′), 66.60 (d, C-4), 63.70 (t, C-8), 62.19 (t, C-6′), 52.42 (q, CO2CH3), 51.20 (q, OCH3), 36.15 (t, C-3), 17.56, 17.52, 17.50, 17.44, 17.31, 17.20, 17.12 [q, 8C, Si-CH-(CH3)2], 14.00, 13.57, 12.93, 12.77 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 873.3880; calcd for C42H66O14Si2Na+: 873.3883.
3.10. 2,3-Di-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl (methyl 3-deoxy-α-d-manno-oct-2-ulopyranosid)onate (15)
TBAF (1 M in THF, 28 μL, 0.028 mmol) was added to a solution of 14 (15.9 mg, 0.019 mmol) in dry THF (1.0 mL) at ambient temperature and stirred for 5 min. Dry MeOH (2.0 mL) was added followed by concentration. The residue was purified by chromatography (EtOAc, then EtOAc/methanol 19:1) to provide 15 (11.0 mg, 97%) as a colorless oil; +50.6 (c 0.52, CHCl3); Rf 0.22 (EtOAc); 1H NMR (CDCl3): δ 7.41–7.30 (m, 10H, Ar), 4.98 (d, 1H, J 11.4 Hz, CHHPh), 4.91 (d, 1H, J 11.6 Hz, CHHPh), 4.85 (d, 1H, J 11.4 Hz, CHHPh), 4.77 (d, 1H, J1′,2′ 3.6 Hz, H-1′), 4.66 (d, 1H, J 11.5 Hz, CHHPh), 4.20 (d, 1H, J 5.8 Hz, OH), 4.06–3.97 (m, 3H, H-4, H-5, H-7), 3.92 (app. t, 1H, J3′,2′ = J3′,4′ 9.3 Hz, H-3′), 3.86–3.78 (m, 6H, CO2CH3, H-5′, H-6′a, H-8a), 3.75–3.68 (m, 2H, H-6′b, H-6), 3.66–3.61 (m, 1 H, H-8b), 3.57 (app. dt, 1H, J4′,5′ 9.3, J4′,OH 3.2 Hz, H-4′), 3.54 (dd, 1H, H-2′), 3.44 (d, 1H, J 11.7 Hz, OH), 3.19 (s, 3H, OCH3), 2.63 (d, 1H, JOH,4′ 3.7 Hz, OH), 2.20 (dd, 1H, J3eq,3ax 12.6, J3eq,4 4.3 Hz, H-3eq), 2.14–2.07 (m, 2H, 2 × OH), 1.73–1.65 (m, 1H, H-3ax); 13C NMR (CDCl3): δ 168.40 (s, C O), 138.17, 136.48 (s, 2C, Ar), 128.89, 128.80, 128.73, 128.63, 128.11, 127.90 (d, 10C, Ar), 100.72 (d, C-1′), 99.04 (s, C-2), 81.33 (d, C-3′), 79.78 (d, C-5), 78.97 (d, C-2′), 75.60 (t, CH2Ph), 74.86 (t, CH2Ph), 73.01 (d, C-5′), 71.90 (d, C-6), 70.69 (d, C-4′), 69.06 (d, C-7), 65.87 (d, C-4), 64.19 (t, C-8), 62.01 (t, C-6′), 52.62 (q, CO2CH3), 51.10 (q, OCH3), 36.20 (t, C-3); ESI-TOF HRMS: m/z = 626.2804; calcd for C30H40O13NH4+: 626.2807.
Deprotection of 15: A solution of 15 (5.6 mg, 0.009 mmol) in dry MeOH (1.0 mL) was hydrogenated for 4 h with 10% Pd–C (1 mg) as described for 13. The suspension was diluted with MeOH and passed through a 0.45 μm syringe filter. Concentration of the filtrate afforded the debenzylated methyl ester which was treated with 0.01 M aq NaOH (1.0 mL) at ambient temperature for 12 h. The solution was made neutral by addition of DOWEX 50 H+ resin. The ion-exchange resin was filtered off and the filtrate was lyophilized. Purification of the residue on Bio-Gel PD10 column (H2O) and freeze-drying of pooled fractions provided 13 (3.8 mg, 95%) as a colorless amorphous solid.
3.11. 2,3,4,6-Tetra-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl (methyl 3-deoxy-α-d-lyxo-2-hept-2-ulopyranosid)onate (16)
A solution of 12 (32.0 mg, 0.041 mmol) in dry CH2Cl2 (3.5 mL) was cooled to −10 °C. Sodium metaperiodate on silica (15 w%, 116 mg, 0.082 mmol) was added and the suspension was stirred for 1 h at −10 °C with exclusion of light. The excess of reagent was destroyed by addition of ethylene glycol (3 w% in water, 84 μL, 0.041 mmol). The mixture was diluted with CHCl3 and extracted with distilled water. The aqueous phase was further extracted with CH2Cl2 (3 × 5 mL) and the combined organic extracts were dried (Na2SO4), filtered, and concentrated. The crude material was dissolved in dry MeOH (1.0 mL), cooled to 0 °C, and treated with sodium borohydride (2.5 mg, 0.065 mmol) for 1 h. Another portion of sodium borohydride (1.0 mg, 0.026 mmol) was added at 0 °C and after 5 min the solution was diluted with EtOAc and aq NH4Cl. The aqueous phase was extracted with EtOAc (3 × 5 mL) and the combined organic layers were dried (Na2SO4). Concentration of the solution gave a crude product which was purified by HP-column chromatography (n-hexane/EtOAc 4:1 → 3:2) to afford 16 (10.4 mg, 34%) as a colorless oil; +40.4 (c 0.96, CHCl3); Rf 0.54 (toluene/EtOAc 1:2); 1H NMR (CDCl3): δ 7.39–7.26 (m, 18H, Ar), 7.18–7.15 (m, 2H, Ar), 4.95–4.91 (m, 2H, CH2Ph), 4.85 (d, 1H, J 11.8 Hz, CHHPh), 4.84 (d, 1H, J 10.6 Hz, CHHPh), 4.74 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 4.64 (d, 1H, J 11.9 Hz, CHHPh), 4.52 (d, 1H, J 11.8 Hz, CHHPh), 4.49 (d, 1H, J 10.7 Hz, CHHPh), 4.45 (d, 1H, J 12.4 Hz, CHHPh), 4.06 (app. t, 1H, J3′,2′ = J3′,4′ 9.4 Hz, H-3′), 4.04–3.98 (m, 2H, H-5′, H-4), 3.92–3.86 (m, 2H, H-5, H-7a), 3.86–3.83 (m, 1H, H-6), 3.82–3.78 (m, 4H, H-7b, CO2CH3), 3.60 (dd, 1H, J6′a,6′b 10.3, J6′a,5′ 2.1 Hz, H-6′a), 3.58–3.54 (m, 2H, H-2′, H-6′b), 3.54–3.48 (m, 2H, H-4′, OH), 3.21 (s, 3H, OCH3), 2.15 (dd, 1H, J3eq,3ax 12.2, J3eq,4 4.5 Hz, H-3eq), 1.71 (app. t, 1H, J3ax,4 12.5 Hz, H-3ax); 13C NMR (CDCl3): δ 168.28 (s, C O), 138.25, 137.63, 137.53, 136.83 (s, 4C, Ar), 128.82, 128.62, 128.51, 128.40, 128.02, 127.99, 127.88, 127.83, 127.80 (d, 20C, Ar), 100.47 (d, C-1′), 99.03 (s, C-2), 81.74 (d, C-3′), 79.80 (d, C-5), 79.22 (d, C-2′), 78.11 (d, C-4′), 75.80 (t, CH2Ph), 75.36 (t, CH2Ph), 74.77 (t, CH2Ph), 73.49 (t, CH2Ph), 71.77 (d, C-5′), 71.59 (d, C-6), 68.49 (t, C-6′), 65.95 (d, C-4), 60.64 (t, C-7), 52.50 (q, CO2CH3), 51.01 (q, OCH3), 36.27 (t, C-3); ESI-TOF HRMS: m/z = 781.3198; calcd for C43H50O12Na+: 781.3194.
3.12. α-d-Glucopyranosyl-(1→5)-sodium (methyl 3-deoxy-α-d-lyxo-hept-2-ulopyranosid)onate (17)
A solution of 16 (8.0 mg, 0.011 mmol) in dry MeOH (0.5 mL) was hydrogenated for 4 h with 10% Pd–C (1 mg) as described for 13. Another portion of catalyst (1 mg) was added and stirring under H2 was continued for 19 h. The suspension was diluted with MeOH and passed through a 0.45 μm syringe filter. Concentration of the filtrate afforded the debenzylated methyl ester which was treated with 0.01 M aq NaOH (1.5 mL) at ambient temperature for 1 h. The solution was made neutral by addition of DOWEX 50 H+ resin. The ion-exchange resin was filtered off and the filtrate was lyophilized. Purification of the residue on Bio-Gel P2 (5% aq EtOH) and freeze-drying of pooled fractions provided 17 (3.4 mg, 79%) as a colorless amorphous solid; +170.7 (c 0.28, D2O); 1H NMR (D2O): 4.89 (d, 1H, J1′,2′ 3.8 Hz, H-1′), 4.10 (ddd, 1H, J3ax,4 12.3, J3eq,4 4.8, J5,4 3.0 Hz, H-4), 4.06 (ddd, 1H, J6′a,5′ 3.5, J6′b,5′ 2.6, J4′,5′ 10.3 Hz, H-5′), 3.96 (dd, 1H, J7a,7b 11.4, J7a,6 8.2 Hz, H-7a), 3.88 (br d, 1H, H-5), 3.81 (dd, 1H, J7b,6 4.7 Hz, H-7b), 3.76-3.68 (m, 4H, H-6, H-3′, H-6′a, H-6′b), 3.48 (dd, 1H, J2′,3′ 10.0 Hz, H-2′), 3.41 (app. t, 1H, J4′,3′ 9.1 Hz, H-4′), 3.14 (s, 3H, OCH3), 2.01–1.99 (m, 1H, H-3eq), 1.82 (app. t, 1H, J3ax,3eq 12.6 Hz, H-3ax); 13C NMR data: see Table 1; ESI-TOF HRMS: m/z = 383.1193; calcd for C14H23O12−: 383.1195.
