

TGF-β signals through Smad-dependent and non-Smad pathways, depending on cell context. In ovarian cancer cells, the clathrin adaptor Dab2 enhances internalization of the type I TGF-β receptor, restricts its lateral mobility, and inhibits TGF-β–mediated, cholesterol-dependent JNK activation.
Abstract
Transforming growth factor-β (TGF-β) ligands activate Smad-mediated and noncanonical signaling pathways in a cell context–dependent manner. Localization of signaling receptors to distinct membrane domains is a potential source of signaling output diversity. The tumor suppressor/endocytic adaptor protein disabled-2 (Dab2) was proposed as a modulator of TGF-β signaling. However, the molecular mechanism(s) involved in the regulation of TGF-β signaling by Dab2 were not known. Here we investigate these issues by combining biophysical studies of the lateral mobility and endocytosis of the type I TGF-β receptor (TβRI) with TGF-β phosphoprotein signaling assays. Our findings demonstrate that Dab2 interacts with TβRI to restrict its lateral diffusion at the plasma membrane and enhance its clathrin-mediated endocytosis. Small interfering RNA–mediated knockdown of Dab2 or Dab2 overexpression shows that Dab2 negatively regulates TGF-β–induced c-Jun N-terminal kinase (JNK) activation, whereas activation of the Smad pathway is unaffected. Moreover, activation of JNK by TGF-β in the absence of Dab2 is disrupted by cholesterol depletion. These data support a model in which Dab2 regulates the domain localization of TβRI in the membrane, balancing TGF-β signaling via the Smad and JNK pathways.
INTRODUCTION
Transforming growth factor-β (TGF-β) ligands mediate an array of physiological and pathological responses (Gordon and Blobe, 2008; Heldin et al., 2009; Massague, 2012). TGF-β signaling is mediated via two receptor serine/threonine kinases, type I (TβRI) and type II (TβRII), sometimes assisted by different coreceptors (Shi and Massague, 2003; Gordon and Blobe, 2008; Bernabeu et al., 2009; Gatza et al., 2010). TGF-β binds to TβRII, which then recruits and phosphorylates TβRI to induce downstream signaling via the canonical Smad pathway as well as via numerous non-Smad pathways, including c-Jun N-terminal kinase (JNK; Shi and Massague, 2003; Moustakas and Heldin, 2009; Zhang, 2009; Ehrlich et al., 2011).
Cholesterol-enriched domains (lipid rafts) at the plasma membrane were proposed to be involved in the regulation of signaling emanating from many receptors, as well as in their sorting and trafficking (Simons and Toomre, 2000; Mukherjee and Maxfield, 2004; Pike, 2009; Collins et al., 2012). For the TGF-β receptor family, there are numerous reports with differing conclusions on the role(s) of partitioning into raft domains/caveolae versus clathrin-coated pits (CCPs) in signaling regulation (Razani et al., 2001; Di Guglielmo et al., 2003; Mitchell et al., 2004; Hartung et al., 2006; Chen, 2009; Shapira et al., 2012). Using CCP endocytosis-defective TβRI mutants to avoid general blockade of internalization pathways, we recently demonstrated that Smad activation by TβRI does not depend on its internalization via CCPs and occurs at the plasma membrane (Shapira et al., 2012). Thus competition between receptor localization to cholesterol-enriched domains versus clathrin-based structures may play important roles in balancing TGF-β signaling pathways. Because membrane proteins are targeted to CCPs via interactions with adaptor proteins (Bonifacino and Traub, 2003; Traub, 2009; Pelkmans et al., 2005), the expression levels of such specific adaptors can be highly relevant to the distribution of a given receptor (i.e., TGF-β receptors) to distinct domains. The endocytic adaptor protein disabled-2 (Dab2; Keyel et al., 2006; Maurer and Cooper, 2006; Chetrit et al., 2009), which interacts with TGF-β receptors and Smads (Hocevar et al., 2001; Itoh et al., 2003; Penheiter et al., 2010), is a candidate regulator of TGF-β receptor signaling and trafficking (Hocevar et al., 2005; Prunier and Howe, 2005; Hannigan et al., 2010; Penheiter et al., 2010).
In the present work, we manipulated the levels of Dab2 in TGF-β–responsive ovarian cell lines (Hirschhorn et al., 2012) to directly assess the role of Dab2 in regulating TβRI dynamics in the plasma membrane, TβRI internalization, and TGF-β signaling via distinct pathways (Smads vs. the non-Smad JNK pathway). We show that Dab2 expression reduces the lateral diffusion of TβRI while augmenting its CCP-mediated internalization. In contrast to TGF-β–mediated Smad2/3 phosphorylation, which is insensitive to Dab2 levels, JNK activation requires cholesterol and is inhibited by Dab2. On the basis of these results, we propose a mechanism for the balancing of distinct TGF-β signaling pathways by Dab2.
RESULTS
Dab2 restricts the lateral mobility of TβRI and enhances its targeting to CCPs
Dab2 and TβRI interact in a number of cell lines (Hocevar et al., 2001; Itoh et al., 2003). ES-2 is an ovarian clear-cell carcinoma cell line that expresses Dab2 (Chetrit et al., 2011) and responds to TGF-β (Hirschhorn et al., 2012). To validate that Dab2 interacts with TβRI in ES-2 cells, we studied the coimmunoprecipitation of transiently expressed myc-TβRI with endogenous Dab2. As shown in Figure 1, TβRI coprecipitated with Dab2 from lysates derived from these cells. To explore whether the Dab2–TβRI interactions identified in vitro by the coimmunoprecipitation experiment occur at the plasma membrane of live cells, we used fluorescence recovery after photobleaching (FRAP) to measure the effects of altered Dab2 expression levels on the lateral diffusion of TβRI. These studies take advantage of the fact that, on one hand, Dab2 is a CCP component (Keyel et al., 2006; Maurer and Cooper, 2006; Chetrit et al., 2009), and, on the other, it binds TβRI (Figure 1). We showed earlier that interactions of membrane proteins, including TGF-β receptors, with CCPs retard their lateral diffusion (Fire et al., 1995; Yao et al., 2002). Because CCPs are laterally immobile on the time scale of the FRAP measurement, the lateral diffusion of the membrane receptor is retarded by association with these immobile structures in a manner that depends on the kinetics of the interactions. Thus transient interactions of a membrane receptor (e.g., TβRI) with CCPs would lead to reduction in its lateral diffusion coefficient (D), with no effect on its mobile fraction (Rf). This occurs because each receptor molecule spends a fraction of the measurement time bound to the immobile CCP but is free to diffuse during the dissociation period. Conversely, interactions that persist longer then the characteristic diffusion time (τD) are reflected in a reduced Rf, since CCP-associated TβRI molecules would remain bound and immobile on the FRAP time scale. To dissect the effect of Dab2 on the lateral diffusion of TβRI, we manipulated Dab2 expression levels in ES-2 cells by transient small interfering RNA (siRNA)–mediated knockdown (Figure 2A) or stable overexpression of Dab2 (the ES-2-Dab2 cell line; Chetrit et al., 2011; Figure 2A). Typical FRAP curves are depicted in Figure 2, B and C, and averaged data from multiple experiments are shown in Figure 2, D–G. Reduction of Dab2 expression by siRNA significantly increased the D value of TβRI without affecting Rf. Such an effect is the hallmark of the loss of transient interactions. Furthermore, enhanced Dab2 expression levels induced a reduction in the Rf value of TβRI (Figure 2F), suggesting that overexpression of Dab2 augments the association of TβRI with immobile structures (presumably CCPs, in line with the concomitant increase in the CCP-mediated endocytosis of TβRI in these cells; see later discussion of Figure 4). Accordingly, knockdown of clathrin by siRNA increased the D value of myc-TβRI without affecting Rf, similar to the effect of silencing Dab2 by siRNA (Figure 3, A and B). These findings support the notion that the mobility-retarding interactions of TβRI mediated by Dab2 are with CCPs.
