

Angiomotin proteins, together with LATS kinase, regulate the Hippo pathway transcriptional coactivator YAP in response to changes in the F-actin cytoskeleton. Competition between F-actin and YAP for binding to angiomotins makes YAP regulation responsive to F-actin levels. Phosphorylation by LATS can switch angiomotins from F-actin to YAP binding.
Abstract
The Hippo pathway regulates the transcriptional coactivator YAP to control cell proliferation, organ size, and stem cell maintenance. Multiple factors, such as substrate stiffness, cell density, and G protein–coupled receptor signaling, regulate YAP through their effects on the F-actin cytoskeleton, although the mechanism is not known. Here we show that angiomotin proteins (AMOT130, AMOTL1, and AMOTL2) connect F-actin architecture to YAP regulation. First, we show that angiomotins are required to relocalize YAP to the cytoplasm in response to various manipulations that perturb the actin cytoskeleton. Second, angiomotins associate with F-actin through a conserved F-actin–binding domain, and mutants defective for F-actin binding show enhanced ability to retain YAP in the cytoplasm. Third, F-actin and YAP compete for binding to AMOT130, explaining how F-actin inhibits AMOT130-mediated cytoplasmic retention of YAP. Furthermore, we find that LATS can synergize with F-actin perturbations by phosphorylating free AMOT130 to keep it from associating with F-actin. Together these results uncover a mechanism for how F-actin levels modulate YAP localization, allowing cells to make developmental and proliferative decisions based on diverse inputs that regulate actin architecture.
INTRODUCTION
The Hippo pathway regulates contact inhibition of cell growth, cell proliferation, apoptosis, stem cell maintenance and differentiation, and the development of cancer in mammals and flies (Yu and Guan, 2013). The core Hippo pathway in mammals consists of the MST1/2 kinases, which activate the LATS1/2 kinases, which in turn phosphorylate and inhibit the homologous transcriptional coactivators YAP and TAZ (hereafter referred to as YAP), causing them to relocalize from the nucleus to the cytoplasm. Nuclear YAP promotes growth, proliferation, and stem cell maintenance. YAP localizes to the nucleus in cells at low density, and at high density YAP exits the nucleus and cells stop proliferation. How YAP is regulated in response to cell density is not known, although recent evidence suggests that the organization of the actin cytoskeleton contributes through an unknown mechanism (Dupont et al., 2011; Fernandez et al., 2011; Sansores-Garcia et al., 2011; Wada et al., 2011; Zhao et al., 2012). In addition, G protein–coupled receptors have been shown to modulate Hippo signaling through F-actin (Miller et al., 2012; Mo et al., 2012; Yu et al., 2012). F-actin can influence YAP activity through both Hippo pathway (LATS)–dependent (Wada et al., 2011; Zhao et al., 2012; Kim et al., 2013) and Hippo pathway–independent mechanisms (Dupont et al., 2011; Aragona et al., 2013). Intriguingly, angiomotin family members AMOT, AMOTL1, and AMOTL2 can also inhibit YAP both in a Hippo pathway–independent manner by binding and sequestering YAP in the cytoplasm and by activating the YAP inhibitory kinase LATS (Hippo dependent; Chan et al., 2011; Paramasivam et al., 2011; Wang et al., 2011; Zhao et al., 2011; Hirate et al., 2013; Leung and Zernicka-Goetz, 2013). Given their ability to associate with actin structures (Ernkvist et al., 2008; Gagne et al., 2009), we hypothesized that angiomotins might mediate the effects of F-actin on YAP. Here we report evidence in support of this hypothesis.
RESULTS
The N-terminal Hippo pathway regulatory domain of angiomotins contains an actin-binding motif
Overexpression of the long isoform of AMOT (AMOT130) causes formation of large F-actin bundles that also contain AMOT130 (Ernkvist et al., 2008; Dai et al., 2013; Figure 1A). When expressed at lower levels, AMOT130 localizes as puncta on stress fibers but does not cause obvious actin bundling (Figure 1B). To determine the significance of AMOT130 localization to the actin cytoskeleton, we sought to identify mutants defective in actin localization and bundling. Deletion analysis revealed that the actin localization domain was contained within an ∼100–amino acid conserved stretch near the amino terminus of all three angiomotin proteins (Figure 1, A, C, and D, and Supplemental Figure S1A). By deleting individual blocks of conserved sequence within this region, we found that actin localization required a short motif (e.g., AMOT130 residues 169–178; Figure 1, C and D). Deletion of this region in full-length AMOT130 (AMOT130-∆AB; AB = actin binding; Figure 1A) or in the actin-localizing fragment of AMOTL2 (Supplemental Figure S1A) disrupts both actin localization and bundling activity. (Note that the AMOT130-∆AB mutant and other forms of AMOT130 that cannot bind F-actin localize to vesicular structures [see Discussion], as observed for AMOT80 [ Heller et al., 2010], a shorter form of AMOT lacking the actin-binding region.) In addition, a small fragment (AMOT130 residues 157–191) centered around the residues deleted in AMOT130-∆AB localized to F-actin structures when fused to green fluorescent protein (GFP; Figure 1A).
FIGURE 1:

AMOT130 associates with F-actin through a domain in its N-terminus. (A) U2OS cells were transfected with plasmids for expression of Myc-tagged full-length AMOT130, amino acids 100–200 of AMOT130 (AMOT130 (100-200)), AMOT130 with a deletion in the actin-binding region (AMOT130-∆AB), or a fragment containing the actin-binding region fused to GFP (AMOT130-(157-191)) and imaged at low densities. Cells were stained for AMOT130 using anti-Myc or GFP antibodies and for F-actin using phalloidin. DNA was stained with DAPI. Bar, 20 μm. (B) U2OS cells were transfected with a plasmid for expression of full-length Myc-tagged AMOT130 and then stained for AMOT130 (using anti-Myc antibodies) and endogenous myosin IIA, which is a marker for stress fibers. Bar, 20 μm. (C) Representation of angiomotin protein features, including the actin-binding region flanked by YAP-binding motifs. (D) An alignment of the amino-terminal region of human AMOT130, AMOTL1, and AMOTL2 is shown. The region containing the actin-binding region (underlined) and LATS phosphorylation site are indicated (box). Numbers correspond to amino acid numbers for AMOT130.