3.13. 2,3-Di-O-benzyl-4,6-O-benzylidene-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (18)
2,3-Dichloro-4,5-dicyano-p-benzoquinone (26.2 mg, 0.116 mmol) was added in four portions during 1 h to a solution of 11 (15.3 mg, 0.014 mmol) in dry CH2Cl2 (0.9 mL) and dry MeOH (0.3 mL). After complete addition the mixture was stirred at ambient temperature for 1.5 h, diluted with CHCl3, and washed with aq NaHCO3. The aqueous phase was extracted with CH2Cl2 (2 × 5 mL) and the combined organic phases were washed with aq NaHCO3, dried (MgSO4), filtered, and concentrated. To remove aromatic impurities the crude product was dissolved in toluene and subjected to chromatography (toluene→toluene/EtOAc 9:1) to afford 18 (11.2 mg, 83%) as a colorless oil; +52.2 (c 0.54, CHCl3); Rf 0.30 (n-hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 7.49–7.47 (m, 2H, Ar), 7.39–7.24 (m, 13H, Ar), 5.54 (s, 1H, CHPh), 5.11 (d, 1H, J1′,2′ 3.9 Hz, H-1′), 4.93 (d, 1H, J 11.2 Hz, CHHPh), 4.90 (d, 1H, J 12.2 Hz, CHHPh), 4.79 (d, 1H, J 11.2 Hz, CHHPh), 4.75 (d, 1H, J 12.5 Hz, CHHPh), 4.47 (app. dt, 1H, J7,6 = J7,8a 8.7, J7,8b 2.1 Hz, H-7), 4.24 (dd, 1H, J6′a,6′b 10.4, J6′a,5′ 4.9 Hz, H-6′a), 4.15–4.07 (m, 5H, H-3′, H-5′, H-4, H-5, H-8a), 3.90 (dd, 1H, J8b,8a 12.9 Hz, H-8b), 3.80 (s, 3H, CO2CH3), 3.75 (d, 1H, H-6), 3.69 (app. t, 1H, J6′b,5′ 10.4 Hz, H-6′b), 3.65 (app. t, 1H, J4′,3′ = J4′,5′ 9.5 Hz, H-4′), 3.62 (dd, 1H, J2′,3′ 9.0 Hz, H-2′), 3.32 (s, 3H, OCH3), 2.85 (bs, 1H, OH), 2.20–2.16 (m, 1H, H-3eq), 1.92 (app. t, 1H, J3ax,3eq = J3ax,4 12.2 Hz, H-3ax), 1.05–0.81 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.53 (s, C O), 138.59, 138.39, 137.28 (s, 3C, Ar), 128.90, 128.27, 128.22, 127.96, 127.56, 127.46, 127.41, 125.97 (d, 15C, Ar), 101.13 (d, CHPh), 100.05 (d, C-1′), 99.07 (s, C-2), 82.09 (d, C-4′), 78.26 (d, C-2′), 78.18 (d, C-3′), 77.25 (d, C-5), 74.84 (t, CH2Ph), 72.57 (t, CH2Ph), 72.32 (d, C-7), 71.49 (d, C-6), 68.75 (t, C-6′), 66.50 (d, C-4), 63.37 (d, C-5′), 63.33 (t, C-8), 52.46 (q, CO2CH3), 51.22 (q, OCH3), 36.28 (t, C-3), 17.62, 17.53, 17.52, 17.49, 17.32, 17.18, 17.11 [q, 8C, Si-CH-(CH3)2], 14.12, 13.58, 12.96, 12.81 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 961.4190; calcd for C49H70O14Si2Na+: 961.4196.
3.14. 2,3,4-Tri-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (19)
A suspension of compound 11 (20.0 mg, 0.019 mmol) and 4 Å molecular sieves (70 mg) in dry CH2Cl2 (0.8 mL) was cooled to −70 °C followed by consecutive addition of triethylsilane (9.8 μL, 0.061 mmol) and dichlorophenylborane (7.2 μL, 0.055 mmol). After 1 h Et3N (44 μL) and dry MeOH (44 μL) were added to the cold solution. The mixture was diluted with CHCl3, allowed to warm up to ambient temperature and washed with aq NaHCO3. The aqueous layer was extracted with CH2Cl2 (3 × 5 mL), the combined organic layers were dried (Na2SO4) and concentrated. The residue was purified by column chromatography (n-hexane/EtOAc 9:1 → 3:1) to give 19 (8.6 mg, 43%) as a colorless oil; +87.6 (c 0.78, CHCl3); Rf 0.31 (n-hexane/EtOAc 3:1); 1H NMR (CDCl3): δ 7.36–7.21 (m, 17H, Ar), 6.84–6.81 (m, 2H, Ar), 5.25 (d, 1H, J1′,2′ 3.7 Hz, H-1′), 4.93 (d, 1H, J 12.0 Hz, CHHPh), 4.88–4.82 (m, 3H, CHHPh, CH2Ph), 4.68 (d, 1H, J 12.0 Hz, CHHPh), 4.63 (d, 1H, J 11.0 Hz, CHHPh), 4.51 (d, 1H, J 11.6 Hz, CHHPh), 4.49 (d, 1H, J 11.8 Hz, CHHPh), 4.40 (app. td, 1H, J7,6 9.3, J7,8a = J7,8b 1.9 Hz, H-7), 4.23–4.21 (m, 1H, H-5), 4.20 (app. td, 1H, J5′,4′ 10.1, J5′,6′a = J5′,6′b 2.8 Hz, H-5′), 4.09 (app. t, 1H, J3′,2′ = J3′,4′ 9.4 Hz, H-3′), 4.01 (dd, 1H, J8a,8b 13.0 Hz, H-8a), 3.83 (ddd, 1H, J4,3ax 11.8, J4,3eq 4.6, J4,5 2.4 Hz, H-4), 3.81 (s, 3H, CO2CH3), 3.77 (s, 3H, PhOCH3), 3.67 (dd, 1H, H-8b), 3.61 (d, 1H, H-6), 3.57–3.51 (m, 2H, H-2′, H-4′), 3.45–3.41 (m, 2H, H-6′a, H-6′b), 3.27 (s, 3H, OCH3), 2.22 (dd, 1H, J3eq,3ax 12.5, H-3eq), 2.15 (app. t, 1H, H-3ax), 1.57–1.53 (m, 1H, OH), 1.04–0.78 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.79 (s, C O), 159.14, 138.77, 138.72, 138.42, 130.22 (s, 5C, Ar), 129.05, 128.40, 128.29, 128.14, 127.98, 127.86, 127.73, 127.44, 127.09, 126.82, 113.77 (d, 19C, Ar), 99.15 (s, C-2), 98.09 (d, C-1′), 81.52 (d, C-3′), 80.24 (d, C-4′), 77.79 (d, C-2′), 75.12 (t, CH2Ph), 74.84 (t, CH2Ph), 73.22 (d, C-4), 72.52 (t, CH2Ph), 71.86 (d, C-7), 71.57 (d, C-5), 71.37 (d, C-6), 70.73 (d, C-5′), 69.81 (t, CH2Ph), 63.02 (t, C-8), 61.43 (t, C-6′), 55.23 (q, PhOCH3), 52.38 (q, CO2CH3), 51.10 (q, OCH3), 33.02 (t, C-3), 17.53, 17.50, 17.49, 17.46, 17.28, 17.17, 17.08 [q, 8C, Si-CH-(CH3)2], 14.17, 13.59, 12.98, 12.78 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 1078.5373; calcd for C57H80O15Si2NH4+: 1078.5374.
3.15. 2,3-Di-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (20)
A solution of 11 (59 mg, 0.056 mmol) and p-toluenesulfonic acid monohydrate (1 mg, 0.006 mmol) in dry MeOH (2.5 mL) was stirred at 40 °C for 2 h. The solution was allowed to cool to ambient temperature followed by treatment with solid NaHCO3 (25 mg) for 2 min. After removal of the solvent, the residue was partitioned between EtOAc and aq NaHCO3, the aqueous layer was washed with EtOAc (2 × 10 mL) and the combined organic phases were dried (Na2SO4), filtered, and concentrated. The residue was purified by chromatography (n-hexane/EtOAc 7:3) providing 20 (49 mg, 91%) as a colorless oil; +86.0 (c 0.44, CHCl3); Rf 0.70 (n-hexane/EtOAc 1:1); 1H NMR (CDCl3): δ 7.38–7.35 (m, 2H, Ar), 7.34–7.22 (m, 10H, Ar), 6.87–6.84 (m, 2H, Ar), 5.27 (d, 1H, J1′,2′ 3.4 Hz, H-1′), 4.89 (d, 1H, J 11.9 Hz, CHHPh), 4.88 (d, 1H, J 11.8 Hz, CHHPh), 4.74 (d, 1H, J 11.3 Hz, CHHPh), 4.71 (d, 1H, J 12.0 Hz, CHHPh), 4.52 (s, 2H, CH2Ph), 4.40–4.37 (m, 1H, H-7), 4.25–4.23 (m, 1H, H-5), 4.14 (app. td, 1H, J5′,4′ 9.9, J5′,6′a = J5′,6′b 3.8 Hz, H-5′), 4.02 (dd, 1H, J8a,8b 13.1, J8a,7 1.9 Hz, H-8a), 3.91 (app. t, 1H, J3′,2′ = J3′,4′ 9.5 Hz, H-3′), 3.86–3.82 (m, 4H, H-4, CO2CH3), 3.80 (s, 3H, PhOCH3), 3.69 (dd, 1H, J8b,7 2.1 Hz, H-8b), 3.63 (d, 1H, J6,7 9.6 Hz, H-6), 3.57 (app. dt, 1H, J4′,OH 2.8 Hz, H-4′), 3.53–3.45 (m, 3H, H-2′, H-6′a, H-6′b), 3.28 (s, 3H, OCH3), 2.25–2.20 (m, 2H, H-3eq, OH), 2.15 (app. t, 1H, J3ax,3eq = J3ax,4 12.1 Hz, H-3ax), 1.67–1.63 (m, 1H, OH), 1.05–0.79 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.84 (s, C O), 159.20, 138.70, 138.51, 130.20 (s, 4C, Ar), 129.05, 128.50, 128.21, 127.99, 127.75, 127.22, 126.89, 113.81 (d, 14C, Ar), 99.18 (s, C-2), 98.06 (d, C-1′), 80.72 (d, C-3′), 79.87 (d, C-2′), 74.68 (t, CH2Ph), 73.19 (d, C-4), 72.30 (t, CH2Ph), 71.96 (d, C-7), 71.50 (d, C-5), 71.38 (d, C-6), 70.94 (d, C-4′), 70.76 (d, C-5′), 69.84 (t, CH2Ph), 63.07 (t, C-8), 62.44 (t, C-6′), 55.26 (q, PhOCH3), 52.40 (q, CO2CH3), 51.14 (q, OCH3), 33.11 (t, C-3), 17.51, 17.48, 17.44, 17.27, 17.17, 17.08 [q, 8C, Si-CH-(CH3)2], 14.14, 13.61, 12.97, 12.78 [d, 4C, Si-CH-(CH3)2]; ESI-TOF HRMS: m/z = 988.4917; calcd for C50H74O15Si2NH4+: 988.4905.
3.16. 2,3-Di-O-benzyl-6-O-(dibenzylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (21) and 2,3-di-O-benzyl-4-O-(dibenzylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (22)
A solution of 20 (47.0 mg, 0.048 mmol) in dry CH2Cl2 (3.0 mL) was flushed with Ar before 1H-tetrazole (6.8 mg, 0.097 mmol) and 4 Å molecular sieves (50 mg) were added. The suspension was stirred for 30 min at ambient temperature and cooled to −5 °C. Dibenzyl N,N-diisopropylphosphoramidite (15.9 μL, 0.048 mmol) in dry CH2Cl2 (1.0 mL) was added dropwise at −5 °C. After 30 min another portion of dibenzyl N,N-diisopropylphosphoramidite (4.0 μL, 0.012 mmol) in dry CH2Cl2 (0.25 mL) was added at −5 °C and after 15 min the solution was treated with m-chloroperbenzoic acid (70 w%, 23.9 mg, 0.097 mmol) for 5 min. The reaction mixture was partitioned between aq NaHCO3 and CH2Cl2, the aqueous phase was extracted with CHCl3 (2 × 10 mL) and the combined organic phases were dried (MgSO4), filtered, and concentrated. Purification by HP-column chromatography (n-hexane/EtOAc 3:1 → 3:2) provided 21 (39.6 mg, 67%) and 22 (5.8 mg, 10%) as colorless oils.
