

Abstract
There is significant interest in the use of primary intestinal epithelial cells in monolayer culture to model intestinal biology. However, it has proven to be challenging to create functional, differentiated monolayers using current culture methods, likely due to the difficulty in expanding these cells. Here, we adapted our recently developed method for the culture of intestinal epithelial spheroids to establish primary epithelial cell monolayers from the colon of multiple genetic mouse strains. These monolayers contained differentiated epithelial cells that displayed robust transepithelial electrical resistance. We then functionally tested them by examining IgA transcytosis across Transwells. IgA transcytosis required induction of polymeric immunoglobulin receptor (pIgR) expression, which could be stimulated by a combination of LPS and inhibition of γ-secretase. In agreement with previous studies using immortalized cell lines, we found that TNFα, IL-1β, IL-17 and heat-killed microbes also stimulated pIgR expression and IgA transcytosis. We used wild-type and knockout cells to establish that amongst these cytokines, IL-17 was the most potent inducer of pIgR expression/IgA transcytosis. IFNγ however did not induce pIgR expression, and instead led to cell death. This new method will allow the use of primary cells for studies of intestinal physiology.
Keywords: IgA transport, IL-17, Transwell, primary cells
INTRODUCTION
The study of intestinal epithelial biology has been made feasible through advances in the field of gastroenterology during the last few decades. Some of these technical advances have included the development of genetically and microbiologically defined animal models, animal models of injury and repair, and immortalized epithelial cell lines for in vitro studies. Most recently, the ability to propagate primary intestinal epithelial cells in vitro has greatly advanced the field1,2.
Prior to the ability to grow primary intestinal epithelial cells, colon cancer cell lines have been widely used to model physiologic and cell biologic intestinal processes in vitro. Studies using these lines have provided initial insights into epithelial biology in many areas. For example, Caco-2, HT-29, and T84 cells can form monolayers of differentiated cells3–5. However, colon cancer cell lines have many well-recognized limitations including prolonged time to attain mature monolayers (~20 days in culture for some lines), aneuploidy, and the presence of numerous undefined DNA mutations. Additional alternatives to human cancer cell lines have been developed and include the use of virally-transformed intestinal epithelial cells such as rat IEC-18 cells, or non-intestinal epithelial cells such as MDCK cells, but these systems also have limitations6,7.
One advantage these various cell lines have over the current primary intestinal epithelial cell culture method is their ability to form polarized monolayers in Transwells. This has provided the means to study many epithelial cell processes including interactions with other cell types, interactions with microbes, drug absorption, and intracellular trafficking6,8,9. One such intestinal epithelial cell process that has been well-dissected and characterized using these cell lines is IgA transcytosis via the polymeric immunoglobulin receptor (pIgR)10–12.
The majority (75%) of the total immunoglobulin produced in the body is made in the intestine, where IgA is the predominant isotype produced13. Humans secrete ~3 g of IgA in the intestine per day. While IgA can be found as a monomer in serum, the polymeric form (pIgA) connected by the J -chain (IgJ) is the predominant form at mucosal sites14.
Plasma cells produce and secrete pIgA locally in the lamina propria. For the IgA to enter the intestinal lumen, pIgA must bind to its receptor pIgR (polymeric immunoglobulin receptor)15,16, which is expressed basolaterally on epithelial cells. The pIgA-bound pIgR is then endocytosed in clathrin-coated vesicles and transcytosed across the cell through several distinct compartments to the apical surface10,12,17. At (or near) the cell surface, as of yet unidentified enzymes cleave the receptor, releasing the pIgA still bound to the extracellular portion of pIgR (known as the secretory component, or SC). This secretory IgA (SIgA), protected by the J -chain as well as the SC, is more resistant to cleavage by intestinal proteases18.
Several studies have shown that the expression of pIgR in the intestinal epithelium is regulated by bacterial stimuli. Germ free mice have a 3-fold increase in pIgR expression in the ileum upon monocolonization with Bacteroides thetaiotamicron19. In vitro studies have shown that stimulation of HT-29 cells with microbial factors (such as LPS, butyrate, and dsRNA) or heat-killed bacteria can upregulate pIgR expression20,21. Pro-inflammatory cytokines produced in response to microbial stimuli, such as IFNγ, TNFα, IL-1, and most recently IL-17 have also been shown to increase pIgR expression in vitro10,11,22–26.
Here for the first time, we establish a system to grow primary intestinal epithelial cell monolayers. This will assist in the study of intestinal epithelial cell processes in vitro using primary cells. We chose to focus on the process of IgA transcytosis using this system. We were able to adapt the previously established three dimensional (3D) primary intestinal epithelial stem cell culture system into a 2D monolayer in a Transwell. These cells are able to express pIgR after stimulation with LPS, and transcytose IgA across the monolayer. TNFα, IL-1β, and IL-17 were able to induce pIgR expression and IgA transcytosis in a dose-dependent manner. Importantly, perhaps demonstrating a distinction with previous methodologies using immortalized cell lines, IFNγ did not enhance pIgR expression. Heat-killed bacteria were also able to stimulate these processes to differing extents. Finally, this system will be readily adaptable for the use with available genetically modified mice to study different genes of interest: primary intestinal epithelial cells from pIgR−/− mice do not show IgA transcytosis into the supernatants, while cells from Tlr4−/− mice have reduced pIgR expression and IgA transcytosis after LPS stimulation compared to wild-type cells.
RESULTS
Developing a Transwell system for mouse primary intestinal epithelial cells
A critical roadblock to understanding intestinal physiology has been the lack of an experimental system to model primary intestinal epithelial cells as a polarized, confluent monolayer. The use of primary cells is of interest due to the differentiation potential of these cells in vitro1 as well as the need to evaluate cells from genetically modified mice. It has been challenging to adapt primary intestinal epithelial cells to Transwell culture as monolayers because this technique requires substantial numbers of viable cells.