Actin binding of AMOT130 is regulated by LATS2 kinase
Of interest, the conserved sequence block in the actin-binding region of angiomotins contains a perfect consensus LATS phosphorylation site (HXRXXS; serine 175 in AMOT130; Figure 1, C and D), suggesting that LATS might regulate the actin-binding properties of angiomotins. Consistent with this idea, expression of LATS2 (but not kinase-dead LATS2) could disrupt both AMOT130 localization to actin fibers and its actin-bundling activity (Figure 2, A–C). Mutation of the putative LATS phosphorylation site in the actin-binding region of AMOT130 or AMOTL2 blocked in vitro phosphorylation of each protein by LATS2 (Supplemental Figure S2A) and blocked the ability of LATS2 to inhibit the actin-bundling and localization activity of AMOT130 (Figure 2, A–C). In contrast, AMOT130-S175E could not localize to or bundle actin (Figure 2, A–C). Thus LATS2 phosphorylation of AMOT130 inhibits its localization to F-actin. Localization of endogenous AMOT130 in 293T cells supported this conclusion. In cells at low density, AMOT130 was observed to colocalize with actin fibers (Figure 2D). In contrast, phospho-AMOT130 (analyzed with phospho-serine 175–specific antibodies; Hirate et al., 2013) did not colocalize with F-actin fibers and was instead observed at regions of cell–cell contact (Figure 2D). As cells became more dense and established more cell–cell contacts, increased phospho-AMOT130 staining was observed at cell–cell junctions (Figure 2E). Endogenous phospho-AMOT130 was only occasionally seen at vesicles, like the phospho-mimetic AMOT130-S175E mutant (see Discussion).
FIGURE 2:

LATS2 inhibits association of AMOT130 with F-actin. (A) U2OS cells were transfected with the indicated AMOT130 and LATS2 plasmids and imaged at low densities. Cells were stained for AMOT130 (Myc), F-actin using phalloidin, and LATS2 or LATS2-KD (FLAG). DNA was stained with DAPI. Bar, 20 μm. (B, C) Quantification of the phenotypes of the cells in A. Graphs represent the average from three experiments (n ≥ 100 each), and error bars indicate SD of the averages. In all cases, brackets on top of bars represent statistical significance (Fisher test, p < 0.00001). (D) Immunostaining of endogenous AMOT130, phospho-AMOT130, and actin. HEK 293T cells were stained with phalloidin to visualize actin and with the indicated antibodies. (E) HEK 293T cells growing at increasing densities were costained with anti-AMOT130 and anti–phospho-AMOT130 (p-AMOT130). DNA was stained with DAPI. Bar, 20 μm.
Because the LATS phosphorylation site is in the middle of the AMOT130 actin-binding region, we hypothesized that just as phosphorylation inhibits AMOT130 actin binding, binding of AMOT130 to F-actin might interfere with phosphorylation by LATS. To test this model in vitro, we first determined whether AMOT130 could bind directly to F-actin in vitro. Consistent with in vivo data, recombinant AMOT130 (Figures 3A and Supplemental Figure S2B), but not AMOT130-S175E (Figure 3A), could bind to F-actin, whereas both AMOT130 and AMOT130-S175E bound recombinant YAP (Figure 3B). Using in vitro kinase assays, we observed that LATS2 could phosphorylate AMOT130 in the absence but not in the presence of F-actin (Figure 3C). This result is consistent with recent observations showing that LATS phosphorylation of AMOT130 in vivo is enhanced by disruption of F-actin (Dai et al., 2013). Thus LATS may act, after perturbations that reduce F-actin levels, to phosphorylate free AMOT130 to keep it from rebinding to F-actin.
FIGURE 3:
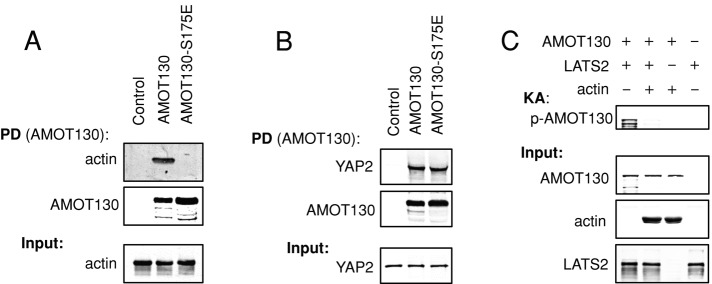
LATS phosphorylation of AMOT130 prevents its association with F-actin, and AMOT130 binding to F-actin inhibits LATS phosphorylation. (A, B) In vitro binding assays between recombinant MBP-AMOT130 or MBP-AMOT130-S175E and purified nonmuscle F-actin (A) or recombinant GST-YAP2 (B). MBP-AMOT130 protein bound to beads was used to pull down (PD) F-actin or GST-YAP2. Levels of bound proteins and input are shown. (C) Kinase assays of recombinant MBP-AMOT130 (preincubated with or without purified nonmuscle F-actin) and LATS2 kinase immunoprecipitated from HEK293 cells. Phosphorylated AMOT130 was detected using a phospho-S175–specific antibody. The levels of bound proteins and input are shown.