Data for 21: +96.7 (c 0.32, CHCl3); Rf 0.19 (n-hexane/EtOAc 2:1, HPTLC); 1H NMR (CDCl3): δ 7.37–7.19 (m, 22H, Ar), 6.84–6.81 (d, 2H, Ar), 5.21 (d, 1H, J1′,2′ 3.6 Hz, H-1′), 5.03–4.89 (m, 6H, 2 × CHHPh, 2 × POCHHPh, 2 × POCHHPh), 4.80 (d, 1H, J 11.2 Hz, CHHPh), 4.64 (d, 1H, J 11.9 Hz, CHHPh), 4.47 (d, 1H, J 11.4 Hz, CHHPh), 4.44 (d, 1H, J 11.4 Hz, CHHPh), 4.42 (app. td, 1H, J7,6 9.8, J7,8a = J7,8b 1.7 Hz, H-7), 4.22–4.20 (m, 1H, H-5), 4.17–4.14 (m, 1H, H-5′), 4.00–3.92 (m, 3H, H-3′, H-6′a, H-8a), 3.85–3.81 (m, 4H, H4, CO2CH3), 3.70 (s, 3H, PhOCH3), 3.66–3.53 (m, 4H, H-4′, H-6′b, H-6, H-8b), 3.49 (dd, 1H, J2′,3′ 9.7 Hz, H-2′), 3.45–3.41 (m, 1H, OH), 3.28 (s, 3H, OCH3), 2.23 (dd, 1H, J3eq,3ax 12.5, J3eq,4 4.1 Hz, H-3eq), 2.11 (app. t, 1H, J3ax,4 12.2 Hz, H-3ax), 1.02–0.72 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.79 (s, C O), 159.20, 138.95, 138.77 (s, 3C, Ar), 135.72 (s, JC,P 6.7 Hz, Ar), 135.62 (s, JC,P 7.3 Hz, Ar), 130.14 (s, Ar), 129.22, 128.52, 128.49, 128.27, 128.07, 127.96, 127.93, 127.88, 127.44, 127.01, 126.81, 113.79 (d, 24C, Ar), 99.11 (s, C-2), 98.76 (d, C-1′), 80.47 (d, C-3′), 79.46 (d, C-2′), 74.89 (t, CH2Ph), 73.32 (d, C-4), 72.22 (t, CH2Ph), 71.94 (d, C-5), 71.58 (d, C-7), 71.30 (d, C-6), 70.24 (d, JC,P 4.8 Hz, C-5′), 69.96 (t, CH2Ph), 69.64 (d, C-4′), 69.47 (t, JC,P 5.4 Hz, POCH2Ph), 69.32 (t, JC,P 5.8 Hz, POCH2Ph), 66.00 (t, JC,P 5.4 Hz, C-6′), 62.83 (t, C-8), 55.14 (q, PhOCH3), 52.37 (q, CO2CH3), 51.10 (q, OCH3), 33.02 (t, C-3), 17.54, 17.48, 17.44, 17.26, 17.14, 17.06 [q, 8C, Si-CH-(CH3)2], 14.16, 13.40, 12.98, 12.74 [d, 4C, Si-CH-(CH3)2]; 31P NMR (CDCl3): δ 0.81; ESI-TOF HRMS: m/z = 1253.5072; calcd for C64H87O18PSi2Na+: 1253.5061.
Data for 22: colorless oil; +72.4 (c 0.53, CHCl3); Rf 0.26 (n-hexane/EtOAc 2:1, HPTLC); 1H NMR (CDCl3): δ 7.35–7.08 (m, 22H, Ar), 6.84–6.81 (m, 2H, Ar), 5.30 (d, 1H, J1′,2′ 3.8 Hz, H-1′), 5.00 (dd, 1H, J 11.8, JH,P 7.9 Hz, POCHHPh), 4.94–4.87 (m, 3H, CHHPh, POCH2Ph), 4.84 (dd, 1H, J 11.9, JH,P 9.8 Hz, POCHHPh), 4.81 (d, 1H, J 12.2 Hz, CHHPh), 4.79 (d, 1H, J 11.9 Hz, CHHPh), 4.65 (d, 1H, J 11.9 Hz, CHHPh), 4.54 (d, 1H, J 11.9 Hz, CHHPh), 4.50 (d, 1H, J 11.8 Hz, CHHPh), 4.44 (app. q, 1H, J4′,3′ = J4′,5′ = J4′,P 9.5 Hz, H-4′), 4.37–4.34 (m, 1H, H-7), 4.27–4.25 (m, 1H, H-5), 4.24–4.20 (m, 1H, H-5′), 4.07 (app. t, 1H, J3′,2′ 9.5 Hz, H-3′), 3.97 (dd, 1H, J8a,8b 13.1, J8a,7 1.7 Hz, H-8a), 3.86–3.78 (m, 5H, CO2CH3, H-4, OH), 3.74 (s, 3H, PhOCH3), 3.62–3.56 (m, 4H, H-2′, H-6′a, H-6, H-8b), 3.46–3.41 (m, 1H, H-6′b), 3.28 (s, 3H, OCH3), 2.23 (dd, 1H, J3eq,3ax 12.4, J3eq,4 4.5 Hz, H-3eq), 2.14 (app. t, J3ax,4 12.2 Hz, H-3ax), 1.03–0.75 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 168.91 (s, C O), 159.17, 138.38 (s, 3C, Ar), 135.65 (s, JC,P 6.4 Hz, Ar), 135.42 (s, JC,P 7.5 Hz, Ar), 130.08 (s, Ar), 128.93, 128.58, 128.55, 128.43, 128.39, 128.14, 128.10, 127.97, 127.87, 127.81, 127.32, 127.13, 126.77, 113.83 (d, 24C, Ar), 99.12 (s, C-2), 97.92 (d, C-1′), 79.85 (d, C-2′), 79.12 (d, JC,P 4.2 Hz, C-3′), 75.04 (d, JC,P 5.8 Hz, C-4′), 74.85 (t, CH2Ph), 73.08 (d, C-4), 72.71 (t, CH2Ph), 71.73 (d, C-7), 71.32 (d, C-5), 71.20 (d, C-6), 70.65 (d, JC,P 3.3 Hz, C-5′), 69.91, 69.86 (t, 2C, CH2Ph, POCH2Ph), 69.55 (t, JC,P 5.5 Hz, POCH2Ph), 62.90 (t, C-8), 60.24 (t, C-6′), 55.19 (q, PhOCH3), 52.37 (q, CO2CH3), 51.14 (q, OCH3), 33.27 (t, C-3), 17.56, 17.49, 17.27, 17.15, 17.07 [q, 8C, Si-CH-(CH3)2], 14.19, 13.51, 12.98, 12.77 [d, 4C, Si-CH-(CH3)2]; 31P NMR (CDCl3): δ 0.67; ESI-TOF HRMS: m/z = 1253.5067; calcd for C64H87O18PSi2Na+: 1253.5061.
3.17. 2,3-Di-O-benzyl-6-O-(diphenylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (23)
A suspension of compound 20 (34.0 mg, 0.035 mmol) and 4-N,N-dimethylaminopyridine (8.6 mg, 0.070 mmol) in dry CH2Cl2 (1.0 mL) containing 4 Å molecular sieves (50 mg) was stirred for 30 min at 0 °C. Diphenyl phosphoryl chloride (7.6 μL, 0.037 mmol) dissolved in dry CH2Cl2 (0.1 mL) was added dropwise to the cold solution. After stirring for 20 min the reactive species was destroyed with dry MeOH (0.1 mL). The solution was stirred for 10 min, filtered over a pad of Celite®, rinsed with CH2Cl2, and concentrated. The residue was purified by chromatography (n-hexane/EtOAc 5:2) to provide 23 (40.6 mg, 96%) as a colorless oil; +76.5 (c 0.81, CHCl3); Rf 0.42 (n-hexane/EtOAc 3:2); 1H NMR (CDCl3): δ 7.36–7.21 (m, 16H, Ar), 7.19–7.11 (m, 6H, Ar), 6.85–6.82 (m, 2H, Ar), 5.21 (d, 1H, J1′,2′ 3.7 Hz, H-1′), 4.92 (d, 1H, J 11.9 Hz, CHHPh), 4.82 (d, 1H, J 11.2 Hz, CHHPh), 4.77 (d, 1H, J 11.3 Hz, CHHPh), 4.64 (d, 1H, J 12.0 Hz, CHHPh), 4.49 (d, 1H, J 11.2 Hz, CHHPh), 4.46 (d, 1H, J 11.2 Hz, CHHPh), 4.42–4.39 (m, 1H, H-7), 4.22 (d, 1H, J5,4 2.3 Hz, H-5), 4.20–4.16 (m, 1H, H-5′), 4.13–4.08 (m, 1H, H-6′a), 3.99 (dd, 1H, J8a,8b 13.1, J8a,7 1.6 Hz, H-8a), 3.91 (app. t, 1H, J3′,2′ = J3′,4′ 9.4 Hz, H-3′), 3.85 (ddd, 1H, J4,3ax 11.9, J4,3eq 4.5, H-4), 3.83 (s, 3H, CO2CH3), 3.76–3.71 (m, 1H, H-6′b), 3.70 (s, 3H, PhOCH3), 3.64–3.61 (m, 2H, H-6, H-8b), 3.52 (app. t, 1H, J4′,5′ 9.7 Hz, H-4′), 3.39 (dd, 1H, H-2′), 3.29 (s, 3H, OCH3), 2.25 (dd, 1H, J3eq,3ax 12.4 Hz, H-3eq), 2.11 (app. t, 1H, H-3ax), 1.04–0.75 (m, 28 H, TIPDS); 13C NMR (CDCl3): δ 168.78 (s, C O), 159.24 (s, Ar), 150.51 (s, JC,P 7.0 Hz, Ar), 150.38 (s, JC,P 7.3 Hz, Ar), 138.93, 138.73, 130.11 (s, 3C, Ar), 129.68, 129.21, 128.28, 128.09, 127.84, 127.46, 127.04, 126.82, 125.43, 125.29 (d, 18C, Ar), 120.25 (d, JC,P 4.6 Hz, 2C, Ar), 120.05 (d, JC,P 5.2 Hz, 2C, Ar), 113.82 (d, 2C, Ar), 99.12 (s, C-2), 98.59 (d, C-1′), 80.30 (d, C-3′), 79.36 (d, C-2′), 74.58 (t, CH2Ph), 73.55 (d, C-4), 72.11 (t, CH2Ph), 71.86 (d, C-5), 71.64 (d, C-7), 71.30 (d, C-6), 70.09 (t, CH2Ph), 70.03 (d, JC,P 4.9 Hz, C-5′), 69.30 (d, C-4′), 67.25 (t, JC,P 5.9 Hz, C-6′), 62.87 (t, C-8), 55.13 (q, PhOCH3), 52.37 (q, CO2CH3), 51.12 (q, OCH3), 33.00 (t, C-3), 17.54, 17.50, 17.48, 17.46, 17.27, 17.16, 17.07 [q, 8C, Si-CH-(CH3)2], 14.17, 13.47, 12.99, 12.76 [d, 4C, Si-CH-(CH3)2]; 31P NMR (CDCl3): δ—10.34; ESI-TOF HRMS: m/z = 1225.4760; calcd for C62H83O18PSi2Na+: 1225.4748.