FIGURE 1:
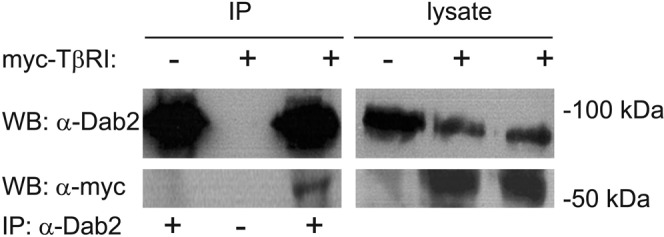
TβRI coprecipitates with endogenous Dab2. ES-2 cells in 10-cm dishes were transfected with vector encoding myc-TβRI (+) or pcDNA3 (–) as described (Materials and Methods). After 48 h, cells were lysed and subjected to either Western blotting (10% of lysate; right) or immunoprecipitation with α-Dab2 (IP; 90% of the lysate) and blotting with α-myc and α-Dab2 antibodies (left). The dash at the bottom (IP: α-Dab2 line) designates a control where protein A–agarose beads without α-Dab2 were used for the immunoprecipitation step.
FIGURE 2:
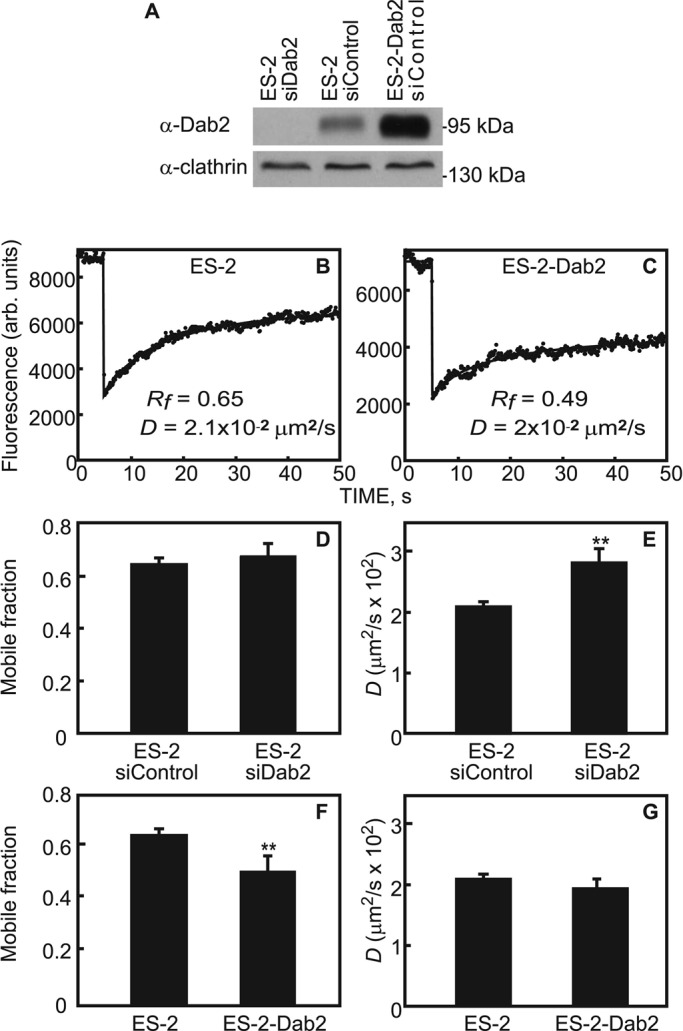
FRAP studies demonstrate that Dab2 reduces the lateral mobility of TβRI. (A) Western blot analysis of Dab2 levels. siRNA transfection (with siDab2 or nontargeting siControl) of ES-2 or ES-2-Dab2 cells and Western blotting for Dab2 and actin were as described in Materials and Methods. (B, C) Typical FRAP curves of the lateral diffusion of myc-TβRI in ES-2 (B) or ES-2-Dabs cells (C). A reduction in Rf of TβRI is seen in Dab2-overexpressing cells (ES-2-Dab2). (D–G) Averaged FRAP data from multiple experiments. Cells were transfected with myc-TβRI alone or siRNA (to Dab2 or control) and subjected to FRAP measurements as described (Materials and Methods). Bars are mean ± SEM of 30–40 measurements. Asterisks indicate significant differences from the paired condition (**p < 0.01, Student's t test). Reducing Dab2 levels in ES-2 cells led to faster diffusion of TβRI, with no effect on its Rf, suggesting a reduction in transient interactions of TβRI with Dab2-containing immobile structures (D, E). Increased Dab2 expression above endogenous levels in ES-2 cells had no further effect on D of TβRI but shifted the effect to a reduction in Rf, suggesting enhanced association of TβRI with the Dab2-containing structures, leading to TβRI immobilization on the FRAP time scale (F, G).
FIGURE 4:
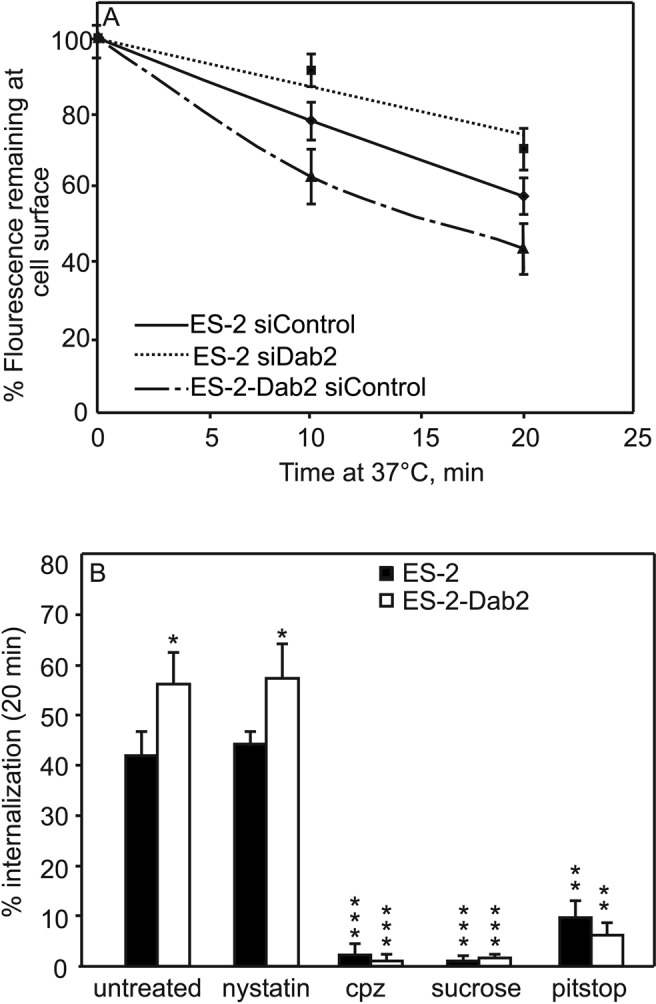
Endocytosis rates of myc-TβRI correlate with expression levels of Dab2. ES-2 or ES-2-Dab2 cells were cotransfected with myc-TβRI and siRNA (nontargeting or siDab2). At 48 h posttransfection, the cell-surface myc-TβRI was labeled at 4°C by mouse α-myc, followed by Alexa 546–GαM Fab′. The cells were warmed to 37°C for the periods shown, returned to 4°C, and fixed (Materials and Methods). The fluorescence of the receptors remaining at the cell surface was measured by the point confocal method (Materials and Methods), focusing the laser beam on defined spots in the focal plane of the plasma membrane, away from vesicular staining. Results are mean ± SEM of 150–200 cells measured in each time point. (A) Internalization of TβRI in cells expressing different Dab2 levels. Intensity at time 0 (i.e., before internalization) for each sample was taken as 100%. The differences between ES-2 siControl and either ES-2 siDab2 or ES-2-Dab2 siControl were significant (p < 0.02 at 10 min and p < 0.01 at 20 min). (B) TβRI internalization in cells with high or low Dab2 levels is abrogated by blocking CCP-mediated endocytosis but not by nystatin. ES-2 or ES-2-Dab2 cells were transfected with myc-TβRI. After 48 h, they were left untreated or treated with CPZ, sucrose (hypertonic medium), Pitstop, or nystatin. The surface receptors were then labeled at 4°C as in A, followed by a 20-min incubation at 37 or 4°C (time 0) in media containing inhibitors where indicated. The cells were fixed, and myc-TβRI endocytosis was measured by the point-confocal method. For each cell type or treatment, the fluorescence intensity of the same sample at time 0 was taken as 100%; the percentage of the fluorescence intensity at 20 min was subtracted to obtain the percentage internalization. Each bar is the mean ± SEM of measurements on 100 cells. Untreated ES-2-Dab2 cells exhibited higher endocytosis than ES-2 cells (*p < 0.05). Each of the CCP internalization-inhibitory treatments blocked TβRI endocytosis in both cell lines (**p < 0.01, ***p < 0.001). Nystatin had no significant inhibitory effect.