Actin binding–deficient mutants of AMOT130 show enhanced YAP inhibition
Previous studies showing that YAP is inhibited by F-actin disruption could be explained if an inhibitor of YAP was kept sequestered by binding to F-actin. If AMOT130 functions in this manner, then mutants that cannot bind F-actin should have enhanced ability to inhibit YAP in vivo. Therefore we tested whether localization to F-actin affected the ability of AMOT130 to inhibit YAP nuclear localization and transcriptional activity. Wild-type and mutant forms of AMOT130 were transfected into U2OS cells, and the localization of endogenous YAP was examined (Figure 4, A–C). In control cells (no AMOT130) YAP remained primarily in the nucleus. Wild-type AMOT130 and AMOT130-S175A were able to cause limited translocation of YAP to the cytoplasm (only in cells with high AMOT130 expression levels; Figure 4C). Of interest, the AMOT130-S175A mutant was less effective than wild-type AMOT130 at bringing YAP to the cytoplasm. In contrast, the mutants that could not bind F-actin (AMOT130-∆AB or AMOT130-S175E) were much more effective at shifting YAP to the cytoplasm (Figure 4, A–C), and in these cases YAP colocalized with AMOT130 on vesicles (Figure 4A), similar to when AMOT130 was coexpressed with LATS2 (Supplemental Figure S3A). Similarly, soon after disruption of F-actin in HEK 293T cells using latrunculin B, endogenous YAP was observed to colocalize with S175-phosphorylated endogenous AMOT130 on structures (possibly vesicles) near the plasma membrane (Figure 4D). When we assayed transcription from a synthetic YAP-dependent promoter (Dupont et al., 2011), although all forms of AMOT130 are expressed similarly (Supplemental Figure S3B) and show inhibition of YAP (probably due to overexpression), we again found that the AMOT130 mutants that could not bind F-actin were more effective at inhibiting YAP (Figure 4E and Supplemental Figure S3C). Together these results show that F-actin binding antagonizes the ability of AMOT130 to inhibit YAP nuclear localization and function.
FIGURE 4:

Actin and YAP compete for binding to AMOT130, and AMOT130 mutants that cannot bind F-actin are more efficient at inhibiting YAP. (A, B) U2OS cells were transfected with either control plasmid or one of the indicated AMOT130 plasmids. The next day, cells were stained for endogenous YAP and scored for the percent of cells with more YAP in the nucleus than the cytoplasm (N > C), more in the cytoplasm than the nucleus (C > N), or equal signal in the cytoplasm and nucleus (C = N). (A) Example images. (B) Average from three experiments (n ≥ 100 each), and the error bars indicate SD of the averages. Brackets on top of bars represent statistical significance (Fisher test, *p < 0.00001, **p < 0.02). Bar, 20 μm. (C) The AMOT130, AMOT130-S175A, AMOT130-S175E, and AMOT130-∆ABD expression levels in single cells were quantified and correlated with endogenous YAP localization. The graphs plot the average AMOT130 levels for individual cells (ordered based on AMOT levels) and are scored for those with more YAP in the nucleus than cytoplasm (N > C, solid symbols) or not (N = C + C > N, open symbols). (D) Endogenous YAP and phospho-AMOT130 (p-AMOT130) staining in HEK193T cells with or without treatment with latrunculin B for 15 min. DNA is stained with DAPI. Bar, 20 μm. (E) U2OS cells were transfected with the same AMOT130 plasmids as in A, as well as with an 8xGTIIC-luciferase YAP-dependent promoter plasmid and a plasmid with the SV40 promoter driving Renilla luciferase. The next day, cell extracts were made, and luciferase activity was measured for each sample. The levels of firefly luciferase (YAP activity) were normalized to the level of Renilla luciferase in each sample. Error bars indicate the SD between triplicates. Brackets on top of bars represent statistical significance (Student's test, *p < 0.005, **p < 0.01). In all cases, the experiments were done in triplicate, and the error bars indicate the SD of the averages. (F) LATS2, YAP, and the indicated AMOT130 plasmids were transfected into HEK293 cells, and the levels of AMOT130, LATS2, YAP, and phospho-YAP were analyzed by Western blotting. The experiment was done in triplicate, and error bars indicate the SD of the averages. (G) Competition between actin and YAP for binding to AMOT130. Recombinant MBP-AMOT130 protein on beads was prebound to F-actin then incubated in the presence or absence of increasing amounts of recombinant GST-YAP2. The levels of bound proteins and input are shown.
F-actin and YAP compete for binding to AMOT130
Binding to F-actin could inhibit the ability of AMOT130 to direct YAP to the cytoplasm by blocking either AMOT130 activation of LATS or binding of AMOT130 to YAP. To address this question, we made AMOT130 mutants that were specifically defective at either activating LATS2 or binding YAP. To disrupt interaction between AMOT130 and YAP, we mutated the three L/PPXY motifs in AMOT130 that are known to mediate interaction between AMOT130 and the WW domains of YAP (Chan et al., 2011; Wang et al., 2011; Zhao et al., 2011; Adler et al., 2013a). Because AMOT130 mutants defective at activating LATS had not been identified, we mutated blocks of conserved residues in the amino terminus of AMOT130, which was known to be required for LATS2 activation (Paramasivam et al., 2011), and tested their ability to promote LATS2 phosphorylation of YAP. Because mutation of residues 13–27 abolished the ability of AMOT130 to activate LATS2 (Figure 4F), this domain was termed the LATS activation domain (LAD). Of interest, both AMOT130-∆AB and wild-type AMOT130 promoted LATS2 phosphorylation of YAP to a similar degree, suggesting that F-actin binding might not regulate AMOT130 activation of LATS2. Next we used these mutants to test how F-actin regulates the ability of AMOT130 to promote cytoplasmic localization of YAP. Expression of different versions of AMOT130-∆AB with deletions in either the YAP-binding motifs or the LAD demonstrated that the enhanced ability of AMOT130-∆AB to translocate YAP to the cytoplasm depends mostly on the L/PPXY motifs, with the LAD making only a minor contribution (Figure 4B). This suggests that F-actin binding primarily interferes with AMOT130 binding to YAP.