3.18. 4-O-Acetyl-2,3-di-O-benzyl-6-O-(diphenylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (24)
Distilled acetic anhydride (9.4 μL, 0.100 mmol) and 4-N,N-dimethylaminopyridine (0.3 mg, 0.003 mmol) were added successively at 0 °C to a solution of 23 (30.0 mg, 0.025 mmol) in dry pyridine (1.0 mL). After stirring for 2 h at 0 °C the reaction was quenched with dry MeOH (100 μL). Concentration of the solution and coevaporation with toluene gave a residue which was purified by chromatography (n-hexane/EtOAc 2:1) to furnish 24 (29.8 mg, 96%) as a colorless oil; +72.8 (c 0.47, CHCl3); Rf 0.25 (n-hexane/EtOAc 2:1); 1H NMR (CDCl3): δ 7.35–7.12 (m, 22H, Ar), 6.82–6.79 (m, 2H, Ar), 5.26 (d, 1H, J1′,2′ 3.7 Hz, H-1′), 5.07 (app. t, 1H, J4′,3′ = J4′,5′ 9.9 Hz, H-4′), 4.90 (d, 1H, J 11.7 Hz, CHHPh), 4.73 (d, 1H, J 11.3 Hz, CHHPh), 4.64 (d, 1H, J 11.8 Hz, CHHPh), 4.60 (d, 1H, J 11.3 Hz, CHHPh), 4.50 (d, 1H, J 11.5 Hz, CHHPh), 4.47 (d, 1H, J 11.5 Hz, CHHPh), 4.42–4.38 (m, 1H, H-5′), 4.38–4.34 (m, 1H, H-7), 4.25 (d, 1H, J5,4 2.2 Hz, H-5), 4.03 (app. t, 1H, J3′,2′ 9.7 Hz, H-3′), 4.01 (dd, 1H, J8a,8b 13.0, J8a,7 1.6 Hz, H-8a), 3.97 (ddd, 1H, J6′a,6′b 11.3, J6′a,P 5.4, J6′a,5′ 2.3 Hz, H-6′a), 3.88–3.83 (m, 4H, CO2CH3, H-4), 3.74–3.69 (m, 4H, H-6′b, PhOCH3), 3.67 (dd, 1H, J8b,7 1.9 Hz, H-8b), 3.64 (d, 1H, J6,7 9.4 Hz, H-6), 3.50 (dd, 1H, H-2′), 3.29 (s, 3H, OCH3), 2.27 (dd, 1H, J3eq,3ax 12.5, J3eq,4 4.6 Hz, H-3eq), 2.13 (app. t, 1H, J3ax,4 12.2 Hz, H-3ax), 1.86 (s, 3H, COCH3), 1.03–0.78 (m, 28H, TIPDS); 13C NMR (CDCl3): δ 169.18 (s, COCH3), 168.94 (s, C-1), 159.23 (s, Ar), 150.70 (s, JC,P 7.2 Hz, Ar), 150.54 (s, JC,P 7.4 Hz, Ar), 138.51, 138.34, 129.97 (s, 3C, Ar), 129.62, 129.57, 129.28, 128.27, 128.16, 128.05, 127.53, 127.16, 126.76, 125.11, 125.02 (d, 18C, Ar), 120.21 (d, JC,P 5.1 Hz, 2C, Ar), 120.11 (d, JC,P 5.1 Hz, 2C, Ar), 113.82 (d, 2C, Ar), 99.15 (s, C-2), 97.84 (d, C-1′), 79.60 (d, C-2′), 78.36 (d, C-3′), 74.19 (t, CH2Ph), 72.99 (d, C-4), 72.59 (t, CH2Ph), 71.73 (d, C-7), 71.19 (d, C-5), 71.17 (d, C-6), 69.95 (t, CH2Ph), 69.25 (d, C-4′), 67.91 (d, JC,P 8.1 Hz, C-5′), 66.64 (t, JC,P 5.5 Hz, C-6′), 62.91 (t, C-8), 55.18 (q, PhOCH3), 52.42 (q, CO2CH3), 51.16 (q, OCH3), 33.11 (t, C-3), 20.83 (q, COCH3), 17.52, 17.48, 17.45, 17.26, 17.15, 17.07 [q, 8C, Si-CH-(CH3)2], 14.21, 13.55, 12.96, 12.79 [d, 4C, Si-CH-(CH3)2]; 31P NMR (CDCl3): δ–12.14; ESI-TOF HRMS: m/z = 1262.5295; calcd for C64H85O19PSi2NH4+: 1262.5299.
3.19. 2,3-Di-O-benzyl-6-O-(dibenzylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-7,8-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-α-d-manno-oct-2-ulopyranosid]onate (25)
A solution of dibenzyl N,N-diisopropylphosphoramidite (22.0 μL, 0.066 mmol) in dry CH2Cl2 (1.0 mL) was added under Ar in two portions to an ice-cold solution of 14 (28.4 mg, 0.033 mmol) and 1H-tetrazole (7.9 mg, 0.113 mmol) in dry CH2Cl2 (2.1 mL) and stirred for 30 min at 0 °C. m-Chloroperbenzoic acid (70 w%, 28.8 mg, 0.117 mmol) was added, and the solution was stirred for 15 min. The solution was diluted with CH2Cl2 and extracted with aq NaHCO3. The aqueous layer was extracted with CH2Cl2 (2 × 5 mL) and the combined organic phases were dried (Na2SO4) and concentrated. The residue was purified by chromatography (1:1 n-hexane/EtOAc containing 0.1% MeOH and 0.1% Et3N) to give 25 (23.4 mg, 63%) as a colorless oil: +56.0 (c 0.72, CHCl3); Rf 0.49 (n-hexane/EtOAc 1:2); 1H NMR (CDCl3): δ 7.35–7.26 (m, 20H, Ar), 5.10 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 5.04–4.96 (m, 4H, 2 × POCH2Ph), 4.87 (d, 1H, J 11.4 Hz, CHHPh), 4.80 (d, 1H, J 12.2 Hz, CHHPh), 4.78 (d, 1H, J 11.5 Hz, CHHPh), 4.74 (d, 1H, J 12.4 Hz, CHHPh), 4.45–4.42 (m, 1H, H-7), 4.25–4.20 (m, 1H, H-6′a), 4.16–4.04 (m, 5H, H-5′, H-6′b, H-4, H-5, H-8a), 3.96 (dd, 1H, J8b,8a 12.8, J8b,7 1.9 Hz, H-8b), 3.87 (app. t, 1H, J3′,2′ = J3′,4′ 9.0 Hz, H-3′), 3.79 (s, 3H, CO2CH3), 3.75 (d, 1H, J6,7 8.3 Hz, H-6), 3.50–3.45 (m, 2H, H-2′, H-4′), 3.31 (s, 3H, OCH3), 2.98 (bs, 1H, OH), 2.14 (dd, 1H, J3eq,3ax 12.8, J3eq,4 4.9 Hz, H-3eq), 1.87 (app. t, J3ax,4 12.3 Hz, H-3ax), 1.04–0.80 (m, 28 H, TIPDS); 13C NMR (CDCl3): δ 168.63 (s, C O), 138.56, 138.23, (s, 2C, Ar), 135.66 (s, JC,P 6.5 Hz, 2C, Ar), 128.58, 128.55, 128.48, 128.35, 128.02, 127.99, 127.97, 127.78, 127.56, 127.32 (d, 20C, Ar), 99.04 (s, C-2), 99.01 (d, C-1′), 80.05 (d, C-3′), 78.41 (d, C-2′), 77.30 (d, C-5), 74.93 (t, CH2Ph), 72.76 (d, C-7), 72.49 (t, CH2Ph), 71.69, 71.63 (d, 2C, C-5′, C-6), 69.60 (t, JC,P 6.1 Hz, POCH2Ph), 69.56 (t, JC,P 7.0 Hz, POCH2Ph), 69.50 (d, C-4′), 66.61 (t, JC,P 7.7 Hz, C-6′), 66.54 (d, C-4), 63.62 (t, C-8), 52.39 (q, CO2CH3), 51.18 (q, OCH3), 35.93 (t, C-3), 17.57, 17.53, 17.50, 17.47, 17.31, 17.20, 17.12 [q, 8C, Si-CH-(CH3)2], 14.03, 13.52, 12.95, 12.78 [d, 4C, Si-CH-(CH3)2]; 31P NMR (CDCl3): δ—0.08; ESI-TOF HRMS: m/z = 1133.4487; calcd for C56H79O17PSi2Na+: 1133.4486.
Alternatively, a solution of 21 (39.9 mg, 0.032 mmol) in CH2Cl2 (2.0 mL) was treated with trifluoroacetic acid (99%, 0.25 mL) at 0 °C for 10 min. Dilution with toluene and azeotropic distillation afforded a crude product which was immediately purified by chromatography (n-hexane/EtOAc 1:1) providing 25 (31.8 mg, 88%) as a colorless oil.
3.20. 2,3-Di-O-benzyl-6-O-(dibenzylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl (methyl 3-deoxy-α-d-manno-2-octulopyranosid)onate (26)
A solution of 25 (6.4 mg, 0.006 mmol) in dry THF (1.2 mL) was treated with TBAF (1 M in THF, 9 μL, 0.009 mmol) at ambient temperature for 15 min. Addition of dry MeOH (1 mL) and concentration provided a crude product which was purified by chromatography (EtOAc) yielding 26 (4.6 mg, 92%) as a colorless oil; +96.0 (c 0.37, MeOH); Rf 0.34 (EtOAc); 1H NMR (MeOD): δ 7.40–7.25 (m, 20H, Ar), 5.18 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 5.07–5.04 (m, 4H, 2 × POCH2Ph), 4.91 (d, 1H, J 11.2 Hz, CHHPh), 4.84–4.82 (m, 1H, CHHPh), 4.75 (d, 1H, J 11.6 Hz, CHHPh), 4.72 (d, 1H, J 11.7 Hz, CHHPh), 4.30–4.27 (m, 1H, H-6′a), 4.26–4.18 (m, 2H, H-5′, H-6′b), 4.10–4.02 (m, 3H, H-4, H-5, H-7), 3.86 (dd, 1H, J3′,2′ 9.6, J3′,4′ 9.0 Hz, H-3′), 3.74 (s, 3H, CO2CH3), 3.70 (dd, 1H, J8a,8b 11.2, J8a,7 2.9 Hz, H-8a), 3.67 (dd, 1H, J6,7 9.3, J6,5 1.1 Hz, H-6), 3.64 (dd, 1H, J8b,7 4.4 Hz, H-8b), 3.51 (app. t, 1H, J4′,5′ 9.3 Hz, H-4′), 3.43 (dd, 1H, H-2′), 3.21 (s, 3H, OCH3), 2.06–2.01 (m, 1H, H-3eq), 1.96 (app. t, 1H, J3ax,3eq = J3ax,4 12.3 Hz, H-3ax); 13C NMR (MeOD): δ 170.39 (s, C O), 140.30, 139.34 (s, 2C, Ar), 135.82 (s, JC,P 6.6 Hz, Ar), 135.81 (s, JC,P 6.6 Hz, Ar), 129.67, 129.66, 129.43, 129.38, 129.24, 129.17, 129.09, 129.07, 128.87, 128.52 (d, 20C, Ar), 100.44 (s, C-2), 100.10 (d, C-1′), 82.37 (d, C-3′), 81.07 (d, C-2′), 77.55 (d, C-5), 76.26 (t, CH2Ph), 74.63 (t, CH2Ph), 73.18 (d, C-6), 72.47 (d, JC,P 7.3 Hz, C-5′), 71.18 (d, C-4′), 70.82 (t, JC,P 5.9 Hz, 2C, 2 × POCH2Ph), 69.98 (d, C-7), 68.23 (t, JC,P 5.5 Hz, C-6′), 67.25 (d, C-4), 64.13 (t, C-8), 52.94 (q, CO2CH3), 51.55 (q, OCH3), 36.47 (t, C-3); 31P NMR (MeOD): δ—1.20; ESI-TOF HRMS: m/z = 891.2966; calcd for C44H53O16PNa+: 891.2963.