We have solved this problem using an in vitro experimental system that allowed for significant expansion of intestinal epithelial stem/progenitor cells2. To obtain cells for a single Transwell, we harvested colonic spheroids from three wells (400–500 spheroids/well) of a 24-well plate that were cultured as spheroids for three days in Matrigel using 50% L-WRN (L-cells expressing Wnt3a, R-spondin3, and Noggin) conditioned media (CM). This produced ~5×105 cells that were seeded onto a single 0.33 cm2 Transwell insert of a 24-well plate. Typically this cell input created a monolayer of ~2.5×105 cells. At the time of seeding cells in Transwells, we used 50% L-WRN CM that also contained 10 μM of the ROCK inhibitor Y-27632 (Figure 1a). The media was maintained for one day post-seeding.
Figure 1. Developing a transwell system using mouse primary intestinal epithelial cells.

(a) Schematic of timeline for Transwell experiments. Wild type cells were treated with +/− 10 μM DAPT +/− 1 μg/ml LPS and were analyzed on day three post-seeding. (b) Cells were fixed and paraffin-embedded on the transwell membranes. Sections were cut and stained with the following: hematoxylin and eosin, anti-ZO-1, anti-villin, and anti-pIgR. Bars = 50 μm. Gene expression analysis was performed by qRT-PCR for pIgR (c), Reg3g (d), and Vil1 (e). All samples were normalized to Gapdh mRNA, and data were presented as fold change relative to untreated (0% CM) cells (mean ± s.e.m.; n ≥ 3 per condition). One-way ANOVA: (c) F = 96.02, P < 0.0001; (d) F = 3.441, P < 0.0376; (e) F = 1.085, P < 0.3762. ***P < 0.001 by Bonferroni's multiple comparison test. (f) Transepithelial electrical resistance was measured on day three. The (resistance × area) is shown for each condition (mean ± s.e.m., n = 6 per group). Statistical analysis by Student's t-test showed no significant difference between the two groups (P < 0.4362).
On day one after seeding, the 50% CM was replaced with 0% CM supplemented with or without specific treatments that were designed to facilitate the study of IgA transcytosis. The treatment included a combination of the γ-secretase inhibitor DAPT to differentiate the cells27,28, and LPS to induce the expression of pIgR (which is known to be regulated by microbial and/or cytokine signaling)19. The cells were treated for two days in this media prior to evaluation for differentiation by histology and gene expression analysis, as well as functional assays such as IgA transcytosis.
We next evaluated the effects of DAPT+LPS on differentiation and lineage allocation of primary epithelial monolayers. Cells were fixed on a Transwell membrane, which was then cut out of the insert and processed for paraffin embedding. Histologic sections were cut and stained with hematoxylin and eosin. We observed that both untreated and DAPT+LPS-treated cells showed a single layer of cells overlying the Transwell membrane (Figure 1b). To confirm that differentiation of enterocytes occurred both with and without DAPT+LPS treatments, we performed immunostaining using antisera against Villin1 and ZO-1. Villin1 marks microvilli29 and ZO-1 is a tight junction marker30. In addition, both of these proteins showed appropriate apical localization throughout the monolayer and this pattern was present regardless of treatment. We also found that a basolateral marker, CD138, showed appropriate localization in Transwell cultures (Supplementary Figure 1a). Taken together, the localization of these markers is consistent with polarized epithelial cells. We stained for markers of additional colonic epithelial lineages including enteroendocrine cells (chromogranin A31) and goblet cells (lectin UEA-132). Whole mount images of immunostained Transwells showed that both of these cell types were present in DAPT+LPS-treated monolayers with lower fractional representation than absorptive enterocytes (Supplementary Figure 1b, c). This lineage allocation is similar to what is observed in vivo for the surface epithelium of the mouse colon (Supplementary Figure 1d–h).
To adapt this experimental system for the study of IgA transcytosis, we evaluated the expression of pIgR both in vivo and in vitro. The colonic epithelium, including surface epithelial cells, expressed pIgR in vivo (Supplementary Figure 1i,j). In vitro, pIgR could be induced in epithelial monolayers that were treated with DAPT+LPS, as shown by immunostaining (Figure 1b). To quantify the relative effects of DAPT and LPS on pIgR expression, we performed qRT-PCR of mRNAs isolated from epithelial cells grown on Transwells for three days. The addition of DAPT+LPS stimulated a robust increase in pIgR mRNA expression compared to untreated cells (~100-fold increase) (Figure 1c). This finding corroborated the effects of DAPT+LPS on protein expression as determined by immunostaining (Figure 1b). Single treatment with either LPS or DAPT did not stimulate pIgR mRNA expression to the extent that was achieved by the combination of these factors (Figure 1c). As a positive control for LPS treatment, expression of RegIIIγ33 was increased after DAPT+LPS treatment (Figure 1d). As a negative control, Villin1 expression (which is not microbially regulated in vivo34) was similar in all groups of treated and untreated cells (Figure 1e). Thus, we were able to show that wild-type primary colonic epithelial cells on Transwells were responsive to DAPT+LPS.
To perform IgA transcytosis experiments, a complete monolayer of cells is required. To demonstrate that the seeded cells formed a functional monolayer, we measured transepithelial electrical resistance (TER) in the Transwells on day three (Figure 1f). For this experiment, we used cells at a density of ~2.5×105 cells/0.33 cm2. The average TER of untreated cells was 3333 Ωcm2. The TER of DAPT+LPS-treated cells was similar to the TER of untreated cells (2877 Ωcm2).
Developing an IgA transcytosis assay using primary mouse Transwell cultures
One of the critical functions of the intestinal epithelium is the transcytosis of IgA from the lamina propria to the lumen of the intestine. This process involves pIgR trafficking across the intestinal epithelium which can occur at a slower rate in the absence of IgA35. To determine if the primary intestinal epithelial monolayers were capable of IgA transcytosis, we developed an assay utilizing these cells. At day three post-plating on Transwell inserts, the cells were washed and placed in wells containing normal mouse IgA in the lower compartment. This media is in contact with the basolateral surface of the epithelial monolayer. Media alone was added to the upper compartment (apical surface). The cells were incubated at 37°C for different periods of time to allow for receptor binding and transcytosis to occur before the media in the upper compartment was sampled for analysis by enzyme-linked immunosorbent assay (ELISA). Colonic cells isolated from pIgR−/− mice were used as a negative control for all transcytosis experiments.