FIGURE 3:
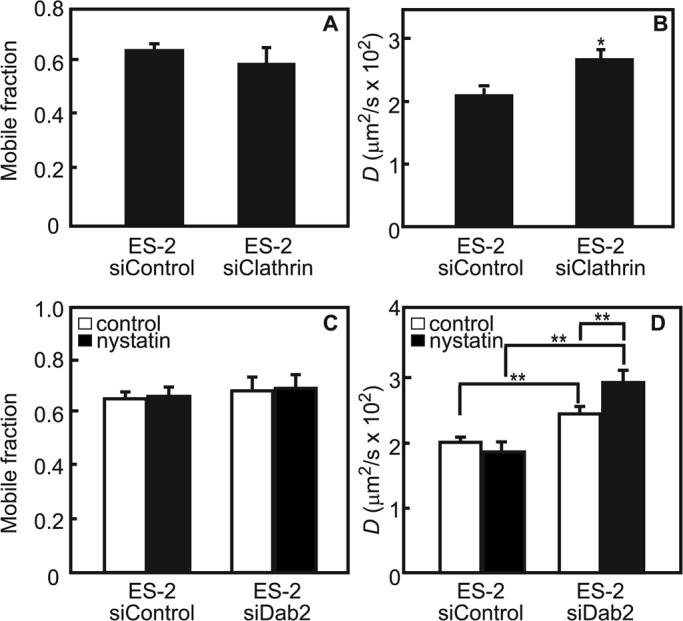
Transient interactions of myc-TβRI with membrane microdomains correlate with Dab2 expression. Cells were cotransfected with myc-TβRI and siRNA (to Dab2, clathrin, or control), treated (or not) with nystatin, and subjected to FRAP measurements as described (Materials and Methods). (A, B) Reducing clathrin levels in ES-2 cells increases TβRI diffusion rate without affecting TβRI Rf, similar to the effect of siRNA for Dab2 (Figure 2). (C, D) Reducing Dab2 levels in ES-2 cells leads to faster diffusion of TβRI, which is further elevated in the present of nystatin with no effect on its Rf, suggesting reduction in transient interactions of TβRI with cholesterol-rich domains. This suggests that the mobility-retarding interactions experienced by TβRI are with CCPs. Bars are mean ± SEM of 30–40 measurements. Asterisks indicate significant differences from the paired condition (*p < 0.05; **p < 0.01, Student's t test).
In view of the reports on interactions of TβRI with rafts/caveolae (Razani et al., 2001; Di Guglielmo et al., 2003), we examined the ability of nystatin, which perturbs cholesterol-dependent raft organization, to modulate TβRI lateral diffusion in the presence or absence of Dab2. As shown in Figure 3, C and D, in cells expressing Dab2 (ES-2 siControl), nystatin had no effect on myc-TβRI mobility. In contrast, depletion of Dab2 (ES-2 siDab2) resulted in nystatin-mediated increase in D (but not in Rf) of TβRI. These results imply that reduced Dab2 levels allow transient interaction of TβRI with cholesterol-rich domains.
The dual interaction of Dab2 with CCPs and TβRI may enhance the association of the latter with CCPs. If that were the case, one would expect the rate of the CCP-mediated endocytosis of TβRI to be affected by the Dab2 expression levels. To test this hypothesis, we used the point-confocal endocytosis assay (Ehrlich et al., 2001; Shapira et al., 2012; described in Materials and Methods) to measure the internalization rate of myc-TβRI. The experiment (Figure 4) is based on cotransfection of siRNA (siDab2 or siControl) and myc-TβRI, followed by fluorescence labeling of cell-surface myc-TβRI in the cold and then incubation at 37°C for defined periods to allow endocytosis (see Materials and Methods). Measurement of the myc-TβRI fluorescence remaining at the cell surface as a function of the incubation time at 37°C revealed a correlation between Dab2 levels and myc-TβRI endocytosis (Figure 4A). Reduction of Dab2 level by siRNA knockdown (ES-2 siDab2) significantly reduced myc-TβRI internalization. Conversely, TβRI endocytosis rate was elevated in ES-2-Dab2 (Dab2 overexpressing) cells. Of note, internalization of TβRI in both ES-2 and ES-2-Dab2 cells, including enhancement by Dab2, was via CCP-mediated endocytosis, as indicated by its abrogation after several treatments known to inhibit the CCP endocytosis pathway (Heuser and Anderson, 1989; Wang et al., 1993; von Kleist et al., 2011; Shapira et al., 2012): hypertonic treatment with sucrose, treatment with chlorpromazine (CPZ), or treatment with Pitstop (Figure 4B). Furthermore, in accord with our previous studies (Shapira et al., 2012), no significant inhibition of TβRI endocytosis was observed after treatment with nystatin (Figure 4B), which is known to perturb caveolar endocytosis.
Dab2 inhibits TGF-β–mediated JNK activation
In view of the foregoing findings on Dab2-TβRI interactions, we next asked whether differences in Dab2 levels are reflected in downstream signaling after TGF-β stimulation. To this end, we transfected ES-2 cells with siDab2 or siControl, stimulated the cells with TGF-β1 (30 or 120 min), and analyzed the activation of the JNK or Smad pathways by immunoblotting for the relevant phosphoproteins (Figure 5A). In siControl-transfected cells, TGF-β1 induced weak transient (not statistically significant) activation (detectable at 30 min, fading at 120 min) of JNK and its downstream effector, c-Jun. Of note, siRNA-mediated reduction of Dab2 strongly and significantly enhanced TGF-β1-induced generation of both phospho-JNK (pJNK) and phospho-c-Jun (pc-Jun). In contrast, the ability of TGF-β1 to activate Smad2/3 (measured by pSmad2/3 levels) was essentially unaffected by siDab2 (Figure 5A). Other non-Smad pathways (Erk, p38, AKT) were not detectably activated by TGF-β1 under either condition of Dab2 expression (unpublished data). We then assessed the effect of a stable increase in the Dab2 level, by exploring the activation of JNK/c-Jun versus Smad in the ES-2-Dab2 cell line, which was also used in the biophysical studies. As shown in Figure 5B, TGF-β–mediated pSmad2/3 formation was readily observed, whereas no JNK activation was detected. To validate this result in a different cell line, we used the Caov3 epithelial ovarian cancer cell line. This choice was based on the negligible amount of endogenous Dab2 expressed in these cells (Figure 5D). Indeed, control cells (transfected with green fluorescent protein [GFP] alone) exhibited JNK activation after TGF-β1 stimulation (30 min), which was abrogated upon transfection with GFP-Dab2. As in Figure 5, A and B, no measurable effects of Dab2 overexpression on pSmad2/3 formation were detected (Figure 5D). Taken together, these results suggest that Dab2 negatively regulates TGF-β–mediated activation of the noncanonical JNK pathway, whereas the canonical Smad pathway is unaffected.