3.21. 6-O-Phosphono-α-d-glucopyranosyl-(1→5)-methyl 3-deoxy-α-d-manno-oct-2-ulopyranosidonic acid (sodium salt) (27)
A solution of 26 (8.2 mg, 0.009 mmol) in dry MeOH (1.0 mL) was hydrogenated for 1 h in the presence of 10% Pd–C (1 mg) as described for 13. The suspension was diluted with MeOH and passed through a 0.45 μm syringe filter. The filtrate was made neutral by adding 0.1 M NaOMe (0.1 M in MeOH, 200 μL). Concentration of the filtrate afforded the debenzylated methyl ester which was saponified with 0.01 M aq NaOH (1.5 mL) at ambient temperature for 1 h. The solution was made neutral by addition of DOWEX 50 H+ resin. The ion exchange resin was filtered off and the filtrate was lyophilized. Purification of the residue on a Bio-Gel PD10 column (H2O) and freeze-drying of pooled fractions provided 27 (5.2 mg, 98%) as a colorless amorphous solid; +97.5 (c 0.49, H2O); 1H NMR (D2O): δ 5.11 (d, 1H, J1′,2′ 4.1 Hz, H-1′), 4.15–4.06 (m, 3H, H-4, H-7, H-5′), 4.05–4.00 (m, 2H, H-5, H-6′a), 3.90 (dd, 1H, J8a,8b 11.8, J8a,7 2.8 Hz, H-8a), 3.78–3.68 (ddd, 1H, J6′b,6′a 12.1, J 4.7, J 1.9 Hz, H-6′b), 3.73 (app. t, 1H, J3′,2′ = J3′,4′ 9.6 Hz, H-3′), 3.62 (dd, 1H, J8b,7 6.1 Hz, H-8b), 3.59 (app. t, 1H, J4′,5′ 9.7 Hz, H-4′), 3.55–3.52 (m, 2H, H-6, H-2′), 3.11 (s, 3H, OCH3), 1.96 (ddd, 1H, J3eq,3ax 12.9, J3eq,4 4.9, J3eq,5 0.7 Hz, H-3eq), 1.82 (app. t, 1H, J3ax,4 12.5 Hz, H-3ax); 13C NMR data: see Table 1; 31P NMR (D2O): δ 4.65; ESI-TOF HRMS: m/z = 493.0966; calcd for C15H26O16P-: 493.0964.
3.22. 4-O-Acetyl-2,3-di-O-benzyl-6-O-(diphenylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-α-d-manno-oct-2-ulopyranosid]onate (28)
A solution of 24 (17.9 mg, 0.014 mmol) in dry CH2Cl2 was treated with triethylamine trihydrofluoride (117 μL, 0.719 mmol) for 4 h at ambient temperature. Another portion of triethylamine trihydrofluoride (117 μL, 0.719 mmol) was added and stirring was continued for 12 h. The colorless solution was added dropwise into ice-cold aq NaHCO3 and extracted with CH2Cl2 (4 × 10 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated. The residue was purified by chromatography (toluene/EtOAc 1:1) which afforded 28 (11.8 mg, 82%) as a colorless oil; +51.6 (c 1.13, CHCl3); Rf 0.24 (toluene/EtOAc 1:1); 1H NMR (CDCl3, ref. to TMS at 0.00 ppm): δ 7.37–7.10 (m, 22H, Ar), 6.84–6.80 (m, 2H, Ar), 5.05 (app. t, 1H, J4′,3′ = J4′,5′ 9.5 Hz, H-4′), 5.04 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 4.83 (d, 1H, J 11.8 Hz, CHHPh), 4.82 (d, 1H, J 11.5 Hz, CHHPh), 4.69 (d, 1H, J 11.5 Hz, CHHPh), 4.61 (d, 1H, J 11.7 Hz, CHHPh), 4.44 (d, 1H, J 11.9 Hz, CHHPh), 4.37 (d, 1H, J 11.8 Hz, CHHPh), 4.19–4.15 (m, 2H, H-5′, H-6′a), 4.12 (bs, 1H, H-5), 4.09–4.03 (m, 2H, H-6′b, H-7), 4.00 (app. t, 1H, J3′,2′ 9.5 Hz, H-3′), 3.85 (ddd, 1H, J4,3ax 11.9, J4,3eq 4.5, J4,5 2.4 Hz, H-4), 3.79 (dd, 1H, J8a,8b 11.1, J8a,7 3.4 Hz, H-8a), 3.76 (s, 3H, CO2CH3), 3.73 (s, 3H, PhOCH3), 3.63 (dd, 1H, J8b,7 4.9 Hz, H-8b), 3.59 (d, 1H, J6,7 9.2 Hz, H-6), 3.52 (dd, 1H, H-2′), 3.18 (s, 3H, OCH3), 2.19 (dd, 1H, J3eq,3ax 12.7 Hz, H-3eq), 1.98 (app. t, 1H, H-3ax), 1.92 (s, 3H, COCH3); 13C NMR (CDCl3): δ 169.44 (s, COCH3), 168.46 (s, C-1), 159.13 (s, Ar), 150.57 (s, JC,P 6.9 Hz, Ar), 150.45 (s, JC,P 6.5 Hz, Ar), 138.04, 136.99, 130.15 (s, 3C, Ar), 129.69, 129.64, 128.85, 128.75, 128.52, 128.49, 128.01, 127.83, 125.22, 125.14 (d, 18C, Ar), 120.16 (d, JC,P 4.6 Hz, 2C, Ar), 120.10 (d, JC,P 4.9 Hz, 2C, Ar), 113.85 (d, 2C, Ar), 99.15 (s, C-2), 98.49 (d, C-1′), 80.60 (d, C-2′), 79.04 (d, C-3′), 75.20 (t, CH2Ph), 74.74 (t, CH2Ph), 73.98 (d, C-5), 72.95 (d, C-4), 72.39 (d, C-6), 69.89 (t, CH2Ph), 69.82 (d, C-4′), 69.02 (d, JC,P 8.3 Hz, C-5′), 68.59 (d, C-7), 67.07 (t, JC,P 5.5 Hz, C-6′), 63.96 (t, C-8), 55.19 (q, PhOCH3), 52.55 (q, CO2CH3), 51.01 (q, OCH3), 32.66 (t, C-3), 20.84 (q, COCH3); 31P NMR (CDCl3): δ–11.95; ESI-TOF HRMS: m/z = 1020.3773; calcd for C52H59O18PNH4+: 1020.3777.
3.23. 4-O-Acetyl-2,3-di-O-benzyl-6-O-(diphenylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-deoxy-4-O-(4-methoxybenzyl)-α-d-lyxo-hept-2-ulopyranosid]onate (29)
A solution of 28 (27.0 mg, 0.027 mmol) in dry CH2Cl2 (3.5 mL) was treated with sodium periodinate on silica (15 w%, 77 mg, 0.054 mmol) for 1.5 h at −10 °C under light protection. Excessive reagent was destroyed with ethylene glycol (3 w% in water, 56 μL, 0.027 mmol). The mixture was diluted with CHCl3 and water, the phases were separated and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were dried (MgSO4), filtered, and concentrated. The residue was treated with sodium borohydride (3.1 mg, 0.081 mmol) in dry MeOH (3.5 mL) at −5 °C for 30 min. The mixture was partitioned between EtOAc and aq NH4Cl, the aqueous phase was extracted with EtOAc (3 × 5 mL) and the combined organic phases were dried (MgSO4). Filtration and concentration afforded a crude product which was purified by HP-column chromatography (toluene/EtOAc 2:1) providing 29 (9.5 mg, 36%) as a colorless oil: +48.0 (c 0.42, CHCl3); Rf 0.32 (toluene/EtOAc 1:1); 1H NMR (CDCl3, ref. to TMS at 0.00 ppm): δ 7.36–7.19 (m, 16H, Ar), 7.18–7.10 (m, 6H, Ar), 6.84–6.81 (m, 2H, Ar), 5.03 (app. t, 1H, J4′,3′ = J4′,5′ 9.8 Hz, H-4′), 4.98 (d, 1H, J1′,2′ 3.4 Hz, H-1′), 4.83 (d, 1H, J 11.5 Hz, CHHPh), 4.79 (d, 1H, J 11.5 Hz, CHHPh), 4.66 (d, 1H, J 11.5 Hz, CHHPh), 4.61 (d, 1H, J 11.8 Hz, CHHPh), 4.47 (d, 1H, J 11.8 Hz, CHHPh), 4.38 (d, 1H, J 11.6 Hz, CHHPh), 4.22–4.18 (m, 1H, H-5′), 4.15 (ddd, J6′a,6′b 11.1, J6′a,P 6.1, J6′a,5′ 2.4 Hz, H-6′a), 4.03–3.96 (m, 3H, H-3′, H-6′b, H-5), 3.90–3.84 (m, 2H, H-4, H-7a), 3.82–3.76 (m, 4H, H-7b, CO2CH3), 3.74–3.70 (m, 4H, H-6, PhOCH3), 3.49 (dd, 1H, J2′,3′ 9.6, H-2′), 3.20 (s, 3H, OCH3), 2.99 (bs, 1H, OH), 2.23 (dd, 1H, J3eq,3ax 12.7, J3eq,4 4.3 Hz, H-3eq), 2.02 (app. t, 1H, J3ax,4 12.2 Hz, H-3ax), 1.90 (s, 3H, COCH3); 13C NMR (CDCl3): δ 169.41 (s, COCH3), 168.56 (s, C-1), 159.16 (s, Ar), 150.60 (s, JC,P 7.5 Hz, Ar), 150.47 (s, JC,P 6.6 Hz, Ar), 138.15, 137.11, 130.14 (s, 3C, Ar), 129.68, 129.63, 128.92, 128.69, 128.47, 128.45, 128.40, 127.95, 127.75, 125.20, 125.12 (d, 18C, Ar), 120.15 (d, 2C, JC,P 5.1 Hz, Ar), 120.09 (d, 2C, JC,P 5.3 Hz, Ar), 113.85 (d, 2C, Ar), 99.15 (s, C-2), 98.22 (d, C-1′), 80.54 (d, C-2′), 78.95 (d, C-3′), 75.20 (t, CH2Ph), 74.56 (t, CH2Ph), 73.59 (d, C-5), 73.09 (d, C-4), 72.70 (d, C-6), 69.99 (t, CH2Ph), 69.66 (d, C-4′), 68.97 (d, JC,P 7.8 Hz, C-5′), 67.04 (t, JC,P 5.5 Hz, C-6′), 60.68 (t, C-7), 55.19 (q, PhOCH3), 52.60 (q, CO2CH3), 51.01 (q, OCH3), 32.71 (t, C-3), 20.81 (q, COCH3); 31P NMR (CDCl3): δ –11.96; ESI-TOF HRMS m/z: 990.3668; calcd for C51H57O17PNH4+: 990.3672.
