

NPM1 is a novel modulator of the BER pathway. NPM1 depletion results in BER protein up-regulation; NPM1 has a stimulatory effect on BER capacity and promotes accumulation of BER proteins within nucleoli. Cisplatin leads to redistribution of NPM1 and BER proteins from nucleoli.
Abstract
Nucleophosmin (NPM1) is a multifunctional protein that controls cell growth and genome stability via a mechanism that involves nucleolar–cytoplasmic shuttling. It is clear that NPM1 also contributes to the DNA damage response, yet its exact function is poorly understood. We recently linked NPM1 expression to the functional activation of the major abasic endonuclease in mammalian base excision repair (BER), apurinic/apyrimidinic endonuclease 1 (APE1). Here we unveil a novel role for NPM1 as a modulator of the whole BER pathway by 1) controlling BER protein levels, 2) regulating total BER capacity, and 3) modulating the nucleolar localization of several BER enzymes. We find that cell treatment with the genotoxin cisplatin leads to concurrent relocalization of NPM1 and BER components from nucleoli to the nucleoplasm, and cellular experiments targeting APE1 suggest a role for the redistribution of nucleolar BER factors in determining cisplatin toxicity. Finally, based on the use of APE1 as a representative protein of the BER pathway, our data suggest a function for BER proteins in the regulation of ribogenesis.
INTRODUCTION
Nucleophosmin (NPM1; B23) is a diverse protein mainly known for its involvement in cell proliferation, primarily through the modulation of the multiple steps of ribosome biogenesis (Herrera et al., 1995; Yu et al., 2006; Murano et al., 2008; Colombo et al., 2011). In accordance with this function, the localization of the protein is largely restricted to the nucleolar compartment, where NPM1 acts as a hub protein. In tumor cells the nucleolar localization of NPM1 facilitates the accumulation of several factors, such as the p14 alternative reading frame tumor suppressor and the apurinic/apyrimidinic endonuclease 1 (APE1) DNA repair protein (Colombo et al., 2005; Apicelli et al., 2008; Vascotto et al., 2013; Antoniali et al., 2014). Continuous shuttling of NPM1 from nucleoli to the cytoplasm has also been described, with this phenomenon being linked to a role as a carrier for ribosomal particles (Yu et al., 2006).
NPM1 takes part in the maintenance of genomic stability, acting as a licensing factor for centrosome replication (Lim and Wang, 2006), and accumulating evidence supports a direct role for NPM1 in the DNA damage response (DDR). NPM1-knockout (KO) studies in mice revealed that this protein is essential for embryonic development and that the absence of NPM1 leads to unrestricted centrosome duplication and overall genomic instability; furthermore, NPM1 haploinsufficiency accelerates oncogenesis (Grisendi et al., 2005). Parallel studies showed elevated DDR markers (i.e., γ-H2AX and phosphorylated ATM) in NPM1 KO embryos, accompanied by higher susceptibility to oncogenic transformation in mouse embryonic fibroblasts (MEFs) derived from KO mice (Colombo et al., 2005). More recently, NPM1 was shown to relocalize to DNA double-strand breaks after phosphorylation (Koike et al., 2010), and pharmacological inhibition of the colocalization between phosphorylated NPM1 and γ-H2AX foci sensitizes cells to ionizing radiation (Sekhar et al., 2011). In addition, different DNA-damaging agents, ultraviolet (UV) irradiation in particular, induce nucleoplasmic relocalization of NPM1, often associated with increased expression of the protein (Wu et al., 2002; Rubbi and Milner, 2003; Kurki et al., 2004; Paron et al., 2004; Burger et al., 2010; Lo et al., 2013). Taken together, these observations suggest a possible role for NPM1 in the DDR, although the precise function(s) of the protein requires more thorough characterization.
A role for NPM1 in the DNA base excision repair (BER) pathway has been put forward. For instance, we demonstrated that NPM1 modulates the AP-site incision activity of APE1 (Vascotto et al., 2009, 2013; Fantini et al., 2010). Accordingly, NPM1 KO MEFs display increased sensitivity to a wide range of genotoxins that elicit a BER response (Vascotto et al., 2013), suggesting that NPM1 might be considered a novel modulator of the BER pathway. Despite the growing body of evidence linking NPM1, the DDR and the BER pathway, a clear picture of the role(s) of NPM1 in coordinating these processes is lacking.
In this study, we demonstrate for the first time that NPM1 plays multiple roles during the DDR, modulating the BER pathway at various levels. We show that NPM1 amount is critical for BER protein stability and demonstrate that lack of NPM1 expression negatively affects overall BER capacity, whereas NPM1 localization appears to be critical for nucleolar retention of a subset of BER enzymes. Moreover, we show that impairment of nucleolar relocalization of a representative BER enzyme (namely APE1) negatively affects cell resistance to cisplatin. In addition, impaired expression of APE1 affects ribosome biogenesis, suggesting an active role for the BER pathway in a correct ribogenesis process. Our results highlight NPM1 multilayered intervention on BER and give new insight into the nucleolar role of DNA repair pathways.
RESULTS
NPM1 depletion leads to BER protein up-regulation independently of p53 status
Earlier work from our lab described and characterized the interaction between NPM1 and APE1 (Vascotto at al., 2009; Fantini et al., 2010). A functional relationship between the two proteins was suggested by the observation that in p53/NPM1 double-KO MEFs (hereafter, NPM1−/−), the APE1 protein level was more abundant than in the isogenic p53 KO control cells (NPM1+/+; Vascotto et al., 2013). We extend these previous studies to investigate here whether the absence of NPM1 has a broader effect on the BER pathway. Western blot analyses carried out on whole-cell extracts from NPM1+/+ and NPM1−/− MEFs reveal that BER protein levels are largely up-regulated upon NPM1 depletion (Figure 1A). This situation is not restricted to the NPM1−/− cells, which exhibit genetic instability that could influence the results, as transient small interfering RNA (siRNA)–mediated knockdown of NPM1 in NPM1+/+ cells recapitulated the BER expression phenotype for many of the proteins tested (Figure 1B). Furthermore, reconstitution of NPM1−/− cells with recombinant human NPM1 largely reverted this phenotype (Figure 1C). Quantitative PCR analyses revealed that most of the BER factors (with DNA polymerase [Polβ] as a clear exception) did not exhibit any significant variation at the transcript level in NPM1−/− MEFs (Figure 1D), indicating that the NPM1 effect on BER proteins is primarily mediated via posttranscriptional mechanisms.