FIGURE 5:

Dab2 reduces JNK activation by TGF-β. (A) Knockdown of Dab2 enhances TGF-β–mediated JNK/c-Jun activation. ES-2 cells were transfected with siRNA (nontargeting or directed against Dab2). At 48 h posttransfection, the cells were serum starved (30 min), stimulated (or not) with 100 pM TGF-β1 (30 or 120 min), and analyzed (see Materials and Methods) by immunoblotting for p-JNK, t-JNK pc-Jun, tc-Jun, pSmad2/3, tSmad2/3, and β-actin. (B) Stable expression of Dab2 abrogates JNK activation. ES-2 and ES-2-Dab2 cells were activated by TGF-β1 (for 30 or 60 min) and analyzed by Western blotting as in A. (C) Quantification of JNK phosphorylation in cells expressing different Dab2 levels (as shown in A and B). Cells transfected with siDab2 demonstrate significantly higher level of pJNK/tJNK ratio compared with ES-2 or ES-2-Dab2 cells. Each bar is the mean ± SEM of three independent experiments (**p < 0.01). (D) Transient overexpression of Dab2 prevents TGF-β stimulation of JNK/c-Jun. Caov3 cells were transfected with GFP (control) or GFP-Dab2. At 24 h posttransfection, the cells were serum starved (60 min), stimulated (or not) with 100 pM TGF-β1 (30 or 60 min), and analyzed as described. All blots shown are of representative experiments (n = 3 in each case).
In view of controversial reports on whether the kinase activity of TβRI is required for the activation of JNK after TGF-β stimulation (Sorrentino et al., 2008; Yamashita et al., 2008; Kim et al., 2009), we investigated the ability of the ALK5 kinase inhibitor SB431542 to perturb TGF-β–mediated pJNK generation in ES-2 cells (treated with siDab2 or siControl). As shown in Figure 6, SB431542 abrogated the formation of only pSmad2/3, but not pJNK or pc-Jun, upon TGF-β stimulation. These findings are in line with reports that TβRI kinase activity is dispensable for activation of the JNK pathway (Sorrentino et al., 2008; Kim et al., 2009).
FIGURE 6:
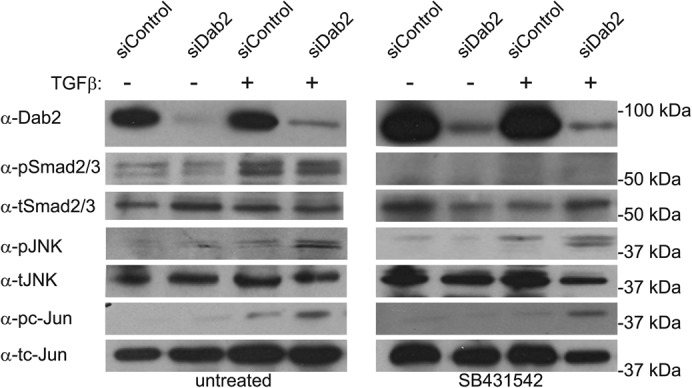
JNK phosphorylation does not require TβRI kinase activity. ES-2 cells were transfected with siRNA (siControl or siDab2). At 48 h posttransfection, the cells were left untreated or treated with SB431542 (10 μM, 1 h), stimulated (or not) with 100 pM TGF-β1 (30 min), and analyzed (see Materials and Methods) by immunoblotting for pJNK, tJNK, pc-Jun, tc-Jun, pSmad2/3, and tSmad2/3. The blot shown is of a representative experiment (n = 3). No significant differences in the fold increase of pJNK/tJNK or of pc-Jun/tc-Jun after TGF-β stimulation were observed between untreated and SB431542-treated cells.
Activation of the JNK pathway by TGF-β is cholesterol dependent
In view of the controversial reports on the regulatory roles of TGF-β receptor localization in cholesterol-rich plasma membrane domains versus CCPs (Razani et al., 2001; Di Guglielmo et al., 2003; Mitchell et al., 2004; Hartung et al., 2006; Chen, 2009; Shapira et al., 2012), we explored the effects of cholesterol-perturbing treatments (metabolic cholesterol depletion or treatment with nystatin) on the ability of Dab2 to modulate TGF-β signaling to the JNK and Smad pathways. The experiments (Figure 7) were conducted on ES-2 cells transfected with either siDab2 or siControl. Reduction of the Dab2 levels by siDab2 enabled the activation of JNK by TGF-β1; however, either cholesterol depletion or nystatin treatment (both of which interfere with raft organization) prevented this activation. As in the other experiments, no effect was observed on Smad2/3 activation (Figure 7). These findings imply opposing roles for Dab2 and cholesterol-rich domains in the activation of the noncanonical JNK pathway by TGF-β (see model in Figure 8).
FIGURE 7:
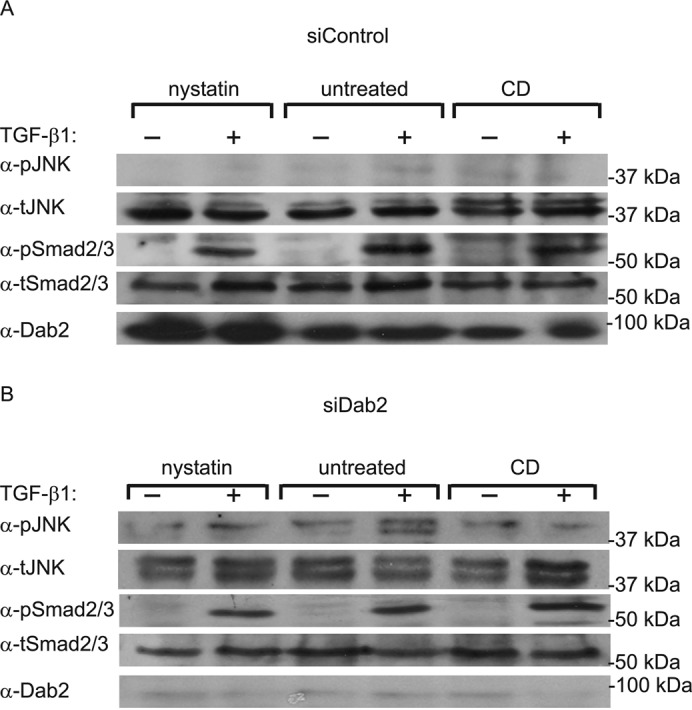
TGF-β-stimulated JNK phosphorylation is cholesterol dependent. ES-2 cells were transfected with siRNA (siControl or siDab2). At 36 h posttransfection, cells were subjected (or not) to metabolic cholesterol depletion (CD; 12 h). Alternatively, cells at 48 h posttransfection were left untreated or treated with nystatin (25 μg/ml, 2 h). In both experimental conditions, cells were then stimulated (or not) with 100 pM TGF-β1 (30 min), and analyzed (see Materials and Methods) by immunoblotting for Dab2, pJNK, tJNK, pSmad2/3, and tSmad2/3. The blot shown is of a representative experiment (n = 3). Although the pJNK/tJNK ratio significantly increased in untreated siDab2 cells after stimulation with TGF-β (p < 0.05), no significant increase was observed in these cells after treatment with nystatin or cholesterol depletion.
FIGURE 8:

A model for regulation of TGF-β signaling by Dab2 and cholesterol. TβRI is distributed in multiple plasma membrane domains, including CCPs and cholesterol-rich domains/caveolae. Although Smad signaling in response to TGF-β is not affected by TβRI localization to either domain, TGF-β stimulation of the JNK pathway is initiated mainly in the cholesterol-rich domains. Dab2, which binds both TβRI and clathrin, targets TβRI preferentially to CCPs, shifting the balance of TGF-β signaling such that Smad signaling is retained while JNK activation is diminished.