3.24. 6-O-Phosphono-α-d-glucopyranosyl-(1→5)-(methyl 3-deoxy-α-d-lyxo-hept-2-ulopyranosid)onic acid (sodium salt) (30)
A suspension of compound 29 (8.0 mg, 0.008 mmol) in dry MeOH was hydrogenated for 36 h and processed as described for 13. The solution obtained upon removal of the catalyst was concentrated. The residue was dried and dissolved in dry MeOH (1.0 mL). PtO2 (1 mg) was added under an Ar atmosphere and hydrogenation was continued for 4 h at rt. The suspension was diluted with MeOH and passed through a 0.45 μm syringe filter. The filtrate was concentrated, and the residue was stirred with 0.01 M aq. NaOH (3.0 mL) at ambient temperature for 3 h. The solution was neutralized by addition of DOWEX 50 H+ resin, the ion-exchange resin was filtered off and the filtrate was lyophilized. Purification of the residue by HILIC (SeQuant ZIC®-HILIC, 5 μm, 250 × 10 mm pre-packed from Merck, 5:1 → 2:3 acetonitrile/water) followed by desalting on a PD10 column (H2O) afforded 30 (2.2 mg, 52%) as an amorphous colorless solid: +69.2 (c 0.39, D2O); 1H NMR (D2O): δ 4.88 (d, 1H, J1′,2′ 4.0 Hz, H-1′), 4.19–4.15 (m, 1H, H-5′), 4.12 (ddd, 1H, J4,3ax 12.3, J4,3eq 4.8, J4,5 3.0 Hz, H-4), 4.05 (ddd, 1H, J6′a,6′b 11.6, J 6.2, J 3.2 Hz, H-6′a), 3.96 (dd, 1H, J7a,7b 11.5, J7a,6 8.3 Hz, H-7a), 3.89–3.85 (m, 2H, H-5, H-6′b), 3.81 (dd, 1H, J7b,6 4.4 Hz, H-7b), 3.75 (br dd, 1H, H-6), 3.72 (app. t, 1H, J3′,2′ = J3′,4′ 9.7 Hz, H-3′), 3.53 (app. t, 1H, J4′,5′ 9.7 Hz, H-4′), 3.51 (dd, 1H, H-2′), 3.14 (s, 3H, OCH3), 2.01 (dd, 1H, J3eq,3ax 13.0, H-3eq), 1.83 (app. t, 1H, H-3ax); 13C NMR data: see Table 1; 31P NMR (D2O): δ 2.40; ESI-TOF HRMS: m/z = 463.0857; calcd for C14H24O15P-: 463.0858.
3.25. 2,3-Di-O-benzyl-6-O-(dibenzylphosphoryl)-α-d-glucopyranosyl-(1→5)-methyl (methyl 3-deoxy-α-d-lyxo-hept-2-ulopyranosid)onate (31)
Sodium metaperiodinate on silica (15 w%, 70 mg, 0.049 mmol) was added to a solution of 26 (21.4 mg, 0.025 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C under light protection. The suspension was allowed to warm up to ambient temperature during 1.5 h. Excessive reagent was destroyed by addition of aq ethylene glycol (3 w%, 51 μL, 0.025 mmol). The product was partitioned between CHCl3 and brine, the aqueous phase extracted with CH2Cl2 (2 × 5 mL), and the combined organic layers were dried (MgSO4) and concentrated. The crude aldehyde was dissolved in dry MeOH (3.0 mL) and treated with sodium borohydride (2.8 mg, 0.074 mmol) at 0 °C for 10 min. The solution was extracted with CH2Cl2 (3 × 10 mL) and aq NH4Cl, and the organic phase was dried (MgSO4), and concentrated. The crude material was purified by HP-column chromatography (n-hexane/EtOAc 1:4 → 0:1) to yield 31 (9.6 mg, 47%) as a colorless oil; +29.2 (c 0.99, CHCl3); Rf 0.59 (EtOAc); 1H NMR (CDCl3): δ 7.42–7.28 (m, 20H, Ar), 5.03–4.95 (m, 4H, 2 × POCH2Ph), 4.93 (d, 1H, J 11.6 Hz, CHHPh), 4.88 (d, 1H, J 11.3 Hz, CHHPh), 4.85 (d, 1H, J 11.7 Hz, CHHPh), 4.81 (d, 1H, J1′,2′ 3.5 Hz, H-1′), 4.65 (d, 1H, J 11.9 Hz, CHHPh), 4.23 (ddd, 1H, J6′a,6′b 11.8, J 8.1, J 5.0 Hz, H-6′a), 4.09 (ddd, 1H, J 9.2, J 2.1 Hz, H-6′b), 4.06–4.00 (m, 1H, H-4), 3.94–3.76 (m, 9H, H-3′, H-5′, H-5, H-6, H-7a, H-7b, CO2CH3), 3.52–3.36 (m, 4H, H-2′, H-4′, 2 × OH), 3.21 (s, 3H, OCH3), 3.21–3.18 (m, 1H, OH), 2.19 (ddd, 1H, J3eq,3ax 12.5, J3eq,4 4.6, J3eq,5 0.6 Hz, H-3eq), 1.72–1.62 (m, 1H, H-3ax); 13C NMR (CDCl3): δ 168.34 (s, C O), 138.35, 136.76 (s, 2C, Ar), 135.53 (s, JC,P 6.5 Hz, Ar), 135.51 (s, JC,P 6.8 Hz, Ar), 128.85, 128.67, 128.63, 128.60, 128.52, 128.03, 128.02, 127.99 (d, 20C, Ar), 100.37 (d, C-1′), 99.02 (s, C-2), 80.50 (d, C-3′), 79.22 (d, C-5), 79.06 (d, C-2′), 75.61 (t, CH2Ph), 74.77 (t, CH2Ph), 72.03 (d, JC,P 5.7 Hz, C-5′), 71.66 (d, C-6), 69.87 (d, C-4′), 69.75 (t, JC,P 5.4 Hz, POCH2Ph), 69.69 (t, JC,P 5.6 Hz, POCH2Ph), 66.27 (t, JC,P 5.1 Hz, C-6′), 66.03 (d, C-4), 60.59 (t, C-7), 52.58 (q, CO2CH3), 51.06 (q, OCH3), 36.32 (t, C-3); 31P NMR (CDCl3): δ 0.04; ESI-TOF HRMS: m/z = 861.2855; calcd for C43H51O15PNa+: 861.2858.
Deprotection of 31: A suspension of compound 31 (7.4 mg, 0.009 mmol) and 10% Pd–C (1 mg) in dry MeOH (1.0 mL) was hydrogenated for 1 h at room temperature as described for 13. The suspension was diluted with MeOH, passed through a 0.45 μm syringe filter and the filtrate was made neutral by addition of 0.1 M NaOMe in MeOH (100 μL). Concentration afforded the debenzylated methyl ester which was saponified with 0.01 M NaOH (1.5 mL) at ambient temperature for 8 h. The solution was neutralized by addition of DOWEX 50 H+ ion-exchange resin. The resin was filtered off and the filtrate was lyophilized. Purification of the residue on BioGel PD10 (H2O) and freeze-drying of pooled fractions provided 30 (3.9 mg, 87%) as a colorless amorphous solid.
3.26. Methyl (methyl 4,5;7,8-di-O-isopropylidene-d-glycero-α-d-talo-oct-2-ulopyranosid)onate (34)
Sodium hydride (0.13 g, 3.28 mmol) was added in small portions to an ice-cold solution of methyl (3-O-acetyl-4,5;7,8-di-O-isopropylidene-d-glycero-α-d-galacto-oct-2-ulopyranosyl)onate 32 (0.85 g, 2.19 mmol) and iodomethane (0.16 mL, 2.63 mmol) in dry DMF (10 mL). After vigorous stirring at 0 °C for 45 min, cleavage of the 3-O-acetyl group was induced by addition of dry MeOH (15 mL) forming NaOMe in situ. After 15 h at ambient temperature the solution was partitioned between EtOAc and aq NH4Cl. The aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic phases were dried (MgSO4), filtered, and concentrated. The crude product was purified by chromatography (toluene/EtOAc 2:1) affording 33 (680 mg, 86%) as a colorless oil with minor impurities.
A solution of 33 (0.67 g, 1.85 mmol) in dry CH2Cl2 (15 mL) was treated with Dess-Martin periodinane (1.57 g, 3.70 mmol) at ambient temperature for 17 h. The mixture was dissolved in Et2O (150 mL) and aq NaHCO3 (50 mL) containing Na2S2O3 (5 g) and was stirred for 30 min. The organic phase was washed with aq NaHCO3, dried (MgSO4), filtered, and concentrated. The residue was dissolved in dry MeOH (15 mL) and treated with borane ammonia complex (0.08 g, 2.50 mmol) at 0 °C for 15 min. After dilution with dry MeOH the solvent was removed. The residue was partitioned between CH2Cl2 and aq NH4Cl and the aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic phases were dried (MgSO4), filtered, and concentrated. Chromatography of the residue (2:1 toluene/EtOAc) gave 34 (0.50 g, 75% based on 33) as a colorless oil; +57.0 (c 1.19, CHCl3); Rf 0.40 (toluene/EtOAc 1:1); 1H NMR (CDCl3): δ 4.46 (dd, 1H, J4,5 6.7, J4,3 4.2 Hz, H-4), 4.44 (ddd, 1H J7,6 7.8, J7,8a 6.1, J7,8b 4.6 Hz, H-7), 4.33 (dd, 1H, J5,6 2.6 Hz, H-5), 4.16 (dd, 1H, J8a,8b 8.8, H-8a), 4.10 (dd, 1H, H-8b), 3.92 (dd, 1H, J3,OH 7.8, H-3), 3.82 (s, 3H, CO2CH3), 3.72 (dd, 1H, H-6), 3.25 (s, 3 H, OCH3), 3.01 (d, 1H, OH), 1.51 [s, 3H, C(CH3)], 1.44 [s, 3H, C(CH3)], 1.38 [s, 6H, 2 × C(CH3)]; 13C NMR (CDCl3): δ 167.98 (s, C O), 109.95 [s, C(CH3)2], 109.41 [s, C(CH3)2], 99.92 (s, C-2), 73.90 (d, C-7), 72.40 (d, C-4), 71.44 (d, C-5), 69.75 (d, C-6), 68.84 (d, C-3), 66.77 (t, C-8), 52.62 (q, CO2CH3), 51.22 (q, OCH3), 26.90, 25.37, 25.33, 25.18 [q, 4C, 4 × C(CH3)]; ESI-TOF HRMS: m/z = 385.1464; calcd for C16H26O9Na+: 385.1469.
3.27. Methyl (methyl 3-O-benzyl-4,5;7,8-di-O-isopropylidene-d-glycero-α-d-talo-oct-2-ulopyranosid)onate (35)
A solution of 34 (184 mg, 0.508 mmol) in dry DMF (8.5 mL) was treated with sodium hydride (41 mg, 1.016 mmol) and stirred for 5 min at 0 °C. Benzyl bromide (121 μL, 1.016 mmol) was added dropwise and the solution was stirred for 40 min at 0 °C. Dry MeOH (1.1 mL) was added to the cold mixture and after 5 min the solution was neutralized with DOWEX 50 H+ resin. The resin was filtered off and washed thoroughly with EtOAc. The filtrate was washed successively with aq NaHCO3 and brine, dried (MgSO4), and concentrated. The crude product was purified by chromatography (toluene/EtOAc 5:1) providing 35 (217 mg, 94%) as a colorless oil: −18.5 (c 1.04, CHCl3); Rf 0.29 (toluene/EtOAc 4:1); 1H NMR (CDCl3): δ 7.37–7.26 (m, 5H, Ar), 4.80 (s, 2H, CH2Ph), 4.42 (dd, 1H, J4,5 7.7, J4,3 3.6 Hz, H-4), 4.40 (ddd, 1H, J7,6 7.3, J7,8a 6.3, J7,8b 4.9 Hz, H-7), 4.30 (dd, 1H, J5,6 2.2 Hz, H-5), 4.12 (dd, 1H, J8a,8b 8.9, Hz, H-8a), 4.02 (dd, 1H, H-8b), 3.77 (s, 3H, CO2CH3), 3.61 (d, 1H, H-3), 3.55 (dd, 1H, H-6), 3.30 (s, 3H, OCH3), 1.54 [s, 3H, C(CH3)], 1.41 [s, 3H, C(CH3)], 1.36 [s, 3H, C(CH3)], 1.34 [s, 3H, C(CH3)]; 13C NMR (CDCl3): δ 167.04 (s, C O), 137.62 (s, Ar), 128.43, 128.30, 127.72 (d, 5C, Ar), 110.60 [s, C(CH3)2], 109.11 [s, C(CH3)2], 101.90 (s, C-2), 75.91 (d, C-3), 73.90 (d, C-4), 73.62 (t, CH2Ph), 73.42 (d, C-7), 72.38 (d, C-5), 70.06 (d, C-6), 66.78 (t, C-8), 52.36 (q, CO2CH3), 50.83 (q, OCH3), 26.84, 25.28, 25.26, 25.07 [q, 4C, 4 × C(CH3)]; ESI-TOF HRMS: m/z = 470.2389; calcd for C23H32O9NH4+: 470.2385.