FIGURE 1:

Depletion of NPM1 positively modulates BER protein amounts. (A) Representative Western blotting analysis on NPM1+/+ and NPM1−/− whole-cell extracts (60 μg), showing the higher levels of BER proteins in the absence of NPM1 expression (left). Antibodies used are indicated on the right-hand side. Right, histogram reporting densitometric quantification of Western blotting signals from at least three independent experiments. Protein amounts are expressed as mean ± SD of the signal, considering NPM1+/+ as reference sample. Either actin or tubulin was used as loading control. (B, C). Representative Western blotting analysis on whole-cell extracts (50 μg) from scrambled siRNA (siScr), NPM1 siRNA (siNPM1)-treated NPM1+/+ cells (B), and mock- or NPM1-reconstituted (pNPM1) NPM1−/− cells (C). Change in BER protein levels upon NPM1 down-regulation or reexpression are indicated on the right-hand side. siScr cells or mock-transfected cells were used as reference, respectively. Values indicated are mean ± SD from at least three independent replicates. Antibodies used are indicated on the right-hand side; tubulin was used as loading control. (D) Real-time PCR analysis comparing the relative amount of transcript for the indicated BER factors between NPM1+/+ and NPM1−/− cells. Among the genes tested, only Polβ showed a statistically significant increment. Values reported are expressed as mean ± SD of the expression level in three independent replicates, considering NPM1+/+ as reference sample.
To gain further insight into the molecular mechanisms by which NPM1 exerts its effect on BER expression, we analyzed the BER protein expression pattern upon NPM1 knockdown in the HeLa human cervical carcinoma cell line. As seen in the MEFs, NPM1 depletion in HeLa cells resulted in a general up-regulation in BER protein expression (Figure 2A). Because p53 function is impaired in the cells studied to this point (Yaginuma and Westphal, 1991; Colombo et al., 2005), we examined the effect of NPM1 depletion in the HepG2 hepatocarcinoma cell line, which harbors wild-type p53 (Vollmer et al., 1999). We found that in the presence of functional p53, NPM1 depletion similarly induces increased BER protein levels (Figure 2B).
FIGURE 2:

NPM1 knockdown positively modulates BER proteins independently of p53 status. (A) Representative Western blotting analysis on HeLa whole-cell extracts (50 μg) from scrambled siRNA (siScr)– or NPM1 siRNA (siNPM1)–treated cells (left). An increased amount of a subset of BER proteins is visible upon NPM1 down-regulation. Antibodies used are indicated on the right-hand side. Right, histogram reporting the quantitative increment of different BER proteins in HeLa cells upon NPM1 down-regulation. Values reported are expressed as mean ± SD of the fold of induction relative to siScr-treated cells in at least three independent replicates. (B) Representative Western blotting analysis on HepG2 whole-cell extracts (50 μg) obtained from siScr or siNPM1 cells (left). NPM1 depletion leads to accumulation of different BER proteins. Antibodies used are indicated on the right-hand side. *Unspecific band. Right, histogram reporting the increase of BER protein levels in HepG2 cells upon NPM1 down-regulation. Values reported are expressed as mean ± SD of the fold of induction relative to siScr-treated cells in at least three independent replicates.
These data imply that NPM1 is a novel, direct modulator of the BER pathway. Moreover, the BER protein up-regulation induced by NPM1 knockdown is a widespread phenomenon independent of cellular p53 status.
BER capacity is positively modulated by NPM1
NPM1−/− MEFs have already been shown to display increased sensitivity to a wide range of DNA-damaging agents that elicit a BER response (e.g., methyl methanesulfonate and oxidizing agents). We previously demonstrated that, despite the presence of an increased amount of APE1 protein, the AP-site incision activity in NPM1−/− cell extracts was significantly impaired (Vascotto et al., 2013). To assess whether other downstream steps of the BER pathway were affected by NPM1 depletion, we compared BER activities at equal amounts of NPM1+/+ and NPM1−/− MEF cell extracts using standard in vitro assays. Surprisingly, despite the higher content of BER proteins in NPM1−/− cell extracts (Figure 1A), both the nick-ligation and the flap-incision activities showed marginal but not significant increase (Supplemental Figure S1A). A small yet significant increased proficiency in gap filling was observed in the NPM1−/− extracts (Supplemental Figure S1A), consistent with elevated Polβ expression (Figure 1A). Of importance, the same repair capacity results were obtained using cell extracts prepared by different methods (i.e., detergent vs. hypotonic extraction), suggesting that differential protein solubility between the two cell lines does not account for our findings (unpublished observations). We also carried out the BER assays at equal (normalized) levels of Polβ, DNA ligase III, or flap endonuclease 1 (FEN1) by appropriately adjusting the amount of the NPM1+/+ and NPM1−/− MEF whole-cell extract used. In this situation, and consistent with the earlier findings, we observed impairment in every enzymatic step in extracts lacking NPM1 (Figure 3A), indicating that NPM1 positively modulates each phase of the BER process in vitro.
FIGURE 3:

NPM1 contributes to the tuning of BER capacity. (A) In vitro BER assays using NPM1+/+ and NPM1−/− whole-cell extracts at equal amounts of DNA Polβ, DNA ligase III (LigIII), or FEN1 reveal impaired gap-filling, nick-ligation, and flap-incision activities, respectively, in the absence of NPM1. For each enzymatic activity we report a representative denaturing gel analysis, along with a Western blot indicating the amount of cell extract used to obtain comparable quantities of each BER factor (left). Graphs (right) describe the percentage of substrate (S) converted to product (P) as a function of time. Values reported are mean ± SD of at least three independent experimental replicates. (B) In vivo BER capacity assessment using a reporter-based system as indicated in Materials and Methods (left). The assays highlight a positive role for NPM1 in the modulation of BER capacity. Histograms show mean ± SD of at least three independent replicates. Luciferase activity for the depurinated plasmid relative to that of undamaged DNA is reported. A statistically significant impairment in BER capacity is observed when comparing NPM1+/+ and NPM1−/− cells. A similar defect in BER-mediated repair is also visible when comparing siScr- and siNPM1-treated NPM1+/+ cells. Reconstitution of NPM1−/− cells with human recombinant NPM1 (pNPM1) instead restored the BER defect. A representative Western blotting analysis (right) shows NPM1 levels in the siScr- and siNPM1-treated NPM1+/+ cells and in the mock- and NPM1-reconstituted (pNPM1) NPM1−/− cells used in the in vivo BER assays. Tubulin was used as loading control.