Because the F-actin–binding domain of AMOT130 is closely flanked by YAP-binding motifs (Figure 1C), we hypothesized that F-actin and YAP might compete for binding to AMOT130, which could allow F-actin levels to modulate the ability of AMOT130 to bind to YAP. Consistent with this idea, overexpression of YAP in U2OS cells blocked localization of coexpressed AMOT130 to F-actin, and both proteins localized to vesicles (Supplemental Figure S3D). We next tested biochemically whether F-actin and YAP compete for binding to AMOT130. AMOT130 (on beads) was allowed to bind F-actin and then incubated in the presence or absence of increasing amounts of YAP (Figure 4G). We observed that high YAP concentrations displaced F-actin from AMOT130, showing that YAP and actin compete for binding to AMOT130. Together these data point toward competition between F-actin and YAP for binding to AMOT130, which could explain how actin modulates AMOT130 regulation of YAP.
Angiomotins mediate the effects of actin perturbation on YAP localization
Various treatments that perturb F-actin (Supplemental Figure S4A) cause YAP to exit the nucleus (Dupont et al., 2011; Fernandez et al., 2011; Sansores-Garcia et al., 2011; Wada et al., 2011; Zhao et al., 2012). Examples include 1) F-actin depolymerization by latrunculin B or cytochalasin D; 2) serum withdrawal, which acts through G protein–coupled receptors to affect the actin cytoskeleton (Miller et al., 2012; Mo et al., 2012; Yu et al., 2012); 3) type 2 myosin inhibition, which affects F-actin stress fibers (Dupont et al., 2011); and 4) increased cell density (Dupont et al., 2011). We found that angiomotins (and LATS) are required for regulation of YAP localization in each case. We used small interfering RNA (siRNA)/short hairpin RNA (shRNA) to knock down AMOT, AMOTL1, and AMOTL2 in HEK293A and MCF10A cells (Supplemental Figure S4B). Although knockdown of individual angiomotins had limited effects, knockdown of all three caused nuclear retention of YAP and maintenance of YAP activity after F-actin depolymerization, type 2 myosin inhibition, serum withdrawal, and increased cell density in HEK293A and MCF10A cells (Figure 5, A–D, and Supplemental Figure S4, C–F). (Note that the effect of triple knockdown in HEK293A cells after latrunculin B treatment or serum starvation could be rescued by overexpression of AMOT130 or AMOTL2; Figure 5, A and B.) In HEK293A cells, triple angiomotin knockdown blocked cytoplasmic accumulation of YAP to a similar degree as LATS1/2 knockdown after latrunculin B treatment but had a significantly stronger effect than LATS1/2 knockdown after starvation (Figure 5, A and B). Combined knockdown of both LATS1/2 and all three angiomotins caused an additive effect after latrunculin B treatment compared with knockdown of LATS1/2 or the three angiomotins alone (Figure 5A). However, after serum starvation, combined LATS1/2 and triple angiomotin knockdown did not significantly enhance YAP nuclear retention compared with triple angiomotin knockdown alone (Figure 5B). The different relative effects of LATS and angiomotin knockdown after latrunculin or serum starvation treatment could be explained if LATS and angiomotin respond somewhat differently to each stimuli. Collectively these results show that LATS and angiomotins are major mediators of various inputs that act through the F-actin cytoskeleton to affect YAP localization.
FIGURE 5:

Angiomotins and LATS are required to efficiently inhibit YAP after F-actin disturbance. (A) HEK293A cells were transfected with control siRNA (luciferase) or siRNA against AMOT130, AMOTL1, AMOTL2, a combination of all three angiomotins (triple KD), or a combination of LATS1 and LATS2 (LATS1+2), as indicated. To test for off-target effects, plasmids for expressing either AMOT130 (R AMOT130) or AMOTL2 (R AMOTL2) were transfected the next day to test for rescue of the triple-knockdown phenotype. Forty-eight hours later, all cells were treated with either latrunculin B (see example images) or blebbistatin (Blebb) and then fixed and stained for localization of endogenous YAP. Cells were scored for the percentage of cells with more YAP in the nucleus than the cytoplasm (N > C), more in the cytoplasm than the nucleus (C > N), or equal signal in the cytoplasm and nucleus (C = N). Brackets on top of bars represent statistical significance (Fisher test, p < 0.0005). (B) HEK293A cells were manipulated as in A, except that instead of drug treatment, cells were shifted to media without serum for 2 h and then fixed and stained for endogenous YAP. Cells were scored as in A. Example images are shown. Brackets on top of bars represent statistical significance (Fisher test, *p < 0.0005, **p < 0.005). (C) Lentiviral infection was used to introduce either control shRNA (directed against luciferase) or shRNA against all three angiomotins (AMOT130, AMOTL1, and AMOTL2; triple knockdown) into MCF10A cells. Sixty hours after infection, cells were left untreated, treated with cytochalasin D (CytoD), or starved of serum for an additional 12 h. Cells were then fixed and stained for endogenous YAP. YAP localization was scored as in A. Example images are shown. (D) HEK293A cells were transfected twice with control or a combination of AMOT130, AMOTL1, and AMOTL2 siRNA (see Materials and Methods). Cells were fixed after 72 h and stained for endogenous YAP. YAP localization was scored as predominantly excluded from the nucleus (excluded) or diffuse throughout the cell (diffuse). Example images are shown. In all cases, the bar graphs represent averages from three experiments (n ≥ 100 each), and the error bars indicate the SD of the averages. Nuclei were visualized with DAPI. Bar, 20 μm. C, cytoplasm; Kd, knockdown; N, nucleus. (E) Model of F-actin–regulated angiomotin (AMOT) inhibition of YAP.