Consistent with the pIgR mRNA expression data, cells treated with DAPT+LPS showed measurable IgA in the apical media whereas the other three groups (untreated, DAPT alone, and LPS alone) had levels at or below the limit of detection (Figure 2a). Apical media from pIgR−/− cells did not contain IgA regardless of treatment. This genetic control indicated that IgA is actively transported through epithelial cells and does not use paracellular transport.
Figure 2. Developing an IgA transcytosis assay.

(a) Wild type and pIgR−/− cells were seeded in Transwells and treated with +/− 10 μM DAPT +/−1 μg/ml LPS as indicated. On day three post-seeding, 40 μl of normal mouse IgA (Santa Cruz) was added to the lower compartment of the Transwells, and supernatants from the upper compartment were taken at six hours for the detection of IgA by ELISA. Time course (b) and IgA dose curve (c) experiments were performed on wild type and pIgR−/− cells treated with 10 μM DAPT and 1μg/ml LPS to determine the optimal conditions for future experiments. An LPS dose curve was performed to determine whether IgA transcytosis (d) and pIgR expression (e) were dose dependent. All LPS-treated cells were also treated with 10 μM DAPT. For IgA transcytosis, results from the ELISA were normalized to the 1 μg/ml LPS treatment group (= 100%). Gene expression analysis by qRT-PCR for pIgR was performed by normalizing to Gapdh, and data are presented as fold change relative to untreated cells. The dotted lines represent the limit of detection by the ELISA. All values are indicated as mean ± s.e.m. One-way ANOVA: (a) F = 573.3, P < 0.0001, n ≥ 3 per group; (c) F = 539.2, P < 0.0001, n ≥ 12 per group; (d) F = 675.7, P < 0.0001, n ≥ 3 per group; (e) F = 46.22, P < 0.0001, n ≥ 6 per group. Means with different letters are significantly different by Bonferroni's multiple comparison test. N.D. = not detected.
To determine the optimal conditions for transcytosis experiments, we performed IgA dose curve and time course experiments using DAPT+LPS-treated cells. One hour after the addition of IgA to the basal compartment, no IgA was detected in the apical media (Figure 2b). Therefore, one hour was not sufficient for IgA in the basal compartment to bind pIgR and transcytose across cells to the apical compartment. This result also showed that the monolayer was intact and thus did not allow the IgA to freely diffuse into the apical media. Because of this result, we utilized the one hour time point as an additional internal control in all subsequent experiments. Additional time points were taken three hours (four hours after the addition of IgA) and six hours later (ten hours after the addition of IgA). Both of these time points showed progressively higher levels of IgA in the apical media (Figure 2b). In subsequent experiments, all three of these time points were evaluated.
In choosing the optimal dose of IgA, the amount of IgA in the apical compartment at three and six hour time points needed to be in linear range of the ELISA. We initially used normal mouse IgA from Santa Cruz Biotechnology at a dose of 40 μl/well (Figure 2c). In later experiments, we used normal mouse IgA from BD Pharmingen at a comparable dose, which allowed for a more precise measurement of IgA concentration and decreased variability between experiments (Supplementary Figure 2). For this source of IgA, we were able to calculate the IgA concentration used to 5 μg/ml, or a total of 3 μg/well.
As it was unclear how sensitive primary wild-type colonic epithelial cells were to LPS, we performed a dose-response curve for LPS using both IgA transcytosis and pIgR expression as readouts (Figure 2d, e). Both transcytosis and pIgR mRNA expression responded in a dose-dependent manner. In all subsequent experiments, we used 1 μg/ml LPS as this was in linear range of the response of wild-type cells.
Although LPS has been used as a standard in the field to efficiently induce pIgR expression in different cell types, there is growing interest in the study of the effects of specific microbes in the intestine and how they stimulate IgA20,21,36–38. To model an example of such host-microbial interactions in vitro, cells were treated with heat-killed E. coli to analyze its ability to induce IgA transcytosis and pIgR expression (Figure 3a, b). A dose equivalent to 107 CFU/ml E. coli was found to induce similar levels of IgA transcytosis and pIgR expression as that demonstrated for to 1 μg/ml LPS.
Figure 3. IgA transcytosis and pIgR expression are induced by heat)killed bacteria.
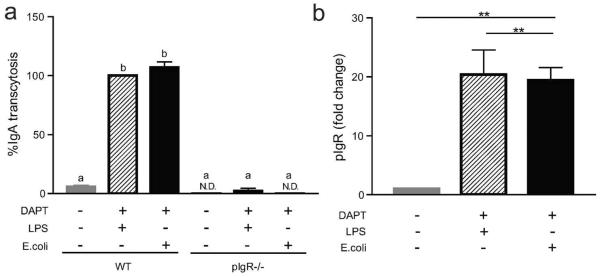
Wild type and pIgR−/− cells were seeded in Transwells and treated with 10 μM DAPT and 1 μg/ml LPS or 107 CFU/ml heat-killed E.coli as indicated. (a) IgA transcytosis was analyzed by ELISA, and results were normalized to the WT+DAPT+LPS group (= 100%). (b) Gene expression analysis by qPT-PCR of pIgR was performed and all samples were normalized to Gapdh. Data are presented as fold change relative to untreated (0% CM) cells. All values are indicated as mean ± s.e.m. One-way ANOVA: (a) F = 426.2, P < 0.0001, n ≥ 6 per group, means with different letters are significantly different by Bonferroni's multiple comparison test; (b) F = 11.47, P < 0.0008, n ≥ 5 per group, **P < 0.01 by Bonferroni's multiple comparison test. N.D. = not detected.