DISCUSSION
Diversity in the molecular mechanisms regulating canonical (Smad) and noncanonical TGF-β signaling pathways potentially establishes a basis for their differential regulation in specific cellular contexts. Altering the balance between these pathways can contribute to the cell-type specificity of the response to TGF-β. Here we used ovarian cancer cells to identify two cellular factors with contrasting influence on TGF-β–mediated activation of the JNK/c-Jun signaling axis. Thus, whereas TGF-β–mediated JNK/c-Jun stimulation is reduced by expression of the clathrin-endocytosis adaptor Dab2, it requires cellular cholesterol and is abrogated by cholesterol depletion. Three major observations led to the model proposed in Figure 8 for the regulation of TGF-β signaling to JNK/c-Jun: 1) localization of Dab2 and cholesterol in distinct plasma membrane domains (CCPs/clathrin platforms vs. cholesterol-rich domains); 2) the here-identified regulation of TβRI dynamics and CCP-mediated endocytosis by Dab2; and 3) the negative regulation by Dab2 of the cholesterol-dependent activation of JNK/c-Jun by TGF-β. We propose that Dab2 expression and/or cholesterol depletion diminish the localization of TβRI to lipid rafts, where activation of the JNK/c-Jun axis preferentially occurs.
In addition to their roles as internalization portals for TGF-β receptors, clathrin endocytosis intermediates and caveolae were proposed to regulate TGF-β–induced Smad signaling. Multiple studies identified clathrin-mediated endocytosis as the major TGF-β receptor internalization pathway, whereas a potential contribution of a cholesterol-dependent mechanism has been contentious (Razani et al., 2001; Hayes et al., 2002; Penheiter et al., 2002; Di Guglielmo et al., 2003; Mitchell et al., 2004; Chen, 2009; Chen et al., 2009; Meyer et al., 2011; Hirschhorn et al., 2012; Shapira et al., 2012). Similarly contentious has been the attribution of a requirement for receptor internalization and to endosomal localization for Smad signaling (Hayes et al., 2002; Penheiter et al., 2002; Di Guglielmo et al., 2003; Mitchell et al., 2004; Chen, 2009; Hirschhorn et al., 2012; Shapira et al., 2012). In view of the reports on interactions of Dab2 with TGF-β receptors and Smads (Hocevar et al., 2001; Itoh et al., 2003; Penheiter et al., 2010), we investigated the ability of Dab2 to regulate TβRI endocytosis, TGF-β signaling to distinct pathways, and the potential involvement of TβRI localization to specific domains in these effects. Using ES-2 ovarian cancer cells as a model, we show that Dab2 interacts with TβRI (Figure 1), restricts its lateral diffusion in the plasma membrane (in line with enhanced association with CCPs; Figure 2), and enhances TβRI endocytosis via CCPs (Figure 4). The concomitant retardation of TβRI mobility and the enhancement of its CCP-mediated endocytosis by Dab2 suggest that the reduced lateral mobility is due to interactions with CCPs. These findings are in accord with our demonstration (Fire et al., 1991, 1995) that interactions of membrane proteins bearing CCP internalization signals with CCPs (which are immobile on the FRAP time scale) reduces their lateral diffusion rates or Rf values, depending on the strength of these interactions. Accordingly, siRNA-mediated Dab2 knockdown augmented the diffusion rate of TβRI, suggesting that interaction of TβRI with CCPs mediated by endogenous Dab2 in ES-2 cells is dynamic (transient) on the FRAP time scale. Overexpression of Dab2 (ES-2-Dab2 cells) stabilized these interactions to the degree that reduction in Rf of TβRI was observed (Figure 2). This behavior is analogous to that detected upon altering the CCP internalization signal on a membrane protein from an intermediate to a strong one (Fire et al., 1995). Of note, alteration of the coated pit structure upon Dab2 overexpression (Chetrit et al., 2009) may also contribute to this last effect. The present results support our recent demonstration that the main internalization pathway for TβRI is CCP-mediated endocytosis (Shapira et al., 2012) and indicate that the enhancement in TβRI endocytosis by Dab2 occurs via the CCP pathway (Figure 4).
Recently we showed that an internalization-defective TβRI mutant lacking its CCP endocytosis motif exhibits enhanced Smad activity (Shapira et al., 2012). Moreover, in ES-2 cells, knockdown of clathrin or α-adaptin under conditions that inhibited transferrin uptake had no effect on Smad3 phosphorylation or nuclear translocation (Hirschhorn et al., 2012). Accordingly, siRNA-mediated reduction of Dab2 expression did not affect TGF-β–mediated Smad signaling in ES-2 cells (Figure 5). We conclude that neither TβRI internalization nor functional CCP endocytosis machinery is required for TGF-β–induced Smad signaling. We therefore turned to explore whether Dab2 affects TGF-β–mediated JNK stimulation, a noncanonical TGF-β signaling pathway (Figures 5–7). Knockdown of Dab2 in ES-2 cells enhanced JNK/c-Jun activation by TGF-β, whereas overexpression of Dab2 in ES-2-Dab2 or Caov3 (and Ovcar3; unpublished data) ovarian cancer cells inhibited such activation. These findings demonstrate that in ovarian cancer cells, Dab2 is a negative regulator of JNK/c-Jun activation by TGF-β. These results differ from those of a previous report (Hocevar et al., 2005) on positive regulation of TGF-β–mediated JNK activation in NIH-3T3 murine fibroblasts and rat aortic smooth muscle cells; such differences likely reflect cellular context diversity. Of note, Dab2 inhibition of JNK/c-Jun activation by TGF-β was not accompanied by any effect on activation of the Smad pathway (Figure 5). Although reductions in Dab2 expression (Santin et al., 2004; Bagadi et al., 2007; Karam et al., 2007), TGF-β signaling (Massague, 2008), and JNK/c-Jun activation (Carey et al., 2010; Vivas-Mejia et al., 2010; Eckhoff et al., 2013) were independently identified as factors predictive of tumor progression and poor prognosis in multiple cancer types, our data provide a mechanism for their functional interconnectivity via Dab2 regulation of the relative levels of activation of JNK/c-Jun versus Smad2/3.
The enhanced Dab2-mediated targeting of TβRI to CCPs raised the possibility that the resulting alteration in TβRI localization to specific plasma membrane domains may be involved in the effects on TGF-β–induced JNK activation. In this context, localization of TGF-β receptors to (and possibly internalization by) caveolae or cholesterol-enriched domains was proposed as a dampening mechanism of TGF-β–mediated Smad signaling (Razani et al., 2001; Di Guglielmo et al., 2003). We therefore examined the hitherto-unknown involvement of these membrane domains in TGF-β signaling to the JNK/c-Jun pathway. Our findings (Figure 7) demonstrate that disruption of lipid rafts/caveolae by either cholesterol depletion or nystatin inhibits JNK activation by TGF-β. These treatments had no effect on TGF-β–mediated Smad activation, indicating that at least in the cells under study, cholesterol-enriched domains serve as a platform for TGF-β–induced JNK signaling and not as a mechanism for negative regulation of the Smad pathway. Taken together, the present findings lead to the following model for the regulation of TGF-β signaling by Dab2 and cholesterol (Figure 8). In this model, TGF-β JNK signaling emanates mainly from cholesterol-rich domains, most likely due to coresidence of accessory proteins that participate in the activation of this pathway; candidate proteins that may fulfill this function are XIAP and TRAF6 (Ha et al., 2003). In contrast, Smad signaling is induced similarly from multiple localizations in the plasma membrane. Dab2 shifts the plasma membrane distribution of TβRI, recruiting it to CCPs, resulting in a parallel loss of TGF-β–mediated JNK activation. This model is in line with a recent report that the balance of activation of Smad versus non-Smad pathways by bone morphogenetic protein receptors (members of the TGF-β receptor superfamily) is affected by their localization in membrane microdomains (Guzman et al., 2012).