Our analyses revealed that little or no difference is seen when comparing the BER capacity of NPM1+/+ and NPM1−/− MEF cell extracts at the same total protein amount (Supplemental Figure S1A). Previous studies, however, demonstrated a higher sensitivity of NPM1−/− cells to genotoxins (Vascotto et al., 2013) and the accumulation of DDR markers in NPM1-KO embryos (Colombo et al., 2005), suggesting that our biochemical approach might not fully capture the dynamics of the BER response in intact cells. To address this issue, we assessed in vivo BER capacity through a reporter-based assay. A luciferase reporter plasmid was heat-depurinated to obtain an AP-DNA substrate and subsequently transfected into either NPM1+/+ or NPM1−/− MEFs; restoration of the luciferase activity was used as an indicator of cellular BER capacity. The heating treatment used was shown to generate mainly AP sites in the reporter DNA, as confirmed through recombinant APE1 incision experiments, although a small amount of spontaneously nicked plasmid DNA (which is also repaired through BER) was also generated during the process (Supplemental Figure S1B). Our measurements confirmed that NPM1-deficient cells have an impaired BER response (Figure 3B), despite their elevated levels of BER proteins, thus explaining the hypersensitivity to genotoxins previously observed (Vascotto et al., 2013). Consistently, transient depletion of NPM1 in NPM1+/+ MEFs resulted in a similar BER impairment (Figure 3B), whereas reconstitution of NPM1−/− cells with an ectopically expressed FLAG-tagged human NPM1 reversed this phenotype (Figure 3B).
NPM1 regulates BER protein localization during nucleolar stress
NPM1 is considered a hub factor, as it mediates the nucleolar retention of several proteins (e.g., p14 alternative reading frame, APE1; Emmott and Hiscox, 2009; Antoniali et al., 2014). Given the dysregulation of BER capacity in the absence of NPM1, we investigated whether the subcellular localization of BER proteins was altered in the NPM1−/− background. Strikingly, whereas immunofluorescence staining for endogenous APE1, FEN1, LigI, or DNA polymerase δ catalytic subunit revealed accumulation of a fraction of these proteins within nucleoli, the lack of NPM1 led to impairment in the nucleolar retention of all of these factors (Figure 4A). Of importance, failure of the BER proteins to accumulate within nucleoli was not a consequence of nucleolar disruption, as NPM1−/− cells did not display abnormalities in the fibrillarin localization (Colombo et al., 2005; Korgaonkar et al., 2005; Supplemental Figure S2). Reconstitution of NPM1−/− MEFs with recombinant NPM1 resulted in restoration of the nucleolar accumulation of the BER proteins (Figure 4B; Vascotto et al., 2013), indicating that NPM1 is indeed crucial for the storage or retention of BER factors within nucleoli. Furthermore, coimmunoprecipitation experiments with HeLa cells transiently transfected with a recombinant FLAG-tagged form of NPM1 confirmed that NPM1 is found in vivo within complexes containing the majority of the BER proteins (Figure 5).
FIGURE 4:

NPM1 promotes the accumulation of BER proteins within nucleoli. (A) Representative immunofluorescence images showing the differential subcellular localization of diverse BER factors in NPM1+/+ and NPM1−/− cells. Each protein analyzed (left) shows nucleolar accumulation (white arrowheads) only in the presence of NPM1. TO-PRO-3 counterstaining was used to highlight nuclei; bars, 16 μm. (B) Representative immunofluorescence analysis on mock- and NPM1-reconstituted (pNPM1) NPM1−/− cells. NPM1 staining (α-FLAG, green) was used to localize cells positively transfected with FLAG-tagged human NPM1; NPM1 reexpression restores the accumulation of BER proteins (red) within nucleoli. Nuclei are counterstained with TO-PRO-3; bars, 16 μm.
FIGURE 5:
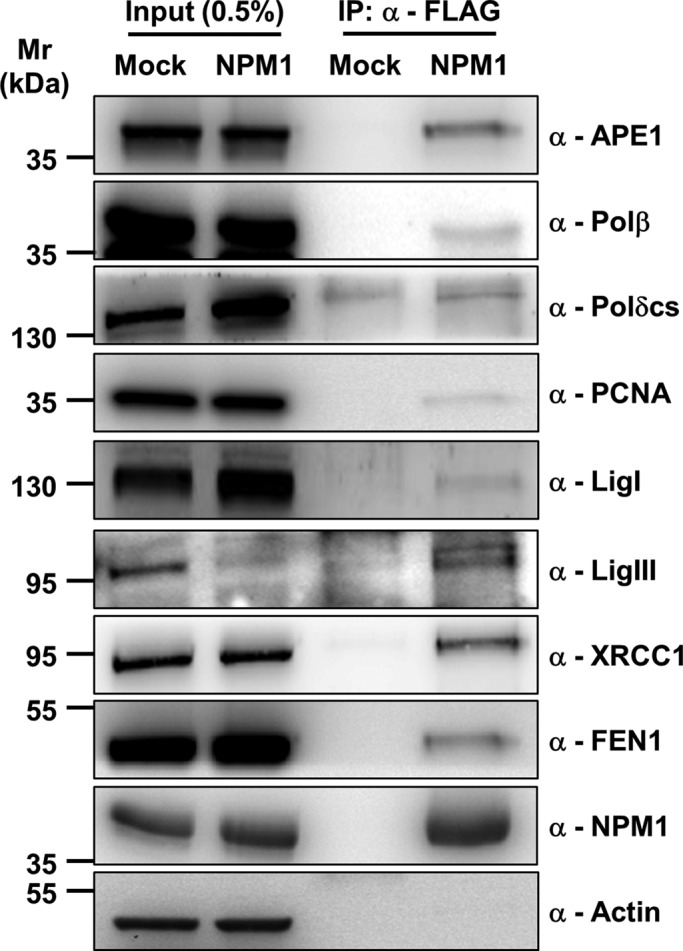
NPM1 is present in complexes containing the majority of the BER proteins. Western blot analysis on coimmunoprecipitated material obtained from HeLa cells either mock-transfected or transfected with FLAG-tagged human NPM1. Immunoprecipitation was performed with an anti-FLAG antibody, and the Western blot analysis was carried out with the indicated antibodies (right). Material coimmunoprecipitated with NPM1 shows enrichment in BER components. Actin was used as loading control for the input fractions.
NPM1 undergoes nucleoplasmic relocalization after diverse stress stimuli, including UV irradiation, RNA polymerase I (Pol I) inhibition, and treatment with several antitumor drugs (Rubbi and Milner, 2003; Burger et al., 2010). We reasoned that if NPM1 is necessary for the nucleolar retention of BER proteins, then a stimulus able to induce NPM1 relocalization from nucleoli should lead to a concomitant change in the localization of the BER enzymes. Cell treatment with cisplatin, daunorubicin, or actinomycin D (a known Pol I inhibitor) led to NPM1 relocalization to the nucleoplasmic compartment (Figure 6 and Supplemental Figure S3, A and B). In accordance with our hypothesis, NPM1 shuttling was associated with a remarkable modification in the localization pattern of several BER proteins, with extensive nucleolar depletion observed for FEN1, LigI, and APE1 (Figure 6 and Supplemental Figure S3, A and B). We note, however, that under conditions that induce DNA damage usually processed by the BER pathway (e.g., H2O2, methyl methanesulfonate), we did not observe any major relocalization of either NPM1 or APE1 (unpublished observations).