3.28. Methyl (methyl 3-O-benzyl-7,8-O-isopropylidene-d-glycero-α-d-talo-oct-2-ulopyranosid)onate (36)
A solution of 35 (101 mg, 0.223 mmol), p-toluenesulfonic acid monohydrate (42 mg, 0.223 mmol) and distilled water (84 μL, 4.460 mmol) in dry acetone (3.0 mL) was stirred at ambient temperature for 15 min. Et3N (160 μL) was added, the solution was stirred for 30 min, and concentrated. The crude product was purified by chromatography (toluene/EtOAc 2:1) affording 36 (65 mg, 71%) as a colorless amorphous solid; +36.8 (c 0.80, CHCl3); Rf 0.41 (toluene/EtOAc 1:1); 1H NMR (CDCl3): δ 7.35–7.27 (m, 3H, Ar), 7.24–7.21 (m, 2H, Ar), 4.81 (d, 1H, J 10.9 Hz, CHHPh), 4.56 (d, 1H, J 11.0 Hz, CHHPh), 4.48 (ddd, 1H, J7,6 7.9, J7,8a 6.3, J7,8b 5.1 Hz, H-7), 4.18 (dd, 1H, J8a,8b 8.8 Hz, H-8a), 4.08 (dd, 1H, J3,4 3.3, J3,5 1.2 Hz, H-3), 4.05 (dd, 1H, H-8b), 3.96 (app. td, 1H, J4,OH 9.8, J4,5 3.4 Hz, H-4), 3.93–3.89 (m, 1H, H-5), 3.71 (s, 3H, CO2CH3), 3.55 (dd, 1H, J6,5 1.2 Hz, H-6), 3.22 (s, 3H, OCH3), 3.06 (d, 1H, JOH,5 11.8 Hz, OH), 2.92 (d, 1H, JOH,4 9.8 Hz, OH), 1.42 [s, 3H, C(CH3)], 1.37 [s, 3H, C(CH3)]; 13C NMR (CDCl3): δ 167.36 (s, C O), 136.93 (s, Ar), 128.55, 128.15, 127.78 (d, 5C, Ar), 109.19 [s, C(CH3)2], 100.70 (s, C-2), 79.86 (d, C-3), 76.55 (t, CH2Ph), 73.35 (d, C-7), 73.28 (d, C-6), 68.69 (d, C-5), 66.96 (d, C-4), 66.77 (t, C-8), 52.44 (q, CO2CH3), 51.09 (q, OCH3), 26.73, 25.31 [q, 2C, 2 × C(CH3)]; ESI-TOF HRMS: m/z = 435.1624; calcd for C20H28O9Na+: 435.1626.
3.29. Methyl [methyl 3-O-benzyl-7,8-O-isopropylidene-4-O-(4-methoxybenzyl)-d-glycero-α-d-talo-oct-2-ulopyranosid]onate (37)
A mixture of 36 (64 mg, 0.155 mmol) and dibutyltin oxide (42 mg, 0.171 mmol) in dry toluene (3.5 mL) was heated to reflux using a Dean-Stark apparatus for 3 h. The solution was allowed to cool to ambient temperature followed by consecutive addition of dry DMF (144 μL, 1.862 mmol), 4-methoxybenzyl chloride (105 μL, 0.776 mmol), and tetrabutylammonium iodide (63 mg, 0.171 mmol). After stirring at 60 °C for 16 h the mixture was diluted with EtOAc and washed successively with HCl (1 M), aq NaHCO3, aq Na2S2O3 (50 g/L), and brine. The organic phase was dried (MgSO4), filtered, and concentrated. Chromatography of the residue (toluene/EtOAc 5:1) gave 37 (67 mg, 81%) as a colorless oil; +28.0 (c 0.90, CHCl3); Rf 0.51 (toluene/EtOAc 2:1); 1H NMR (CDCl3): δ 7.35–7.24 (m, 5H, Ar), 7.22–7.20 (m, 2H, Ar), 6.91–6.88 (m, 2H, Ar), 4.97 (d, 1H, J 10.7 Hz, CHHPh), 4.81 (d, 1H, J 11.3 Hz, CHHPh), 4.56–4.51 (m, 1H, H-7), 4.52 (d, 1H, J 11.2 Hz, CHHPh), 4.49 (d, 1H, J 10.7 Hz, CHHPh), 4.19 (dd, 1H, J8a,8b 8.8, J8a,7 6.3 Hz, H-8a), 4.17–4.14 (m, 2H, H-3, H-5), 4.04 (dd, 1H, J8b,7 5.4 Hz, H-8b), 3.82–3.80 (m, 1H, H-4), 3.81 (s, 3H, PhOCH3), 3.68 (s, 3H, CO2CH3), 3.68 (d, 1H, JOH,5 10.1 Hz, OH), 3.46 (dd, 1H, J6,7 7.7, J6,5 1.3 Hz, H-6), 3.19 (s, 3 H, OCH3), 1.43 [s, 3H, C(CH3)], 1.37 [s, 3H, C(CH3)]; 13C NMR (CDCl3): δ 167.47 (s, C O), 159.27, 137.22, 130.03 (s, 3C, Ar), 129.20, 128.36, 128.19, 127.95, 113.86 (d, 9C, Ar), 109.07 [s, C(CH3)2], 100.79 (s, C-2), 78.20 (d, C-3), 75.99 (t, CH2Ph), 74.04 (d, C-6), 73.62, 73.60 (d, 2C, C-4, C-7), 69.36 (t, CH2Ph), 66.87 (t, C-8), 65.84 (d, C-5), 55.26 (q, PhOCH3), 52.44 (q, CO2CH3), 51.07 (q, OCH3), 26.68, 25.40 [q, 2C, 2 × C(CH3)]; ESI-TOF HRMS: m/z = 550.2641; calcd for C28H36O10NH4+: 550.2647.
3.30. 2,3-Di-O-benzyl-4,6-O-benzylidene-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-O-benzyl-7,8-O-isopropylidene-4-O-(4-methoxybenzyl)-d-glycero-α-d-talo-oct-2-ulopyranosid]onate (38)
A suspension of predried compounds 37 (34.6 mg, 0.065 mmol) and 10 (52.3 mg, 0.084 mmol) in dry CH2Cl2 (1.0 mL) containing 4 Å molecular sieves was stirred at ambient temperature for 1 h. At −30 °C TMSOTf (0.6 μL, 0.004 mmol) in dry CH2Cl2 (0.2 mL) was added in two portions within an interval of 1 h. The mixture was kept at −30 °C for 10 min after complete addition. The promoter was destroyed by addition of Et3N (19 μL, 0.130 mmol) in dry CH2Cl2 (0.3 mL). The suspension was allowed to warm up to ambient temperature, filtered over a pad of Celite®, rinsed with CH2Cl2, and the filtrate was concentrated. Product 38 (26.0 mg, 42%) was isolated by chromatography (n-hexane/EtOAc 3:1) as a colorless oil; +27.8 (c 0.68, CHCl3); Rf 0.45 (n-hexane/EtOAc 3:2, HPTLC); 1H NMR (CDCl3): δ 7.41–7.32 (m, 9H, Ar), 7.31–7.20 (m, 10H, Ar), 7.08–7.03 (m, 3H, Ar), 6.91–6.88 (m, 2H, Ar), 5.43 (s, 1H, CHPh), 5.20 (d, 1H, J1′,2′ 3.8 Hz, H-1′), 5.18 (d, 1H, J 11.9 Hz, CHHPh), 4.91 (d, 1H, J 11.6 Hz, CHHPh), 4.78 (ddd, 1H, J7,6 9.6, J7,8b 6.1, J7,8a 3.5 Hz, H-7), 4.73 (d, 1H, J 12.0 Hz, CHHPh), 4.69 (d, 1H, J 11.8 Hz, CHHPh), 4.62 (d, 1H, J 11.6 Hz, CHHPh), 4.54 (d, 1H, J 11.3 Hz, CHHPh), 4.49 (app. dt, 1H, J5′,6′b = J5′,4′ 9.9, J5′,6′a 5.1 Hz, H-5′), 4.48 (d, 1H, J 11.7 Hz, CHHPh), 4.20 (d, 1H, J 11.2 Hz, CHHPh), 4.13–4.12 (m, 2H, H-3, H-5), 3.93 (dd, 1H, J8a,8b 8.9 Hz, H-8a), 3.91 (dd, 1H, J6′a,6′b 10.0 Hz, H-6′a), 3.88 (app. t, 1H, J3′,2′ = J3′,4′ 9.4 Hz, H-3′), 3.84 (dd, 1H, H-8b), 3.78 (s, 3H, PhOCH3), 3.78–3.77 (m, 1H, H-4), 3.70 (s, 3H, CO2CH3), 3.57 (dd, 1H, H-2′), 3.51 (app. t, 1H, H-6′b), 3.48 (app. t, 1H, H-4′), 3.38 (dd, 1H, J6,5 1.1 Hz, H-6), 3.18 (s, 3H, OCH3), 1.36 [s, 3H, C(CH3)], 1.23 [s, 3H, C(CH3)]; 13C NMR (CDCl3): δ 167.72 (s, C O), 159.33, 139.12, 138.75, 138.62, 137.97, 129.74 (s, 6C, Ar), 129.11, 128.56, 128.13, 128.11, 128.08, 127.94, 127.83, 127.75, 127.31, 127.29, 127.22, 127.02, 126.16, 114.05 (d, 24C, Ar), 109.19 [s, C(CH3)2], 101.59 (s, C-2), 101.02 (d, CHPh), 99.98 (d, C-1′), 83.01 (d, C-4′), 78.87 (d, C-2′), 77.55 (d, C-3′), 76.96 (d, C-3), 75.38 (t, CH2Ph), 75.22 (d, C-4), 74.64 (t, CH2Ph), 73.83 (d, C-6), 72.79 (d, C-5), 71.96 (d, C-7), 71.75 (t, CH2Ph), 70.93 (t, CH2Ph), 69.16 (t, C-6′), 67.16 (t, C-8), 62.61 (d, C-5′), 55.23 (q, PhOCH3), 52.36 (q, CO2CH3), 51.11 (q, OCH3), 27.37, 25.34 [q, 2C, 2 × C(CH3)]; ESI-TOF HRMS: m/z = 985.3979; calcd for C55H62O15Na+: 985.3981.