Cell density affects IgA transcytosis and pIgR expression in Transwell cultures
We noticed that there was occasional experiment-to-experiment variability of the amount of IgA transcytosed. Further scrutiny of the Transwells utilized in these experiments led us to hypothesize that the cellular density plays a role in the level of IgA transcytosis in vitro. To test this idea, we performed two-fold serial dilutions of the cells seeded in the Transwells (Figure 4a). Both the undiluted (using ~5×105 cells as above) and a 1:2 dilution showed similar amounts of transcytosed IgA. However, a 1:4 dilution reproducibly showed significantly less transcytosed IgA. Furthermore, apical media collected from a 1:8 dilution showed IgA at levels below the limit of detection. This result suggested that there was an intact monolayer at this cell density but no detectable IgA was transcytosed. At a 1:16 dilution, we could not reproducibly achieve a monolayer, as in some experiments, IgA was readily detected in the apical compartment at levels similar to the basolateral chamber. TER measurements corroborated these findings. Higher cell densities all showed resistance values of >2500 Ωcm2 (Supplementary Figure 3). The TER for the 1:8 cell dilution was more variable, with average values of ~1240 Ωcm2. The TER for the 1:16 dilution was near baseline values.
Figure 4. Cell density affects differentiation state of IECs in transwells.

Two-fold serial dilutions of wild type cells were seeded into Transwells and treated with 10 μM DAPT and 1 μg/ml LPS. (a) IgA transcytosis was performed on day three post-seeding, and measurement of IgA in the supernatants at the six hour time point is shown. (b, c) Cells were fixed and stained on the Transwell membranes (c) with anti-pIgR (green) and bis-benzamide dye (blue). Bars = 200 μm. Quantification of cell density of cells on Transwells was performed using ImageJ software (b). Gene expression analysis was performed by qRT-PCR for pIgR (d), Muc2 (e), Atoh1 (f), and Vil1 (g). All samples were normalized to Gapdh, and data are presented as fold change relative to untreated (0% CM) cells. The dotted lines represent the limit of detection by the ELISA. All values are indicated as mean ± s.e.m. One-way ANOVA: (a) F = 53.11, P < 0.0001, n ≥ 6 per group; (b) F = 183.2, P < 0.0001, n ≥ 15 per group; (d) F = 39.02, P < 0.0001, n ≥ 3 per group; (e) F = 11.07, P < 0.0001, n ≥ 3 per group; (f) F = 3.436, P < 0.0183, n ≥ 3 per group; (g) F = 2.186 , P < 0.0894, n ≥ 3 per group. Means with different letters are significantly different by Bonferroni's multiple comparison test.
The cell density was quantified for each dilution by nuclei counts of whole mount images of bis-benzimide-stained Transwell membranes (Figure 4b, c). A 1:2 dilution of input cells resulted in only a small change in the number of seeded cells on the Transwell, indicating that maximum cell numbers were seeded at these two input densities. Each successive two-fold dilution showed a ~two-fold decrease in cell density. We next tested whether the correlation of cell density to IgA transcytosis was due to cell number and/or pIgR expression. Co-staining of cells with anti-pIgR antisera showed detectable staining in the majority of cells in Transwells with higher cell densities (no dilution, 1:2 and 1:4). We observed a substantial decrease in the number of cells that were positive for pIgR in 1:8 dilution samples. There was no detectable staining for pIgR in the 1:16 dilution samples. To confirm this observation, we performed gene expression analysis for pIgR at each dilution (Figure 4d). The highest relative expression of pIgR was observed at the 1:2 and 1:4 dilution samples. We observed a threshold effect whereby pIgR expression was substantially lower in the 1:8 and 1:16 dilution samples. Our interpretation is that the decrease in IgA transcytosis that occurred between the 1:2 and the 1:4 dilutions is likely due to changes in cell density while the decrease in IgA trancytosis in the 1:8 dilution is likely further driven by diminished pIgR expression. One other possibility is that global differentiation is altered by decreased density. We found that markers of secretory lineages (Muc2 and Atoh1) were diminished in a density-dependent manner while a general marker of epithelial cells (Vil1) was not altered (Figure 4e–g).
Cytokines induce pIgR expression and IgA transcytosis in the primary Transwell cultures
Several different cytokines were previously shown to induce pIgR expression in tumor cell lines including IL-1β, TNFα, IFNγ, and most recently, IL-1710,11,22–24,26. We wanted to test if primary intestinal epithelial cells responded similarly to these cytokines, and if so, what was the relative potency. To do this, we performed a dose titration using these four cytokines with IgA transcytosis and pIgR expression as readouts (Figure 5). IL-17 was the most potent cytokine tested. A dose of 0.5 ng/ml resulted in higher levels of IgA transcytosis than LPS treatment (Figure 5a). IL-1β and TNFα were both significantly less potent than IL-17, as a 100 ng/ml dose of these cytokines only induced half the levels of IgA transcytosis as LPS (Figure 5b, c). Apical media from pIgR−/− cells at all doses of IL-1β, TNFα, and IL-17 contained no IgA (Figure 5a–c). For all three of these cytokines the dose response curves for pIgR expression mirrored the IgA transcytosis (Figure 5d–f).
Figure 5. IgA transcytosis and pIgR expression in response to IL-17, IL-1β, and TNFα.

Wild type and pIgR−/− cells were treated with 10 μM DAPT and varying doses of IL-17 (a,d), IL-1β (b,e), or TNFα (c,f). IgA transcytosis was analyzed by ELISA (a–c), and results were normalized to the WT+DAPT+LPS group (= 100%). Gene expression analysis of pIgR was performed (d–f) and all samples were normalized to Gapdh. Data are presented as fold change relative to untreated (0% CM) cells. All values are indicated as mean ± s.e.m. One-way ANOVA: (a) F = 152.3, P < 0.0001, n ≥ 3 per group; (b) F = 187.9, P < 0.0001, n ≥ 3 per group; (c) F = 376.7, P < 0.0001, n ≥ 3 per group; (d) F = 119.5, P < 0.0001, n ≥ 4 per group. (e) F = 49.92, P <0.0001, n ≥ 5 per group; (f) F = 28.29, P < 0.0001, n ≥ 4 per group; Means with different letters are significantly different by Bonferroni's multiple comparison test. N.D. = not detected.