DISCUSSION
The F-actin cytoskeleton is a major regulator of the Hippo pathway target YAP, mediating signals triggered by substrate stiffness, cell density, and cell detachment, as well as signaling from G protein–coupled receptors (Dupont et al., 2011; Sansores-Garcia et al., 2011; Wada et al., 2011; Miller et al., 2012; Mo et al., 2012; Yu et al., 2012; Zhao et al., 2012). We show here that angiomotin proteins connect F-actin organization to YAP regulation. The AMOT130 protein binds purified F-actin in vitro, and we observe it on stress fibers in cells. This fits with studies suggesting that F-actin structures that respond to mechanical forces such as stress fibers are involved in YAP regulation (Dupont et al., 2011; Wada et al., 2011). Although we show that AMOT130 can bind F-actin in vitro, it will be important in future studies to determine whether AMOT130 can distinguish between types of F-actin structures in vivo. A direct competition for binding to AMOT130 between F-actin and YAP appears to underlie the ability of F-actin to keep AMOT130 from binding and sequestering YAP in the cytoplasm. Angiomotins are major mediators of the effects of F-actin on YAP, since they are required for the cytoplasmic retention of YAP that occurs when F-actin is disrupted. Together these results suggest a model (Figure 5E) in which AMOT130 is sequestered on F-actin structures and stimuli that cause loss of these structures, such as increased cell density, result in release of AMOT130, allowing it to bind and inhibit YAP.
This simple model may actually be more complex. For example, in overexpression studies, we observe that the phosphomimetic form of AMOT130, which does not bind F-actin and has enhanced ability to keep YAP out of the nucleus, colocalizes with YAP in vesicular structures in the cytoplasm. This raises the possibility that membrane/vesicular localization could play an additional role in YAP regulation. It is worth noting that we only observe localization of endogenous phospho-AMOT130 and YAP to possible vesicular structures soon after F-actin disruption. In other situations phospho-AMOT130 colocalizes with YAP at cell junctions. One explanation for these results is that overexpression of AMOT130-S175E may cause accumulation of vesicular intermediates that would normally be sent on to the plasma membrane. Consistent with this notion, overexpression of AMOT80, a short form of AMOT lacking the F-actin–binding domain, causes accumulation of large endosomal-like compartments (Heller et al., 2010). In future studies it will be important to determine whether localization of AMOT130-YAP complexes to vesicles and the plasma membrane plays a role in YAP regulation.
There has been some question about the importance of LATS for F-actin–dependent regulation of YAP (Dupont et al., 2011; Yu et al., 2012; Zhao et al., 2012; Aragona et al., 2013). Our work, together with other studies, suggests that LATS functions together with angiomotins to regulate YAP in response to F-actin perturbation. We show that LATS contributes to cytoplasmic retention of YAP after F-actin disruption and serum withdrawal, and several reports have shown that LATS becomes activated and inhibits YAP by direct phosphorylation when F-actin is disrupted (Wada et al., 2011; Zhao et al., 2012; Aragona et al., 2013). Our work indicates that activated LATS can also act through angiomotins to inhibit YAP. LATS phosphorylation of AMOT130 is enhanced by F-actin disruption in vivo (Dai et al., 2013), and we show that the ability of LATS2 to phosphorylate AMOT130 in vitro is increased in the absence of F-actin. From this study, as well as from several recent reports, it is clear that LATS phosphorylation of AMOT130 inhibits its ability to bind F-actin (Adler et al., 2013b; Chan et al., 2013; Dai et al., 2013; Hirate et al., 2013). We show that LATS phosphorylation blocks AMOT130 binding to F-actin, allowing it to bind YAP and sequester it in the cytoplasm. LATS phosphorylation of AMOT130 appears to have additional functions. A recent study indicates that AMOT130 phosphorylation could also enhance AMOT130 binding to the WW domain–containing E3 ubiquitin ligase AIP4, which can both stabilize AMOT130 and promote YAP degradation (Adler et al., 2013a, b). It remains to be determined whether AIP4, like YAP, directly competes with F-actin for binding to AMOT130. Recent studies also suggest that AMOT130 phosphorylation by LATS could enhance the AMOT130–LATS interaction (Hirate et al., 2013) and have effects on the actin cytoskeleton (Dai et al., 2013). Thus LATS can promote cytoplasmic localization of YAP in response to F-actin depolymerization by phosphorylating AMOT130 in addition to its well-characterized function in phosphorylating YAP (Figure 5E).
The competition between F-actin and YAP for binding to AMOT130 could also provide a LATS-independent mechanism for F-actin–dependent regulation of YAP. The LATS-dependent and -independent mechanisms could allow for combinatorial regulation of YAP activity based on both inputs that affect the actin cytoskeleton, such as cell density, and inputs that affect LATS activity, such as cell–cell contacts (Kim et al., 2011), as was recently suggested (Aragona et al., 2013). Together this work shows that F-actin, angiomotins, and LATS form a regulatory module that controls YAP in response to diverse inputs such as changes in cell density, substrate stiffness, and G protein–coupled receptor signaling (Halder et al., 2012).