MATERIALS AND METHODS
Reagents
Recombinant TGF-β1 was from PeproTech (Rocky Hill, NJ). Fatty acid–free bovine serum albumin (BSA; fraction V), CPZ, and protease inhibitor cocktail (P8340) were from Sigma-Aldrich (St. Louis, MO). Hank's balanced salts solution (HBSS) and nystatin suspension were from Biological Industries (Kibbutz Beit HaEmek, Israel). Anti-myc tag (α-myc) 9E10 mouse ascites (Evan et al., 1985) was purchased from Covance Research Products (Denver, PA). Goat anti-mouse (GαM) F(ab′)2 conjugated to Alexa 546 was from Invitrogen-Molecular Probes (Eugene, OR). Fluorescent F(ab′)2 was converted to monovalent Fab′ as described (Henis et al., 1994). Normal goat γ-globulin (NGG), peroxidase-conjugated goat anti-rabbit, and GαM immunoglobulin G's (IgGs) were from Jackson ImmunoResearch (West Grove, PA). Rabbit IgG against Smad3 (reactive with Smad3 and Smad2; α-tSmad2/3), rabbit anti-Dab2 (α-Dab2), and anti-GFP (FL; v-GFP) were from Santa Cruz Biotechnology (Santa Cruz, CA). Fluorescent mounting medium was from Golden Bridge International (Mukilteo, WA). Mouse anti-actin (α-actin) was from MP Biomedicals (Solon, OH). Rabbit IgG against total JNK (α-tJNK), phospho-JNK (α-pJNK), phospho-c-Jun (pc-Jun), and mouse anti–total c-Jun (α-tc-Jun) were from Cell Signaling (Beverly, MA). Mouse anti-clathrin (α-clathrin) was from Novus Biologicals (Littleton, CO). Monoclonal mouse anti-Dab2 was described by us earlier (Chetrit et al., 2011). The TβRI kinase inhibitor SB431542 was from Sigma-Aldrich and used at a concentration of 10 μM.
Plasmids
The expression vector encoding human TβRI (in pcDNA3) with an extracellular myc epitope tag was described by us earlier (Ehrlich et al., 2001). Expression vectors encoding GFP-Dab2 and myc-Dab2-N227S were described previously (Chetrit et al., 2011).
Cell culture
ES-2 human ovarian cancer cells (CRL1978; American Type Culture Collection) were grown in DMEM supplemented with 10% fetal calf serum (FCS), penicillin (25 μg/ml), streptomycin (40 μg/ml), and glutamine (5 mM), all from Biological Industries. Caov3 cells (HTB75; American Type Culture Collection) were grown in DMEM supplemented with 20% FCS, 1 mM sodium pyruvate. and the medium supplements mentioned earlier. ES-2-Dab2 cells were generated from ES-2 cells by stable expression of a myc-tagged rat Dab2 (p82) construct mutated at Asp 227 to Ser to better resemble the human protein. They were maintained in the same growth medium as ES-2 supplemented with puromycin (2 ng/ml) and neomycin (1.5 mg/ml).
Transient transfections were carried out using jetPRIME (Polyplus, Illkirch, France).
siRNA-mediated expression knockdown experiments
ES-2 or ES-2-Dab2 cells were grown on 60-cm plates (for biochemical experiments) or on glass coverslips placed in six-well plates (for FRAP or endocytosis studies). They were transfected by jetPRIME (Polyplus) with 50 nM ON-TARGETplus human Dab2 SMART pool siRNA (Dharmacon, Lafayette, CO), target sequences AAACUGAAAUCGGGUGUUG, GAUCUAAACUCGAAAUCG, CAAAGGAUCUGGGUCAACA, and GAACCAGCCUUCACCCUUU; with 50 nM siRNA against human clathrin heavy chain, target sequence GCAATGAGCTGTTTGAAGA (Dharmacon); or with 50 nM ON-TARGETplus nontargeting pool siRNA (negative control; Dharmacon). Immunoblotting analysis and FRAP and endocytosis studies were performed 48 h posttransfection as described in the following relevant sections.
Immunoblotting
ES-2 or Caov3 cells were cultured in 60-mm plates and transfected as described with siRNA (ES-2 cells) GFP or GFP-Dab2 (Caov3 cells, 2 μg/plate). After 48 h, cells were starved in serum-free medium (30 min, 37°C) and stimulated (or not) with 200 pM TGF-β1 (30, 60. or 120 min), followed by lysis on ice (30 min) with lysis buffer (420 mM NaCl, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 5 mM EDTA, 1% NP-40, 3 mM dithiothreitol, protease inhibitor cocktail, and 0.1 mM Na3VO4). After low-speed centrifugation to remove nuclei and cell debris, the lysates were subjected to SDS–PAGE (10% polyacrylamide) and immunoblotting as described previously (Kfir et al., 2005). The blots were then probed (12 h, 4°C) by primary antibodies, followed by peroxidase-coupled goat anti-rabbit or GαM IgG (1:5000 for 1 h at 22°C). The bands were visualized by enhanced chemiluminescence (Amersham, Piscataway, NJ). Different treatments (nystatin, cholesterol depletion, TβRI kinase inhibition) are described in the following relevant sections and the figure legends.
Coimmunoprecipitation
ES-2 cells were cultured in three 100-mm plates and transfected with 4 μg/plate of myc-TβRI using jetPRIME. At 48 h posttransfection, cells were lysed (as described). Ten percent (vol/vol) of the lysate was taken for SDS–PAGE and Western blotting to probe total myc-TβRI (0.6 μg/ml mouse α-myc), followed by peroxidase-GαM (1:5000). The remainder of each lysate was subjected to immunoprecipitation with 2 μg of rabbit α-Dab2 antibodies overnight, followed by precipitation with protein A–Sepharose (50 μl, 2 h, 4°C). Immunoprecipitates were rinsed and subjected to SDS–PAGE (10% gel) and blotting using mouse α-myc and mouse α-Dab2. The blots were then probed as described for immunoblotting.
Fluorescence recovery after photobleaching
ES-2 and ES-2-Dab2 cells were grown on glass coverslips in 35-mm dishes and transfected with 1 μg of myc-TβRI or cotransfected with 1 μg of myc-TβRI together with siRNA (nontargeting or directed against Dab2). After 48 h, the cells were washed with cold HBSS/HEPES/BSA, blocked with NGG in the same buffer (200 μg/ml, 30 min, 4°C), and labeled in the cold with monovalent α-myc Fab′, followed by Alexa546 GαM-Fab′ (each incubation with 50 μg/ml Fab′, 45 min). After three washes, the coverslips were mounted over a chamber containing HBSS/HEPES/BSA and subjected to FRAP measurements at 18°C, replacing samples within 15 min to minimize internalization during the measurement. FRAP experiments were conducted as described earlier (Fire et al., 1991). An argon-ion laser beam (Innova 70; Coherent, Santa Clara, CA) was focused through a florescence microscope (Axioimager.D1; Carl Zeiss MicroImaging, Jena, Germany) to a spot with a Gaussian radius of 0.77 ± 0.03 μm (63×/1.4 numerical aperture oil-immersion objective). After a brief measurement at monitoring intensity (528.7 nm, 1 mW), a 5-mW pulse (20 ms) bleached 60–75% of the fluorescence in the illuminated region, and fluorescence recovery was followed by the monitoring beam. Values of D and Rf were extracted from the FRAP curves by nonlinear regression analysis, fitting to a lateral diffusion process (Henis et al., 2006). FRAP assays on cells subjected to nystatin treatment were conducted similarly, except that 15 min before and during labeling, the cells were subjected (or not) to nystatin treatment (25 μg/ml).
Internalization measurements
ES-2 and ES-2-Dab2 cells grown on glass coverslips in six-well plates were transfected as described for FRAP experiments. After 48 h, cells were incubated (30 min, 37°C) in serum-free medium, washed with cold HBSS/HEPES/BSA (20 mM HEPES, pH 7.2, 2% BSA), blocked with NGG (200 μg/ml, 30 min, 4°C), and labeled with α-myc (20 μg/ml, 45 min, 4°C), followed by Alexa 546–GαM Fab′ (40 μg/ml, 30 min, 4°C), all in HBSS/HEPES/BSA. The internalization of the myc-tagged receptors was quantified by the point confocal method employing the FRAP setup under nonbleaching illumination conditions as described by us earlier (Ehrlich et al., 2001; Shapira et al., 2012). Labeled cells were either fixed immediately with 4% paraformaldehyde or warmed to 37°C for the indicated periods to allow endocytosis; they were then transferred back to 4°C, fixed, and mounted for immunofluorescence as described. Endocytosis was quantified by measuring the reduction in the fluorescence intensity levels at the plasma membrane, focusing the laser beam through the 63× objective at defined spots (1.86 μm2) in the focal plane of the plasma membrane, away from vesicular staining, passing the fluorescence through a pinhole in the image plane to make it a true confocal measurement (Ehrlich et al., 2001).