Of note, the results for IFNγ treatment were distinct from that of the other cytokines as well as previously published literature using immortalized cell lines (Supplementary Figure 4). At 0.1–1 ng/ml, the epithelial monolayer did not remain intact, as apical media from wild-type and pIgR−/− cells both contained IgA levels similar to the media in the lower compartment. Furthermore, TER measurements of cytokine-treated cells showed no measureable resistance after IFNγ treatment while treatment with IL-1β, TNFα, and IL-17 showed comparable TER values with LPS-treated cells (Supplementary Figure 4). At lower doses (0.001–0.01 ng/ml), less IgA was present in the apical media, however none of these conditions led to an increase of pIgR mRNA expression. Our conclusion is that IgA uses a paracellular route in IFNγ-treated cells that could either be due to cell death or leaky junctions.
Colonic epithelial cells generated from Tlr4−/− mice show reduced IgA transcytosis and pIgR expression after LPS but not IL-17 treatment
One advantage of this system is the ability to isolate cells directly from different genetically modified mice for experiments. As a proof of principle, cells were harvested from Tlr4−/− mice to generate colonic spheroids. These cells lack the LPS receptor, Tlr4, and therefore should have a reduced response to LPS stimulation. As expected, IgA transcytosis and pIgR expression was reduced to ~50% of that of wild-type cells (Figure 6a, c). The ability of these cells to respond to other stimuli for IgA transcytosis was not affected, as IL-17 treatment of Tlr4−/− cells showed no difference from wild-type cells (Figure 6b, d).
Figure 6. Cells from Tlr4−/− mice have an impaired IgA transcytosis response to LPS but not IL-17.

(a,c) Wild type Tlr4 and Tlr4−/− were treated with 10 μM DAPT and 1 μg/ml LPS. (a) IgA transcytosis was measured by ELISA and results were normalized to the WT group (= 100%). Values are indicated as mean ± s.e.m.; n = 8 per group. Wilcoxon signed rank test: P = 0.0039. (b,d) Wild type Tlr4 and Tlr4−/− cells were treated with 10 μM DAPT and varying doses of IL-17. (b) IgA transcytosis was analyzed by ELISA and results were normalized to the WT+D+L group (= 100%). Gene expression analysis of pIgR was performed (c–d) and all samples were normalized to Gapdh. Data are presented as fold change relative to untreated (0% CM) cells. All values are indicated as mean ± s.e.m. One-way ANOVA: (b) F = 116.9, P < 0.0001, n ≥ 3 per group; (c) F = 40.92, P < 0.0001, n ≥ 7 per group; (d) F = 6.364, P < 0.0004, n ≥ 3 per group. Means with different letters are significantly different by Bonferroni's multiple comparison test. N.D. = not detected.
DISCUSSION
We have established a new method to grow primary intestinal epithelial cells in a functional monolayer. To do this, we adapted the previously established 3D primary intestinal epithelial stem cell culture system2 to a 2D monolayer in a Transwell. The system we have developed has three important components. Firstly, the use of the L-WRN CM system allowed us to attain large cell numbers that are required to form monolayers in Transwells. Secondly, cells only needed to be grown in Matrigel culture for three days prior to recovery and seeding into Transwells. Lastly, seeded cells quickly formed polarized monolayers and demonstrated high TER values within three days. In contrast, previously established intestinal epithelial monolayer systems that utilize immortalized cell lines require as many as twenty days in culture to form differentiated, polarized monolayers3–5.
This system will be useful for the study of many different physiologic processes. Here, we chose to focus on IgA transcytosis. For this process to occur, epithelial cells must express the IgA receptor pIgR, which has been shown to be regulated by microbial products19. LPS has been used as a standard in the field to induce pIgR expression in vitro. Typically immortalized cell lines have been used. Here we showed that LPS-treated primary intestinal epithelial cells were also able to induce pIgR expression. In addition, we found that maximal pIgR expression required DAPT treatment in addition to LPS, suggesting that the intestinal epithelial cells require differentiation for efficient pIgR expression. To demonstrate the robustness of primary intestinal epithelial cells for IgA transcytosis, we carefully analyzed several key factors including time, IgA dose, and LPS dose. We titrated the assay and chose to use 10 μM DAPT, 1 μg/ml LPS, and 40 μg/ml IgA as our optimal conditions. The time course of a typical experiment is 10 hours with multiple samples taken from each Transwell.
Interestingly, we found that cell density also affected IgA transcytosis. After recovery from the Matrigel, two-fold serial dilutions were made before seeding the cells in the Transwells. As it might be expected, we observed a cell density-dependent decrease in the amount of IgA transcytosed. However, this decrease in IgA was not just due to the decrease in cell number. Gene expression analysis of pIgR showed a substantial decrease at the lowest cell densities. Immunofluorescence staining for pIgR correlated with the mRNA expression data, showing fewer pIgR-positive cells at the lowest cell densities. We hypothesized the lower density cells may be in a less differentiated state, which has been shown in other in vitro cell systems39–41. We therefore tested the expression of other cell differentiation markers, including goblet cell marker Muc2 and a secretory cell lineage marker Atoh1, and found both to show a correlation of expression with cell density. This suggests that cell-to-cell endogenous signaling, and not solely exogenous chemical stimuli, regulate colonic cell differentiation in this culture system.
While LPS robustly induces pIgR expression, it is difficult to translate in vitro quantities to the biomass of intestinal microbes that generate this molecule. To make this system more applicable to specific bacteria, we utilized heat-killed bacteria. We therefore treated the cells with heat-killed E. coli and found that a dose of 107 CFU/ml stimulated IgA transcytosis and pIgR expression to a similar extent as 1μg/ml LPS. We feel that this system will be useful for future studies looking at host-microbial interactions in vitro.