3.31. 2,3-Di-O-benzyl-α-d-glucopyranosyl-(1→5)-methyl [methyl 3-O-benzyl-4-O-(4-methoxybenzyl)-d-glycero-α-d-talo-oct-2-ulopyranosid]onate (39)
A suspension of 38 (26.4 mg, 0.027 mmol) and p-toluenesulfonic acid monohydrate (0.9 mg, 0.005 mmol) in dry MeOH (1.5 mL) was kept at 0 °C for 1 h followed by stirring at ambient temperature for 24 h. Et3N (10 μL) was added to the mixture, stirring was continued for 10 min, and the solution was concentrated. Chromatography of the residue afforded 39 (16.0 mg, 70%) as a colorless oil; +53.8 (c 0.71, CHCl3); Rf 0.16 (n-hexane/EtOAc 1:4); 1H NMR (CDCl3): δ 7.39–7.27 (m, 16H, Ar), 7.25–7.21 (m, 1H, Ar), 6.91–6.89 (m, 2H, Ar), 5.01 (d, 1H, J 12.0 Hz, CHHPh), 4.92 (d, 1H, J 11.5 Hz, CHHPh), 4.81 (d, 1H, J 11.5 Hz, CHHPh), 4.80 (d, 1H, J1′,2′ 3.4 Hz, H-1′), 4.79 (d, 1H, J 11.0 Hz, CHHPh), 4.61 (d, 1H, J 12.4 Hz, CHHPh), 4.59 (d, 1H, J 11.9 Hz, CHHPh), 4.54 (d, 1H, J 11.8 Hz, CHHPh), 4.52 (d, 1H, J 11.9 Hz, CHHPh), 4.20–4.15 (m, 1H, H-7), 4.12 (d, 1H, JOH,7 6.0 Hz, OH), 4.02–3.99 (m, 2H, H-3, H-5), 3.83–3.79 (m, 4H, PhOCH3, H-5′), 3.78–3.72 (m, 3H, H-3′, H-4, H-8a), 3.68 (s, 3H, CO2CH3), 3.66–3.62 (m, 1H, H-8b), 3.61 (dd, 1H, J6,7 9.2, J6,5 1.7 Hz, H-6), 3.44 (dd, 1H, J2′,3′ 9.6 Hz, H-2′), 3.33–3.28 (m, 1H, H-4′), 3.26–3.19 (m, 2H, H-6′a, H-6′b), 3.16 (s, 3H, OCH3), 1.85 (bt, 1H, JOH,8a = JOH,8b 6.5 Hz, OH), 1.80 (bt, 1H, JOH,6′a = JOH,6′b 6.0 Hz, OH), 0.88 (d, 1H, JOH,4′ 7.5 Hz, OH); 13C NMR (CDCl3): δ 167.81 (s, C O), 159.47, 139.09, 138.59, 137.05, 129.53 (s, 5C, Ar), 129.27, 128.72, 128.71, 128.51, 128.48, 128.32, 127.99, 127.74, 127.35, 126.29, 113.99 (d, 19C, Ar), 101.25 (d, C-1′), 101.10 (s, C-2), 82.07 (d, C-3′), 80.31 (d, C-2′), 76.56, 75.82 (d, 2C, C-3, C-5), 75.26 (t, CH2Ph), 75.07 (d, C-4), 75.00 (t, CH2Ph), 74.39 (t, CH2Ph), 72.55 (d, C-4′), 71.79 (d, C-6), 71.61 (d, C-5′), 70.85 (t, CH2Ph), 68.05 (d, C-7), 64.12 (t, C-8), 62.45 (t, C-6′), 55.27 (q, PhOCH3), 52.46 (q, CO2CH3), 51.04 (q, OCH3); ESI-TOF HRMS: m/z = 857.3351; calcd for C45H54O15Na+: 857.3355.
3.32. α-d-Glucopyranosyl-(1→5)-sodium (methyl d-glycero-α-d-talo-oct-2-ulopyranosid)onate (40)
A suspension of 39 (5.9 mg, 0.007 mmol) in dry MeOH (1 mL) was hydrogenated for 4 h with 10% Pd–C (1 mg) as described for 13. Fresh catalyst (1 mg) was added and stirring was continued under H2 for 14 h. The suspension was diluted with MeOH and passed through a 0.45 μm syringe filter. The filtrate was concentrated and the residue was treated with 0.01 M aq. NaOH (2 mL) at ambient temperature for 3 h. The solution was neutralized by addition of DOWEX 50 H+ resin. The ion-exchange resin was filtered off and the filtrate was lyophilized. Purification of the residue on a PD10 column (H2O) and freeze-drying of pooled fractions gave 40 (2.9 mg, 92%) as a colorless amorphous solid; +128.1 (c 0.25, H2O); 1H NMR (D2O): δ 5.13 (d, 1H, J1′,2′ 3.9 Hz, H-1′), 4.21–4.19 (m, 1H, H-5), 4.15–4.10 (m, 2H, H-5′, H-7), 4.01 (app. t, 1H, J4,3 = J4,5 3.3 Hz, H-4), 3.95 (dd, 1H, J8a,8b 11.8, J8a,7 2.8 Hz, H-8a), 3.85 (dd, 1H, J3,5 0.9 Hz, H-3), 3.81 (dd, 1H, J6′a,6′b 12.5, J6′a,5′ 3.6 Hz, H-6′a), 3.75 (dd, 1H, J6′b,5′ 2.4 Hz, H-6′b), 3.69 (dd, 1H, J8b,7 5.9 Hz, H-8b), 3.64 (dd, 1H, J6,7 9.9, J6,5 1.0 Hz, H-6), 3.59 (dd, 1H, J3′,2′ 10.1, J3′,4′ 9.2 Hz, H-3′), 3.52 (dd, 1H, H-2′), 3.44 (dd, 1H, J4′,5′ 10.2 Hz, H-4′), 3.15 (s, 3H, OCH3); 13C NMR data: see Table 1; ESI-TOF HRMS: m/z = 453.1210; calcd for C15H26O14Na+: 453.1215.
Acknowledgments
The authors thank the Austrian Science Fund FWF for financial support (Grant P 24921) and Dr. Andreas Hofinger for recording NMR spectra.
References
- 1.Peleg A.Y., Seifert H., Paterson D.L. Clin. Microbiol. Rev. 2008;21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dijkshoorn L., Nemec A., Seifert H. Nat. Rev. Microbiol. 2007;5:939–951. doi: 10.1038/nrmicro1789. [DOI] [PubMed] [Google Scholar]
- 3.(a) Holst O. In: Endotoxins in Health and Disease. Brade H., Opal S.M., Vogel S.N., Morrison D.C., editors. Marcel Dekker Inc.; New York, Basel: 1999. pp. 115–154. [Google Scholar]; (b) Holst O. FEMS Microbiol. Lett. 2007;271:3–11. doi: 10.1111/j.1574-6968.2007.00708.x. [DOI] [PubMed] [Google Scholar]; (c) Holst O., Molinaro A. In: Microbial Glycobiology. Moran A., Holst O., Brennan P.J., von Itzstein M., editors. Elsevier; Amsterdam: 2009. pp. 29–55. [Google Scholar]
- 4.Kawahara K., Brade H., Rietschel E.T.H., Zähringer U. Eur. J. Biochem. 1987;163:489–495. doi: 10.1111/j.1432-1033.1987.tb10895.x. [DOI] [PubMed] [Google Scholar]
- 5.Vinogradov E.V., Bock K., Petersen B.O., Holst O., Brade H. Eur. J. Biochem. 1997;243:122–127. doi: 10.1111/j.1432-1033.1997.0122a.x. [DOI] [PubMed] [Google Scholar]
- 6.Vinogradov E.V., Müller-Loennies S., Petersen B.O., Meshkov S., Thomas-Oates J.E., Holst O., Brade H. Eur. J. Biochem. 1997;247:82–90. doi: 10.1111/j.1432-1033.1997.00082.x. [DOI] [PubMed] [Google Scholar]
- 7.Zähringer U., Kawahara K., Kosma P. Carbohydr. Res. 2013;378:63–70. doi: 10.1016/j.carres.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Brade L., Brade H. Eur. J. Biochem. 1985;50:687–694. [Google Scholar]
- 9.Brade L., Brade H., Fischer W. Microb. Pathogenesis. 1990;9:355–362. doi: 10.1016/0882-4010(90)90069-3. [DOI] [PubMed] [Google Scholar]
- 10.Brade L., Brade H. Infect. Immun. 1985;50:687–694. doi: 10.1128/iai.50.3.687-694.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Müller-Loennies, S. personal communication.
- 12.Ngyuen H.P., Seto N.O.L., MacKenzie C.R., Brade L., Kosma P., Brade H., Evans S.V. Nat. Struct. Biol. 2003;10:1019–1025. doi: 10.1038/nsb1014. [DOI] [PubMed] [Google Scholar]
- 13.Blackler R.J., Müller-Loennies S., Brade L., Kosma P., Brade H., Evans S.V. In: Anticarbohydrate Antibodies—From Molecular Basis to Clinical Application. Kosma P., Müller-Loennies S., editors. Springer-Verlag; Wien-New York: 2012. Antibody Recognition of Chlamydia LPS: Structural Insights of Inherited Immune Responses; pp. 75–120. [Google Scholar]
- 14.Gomery K., Müller-Loennies S., Brooks C.L., Brade L., Kosma P., Di Padova F., Brade H., Evans S.V. Proc. Natl. Acad. Sci. U.S.A. 2012;109:20877–20882. doi: 10.1073/pnas.1209253109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H., Head J., Kosma P., Brade H., Müller-Loennies S., Sheikh S., McDonald B., Smith K., Cafarella T., Seaton B., Crouch E. Biochemistry. 2008;47:710–720. doi: 10.1021/bi7020553. [DOI] [PubMed] [Google Scholar]
- 16.Unger F.M., Stix D., Schulz G. Carbohydr. Res. 1980;80:191–195. [Google Scholar]
- 17.Van Boeckel C.A.A., van Boom J.H. Tetrahedron Lett. 1980;21:3705–3708. [Google Scholar]
- 18.Paulsen H., Heitmann A. Liebigs Ann. Chem. 1989:655–663. [Google Scholar]
- 19.Ekelöf K., Oscarson S. Carbohydr. Res. 1995;278:289–300. doi: 10.1016/0008-6215(95)00269-3. [DOI] [PubMed] [Google Scholar]
- 20.Yu B., Tao H. Tetrahedron Lett. 2001;42:2405–2407. [Google Scholar]
- 21.Huchel U., Tiwari P., Schmidt R.R. J. Carbohydr. Chem. 2010;29:61–75. [Google Scholar]
- 22.Yu B., Jiansong S. Chem. Commun. 2010:4668–4679. doi: 10.1039/c0cc00563k. [DOI] [PubMed] [Google Scholar]
- 23.Crich D., Cai W. J. Org. Chem. 1999;64:4926–4930. doi: 10.1021/jo990243d. [DOI] [PubMed] [Google Scholar]
- 24.Reed L.A.I.I.I., Ito Y., Masamune S., Sharpless K.B. J. Am. Chem. Soc. 1982;104:6468–6470. [Google Scholar]
- 25.Liotta L.J., Capotosto R.D., Garbitt R.A., Horan B.M., Kelly P.J., Koleros A.P., Brouillette L.M., Kuhn A.M., Targontsidis S. Carbohydr. Res. 2001;331:247–253. doi: 10.1016/s0008-6215(01)00044-1. [DOI] [PubMed] [Google Scholar]
- 26.Ogilvie K.K., Beaucage S.L. Nucleic Acids Res. 1979;7:805–823. doi: 10.1093/nar/7.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiner M., Schmidt R. Tetrahedron: Asymmetry. 2000;11:319–335. [Google Scholar]
- 28.(a) Ferrier R.J., Furneaux R.H. Carbohydr. Res. 1976;52:63–68. The hemiacetal was prepared via the corresponding thioglycoside and final anomeric deprotection following reported procedures: [Google Scholar]; (b) Basu N., Maity S.K., Roy S., Singha S., Ghosh R. Carbohydr. Res. 2011;346:534–539. doi: 10.1016/j.carres.2011.01.003. [DOI] [PubMed] [Google Scholar]; (c) Thomann J.-S., Monneaux F., Creusat G., Spanedda M.V., Heurtault B., Habermacher C., Schuber F., Bourel-Bonnet L., Frisch B. Eur. J. Med. Chem. 2012;51:174–183. doi: 10.1016/j.ejmech.2012.02.039. [DOI] [PubMed] [Google Scholar]