Treatments affecting internalization
Endocytosis assays were conducted in HBSS/HEPES/BSA; all treatments were initiated by a 15-min preincubation (37°C) with the inhibitory drug/medium. The cells were kept under the inhibitory condition throughout the labeling and internalization measurement. Hypertonic treatment to disrupt the structure of clathrin-coated pits (Heuser and Anderson, 1989) was conducted in HBSS/HEPES/BSA supplemented with 0.45 M sucrose (Heuser and Anderson, 1989; Ehrlich et al., 2001).Treatment with CPZ, which inhibits CCP-mediated endocytosis and redistributes AP2 from the plasma membrane to endosomes (Wang et al., 1993; Shapira et al., 2012), used 50 μM CPZ. Nystatin treatment to inhibit caveolar endocytosis (Schnitzer et al., 1994; Di Guglielmo et al., 2003; Mitchell et al., 2004) used 25 μg/ml drug; treatment with the clathrin inhibitor Pitstop used 30 μM Pitstop (von Kleist et al., 2011).
Cholesterol depletion and cholesterol sequestration by nystatin
At 24 h posttransfection, cells were subjected to metabolic cholesterol depletion by incubation (18 h) with 50 μM compactin and 50 μM mevalonate (both from Sigma-Aldrich) in medium supplemented with 10% lipoprotein-deficient fetal calf serum following established procedures (Lin et al., 1998; Shvartsman et al., 2006). This treatment reduces cholesterol by 30–33% (Eisenberg et al., 2006; Shvartsman et al., 2006; present results), leading to a selective increase in the lateral diffusion of raft-associated proteins without affecting the general biophysical properties of the plasma membrane. Nystatin treatment (25 μg/ml) was conducted in HBSS/HEPES/BSA and initiated 60 min (37°C) before treatment with TGF-β1 (Schnitzer et al., 1994; Di Guglielmo et al., 2003; Mitchell et al., 2004).
Acknowledgments
This work was supported by grants from the Israel Science Foundation (grant 148/13 to Y.I.H. and grant 1529/11 to M.E.) and the Israel Cancer Research Fund (to Y.I.H.). Y.I.H. is an incumbent of the Zalman Weinberg Chair in Cell Biology.
Abbreviations used:
- CCP
clathrin-coated pit
- CPZ
chlorpromazine
- Dab2
disabled-2
- FRAP
fluorescence recovery after photobleaching
- GαM
goat anti-mouse
- JNK
c-Jun N-terminal kinase
- NGG
normal goat γ-globulin
- pc-Jun
phosphor-c-Jun
- pJNK
phospho-JNK
- TβRI
type I TGF-β receptor
- TGF-β
transforming growth factor β
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-09-0537) on March 19, 2014.
REFERENCES
- Bagadi SA, Prasad CP, Srivastava A, Prashad R, Gupta SD, Ralhan R. Frequent loss of Dab2 protein and infrequent promoter hypermethylation in breast cancer. Breast Cancer Res Treat. 2007;104:277–286. doi: 10.1007/s10549-006-9422-6. [DOI] [PubMed] [Google Scholar]
- Bernabeu C, Lopez-Novoa JM, Quintanilla M. The emerging role of TGF-b superfamily coreceptors in cancer. Biochim Biophys Acta. 2009;1792:954–973. doi: 10.1016/j.bbadis.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Carey MS, et al. Functional proteomic analysis of advanced serous ovarian cancer using reverse phase protein array: TGF-beta pathway signaling indicates response to primary chemotherapy. Clin Cancer Res. 2010;16:2852–2860. doi: 10.1158/1078-0432.CCR-09-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Hou WH, Liu IH, Hsiao G, Huang SS, Huang JS. Inhibitors of clathrin-dependent endocytosis enhance TGFb signaling and responses. J Cell Sci. 2009;122:1863–1871. doi: 10.1242/jcs.038729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG. Endocytic regulation of TGF-b signaling. Cell Res. 2009;19:58–70. doi: 10.1038/cr.2008.315. [DOI] [PubMed] [Google Scholar]
- Chetrit D, Barzilay L, Horn G, Bielik T, Smorodinsky NI, Ehrlich M. Negative regulation of the endocytic adaptor disabled-2 (Dab2) in mitosis. J Biol Chem. 2011;286:5392–5403. doi: 10.1074/jbc.M110.161851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetrit D, Ziv N, Ehrlich M. Dab2 regulates clathrin assembly and cell spreading. Biochem J. 2009;418:701–715. doi: 10.1042/BJ20081288. [DOI] [PubMed] [Google Scholar]
- Collins BM, Davis MJ, Hancock JF, Parton RG. Structure-based reassessment of the caveolin signaling model: do caveolae regulate signaling through caveolin-protein interactions. Dev Cell. 2012;23:11–20. doi: 10.1016/j.devcel.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-b receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- Eckhoff K, Flurschutz R, Trillsch F, Mahner S, Janicke F, Milde-Langosch K. The prognostic significance of Jun transcription factors in ovarian cancer. J Cancer Res Clin Oncol. 2013;139:1673–1680. doi: 10.1007/s00432-013-1489-y. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Horbelt D, Marom B, Knaus P, Henis YI. Homomeric and heteromeric complexes among TGF-b and BMP receptors and their roles in signaling. Cell Signal. 2011;23:1424–1432. doi: 10.1016/j.cellsig.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Shmuely A, Henis YI. A single internalization signal from the di-leucine family is critical for constitutive endocytosis of the type II TGF-b receptor. J Cell Sci. 2001;114:1777–1786. doi: 10.1242/jcs.114.9.1777. [DOI] [PubMed] [Google Scholar]
- Eisenberg S, Shvartsman DE, Ehrlich M, Henis YI. Clustering of raft-associated proteins in the external membrane leaflet modulates internal leaflet H-Ras diffusion and signaling. Mol Cell Biol. 2006;26:7190–7200. doi: 10.1128/MCB.01059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire E, Gutman O, Roth MG, Henis YI. Dynamic or stable interactions of influenza hemagglutinin mutants with coated pits. Dependence on the internalization signal but not on aggregation. J Biol Chem. 1995;270:21075–21081. doi: 10.1074/jbc.270.36.21075. [DOI] [PubMed] [Google Scholar]
- Fire E, Zwart DE, Roth MG, Henis YI. Evidence from lateral mobility studies for dynamic interactions of a mutant influenza hemagglutinin with coated pits. J Cell Biol. 1991;115:1585–1594. doi: 10.1083/jcb.115.6.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatza CE, Oh SY, Blobe GC. Roles for the type III TGF-b receptor in human cancer. Cell Signal. 2010;22:1163–1174. doi: 10.1016/j.cellsig.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon KJ, Blobe GC. Role of transforming growth factor-b superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Guzman A, Zelman-Femiak M, Boergermann JH, Paschkowsky S, Kreuzaler PA, Fratzl P, Harms GS, Knaus P. SMAD versus non-SMAD signaling is determined by lateral mobility of bone morphogenetic protein (BMP) receptors. J Biol Chem. 2012;287:39492–39504. doi: 10.1074/jbc.M112.387639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha H, Kwak HB, Le SW, Kim HH, Lee ZH. Lipid rafts are important for the association of RANK and TRAF6. Exp Mol Med. 2003;35:279–284. doi: 10.1038/emm.2003.38. [DOI] [PubMed] [Google Scholar]