MATERIALS AND METHODS
Cell culture
Human HEK 293, HEK293A, HeLa, and U2OS cell lines were grown in DMEM (GIBCO, Grand Island, NY) supplemented with 10% (vol/vol) fetal bovine serum (GIBCO) and 1% (vol/vol) penicillin/streptomycin (Invitrogen, Grand Island, NY). Human mammary epithelial MCF10A cells were cultured in MEGM BulletKit (Lonza, Hopkinton, MA) with all additives except for the gentamicin–amphotericin B mix. Media was also complemented with 100 ng/ml cholera toxin (Sigma-Aldrich, St. Louis, MO) and 1% penicillin and streptomycin (Invitrogen). All cell lines were cultured in a humidified incubator at 37°C with 5% CO2.
In vitro kinase assays and luciferase assays
For detection of LATS2-mediated phosphorylation of angiomotins with P-32, HEK 293 cells were transfected in 12-well plates with LATS2, various angiomotin constructs, and LATS activators (MST1, SAV, and MOB1), using Lipofectamine 2000 (Invitrogen). Forty hours after transfection, cells were lysed in immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1.0% Nonidet P-40, 2% glycerol) supplemented with 1× protease inhibitor cocktail (Sigma-Aldrich), 100 nM sodium vanadate (Sigma-Aldrich), and 50 mM sodium fluoride (Sigma-Aldrich), and lysates were cleared by centrifugation at 13,000 rpm for 10 min at 4°C. Protein lysate (300 μg) was processed for immunoprecipitation as described previously (Paramasivam et al., 2011). Both LATS2 and angiomotin proteins were immunoprecipitated together on the same beads. Kinase assays and Western blotting were carried out as previously described (Paramasivam et al., 2011).
For kinase assays in the presence of F-actin, LATS2-FLAG was transfected in HEK293 cells together with its activators, MST1 and MOB1. After 24 h, LATS2 was purified in phosphate buffer using anti-FLAG M2 antibody (Sigma-Aldrich) and magnetic protein G beads (Invitrogen) following the manufacturers’ directions. Maltose-binding protein (MBP)–AMOT130 was expressed and purified as described and eluted with 20 mM maltose in supplemented actin buffer (5 mM Tris-Cl, pH 8.0, 0.2 mM CaCl2, 50 mM KCl, 2 mM MgCl2, and 1 mM ATP; Cytoskeleton, Denver, CO) for 30 min at 4ºC. Eluted AMOT130 (10 μl, ∼0.5 μg) was then preincubated with or without 10 μl of F-actin (see prior description, 5 μM final concentration) for 15 min at room temperature. Control reactions were taken to 20 μl with supplemented actin buffer. For kinase reactions the AMOT130/F-actin mix was added to LATS2-bound beads prerinsed with supplemented actin buffer. After incubation at 30°C for 30 min, kinase reactions were stopped by boiling in SDS sample buffer. Samples were then subjected to SDS–PAGE, and phospho-AMOT130 was detected by Western blotting using a phosphospecific antibody.
Luciferase assays were performed in U2OS and HeLa cells 24 h after transfection. All transfections were performed in 12-well plates using Lipofectamine 2000 and a combination of 300 ng of GTIIC-Luc (34615; Addgene, Cambridge, MA), 20 ng of pRL-SV40P (referred to as renilla, 27163; Addgene), and the described AMOT130 plasmid (300 ng for U2OS and 25 ng for HeLa cells). Cells lysates were generated and reactions performed following directions described in the Dual Luciferase Reporter Assay System (Promega, Madison, WI).
Cell starvation and drug treatments
HEK293A cells were starved for 2 h in DMEM without serum. MCF10A cells were starved overnight in DMEM/F12 supplemented with 100 ng/ml cholera toxin (Sigma-Aldrich) and 1% penicillin and streptomycin (Invitrogen). Latrunculin B and cytochalasin D were used at 1 μM for 1 h, except for the phospho-AMOT130/YAP staining (Figure 4D), for which cells were incubated for only 15 min. Note that cytochalasin D was used to disrupt F-actin in MCF10A cells because latrunculin B was too toxic in these cells. Blebbistatin was used at 25 μM for 1 h.
Immunocytochemistry
U2OS, HeLa, and MCF01A cells cultured on coverslips were fixed in phosphate-buffered saline (PBS)/4% paraformaldehyde for 10 min and permeabilized/blocked with 0.1% Triton X-100 and 5% normal goat serum (Invitrogen) for 30 min. Cells were subsequently incubated with appropriate primary antibodies for 1–2 h at room temperature. They were washed three times in PBS with 0.1% Triton X-100 and incubated with Alexa Fluor–conjugated secondary antibodies (Molecular Probes, Grand Island, NY) for 1 h at room temperature. 4′,6-Diamidino-2-phenylindole (DAPI) staining and Alexa-conjugated phalloidin (488 or 568; Invitrogen) were also added to the secondary antibody solution when appropriate. After three washes, coverslips were mounted on slides using Vectashield (Vecta Laboratories, Burlingame, CA) and viewed using fluorescent microscopy (Nikon Eclipse E600). Images were acquired using a cooled charge-coupled device camera (ORCA-ER; Hamamatsu, Bridgewater, NJ). Image processing and analysis were carried out with IPLab Spectrum software (Signal Analytics, Vienna, VA) and ImageJ software (Schneider et al., 2012).