FIGURE 6:

NPM1 modulates BER protein localization during nucleolar stress. Representative immunofluorescence analysis on the indicated proteins underlines the dramatic change in subcellular localization upon nucleolar stress induced by cisplatin treatment (100 μM for 6 h) in HeLa cells. BER proteins, as well as NPM1, relocalize from nucleoli to the nucleoplasm; for some BER factors (APE1, FEN1, LigI) nucleolar emptying is also evident. TO-PRO-3 counterstaining was used to highlight nuclei; bars, 16 μm.
Taken together, our findings suggest that NPM1 can modulate nucleolar accumulation of various BER proteins under normal physiological conditions. On triggering of nucleolar stress, through either a specific DNA-damaging drug or Pol I inhibition, NPM1 relocalization to the nucleoplasm coincides with simultaneous release of BER factors from nucleoli.
Active redistribution of APE1 from nucleoli after cisplatin-induced stress
The onset of a nucleolar stress response is often associated with extensive reorganization of the nucleolar proteome, which affects the ribosome synthesis rate, as well as the distribution of DNA repair proteins (Rubbi and Milner, 2003; Burger et al., 2010; Moore et al., 2011; Antoniali et al., 2014). The cisplatin treatment used to induce the relocalization of NPM1 and BER enzymes leads to the formation of fibrillarin caps (Shav-Tal et al., 2005), along with a drop in the 47S rRNA transcription rate (Supplemental Figure S4A). To examine whether redistribution of BER proteins is merely a passive consequence of nucleolar disassembly, we analyzed the behavior of APE1 as a representative enzyme of the pathway. We recently characterized a set of critical lysine residues within the APE1 N-terminal domain (K27, K31, K32, and K35), conversion of which to alanine generates a mutant protein (APE1K4pleA) endowed with higher repair endonuclease activity that accumulates less efficiently within nucleoli and strongly impairs cell proliferation (Lirussi et al., 2012). Substitution of the same residues with arginine renders the APE1 protein (APE1K4pleR) refractory to acetylation but does not affect its nucleolar accumulation, the cell growth rate, or its ability to interact with NPM1 (Lirussi et al., 2012). Although we observed prompt relocalization of APE1WT from nucleoli upon cisplatin treatment, the nonacetylatable APE1K4pleR mutant was incapable of such redistribution, and the APE1K4pleA mutant exhibited no change in its localization pattern (Figure 7A); expression of either APE1 mutant did not affect the cisplatin-induced relocalization of NPM1 (unpublished observations). These results indicate that other factors, in addition to NPM1, may contribute to APE1 retention within nucleoli. In the case of cisplatin-induced nucleolar stress, APE1 does not freely relocalize as a consequence of nucleolar breakdown, nor is the protein simply towed to the nucleoplasm by NPM1, but instead, some kind of posttranslational modification (PTM; e.g., acetylation [ Lirussi et al., 2012] or ubiquitination [ Busso et al., 2009; Meisenberg et al., 2011]) is seemingly required for its redistribution.
FIGURE 7:

APE1 redistribution upon cisplatin requires active modification of lysine residues and protects cells from cisplatin cytotoxicity. (A) Representative immunofluorescence analysis on HeLa cells expressing different FLAG-tagged APE1 forms. On cisplatin treatment (100 μM for 6 h) only APE1WT redistributes from nucleoli (white arrowheads), whereas the nonacetylatable APE1K4pleR does not display major relocalization. The APE1K4pleA form fails to accumulate within nucleoli, regardless of the cisplatin treatment. TO-PRO-3 counterstaining was used to highlight nuclei; bars, 16 μm. (B) Viability assay on HeLa cell clones expressing the indicated APE1 forms. Cells were treated with doxycycline to induce endogenous APE1 knockdown (Lirussi et al., 2012) and challenged with increasing amounts of cisplatin for 24 h. The histogram reports the mean ± SD of at least three independent experimental replicates. The APE1K4pleR-expressing clone is hypersensitive to cisplatin, indicating that modification on critical APE1 lysine residues and redistribution of the protein from nucleoli are important to cell survival. The APE1K4pleA-expressing cells, conversely, show a protective phenotype linked to a decreased cell proliferation rate. (C) Representative Western blot analysis on HeLa cell clones used for the experiments in B. HeLa whole-cell extracts (20 μg) from the indicated clones were probed with an anti-APE1 antibody. The analysis shows comparable expression of the ectopic APE1 forms (ecto) and minimum residual of the endogenous protein (endo). Tubulin was used as loading control.
To examine whether the ability of APE1 to translocate from nucleoli affects cellular response to cisplatin, we exploited a HeLa cell model that, through a doxycycline-dependent inducible expression of specific short hairpin RNAs, allows us to knock down the endogenous form of APE1 and substitute ectopically expressed siRNA-resistant wild-type or mutant APE1 forms (Vascotto et al., 2009; Lirussi et al., 2012). The APE1K4pleR-expressing clone showed significantly increased sensitivity to cisplatin, suggesting that correct shuttling of APE1 from the nucleoli to the nucleoplasm is essential for cellular resistance to cisplatin (Figure 7B and C). Moreover, expression of the nucleoplasmic APE1K4pleA mutant form is protective at low doses of cisplatin (Figure 7B and C). Because inhibition of the APE1 catalytic activity via an indirect inhibitor, methoxyamine, or a direct small-molecule inhibitor, compound #3, in the APE1K4pleA cells did not affect the resistance phenotype to cisplatin (Supplemental Figure S4, B and C), the lower proliferation rate of the APE1K4pleA-expressing clone likely explains the protective effect, not the higher AP-site incision capacity of the APE1 mutant.
In summary, these results suggest that, upon nucleolar stress, APE1 redistribution (and possibly that of other BER proteins) is not a passive event but is a tightly regulated phenomenon that likely involves PTMs of specific lysine residues in the protein. Furthermore, it is clear that interfering with cisplatin-induced BER protein relocalization has a major effect on cell survival.