- Hannigan A, et al. Epigenetic downregulation of human disabled homolog 2 switches TGF-beta from a tumor suppressor to a tumor promoter. J Clin Invest. 2010;120:2842–2857. doi: 10.1172/JCI36125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Borgermann JH, Hassel S, Henis YI, Knaus P. Different routes of BMP receptor endocytosis influence BMP signaling. Mol Cell Biol. 2006;26:7791–7805. doi: 10.1128/MCB.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes S, Chawla A, Corvera S. TGF-b receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158:1239–1249. doi: 10.1083/jcb.200204088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-b signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- Henis YI, Moustakas A, Lin HY, Lodish HF. The types II and III transforming growth factor-b receptors form homo-oligomers. J Cell Biol. 1994;126:139–154. doi: 10.1083/jcb.126.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis YI, Rotblat B, Kloog Y. FRAP beam-size analysis to measure palmitoylation-dependent membrane association dynamics and microdomain partitioning of Ras proteins. Methods. 2006;40:183–190. doi: 10.1016/j.ymeth.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Anderson RGW. Hypertonic media inhibit receptor mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn T, Barizilay L, Smorodinsky NI, Ehrlich M. Differential regulation of Smad3 and of the type II transforming growth factor-beta receptor in mitosis: implications for signaling. PLoS One. 2012;7:e43459. doi: 10.1371/journal.pone.0043459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocevar BA, Prunier C, Howe PH. Disabled-2 (Dab2) mediates transforming growth factor beta (TGFbeta)-stimulated fibronectin synthesis through TGFbeta-activated kinase 1 and activation of the JNK pathway. J Biol Chem. 2005;280:25920–25927. doi: 10.1074/jbc.M501150200. [DOI] [PubMed] [Google Scholar]
- Hocevar BA, Smine A, Xu XX, Howe PH. The adaptor molecule Disabled-2 links the transforming growth factor b receptors to the Smad pathway. EMBO J. 2001;20:2789–2801. doi: 10.1093/emboj/20.11.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh S, Thorikay M, Kowanetz M, Moustakas A, Itoh F, Heldin CH, ten Dijke P. Elucidation of Smad requirement in transforming growth factor-b type I receptor-induced responses. J Biol Chem. 2003;278:3751–3761. doi: 10.1074/jbc.M208258200. [DOI] [PubMed] [Google Scholar]
- Karam JA, Shariat SF, Huang HY, Pong RC, Ashfaq R, Shapiro E, Lotan Y, Sagalowsky AI, Wu XR, Hsieh JT. Decreased DOC-2/DAB2 expression in urothelial carcinoma of the bladder. Clin Cancer Res. 2007;13:4400–4406. doi: 10.1158/1078-0432.CCR-07-0287. [DOI] [PubMed] [Google Scholar]
- Keyel PA, Mishra SK, Roth R, Heuser JE, Watkins SC, Traub LM. A single common portal for clathrin-mediated endocytosis of distinct cargo governed by cargo-selective adaptors. Mol Biol Cell. 2006;17:4300–4317. doi: 10.1091/mbc.E06-05-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfir S, Ehrlich M, Goldshmid A, Liu X, Kloog Y, Henis YI. Pathway- and expression level-dependent effects of oncogenic N-Ras: p27Kip1 mislocalization by the Ral-GEF pathway and Erk-mediated interference with Smad signaling. Mol Cell Biol. 2005;25:8239–8250. doi: 10.1128/MCB.25.18.8239-8250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SI, Kwak JH, Na HJ, Kim JK, Ding Y, Choi ME. Transforming growth factor-b (TGF-b1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-b receptor kinase activity in mesangial cells. J Biol Chem. 2009;284:22285–22296. doi: 10.1074/jbc.M109.007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Naim HY, Rodriguez AC, Roth MG. Mutations in the middle of the transmembrane domain reverse the polarity of transport of the influenza virus hemagglutinin in MDCK epithelial cells. J Cell Biol. 1998;142:51–57. doi: 10.1083/jcb.142.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFb in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGFb signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer ME, Cooper JA. The adaptor protein Dab2 sorts LDL receptors into coated pits independently of AP-2 and ARH. J Cell Sci. 2006;119:4235–4246. doi: 10.1242/jcs.03217. [DOI] [PubMed] [Google Scholar]
- Meyer C, Godoy P, Bachmann A, Liu Y, Barzan D, Ilkavets I, Maier P, Herskind C, Hengstler JG, Dooley S. Distinct role of endocytosis for Smad and non-Smad TGF-b signaling regulation in hepatocytes. J Hepatol. 2011;55:369–378. doi: 10.1016/j.jhep.2010.11.027. [DOI] [PubMed] [Google Scholar]
- Mitchell H, Choudhury A, Pagano RE, Leof EB. Ligand-dependent and -independent TGF-b receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell. 2004;15:4166–4178. doi: 10.1091/mbc.E04-03-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. The regulation of TGFb signal transduction. Development. 2009;136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Maxfield FR. Membrane domains. Annu Rev Cell Dev Biol. 2004;20:839–866. doi: 10.1146/annurev.cellbio.20.010403.095451. [DOI] [PubMed] [Google Scholar]
- Pelkmans L, Fava E, Grabner H, Hannus M, Habermann B, Krausz E, Zerial M. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436:78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- Penheiter SG, Mitchell H, Garamszegi N, Edens M, Dore JJ, Jr, Leof EB. Internalization-dependent and -independent requirements for transforming growth factor b receptor signaling via the Smad pathway. Mol Cell Biol. 2002;22:4750–4759. doi: 10.1128/MCB.22.13.4750-4759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penheiter SG, Singh RD, Repellin CE, Wilkes MC, Edens M, Howe PH, Pagano RE, Leof EB. Type II transforming growth factor-b receptor recycling is dependent upon the clathrin adaptor protein Dab2. Mol Biol Cell. 2010;21:4009–4019. doi: 10.1091/mbc.E09-12-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. The challenge of lipid rafts. J Lipid Res. 2009;50(suppl):S323–S328. doi: 10.1194/jlr.R800040-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunier C, Howe PH. Disabled-2 (Dab2) is required for transforming growth factor b-induced epithelial to mesenchymal transition (EMT) J Biol Chem. 2005;280:17540–17548. doi: 10.1074/jbc.M500974200. [DOI] [PubMed] [Google Scholar]
- Razani B, Zhang XL, Bitzer M, von Gersdorff G, Bottinger EP, Lisanti MP. Caveolin-1 regulates transforming growth factor (TGF)-b/SMAD signaling through an interaction with the TGF-b type I receptor. J Biol Chem. 2001;276:6727–6738. doi: 10.1074/jbc.M008340200. [DOI] [PubMed] [Google Scholar]
- Santin AD, et al. Gene expression profiles in primary ovarian serous papillary tumors and normal ovarian epithelium: identification of candidate molecular markers for ovarian cancer diagnosis and therapy. Int J Cancer. 2004;112:14–25. doi: 10.1002/ijc.20408. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira KE, Gross A, Ehrlich M, Henis YI. Coated pit-mediated endocytosis of the type I transforming growth factor-b (TGF-b) receptor depends on a di-leucine family signal and is not required for signaling. J Biol Chem. 2012;287:26876–26889. doi: 10.1074/jbc.M112.362848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-b signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shvartsman DE, Gutman O, Tietz A, Henis YI. Cyclodextrins but not compactin inhibit the lateral diffusion of membrane proteins independent of cholesterol. Traffic. 2006;7:917–926. doi: 10.1111/j.1600-0854.2006.00437.x. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. The type I TGF-b receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10:1199–1207. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- Traub LM. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- Vivas-Mejia P, et al. c-Jun-NH2-kinase-1 inhibition leads to antitumor activity in ovarian cancer. Clin Cancer Res. 2010;16:184–194. doi: 10.1158/1078-0432.CCR-09-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107–1117. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-b. Mol Cell. 2008;31:918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Ehrlich M, Henis YI, Leof EB. Transforming growth factor-b receptors interact with AP2 by direct binding to b2 subunit. Mol Biol Cell. 2002;13:4001–4012. doi: 10.1091/mbc.02-07-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF-b signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]