In vitro protein-binding assays
AMOT130 and AMOT130-S175E were cloned in pDEST-MBP (provided by Marian Walhout's lab) using Gateway (Invitrogen) standard procedures. MBP-AMOT130 and MBP-AMOT130-S175E were expressed with 1 mM isopropyl-β-d-thiogalactoside (IPTG) for 4 h at 25°C and shaking. MBP fusion proteins were purified with maltose beads (NEB, Ipswich, MA) in phosphate buffer (50 mM NaH2PO4, 150 mM NaCl, 10 mM β-mercaptoethanol, 0.1% Triton, and 1 mM phenylmethylsulfonyl fluoride) following the manufacturer's directions. Expression of glutathione S-transferase (GST)–YAP2 (pGEX-5X-2 vector; GE Healthcare, Piscataway, NJ) was induced by addition of 1 mM IPTG for 2 h at 25°C, and then GST-YAP2 was purified with glutathione beads (GE Healthcare) in phosphate buffer and eluted with 20 mM glutathione for 30 min. Nonmuscle actin was purchased as part of the Actin Binding Protein Kit (Cytoskeleton) and was polymerized for 1 h at 25°C following the manufacturer's directions. For the in vitro pull-down experiments, bead-bound AMOT130 and AMOT130-S175E were incubated for 30 min at room temperature with eluted GST-YAP2 and/or ∼5 μM F-actin in phosphate buffer containing 2 mM ATP and 2 mM MgCl2 to keep F-actin stable (Actin Binding Protein Kit manual). Competition assays were assembled as follows. First, a constant amount of actin was incubated with MBP-AMOT130 beads for 15 min at room temperature. Then a constant volume of either GST elution buffer or increasing amounts of eluted GST-YAP2 were added as indicated in Figure 3F. Samples were then incubated for an additional 30 min. In all cases, beads were washed once with phosphate buffer and boiled in SDS–PAGE sample buffer. For the cosedimentation experiment, MBP-AMOT130 was eluted from maltose beads with 10 mM maltose for 30 min and incubated with actin as for 30 min at room temperature. Samples were then centrifuged at 150,000 × g in a Beckman TLX bench-top ultracentrifuge for 1.5 h. Pellets were suspended in the same volume as the supernatants and boiled in SDS–PAGE loading buffer. Protein samples were the subjected to SDS–PAGE and Western blotting with the specified antibodies.
Plasmids
Sources for plasmids used in this study were described previously (Paramasivam et al., 2011). All AMOT130, AMOTL1, and AMOTL2 constructs were expressed from pCDNA4-Myc-His. Large deletion mutants in AMOT130, AMOTL1, and AMOTL2 were constructed using PCR followed by subcloning. Point and small deletion mutations in AMOT130 and AMOTL2 were made using the Quick-Change II Site mutagenesis kit (Stratagene, Santa Clara, CA). All localization studies were performed in a 12-well format. The various angiomotin plasmids were transfected at 600 ng/well, and LATS2 constructs (pcDNA3.1-LATS2-FLAG and pcDNA3.1-LATS2-KD-FLAG) were transfected at 400 ng/well.
Antibodies
Mouse anti-tubulin and mouse anti-FLAG (M2) were purchased from Sigma-Aldrich. The rabbit-anti YAP (sc15407), mouse anti-YAP (sc10199), rabbit anti-Myc (sc789), mouse anti-Myc 9E10 (sc46), mouse anti-GFP (9996), mouse anti-AMOT130 B-4 (sc-166924), and goat anti-AMOTL2 (82501) were from Santa Cruz Biotechnology (Dallas, TX). Myosin IIa was purchased from Cell Signaling Technology (3403; Beverly, MA). The rabbit anti-AMOT antibody was generated by the Fernandes lab (CHUQ-CHUL Research Center, Université Laval, Quebec City, Canada). Rabbit anti-AMOTL1 was provided by Anthony Schmitt (Pennsylvania State University, State College, PA). AMOT130-S175 phospho-specific antibody was from Hiroshi Sasaki (Kumamoto University, Kumamoto, Japan).
siRNA/shRNA transfection
Knockdowns in HEK293A cells were performed using 30 nM control siRNA or SMARTpool siRNA (Dharmacon, Lafayette, CO) and 3 μl of RNAiMAX Lipofectamine (Invitrogen). Cells were cultured for 48 h after transfection. The only exceptions were experiments with cells at high densities, for which siRNAs were transfected twice at 40 nM (second transfection after 24 h), and cells were fixed after 72 h of the first transfection. For rescuing experiments, plasmids for protein expression were transfected after 24 h of knockdown with Lipofectamine 2000. Silencing reagents were as follows. Control siRNA (firefly luciferase 5′CGUACGCGGAAUACUUCGA3′, referred to as GL2), AMOT SMARTpool siRNA (targeting both AMOT80 and AMOT130; M-015417), AMOTL1 SMARTpool siRNA (M-017595), AMOTL2 SMARTpool siRNA (M-013232), LATS1 SMARTpool siRNA (M- 004632), and LATS2 SMARTpool siRNA (M-003865). MCF10A-cell knockdowns were done using lentiviral infection of shRNA, and cells were collected after 3 d. For the studies with AMOTL2 knockdown alone, MCF10A with integrated constructs for stably knocking down AMOTL2 and control (luciferase) were used (Paramasivam et al., 2011). To generate a triple knockdown, stable AMOTL2 knockdown cells were infected with a combination of AMOT130 and AMOTL1 lentiviral supernatants. At the same time, stable control cells were infected with control viral supernatant as a control. Viral supernatants were generated by the shRNA Core Facility, University of Massachusetts Medical School (Worcester, MA), to target GCCATGAGAAACAAATTGG (AMOTL1) or TGGTGGAATATCTCATCTA (AMOT130).