The expression of APE1 affects ribosome biogenesis
The effect observed on BER protein localization upon cisplatin, along with the concurrent impairment in nucleolar transcription, points to a major role for BER components in the maintenance of ribosome biogenesis. We previously demonstrated that the accumulation of APE1 within nucleoli is linked to the efficiency of the rRNA quality control mechanisms (Vascotto et al., 2009). To gain further insights into the function of nucleolar BER enzymes, we transiently depleted cells of APE1 and measured the rRNA transcription rate through two complementary assays. First, we assessed the nucleolar incorporation of fluorouridine (FUrd), a fluorinated UTP analogue, into nascent rRNA transcripts (Kruhlak et al., 2007; Percipalle and Louvet, 2012). Of note, depletion of APE1 resulted in a strong impairment in nucleolar FUrd uptake (Figure 8, A and B). Second, we corroborated this observation by metabolic [32P]orthophosphate-labeling experiments, which confirmed the reduced synthesis rate of the 47S rRNA precursor in APE1-depleted cells (Figure 8, A and C). Together, these data show that the expression of APE1 is crucial for efficient transcription of ribosomal genes. These data, coupled with our previous evidence (Vascotto et al., 2009), allow us to conclude that nucleolar APE1 and possibly other BER enzymes within the nucleolus play an active role in ribogenesis.
FIGURE 8:

Depletion of APE1 leads to nucleolar impairment. (A) Representative Western blot analysis on HeLa cells treated with either a scrambled siRNA (siScr) or an APE1-targeting siRNA (siAPE1) shows the efficiency of APE1 knockdown. Tubulin was used as loading control. (B) FUrd labeling of siScr- and siAPE1-treated HeLa cells. Left, representative immunofluorescence showing the preferential accumulation of FUrd within transcriptionally active nucleoli only in APE1-proficient cells (white arrowheads). Bars, 8 μm. Right, histogram reporting the average FUrd nucleolar incorporation efficiency in siScr and siAPE1 cells. Values reported express the average amount of FUrd-positive cells ± SD from at least three independent experiments. (C) [32P]phosphate metabolic labeling of nascent rRNA transcripts in HeLa cells treated with siScr, siAPE1, or actinomycin D (as positive control) reveals the nucleolar impairment in APE1-depleted cells. Cells were transfected with siRNAs and 48 h later pulsed for 1 h with [32P]orthophosphate as described in Materials and Methods. Left panel, total RNA was extracted and the amount of labeled rRNA precursors was measured through autoradiography. Ethidium bromide staining was used as loading control. Right, histogram reporting the average [32P]phosphate incorporation into the 47S precursor in siScr and siAPE1 cells. Values reported express the mean incorporation ± SD from at least three independent experiments. siScr was used as reference sample.
DISCUSSION
The data presented here support a novel role for NPM1 as a multilevel modulator of the BER pathway. NPM1 is a well-known nucleolar stress marker that undergoes nucleolus-to-nucleoplasm relocalization upon diverse DNA-threatening cues or after inhibition of Pol I–mediated transcription (Rubbi and Milner, 2003). On the basis of our results, we suggest that NPM1 operates on the BER pathway at (at least) three different levels:
NPM1 affects the stability of BER proteins. Our data demonstrate that lack of or reduced NPM1 expression results in an accumulation of several BER factors through a mechanism that is independent of the p53 status. It is worth pointing out that some of the expression changes that we observed are quantitatively small and may be indirect, in that increased expression of one BER protein may lead to higher levels of another pathway component. The coordinated modulation of so many factors, however, might play a significant role in overall repair capacity.
NPM1 promotes BER protein nucleolar retention. The presence of DNA-repair proteins within nucleoli is well established (Moore et al., 2011; Antoniali et al., 2014). In fact, several BER proteins, including SMUG1, APE1, FEN1, XRCC1, aprataxin, and PARP1/2, have been reported to accumulate within nucleoli, where they may carry out functions that differ from their “canonical” roles in DNA repair (e.g., rRNA processing; Meder et al., 2005; Becherel et al., 2006; Guo et al., 2008; Vascotto et al., 2009, 2013; Rancourt and Satoh, 2009; Jobert et al., 2013). To the best of our knowledge, this is the first report showing that a fraction of LigI and DNA polymerase δ catalytic subunit also accumulates within nucleoli in an NPM1-dependent manner, suggesting a prominent role for NPM1 in modulating nucleolar BER accumulation.
NPM1 regulates the efficiency of each BER step. We previously demonstrated that NPM1 modulates APE1 endonuclease activity through direct protein–protein interaction (Vascotto et al., 2009, 2013). Here we show that NPM1 affects not only AP-site incision, but also each enzymatic step of BER after APE1 cleavage, with a reduction of in gap-filling, flap-incision, and nick-ligation activities after NPM1 depletion, despite increases in several BER protein levels. Moreover, in vivo repair assays point to significant dysregulation in BER pathway dynamics in the absence of NPM1. A possible explanation for the observed dysfunction is that NPM1 may enhance overall BER capacity through its chaperone-like activity, acting through protein–protein or protein–DNA interactions, as already described for histones (Gadad et al., 2011). Attempts to restore each BER enzymatic step in NPM1−/− cell extracts with purified recombinant NPM1 were unsuccessful (unpublished observations), suggesting that PTMs either of, or induced by, NPM1 may be required for BER modulation. On the other hand, the measured BER impairment may reflect overall dysregulation in BER enzyme expression, which might result in an uncoordinated BER response. Although future studies aimed at elucidating molecular mechanisms underlying NPM1-mediated BER stimulation are needed, the BER impairment measured in vivo likely reflects a profound role for NPM1 in stimulating BER capacity.
A peculiar aspect of our observations is the extensive relocalization of BER proteins from nucleoli upon DNA damage or Pol I inhibition. If we consider the nucleolus as a storage depot, our data would point to the redistribution of BER proteins as a means of increasing the nucleoplasmic repair capacity. A possible contribution of the BER pathway in repairing damage induced by cisplatin (as already suggested; Wilson and Seidman, 2010; Kothandapani and Patrick, 2013), daunorubicin, or actinomycin D is conceivable, as all of these agents have been reported to promote accumulation of oxidative stress and markers of DNA strand breaks (Ali et al., 2002; Mischo et al., 2005; Cariño-Cortés et al., 2010). Despite numerous studies reporting NPM1 relocalization upon nucleolar stress, there is little evidence supporting this model for classic BER-related agents (e.g., alkylating compounds or H2O2). Furthermore, crude estimates on nucleoli biochemically purified from HeLa cells under basal conditions revealed that the amount of BER proteins retained in nucleoli is only a small fraction of the whole nuclear content (0.5–9%; M. Poletto unpublished data). Because relocalization involves such a tiny amount of protein, it is very unlikely that redistribution per se is affecting overall BER capacity in a relevant manner. What is more significant, however, is the dramatic drop in nucleolar BER protein amount upon nucleolar stress. Nucleolar BER proteins have been suggested to exert functions related to rRNA quality control and cellular proliferation in addition to DNA repair (Guo et al., 2008; Vascotto et al., 2009; Jobert et al., 2013). SMUG1 enzymatic activity, for instance, has been implicated in rRNA quality control mechanisms (Jobert et al., 2013), whereas interfering with FEN1 nucleolar retention causes reduced survival (Guo et al., 2008). We previously showed that impairment of nucleolar accumulation of APE1 reduces cell proliferation (Lirussi et al., 2012). Data presented here suggest that APE1’s ability to redistribute from nucleoli also affects the cellular response to cisplatin. Moreover, we show here that the expression of APE1 is critical for transcription of ribosomal genes within nucleoli, and it seems likely that other BER genes will also play an important role during ribogenesis. Thus we suggest that the BER protein relocalization observed upon stimuli known to affect the cell cycle (e.g., cisplatin, daunorubicin, actinomycin D) but not to activate a BER response reflects the onset of cell proliferation arrest. This hypothesis, which requires further investigation, would imply a leading role for nucleolar BER proteins in controlling cell proliferation.