Previous studies found that several host cytokines including IL-1β, TNFα, IFNγ, and IL-17 play a role in pIgR expression11,22–26. Similar to what has been shown in immortalized cell lines, IL-1β, TNFα, and IL-17 showed a dose-dependent induction of pIgR expression, with IL-17 being the most potent inducer of IgA transcytosis and pIgR expression of these cytokines. However, an important difference we found between our primary mouse intestinal epithelial cells and human cancer cell lines is the ability of IFNγ to induce pIgR expression. IFNγ treatment did not lead to pIgR expression or IgA transcytosis at any dose tested, and actually resulted in cell death at higher doses. This may be a result that would otherwise be obscured by using immortalized cell lines, which can be resistant to cytokine-induced cell death. Furthermore, this does not appear to be species-specific phenomenon, as the mouse promoter for pIgR is highly conserved to the human promoter and also contains the IRF1 binding site42.
One final important advantage of this system is the ability to readily use primary intestinal epithelial cells taken from different genetically modified mice. In this study, we utilized cells obtained from available lines of knockout mice for the pIgR and Tlr4 genes. Monolayers of epithelial cells derived from these mice allowed us to directly address questions using cells with complete loss-of-function, as compared to the usual incomplete loss of function with shRNA knockdowns (i.e. ref. 2).
Prospectus
In summary, we developed a new system to culture primary intestinal epithelial cells in monolayer culture. While we have highlighted the capability of this system to be utilized for studies of IgA transcytosis, there are many other experimental avenues where this system should prove to be useful. These areas include barrier function, host-microbial interactions and epithelial-immune/stromal cell interactions. We propose that not only will this system be useful for mechanistic studies, but also for large-scale chemical biology screens in these arenas.
METHODS
Mice
Animal protocols were approved by the Washington University Animal Studies Committee. All mice were maintained in a specific pathogen-free barrier facility. pIgR−/− mice (B6.129P2-Pigrtm1Fejo/Mmmh43) were initially obtained from Mutant Mouse Regional Resource Center (Columbia, MO). Tlr4−/− mice were provided by the laboratory of Dr. William F. Stenson (B6.B10ScN-TLR4lps-del/JthJ from Jackson Laboratory44). All mice used were 8–10 weeks of age.
3D spheroid cell culture
Primary colonic epithelial stem cells were isolated, grown, and maintained as 3D spheroid cultures in Matrigel (BD Biosciences; San Jose, CA) as described in Miyoshi et al2. Cells were kept in 50% L-WRN conditioned media (CM). Media was changed every two days, and cells were passaged every three days (1:3 split).
Formation of Transwell monolayers
To form monolayers of intestinal epithelial cells, spheroids were taken from three-day-old 3D cultures for plating in Transwells (Corning Costar 3413; Tewksbury, MA). The Transwells were coated in 0.1% gelatin for ≥1 hour at 37°C. Spheroids were recovered from Matrigel by first washing in a solution of 0.5 mM EDTA, and then dissociated for 4 minutes at 37°C using a solution of 0.05% Trypsin/0.5 mM EDTA. The trypsin was then inactivated using DMEM/F12media containing 10% FBS. The spheroids were then dissociated by vigorous pipetting (using a 1000 μl pipet). The cells were then passed through a 40 μm cell strainer (BD Biosciences) and re-suspended in 50% L-WRN CM containing 10 μM Y-27632 (ROCK inhibitor; Tocris Bioscience and R&D Systems; Minneapolis, MN). On average, spheroids from three wells of a 24-well plate were plated into the upper compartment of a single Transwell in 100μl of media. An additional 600 μl of media was added to the lower compartment of the Transwells.
Cell treatments
On day one (24 hours after seeding the Transwells) the 50% L-WRN CM supplemented with Y-27632 was removed and replaced with 0% CM (Advanced DMEM/F12 containing 20% FBS, 100 units of Penicillin, 0.1 mg/ml Streptomycin and 2mM L-glutamine). At this time, any additional treatments were also administered to the cells: LPS (Sigma L4391; Saint Louis, MO), DAPT γ-secretase inhibitor (Millipore 565784; Billerica, MA), recombinant mouse IL-1β (R&D Systems 401-ML), recombinant mouse IFNγ (R&D Systems 485-ML), recombinant mouse TNFα (R&D Systems 410-MT), recombinant mouse IL-17 (R&D Systems 421-ML), and heat-killed E. coli (lab stocks, mouse adapted strain)45. Cells were given fresh media with the respective treatments on day two, and were treated for a total of 48 hours before being used for IgA transcytosis, histology, or RNA extraction on day three.
Transepithelial electrical resistance (TER) measurements
TER was measured for cells in Transwells using an epithelial volt-ohm meter (World Precision Instruments, Inc.; Sarasota, FL). Resistance of the intestinal epithelial cell monolayer was calculated by subtracting the resistance of the (membrane + media) from the resistance of the (membrane + media + cells). Each Transwell was measured in triplicate and the average value was taken. This value was then multiplied by the area of the Transwell membrane (0.33 cm2) to obtain a final value in Ωcm2.
IgA transcytosis assay
On day three, the Transwells were removed from the various treatment conditions and washed with 0% CM. 600 μl of 0% CM containing mouse IgA (for early studies: Santa Cruz Biotechnology sc-3900; Dallas, TX; later studies: BD Pharmingen 553476) was added to the lower compartment. Both sources of IgA contained a mixture of monomeric and polymeric IgA, according to each company. For the lot of BD Pharmingen IgA used in these studies, the amount of dimeric IgA was ~85% according to the manufacturer. 100 μl of 0% CM alone was added to the upper compartment and collected at different time points to evaluate the amount of IgA transcytosed by ELISA (Immunology Consultants Labs E-90A; Portland, OR).