Real-time quantitative PCR
After appropriate treatments to cells on 6-well (MCF10A) or 12-well plates (HEK293A), media was aspirated off and cells were lysed with TRIzol (Life Technologies, Grand Island, NY) and processed for total RNA isolation according to the manufacturer's protocol. cDNA was prepared by oligo-dT (Promega) using SuperScript II Reverse Transcriptase (Invitrogen). Real-time quantitative PCR was performed using KAPA SYBR Fast-Master Mix Universal kit (Kapa Biosystems, Wilmington, MA). Target mRNA levels were measured relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels. The following primers were used. GAPDH-F, CTCCTGCACCACCAACTGCT, and GAPDH-R, GGGCCATCCACAGTCTTCTG; CTGF-F, AGGAGTGGGTGTGTGACGA, and CTGF-R, CCAGGCAGTTGGCTCTAATC; AMOT-F2, ACTACCACCACCTCCAGTCA, and AMOT-R2, ACAAGGTGACGACTCTCTGC; AMOTL1-F1, GCAGACAGGAAAACTGAGGA, and AMOTL1-R1, AAATGTGGTGGGAACAGAGA; and AMOTL2-F1, GCTACTGGGGTAGCAACTGA, and AMOTL2-R1, GAAGGCAGTGAGGAACTGAA. AMOT, AMOTL1, and AMOTL2 primers were ordered from Real Time Primers (Elkins Park, PA).
Supplementary Material
Acknowledgments
We thank Clark Wells and Bin Zhao for communication of unpublished results; Anthony Schmitt, Maria Fernandes, and Hiroshi Sasaki for antibodies; Elizabeth Luna for technical advice; and Peter Pryciak for comments on the manuscript. This work was supported by National Institutes of Health Grant GM058406-14 to D.M.
Abbreviations used:
- AB
actin binding
- BSA
bovine serum albumen
- DAPI
4′,6-diamidino-2-phenylindole
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- IgG
immunoglobulin G
- PBS
phosphate-buffered saline
- MBP
maltose-binding protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-11-0701) on March 19, 2014.
REFERENCES
- Adler JJ, Heller BL, Bringman LR, Ranahan WP, Cocklin RR, Goebl MG, Oh M, Lim HS, Ingham RJ, Wells CD. Amot130 adapts atrophin-1 interacting protein 4 to inhibit Yes-associated protein signaling and cell growth. J Biol Chem. 2013a;288:15181–15193. doi: 10.1074/jbc.M112.446534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler JJ, Johnson DE, Heller BL, Bringman LR, Ranahan WP, Conwell MD, Sun Y, Hudmon A, Wells CD. Serum deprivation inhibits the transcriptional co-activator YAP and cell growth via phosphorylation of the 130-kDa isoform of angiomotin by the LATS1/2 protein kinases. Proc Natl Acad Sci USA. 2013b;110:17368–17373. doi: 10.1073/pnas.1308236110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J Biol Chem. 2011;286:7018–7026. doi: 10.1074/jbc.C110.212621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Lim CJ, Guo F, Tan I, Leung T, Hong W. Actin-binding and cell proliferation activities of angiomotin family members are regulated by Hippo pathway-mediated phosphorylation. J Biol Chem. 2013;288:37296–37307. doi: 10.1074/jbc.M113.527598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, et al. Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration and angiogenesis. J Biol Chem. 2013;288:34041–34051. doi: 10.1074/jbc.M113.518019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Ernkvist M, Birot O, Sinha I, Veitonmaki N, Nystrom S, Aase K, Holmgren L. Differential roles of p80- and p130-angiomotin in the switch between migration and stabilization of endothelial cells. Biochim Biophys Acta. 2008;1783:429–437. doi: 10.1016/j.bbamcr.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Fernandez BG, Gaspar P, Bras-Pereira C, Jezowska B, Rebelo SR, Janody F. Actin-capping protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development. 2011;138:2337–2346. doi: 10.1242/dev.063545. [DOI] [PubMed] [Google Scholar]
- Gagne V, Moreau J, Plourde M, Lapointe M, Lord M, Gagnon E, Fernandes MJ. Human angiomotin-like 1 associates with an angiomotin protein complex through its coiled-coil domain and induces the remodeling of the actin cytoskeleton. Cell Motil Cytoskeleton. 2009;66:754–768. doi: 10.1002/cm.20405. [DOI] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13:591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- Heller B, Adu-Gyamfi E, Smith-Kinnaman W, Babbey C, Vora M, Xue Y, Bittman R, Stahelin RV, Wells CD. Amot recognizes a juxtanuclear endocytic recycling compartment via a novel lipid binding domain. J Biol Chem. 2010;285:12308–12320. doi: 10.1074/jbc.M109.096230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirate Y, et al. Polarity-dependent distribution of angiomotin localizes hippo signaling in preimplantation embryos. Curr Biol. 2013;23:1181–1194. doi: 10.1016/j.cub.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci USA. 2011;108:11930–11935. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Lee S, Kuninaka S, Saya H, Lee H, Lim DS. cAMP/PKA signalling reinforces the LATS-YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J. 2013;32:1543–1555. doi: 10.1038/emboj.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CY, Zernicka-Goetz M. Angiomotin prevents pluripotent lineage differentiation in mouse embryos via Hippo pathway-dependent and -independent mechanisms. Nat Commun. 2013;4:2251. doi: 10.1038/ncomms3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM, Liu J, Peters EC, Wu X. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol. 2012;19:955–962. doi: 10.1016/j.chembiol.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs) Genes Dev. 2012;26:2138–2143. doi: 10.1101/gad.197582.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramasivam M, Sarkeshik A, Yates JR, 3rd, Fernandes MJ, McCollum D. Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol Biol Cell. 2011;22:3725–3733. doi: 10.1091/mbc.E11-04-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H, Halder G. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011;30:2325–2335. doi: 10.1038/emboj.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138:3907–3914. doi: 10.1242/dev.070987. [DOI] [PubMed] [Google Scholar]
- Wang W, Huang J, Chen J. Angiomotin-like proteins associate with and negatively regulate YAP1. J Biol Chem. 2011;286:4364–4370. doi: 10.1074/jbc.C110.205401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011;25:51–63. doi: 10.1101/gad.2000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26:54–68. doi: 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.