Based on our findings, the effect of NPM1 on the BER pathway during stress response might be summarized as follows: under basal conditions, NPM1 functions as a nucleolar gatekeeper, retaining a small subpopulation of BER factors within nucleoli. Nucleoplasmic BER proteins are homeostatically maintained at sufficient concentration and activity to grant a steady-state DNA repair level (Parsons and Dianov, 2013), limiting accumulation of DNA repair factors that could otherwise increase genomic instability (Parsons et al., 2008). On triggering of nucleolar stress, NPM1 is released to the nucleoplasm along with BER proteins. This event may foster p53-dependent cell cycle arrest (where p53 stabilization is achieved through NPM1-mediated HDM2 sequestration; Kurki et al., 2004). BER proteins exiting from nucleoli might contribute to the arrest of cell proliferation, at least through the impairment of normal ribosome biogenesis. This cascade of events should maintain a cell cycle delay sufficient to complete DNA repair, while at the same time, nucleoplasmic NPM1-mediated coordination of BER enzymes could promote faster DNA repair. A similar involvement of NPM1 in the fine tuning of additional DNA repair pathways is not to be excluded, and evidence of the nucleolar exit of other DNA repair proteins in response to DNA damage has been reviewed (Antoniali et al., 2014, and references therein). Further comprehension of the precise functional significance of DNA repair protein shuttling between the nucleolus and the nucleoplasm, however, deserves more extensive investigation.
MATERIALS AND METHODS
Cell culture, treatments, and siRNA transfection
p53−/− (NPM1+/+) and p53−/−/NPM1−/− (NPM1−/−) MEFs (Colombo et al., 2005) and all the cell lines used were grown in DMEM (GE Healthcare, Piscataway, NJ) supplemented with 10% fetal bovine serum (FBS; GE Healthcare), 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. HeLa cells reconstituted with mutant APE1 forms were grown as indicated (Lirussi et al., 2012). Daunorubicin was from Bosche Scientific; cisplatin was purchased from Sigma-Aldrich (Milan, Italy) and freshly resuspended in dimethylformamide before each use. For siRNA experiments, 1 × 105 cells were seeded onto six-well plates and transfected 24 h later with 50 pmol of either a scrambled control siRNA or the target-specific siRNA by using the Oligofectamine Reagent (Invitrogen, Carlsbad, CA) as per manufacturer's indications. All siRNAs were from Thermo Scientific (Lafayette, CO); ON-TARGETplus SMARTpools were used for NPM1 down-regulation in human and mouse cells, and the oligonucleotide sequence used for APE1 was 5′-UACUCCAGUCGUACCAGACCU-3′. Cells were harvested 48 or 72 h after transfection.
Preparation of cell extracts and Western blotting
Whole cell extracts (WCEs) for Western blotting analyses were prepared as described (Vascotto et al., 2009). Cell extracts for in vitro BER assays were prepared by resuspending cells in hypotonic buffer (50 mM Tris pH 7.4, 1 mM EDTA, 0.5 mM dithiothreitol [DTT], Protease Inhibitor Cocktail [Sigma-Aldrich], 1 mM phenylmethylsulfonyl fluoride [PMSF]); the cell suspension was freeze-thawed, and KCl was added to a final concentration of 222 mM. After 30 min on ice, the cell lysate was cleared by centrifugation and supplemented with glycerol (10% final concentration); protein concentration was determined using the Protein Assay reagent (Bio-Rad, Hercules, CA). Western blotting procedures were carried out as already described (Vascotto et al., 2009). Images were acquired and quantified using a Chemidoc XRS video-densitometer. A complete list of the antibodies used is given in the Supplemental Materials and Methods.
Coimmunoprecipitation
Cells transfected with FLAG-tagged NPM1 were lysed in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 supplemented with Protease Inhibitor Cocktail, 0.5 mM PMSF, 1 mM NaF, and 1 mM Na3VO4 under gentle rocking for 20 min at 4°C. The cell extract was clarified by centrifugation, and coimmunoprecipitation was carried out for 3 h using the anti-FLAG M2 affinity gel (Sigma-Aldrich) as per the manufacturer's indications. Coimmunoprecipitated material was eventually eluted by incubation with 0.15 mg/ml FLAG peptide in Tris-buffered saline (TBS) and analyzed as indicated.
Immunofluorescence and confocal microscopy
For immunofluorescence analyses, cells grown on glass coverslips were fixed with 4% paraformaldehyde for 20 min and extracted for 5 min in PBS/Triton X-100, 0.1%. Cells were saturated with 10% fetal bovine serum in TBS/Tween-20, 0.1%, and incubated overnight with primary antibodies. A complete list of the antibodies used is given in the Supplemental Materials and Methods. Rhodamine red–, DyLight 549–, or Alexa Fluor 488–conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were used for detection. Cells were visualized through a Leica TCS SP laser-scanning confocal microscope (Leica Microsystems, Wetzlar, Germany) equipped with a 488-nm argon laser, a 543-nm HeNe laser, and a 63× oil immersion objective (HCX PL APO CS 63×/1.32–0.60; Leica). Data were acquired at room temperature (23°C) using the integrated Leica Confocal Software package; multicolor images were captured through sequential scanning.
Viability assays
Cell viability was measured through the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) as per the manufacturer's instructions.
In vitro BER assays.
WCE-based BER assays were carried out by using standard 32P-radiolabeled oligonucleotide substrates essentially as described in McNeill et al. (2004), with minor modifications. Briefly, all reactions were performed in the same reaction buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 100 mM KCl, 10% glycerol, 1 mM DTT) at 37°C for the indicated time; oligonucleotide substrates have been described (McNeill et al., 2004). Flap-incision reactions were carried out using 1 μg of WCE; gap-filling reactions were carried out using 3 μg of WCE, and nick-ligation assays were completed with 5 μg of WCE. Gap-filling reactions were supplemented with 0.5 mM dCTP; nick-ligation reactions were instead supplemented with 1 mM ATP. Reactions were stopped with 10 μl of 96% formamide and 10 mM EDTA and analyzed by electrophoresis on 15 or 20% sequencing (for gap-filling reactions) denaturing polyacrylamide gel. The percentage of substrate converted to product was determined after background subtraction using standard phosphorimager analysis and ImageQuant software.