Immunostaining and histologic analysis
Cells in the Transwells were washed with PBS and fixed in either 10% formalin or Bouins fixative for 10 minutes. The cells were then washed three times in 70% ethanol and the Transwell membranes were cut out from the Transwell inserts using a surgical blade. The membranes were processed for paraffin embedding. 5 μm thick transverse sections were cut for hematoxylin and eosin staining and immunostaining. For this procedure, the sections were de-paraffinized, hydrated, boiled in Trilogy solution (Cell Marque; Rocklin, CA) for 20 minutes, rinsed in PBS, blocked with 1% bovine serum albumin/0.1% Tritin-X100 for 30 minutes, and incubated with primary antibody at 4°C overnight. Primary antibodies include: rabbit anti-ZO-1 (1:100, Invitrogen/Life Technologies; Grand Island, NY), mouse anti-chicken Villin1 (1:100, AbDSerotec; Raleigh, NC), and goat anti-pIgR (1:500, R&D Systems). The slides were rinsed three times in PBS and then incubated with AlexaFluor594- or AlexaFluor488-conjugaed species-specific secondary antibodies for one hour at room temperature (1:500, Invitrogen/Life Technologies). Slides were washed three times in PBS and stained with bis-benzimide (Hoescht 33258, Invitrogen/Life Technologies) to visualize nuclei and mounted with a 1:1 PBS:glycerol solution. Staining was visualized with a Zeiss (Oberkochen, Germany) Axiovert 200 microscope with an Axiocam MRM digital camera.
For whole mount immunostaining, cells were fixed and washed in the Transwells as described above, then washed three times with PBS before they were cut out from the Transwell inserts and placed in the wells of a 24-well plate. The membranes were then blocked and stained as described above. Nuclei counts were made using ImageJ46.
For whole tissue immunostaining, mouse colons were harvested and prepared as previously described47.
Gene expression analysis
RNA was isolated from cells in the Transwells on day three after seeding using the NucleoSpin RNA II isolation kit (Macherey-Nagel; Bethlehem, PA). Complementary DNA (cDNA) synthesis was performed using 0.2 ug of RNA and the SuperScript III reverse transcriptase (Life Technologies). Quantitative PCR reactions (qPCR) were performed with SYBR Advantage qPCR Premix (Clontech; Mountain View, CA). Expression levels were determined in triplicate per sample and normalized to the expression of glyceraldehyde 3-phosphate dehydrogenase (Gapdh). Primers used include: Gapdh for 5'-AGG TCG GTG TGA ACG GAT TTG-3', Gapdh rev 5'- TGT AGA CCA TGT AGT TGA GGT CA- 3', pIgR for 5'-ATG AGG CTC TAC TTG TTC ACG C-3', pIgR rev 5'-CGC CTT CTA TAC TAC TCA CCT CC-3', Villin1 for 5'-ATG ACT CCA GCT GCC TTC TCT-3', Villin1 rev 5'- GCT CTG GGT TAG AGC TGT AAG-3', Reg3g for 5'-CATCAACTGGGAGACGAATCC-3', Reg3g rev 5'-CAGAAATCCTGAGGCTCTTGACA-3', Muc2 for 5'-ATG CCC ACC TCC TCA AAG AC-3', Muc2 rev 5'-GTA GTT TCC GTT GGA ACA GTG AA-3', Atoh1 for 5'- GAG TGG GCT GAG GTA AAA GAG T-3', and Atoh1 rev 5'-GGT CGG TGC TAT CCA GGA G-3'.
Supplementary Material
Acknowledgements
We thank Gerard E. Kaiko for comments on the manuscript. This work was supported by AI08488702, the CCFA Genetics initiative and the Helmsley Charitable Trust. C.M. and K.L.V. were supported by an NIH training grant (T32 AI007163). The Washington University Digestive Disease Research Core Center is supported by a grant from the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) (P30DK052574).
Footnotes
Conflict of Interest: No conflict
REFERENCES
- 1.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 2.Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sambuy Y, et al. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- 4.Meunier V, Bourrie M, Berger Y, Fabre G. The human intestinal epithelial cell line Caco-2; pharmacological and pharmacokinetic applications. Cell Biol Toxicol. 1995;11:187–194. doi: 10.1007/BF00756522. [DOI] [PubMed] [Google Scholar]
- 5.Rousset M. The human colon carcinoma cell lines HT-29 and Caco-2: two in vitro models for the study of intestinal differentiation. Biochimie. 1986;68:1035–1040. doi: 10.1016/s0300-9084(86)80177-8. [DOI] [PubMed] [Google Scholar]
- 6.Duizer E, Penninks AH, Stenhuis WH, Groten JP. Comparison of permeability characteristics of the human colonic Caco-2 and rat small intestinal IEC-18 cell lines. J Control Release. 1997;49:39–49. doi:Doi 10.1016/S0168-3659(97)00058-8. [Google Scholar]
- 7.Irvine JD, et al. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J Pharm Sci. 1999;88:28–33. doi: 10.1021/js9803205. doi:10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- 8.Kuratnik A, Giardina C. Intestinal organoids as tissue surrogates for toxicological and pharmacological studies. Biochem Pharmacol. 2013;85:1721–1726. doi: 10.1016/j.bcp.2013.04.016. doi:DOI 10.1016/j.bcp.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Sun H, Chow ECY, Liu S, Du Y, Pang KS. The Caco-2 cell monolayer: usefulness and limitations. Expert Opin Drug Met. 2008;4:395–411. doi: 10.1517/17425255.4.4.395. doi:Doi 10.1517/17425255.4.4.395. [DOI] [PubMed] [Google Scholar]
- 10.Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. 2005;206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x. [DOI] [PubMed] [Google Scholar]
- 11.Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011;4:598–602. doi: 10.1038/mi.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rojas R, Apodaca G. Immunoglobulin transport across polarized epithelial cells. Nat Rev Mol Cell Biol. 2002;3:944–955. doi: 10.1038/nrm972. [DOI] [PubMed] [Google Scholar]
- 13.Hooper LV. OPINION Do symbiotic bacteria subvert host immunity? Nat Rev Microbiol. 2009;7:367–374. doi: 10.1038/nrmicro2114. [DOI] [PubMed] [Google Scholar]
- 14.Hamburger AE, Bjorkman PJ, Herr AB. Structural Insights into Antibody-Mediated Mucosal Immunity. Current Topics in Microbiology and Immunology. 2006;308:173–204. doi: 10.1007/3-540-30657-9_8. [DOI] [PubMed] [Google Scholar]
- 15.Brandtzaeg P, Prydz H. Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature. 1984;311:71–73. doi: 10.1038/311071a0. [DOI] [PubMed] [Google Scholar]