In vivo BER capacity measurements.
For reporter-based BER assays, a pCMV/Luc plasmid was in vitro depurinated by incubation at 65°C for 40 min in 0.1 M sodium citrate and 0.5 M NaCl. DNA was ethanol precipitated and resuspended in water for transfection. We seeded 1 × 104 cells onto 96-well plates and transfected them with 200 ng of either damaged or undamaged pCMV/Luc along with 8 ng of intact pCMV/RL (encoding for the Renilla luciferase, as a reference for transfection efficiency) using Lipofectamine2000 Reagent (Invitrogen) as per manufacturer's indications. Luciferase activity was measured 8 h after transfection by the Dual-Glo Luciferase Assay System (Promega); repair efficiency was expressed as percentage of luciferase signal normalized to undamaged plasmid signal.
Assessment of nucleolar transcription
Fluorouridine incorporation was assessed as described previously (Kruhlak et al., 2007; Percipalle and Louvet, 2012), with minor modifications. Briefly, cells were transfected with the indicated siRNA for 48 h, washed in warm DMEM, and incubated with 2 mM FUrd (Sigma-Aldrich) in DMEM for 15 min at 37°C. Cells were then washed with ice-cold phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 10 min at room temperature, and washed with PBS. FUrd was visualized after cell permeabilization with 0.5% Triton X-100 for 5 min at room temperature; cells were incubated with a monoclonal anti-BrdU antibody (Sigma-Aldrich) for 1 h, washed twice with PBS, and then stained for 1 h with an Alexa Fluor 488–conjugated anti-mouse secondary antibody (Jackson ImmunoResearch).
[32P]orthophosphate metabolic labeling was performed essentially as described previously (Burger et al., 2010). Briefly, 48 h after siRNA transfection cells were harvested by trypsinization and phosphate depleted in phosphate-free DMEM (Invitrogen) supplemented with 10% dialyzed FBS for 1 h under rocking. [32P]orthophosphate labeling was then carried out in phosphate-free DMEM supplemented with 15 μCi [32P]orthophosphate (PerkinElmer, Waltham, MA) for 1 h. Total RNA was eventually isolated using the TRIzol Reagent (Invitrogen) as per manufacturer's indications. Samples were then separated onto a 1% agarose-formaldehyde gel, dried, and subjected to autoradiography; ethidium bromide staining of the 28S species was used as loading control. Actinomycin D (1 μg/ml) was used as positive control and was introduced for 1 h during phosphate depletion.
Statistical analyses
Statistical analyses were performed by using the Student's t test. p < 0.05 was considered as statistically significant.
Supplementary Material
Acknowledgments
We acknowledge Bruce Demple and Alex Pines for critical comments on the manuscript and the National Center for Advancing Translational Sciences for providing the APE1-specific inhibitor (compound #3). This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (IG10269 and IG14038) to G.T. and in part by the Intramural Research Program at the National Institutes of Health, National Institute on Aging, to D.M.W.
Abbreviations used:
- APE1
apurinic/apyrimidinic endonuclease 1
- BER
base excision repair
- DDR
DNA damage response
- FEN1
flap endonuclease 1
- FUrd
fluorouridine
- LigI
DNA ligase I
- NPM1
nucleophosmin
- Pol I
RNA polymerase I
- Polβ
DNA polymerase β
- PTM
posttranslational modification
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-12-0717) on March 19, 2014.
The authors declare that they have no conflict of interest.
REFERENCES
- Ali MM, Frei E, Straub J, Breuer A, Wiessler M. Induction of metallothionein by zinc protects from daunorubicin toxicity in rats. Toxicology. 2002;179:85–93. doi: 10.1016/s0300-483x(02)00322-0. [DOI] [PubMed] [Google Scholar]
- Antoniali G, Lirussi L, Poletto M, Tell G. Emerging roles of the nucleolus in regulating the DNA damage response: the non-canonical DNA repair enzyme APE1/Ref-1 as a paradigmatical example. Antioxid Redox Signal. 2014;20:621–639. doi: 10.1089/ars.2013.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicelli AJ, et al. A non-tumor suppressor role for basal p19ARF in maintaining nucleolar structure and function. Mol Cell Biol. 2008;28:1068–1080. doi: 10.1128/MCB.00484-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel OJ, Gueven N, Birrell GW, Schreiber V, Suraweera A, Jakob B, Taucher-Scholz G, Lavin MF. Nucleolar localization of aprataxin is dependent on interaction with nucleolin and on active ribosomal DNA transcription. Hum Mol Genet. 2006;15:2239–2249. doi: 10.1093/hmg/ddl149. [DOI] [PubMed] [Google Scholar]
- Burger K, et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010;16:12416–12425. doi: 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso CS, Iwakuma T, Izumi T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene. 2009;28:1616–1625. doi: 10.1038/onc.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariño-Cortés R, Alvarez-González I, Martino-Roaro L, Madrigal-Bujaidar E. Effect of naringin on the DNA damage induced by daunorubicin in mouse hepatocytes and cardiocytes. Biol Pharm Bull. 2010;33:697–701. doi: 10.1248/bpb.33.697. [DOI] [PubMed] [Google Scholar]
- Colombo E, Alcalay M, Pelicci PG. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene. 2011;30:2595–2609. doi: 10.1038/onc.2010.646. [DOI] [PubMed] [Google Scholar]
- Colombo E, Bonetti P, Lazzerini Denchi E, Martinelli P, Zamponi R, Marine JC, Helin K, Falini B, Pelicci PG. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol Cell Biol. 2005;25:8874–8886. doi: 10.1128/MCB.25.20.8874-8886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep. 2009;10:231–238. doi: 10.1038/embor.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini D, et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010;38:8239–8256. doi: 10.1093/nar/gkq691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadad SS, et al. The multifunctional protein nucleophosmin (NPM1) is a human linker histone H1 chaperone. Biochemistry. 2011;50:2780–2789. doi: 10.1021/bi101835j. [DOI] [PubMed] [Google Scholar]
- Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- Guo Z, Qian L, Liu R, Dai H, Zhou M, Zheng L, Shen B. Nucleolar localization and dynamic roles of flap endonuclease 1 in ribosomal DNA replication and damage repair. Mol Cell Biol. 2008;28:4310–4319. doi: 10.1128/MCB.00200-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera JE, Savkur R, Olson MO. The ribonuclease activity of nucleolar protein B23. Nucleic Acids Res. 1995;23:3974–3979. doi: 10.1093/nar/23.19.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobert L, Skjeldam HK, Dalhus B, Galashevskaya A, Vågbø CB, Bjørås M, Nilsen H. The human base excision repair enzyme SMUG1 directly interacts with DKC1 and contributes to RNA quality control. Mol Cell. 2013;49:339–345. doi: 10.1016/j.molcel.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Koike A, Nishikawa H, Wu W, Okada Y, Venkitaraman AR, Ohta T. Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res. 2010;70:6746–6756. doi: 10.1158/0008-5472.CAN-10-0382. [DOI] [PubMed] [Google Scholar]
- Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW, Quelle DE. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol. 2005;25:1258–1271. doi: 10.1128/MCB.25.4.1258-1271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothandapani A, Patrick SM. Evidence for base excision repair processing of DNA interstrand crosslinks. Mutat Res. 2013;743–744:44–52. doi: 10.1016/j.mrfmmm.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak M, Crouch EE, Orlov M, Montaño C, Gorski SA, Nussenzweig A, Misteli T, Phair RD, Casellas R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007;447:730–734. doi: 10.1038/nature05842. [DOI] [PubMed] [Google Scholar]
- Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–475. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- Lim MJ, Wang XW. Nucleophosmin and human cancer. Cancer Detect Prev. 2006;30:481–490. doi: 10.1016/j.cdp.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lirussi L, et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol Biol Cell. 2012;23:4079–4096. doi: 10.1091/mbc.E12-04-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SJ, et al. A novel interaction of nucleophosmin with BCL2-associated X protein regulating death evasion and drug sensitivity in human hepatoma cells. Hepatology. 2013;57:1893–1905. doi: 10.1002/hep.26209. [DOI] [PubMed] [Google Scholar]
- McNeill DR, Narayana A, Wong HK, Wilson DM., 3rd Inhibition of Ape1 nuclease activity by lead, iron, and cadmium. Environ Health Perspect. 2004;112:799–804. doi: 10.1289/ehp.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meder VS, Boeglin M, de Murcia G, Schreiber V. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci. 2005;118:211–222. doi: 10.1242/jcs.01606. [DOI] [PubMed] [Google Scholar]
- Meisenberg C, et al. Ubiquitin ligase UBR3 regulates cellular levels of the essential DNA repair protein APE1 and is required for genome stability. Nucleic Acids Res. 2011;40:701–711. doi: 10.1093/nar/gkr744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, Hemmerich P, Grosse F, Zhang S. Actinomycin D induces histone gamma-H2AX foci and complex formation of gamma-H2AX with Ku70 and nuclear DNA helicase II. J Biol Chem. 2005;280:9586–9594. doi: 10.1074/jbc.M411444200. [DOI] [PubMed] [Google Scholar]
- Moore HM, Bai B, Boisvert FM, Latonen L, Rantanen V, Simpson JC, Pepperkok R, Lamond AI, Laiho M. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol Cell Proteomics. 2011;10:M111.009241. doi: 10.1074/mcp.M111.009241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murano K, Okuwaki M, Hisaoka M, Nagata K. Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol Cell Biol. 2008;28:3114–3126. doi: 10.1128/MCB.02078-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paron I, D'Elia A, D'Ambrosio C, Scaloni A, D'Aurizio F, Prescott A, Damante G, Tell G. A proteomic approach to identify early molecular targets of oxidative stress in human epithelial lens cells. Biochem J. 2004;378:929–937. doi: 10.1042/BJ20031190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JL, Dianov GL. Co-ordination of base excision repair and genome stability. DNA Repair (Amst) 2013;12:326–333. doi: 10.1016/j.dnarep.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Parsons JL, Tait PS, Finch D, Dianova II, Allinson SL, Dianov GL. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol Cell. 2008;29:477–487. doi: 10.1016/j.molcel.2007.12.027. [DOI] [PubMed] [Google Scholar]
- Percipalle P, Louvet E. In vivo run-on assays to monitor nascent precursor RNA transcripts. Methods Mol Biol. 2012;809:519–533. doi: 10.1007/978-1-61779-376-9_34. [DOI] [PubMed] [Google Scholar]
- Rancourt A, Satoh MS. Delocalization of nucleolar poly(ADP-ribose) polymerase-1 to the nucleoplasm and its novel link to cellular sensitivity to DNA damage. DNA Repair (Amst) 2009;8:286–297. doi: 10.1016/j.dnarep.2008.11.018. [DOI] [PubMed] [Google Scholar]
- Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003;22:6068–6077. doi: 10.1093/emboj/cdg579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhar KR, et al. The novel chemical entity YTR107 inhibits recruitment of nucleophosmin to sites of DNA damage, suppressing repair of DNA double-strand breaks and enhancing radiosensitization. Clin Cancer Res. 2011;17:6490–6499. doi: 10.1158/1078-0432.CCR-11-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y, Blechman J, Darzacq X, Montagna C, Dye BT, Patton JG, Singer RH, Zipori D. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol Biol Cell. 2005;16:2395–2413. doi: 10.1091/mbc.E04-11-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascotto C, et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol. 2009;29:1834–1854. doi: 10.1128/MCB.01337-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascotto C, Lirussi L, Poletto M, Tiribelli M, Damiani D, Fabbro D, Damante G, Demple B, Colombo E, Tell G. Functional regulation of the apurinic/apyrimidinic endonuclease APE1 by nucleophosmin (NPM1): impact on tumor biology. Oncogene. 2013 DOI:10.1038/onc.2013.251. [Google Scholar]
- Vollmer CM, et al. p53 selective and nonselective replication of an E1B-deleted adenovirus in hepatocellular carcinoma. Cancer Res. 1999;59:4369–4374. [PubMed] [Google Scholar]
- Wilson DM, 3rd, Seidman MM. A novel link to base excision repair. Trends Biochem Sci. 2010;35:247–252. doi: 10.1016/j.tibs.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MH, Chang JH, Chou CC, Yung BY. Involvement of nucleophosmin/B23 in the response of HeLa cells to UV irradiation. Int J Cancer. 2002;97:297–305. doi: 10.1002/ijc.1606. [DOI] [PubMed] [Google Scholar]
- Yaginuma Y, Westphal H. Analysis of the p53 gene in human uterine carcinoma cell lines. Cancer Res. 1991;51:6506–6509. [PubMed] [Google Scholar]
- Yu Y, Maggi LB, Jr, Brady SN, Apicelli AJ, Dai MS, Lu H, Weber JD. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol. 2006;26:3798–3809. doi: 10.1128/MCB.26.10.3798-3809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.