- 16.Mostov KE, Kraehenbuhl JP, Blobel G. Receptor-mediated transcellular transport of immunoglobulin: synthesis of secretory component as multiple and larger transmembrane forms. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:7257–7261. doi: 10.1073/pnas.77.12.7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mostov KE, Verges M, Altschuler Y. Membrane traffic in polarized epithelial cells. Curr Opin Cell Biol. 2000;12:483–490. doi: 10.1016/s0955-0674(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 18.Lindh E. Increased Resistance of Immunoglobulin a Dimers to Proteolytic Degradation after Binding of Secretory Component. J Immunol. 1975;114:284–286. [PubMed] [Google Scholar]
- 19.Hooper LV, et al. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 20.Bruno ME, et al. Regulation of the polymeric immunoglobulin receptor in intestinal epithelial cells by Enterobacteriaceae: implications for mucosal homeostasis. Immunol Invest. 2010;39:356–382. doi: 10.3109/08820131003622809. [DOI] [PubMed] [Google Scholar]
- 21.Schneeman TA, et al. Regulation of the polymeric Ig receptor by signaling through TLRs 3 and 4: Linking innate and adaptive immune responses. J Immunol. 2005;175:376–384. doi: 10.4049/jimmunol.175.1.376. [DOI] [PubMed] [Google Scholar]
- 22.Blanch VJ, Piskurich JF, Kaetzel CS. Cutting edge: coordinate regulation of IFN regulatory factor-1 and the polymeric Ig receptor by proinflammatory cytokines. J Immunol. 1999;162:1232–1235. [PubMed] [Google Scholar]
- 23.Piskurich JF, et al. Transcriptional regulation of the human polymeric immunoglobulin receptor gene by interferon-gamma. Mol Immunol. 1997;34:75–91. doi: 10.1016/s0161-5890(96)00079-x. [DOI] [PubMed] [Google Scholar]
- 24.Schjerven H, Brandtzaeg P, Johansen FE. A novel NF-kappa B/Rel site in intron 1 cooperates with proximal promoter elements to mediate TNF-alpha-induced transcription of the human polymeric Ig receptor. J Immunol. 2001;167:6412–6420. doi: 10.4049/jimmunol.167.11.6412. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi M, et al. The polymeric immunoglobulin receptor (secretory component) in a human intestinal epithelial cell line is up-regulated by interleukin-1. Immunology. 1997;92:220–225. doi: 10.1046/j.1365-2567.1997.00341.x. doi:DOI 10.1046/j.1365-2567.1997.00341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao AT, Yao S, Gong B, Elson CO, Cong Y. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol. 2012;189:4666–4673. doi: 10.4049/jimmunol.1200955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.VanDussen KL, et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139:488–497. doi: 10.1242/dev.070763. doi:10.1242/dev.070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. doi:Doi 10.1038/Nature03659. [DOI] [PubMed] [Google Scholar]
- 29.Bretscher A, Weber K. Villin: the major microfilament-associated protein of the intestinal microvillus. Proc Natl Acad Sci U S A. 1979;76:2321–2325. doi: 10.1073/pnas.76.5.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103:755–766. doi: 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Connor DT, Burton D, Deftos LJ. Chromogranin A: immunohistology reveals its universal occurrence in normal polypeptide hormone producing endocrine glands. Life Sci. 1983;33:1657–1663. doi: 10.1016/0024-3205(83)90721-x. [DOI] [PubMed] [Google Scholar]
- 32.Falk P, Roth KA, Gordon JI. Lectins are sensitive tools for defining the differentiation programs of mouse gut epithelial cell lineages. Am J Physiol. 1994;266:G987–1003. doi: 10.1152/ajpgi.1994.266.6.G987. [DOI] [PubMed] [Google Scholar]
- 33.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maunoury R, et al. Villin Expression in the Visceral Endoderm and in the Gut Anlage during Early Mouse Embryogenesis. Embo J. 1988;7:3321–3329. doi: 10.1002/j.1460-2075.1988.tb03203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song W, Bomsel M, Casanova J, Vaerman JP, Mostov K. Stimulation of transcytosis of the polymeric immunoglobulin receptor by dimeric IgA. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:163–166. doi: 10.1073/pnas.91.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diebel LN, Liberati DM. Disparate effects of bacteria and Toll-like receptor-dependant bacterial ligand stimulation on immunoglobulin A transcytosis. J Trauma. 2011;70:691–700. doi: 10.1097/TA.0b013e31820c780e. [DOI] [PubMed] [Google Scholar]
- 37.Frantz AL, et al. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 2012;5:501–512. doi: 10.1038/mi.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura Y, et al. Upregulation of Polymeric Immunoglobulin Receptor Expression by the Heat-Inactivated Potential Probiotic Bifidobacterium bifidum OLB6378 in a Mouse Intestinal Explant Model. Scand J Immunol. 2012;75:176–183. doi: 10.1111/j.1365-3083.2011.02645.x. [DOI] [PubMed] [Google Scholar]
- 39.Ezeonu I, Wang M, Kumar R, Dutt K. Density-dependent differentiation in nontransformed human retinal progenitor cells in response to basic fibroblast growth factor- and transforming growth factor-alpha. DNA Cell Biol. 2003;22:607–620. doi: 10.1089/104454903770238085. doi:10.1089/104454903770238085. [DOI] [PubMed] [Google Scholar]
- 40.Lee YS, Yuspa SH, Dlugosz AA. Differentiation of cultured human epidermal keratinocytes at high cell densities is mediated by endogenous activation of the protein kinase C signaling pathway. J Invest Dermatol. 1998;111:762–766. doi: 10.1046/j.1523-1747.1998.00365.x. doi:10.1046/j.1523-1747.1998.00365.x. [DOI] [PubMed] [Google Scholar]
- 41.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 42.Mestecky J, et al. Mucosal Immunology. Academic Press; San Diego: 2005. [Google Scholar]
- 43.Johansen FE, et al. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J Exp Med. 1999;190:915–921. doi: 10.1084/jem.190.7.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 45.Bloom SM, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang SS, et al. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med. 2008;5:e41. doi: 10.1371/journal.pmed.0050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.