

Abstract
Although angiotensin II (Ang II) and its receptor AT1 have been implicated in abdominal aortic aneurysm (AAA) formation, the proximal signaling events primarily responsible for AAA formation remain uncertain. Caveolae are cholesterol-rich membrane microdomains that serve as a signaling platform to facilitate the temporal and spatial localization of signal transduction events including those stimulated by Ang II. Caveolin-1 (Cav1) enriched caveolae in vascular smooth muscle cells mediate ADAM17-dependent epidermal growth factor receptor (EGFR) transactivation, which is linked to vascular remodeling induced by Ang II. Here, we have tested our hypothesis that Cav1 plays a critical role for development of AAA at least in part via its specific alteration of Ang II signaling within caveolae. Cav1−/− mice and the control wild-type mice were co-infused with Ang II and β-aminopropionitrile to induce AAA. We found that Cav1−/− mice with the co-infusion did not develop AAA compared to control mice in spite of hypertension. We found an increased expression of ADAM17 and enhanced phosphorylation of EGFR in AAA. These events were markedly attenuated in Cav1−/− aortae with the co-infusion. Furthermore, Cav1−/− mice aortae with the co-infusion showed less endoplasmic reticulum stress, oxidative stress and inflammatory responses compared to aortae from control mice. Cav1 silencing in cultured vascular smooth muscle cells prevented Ang II-induced ADAM17 induction and activation. In conclusion, Cav1 appears to play a critical role in the formation of AAA and associated endoplasmic reticulum/oxidative stress presumably through the regulation of caveolae compartmentalized signals induced by Ang II.
Keywords: abdominal aortic aneurysm, hypertension, mouse model, vascular smooth muscle, signaling mechanism, caveolae
INTRODUCTION
Abdominal aortic aneurysm (AAA) is a significant cause of mortality for adults aged >65 years. No established pharmacological treatment is currently available to prevent AAA advancement and rupture [1, 2]. Accumulating evidence suggests the involvement of angiotensin II (Ang II) and its receptor AT1 in AAA formation [3]. Although there is a large gap in our knowledge regarding the Ang II-sensitive proximal signal transduction events primarily responsible for AAA development, enhanced oxidative stress, inflammation and extracellular matrix protein degradation seem to be crucial for Ang II-induced AAA in mice [3–5].
Caveolae are cholesterol-rich membrane microdomains that serve as a signaling platform to facilitate the temporal and spatial localization of signal transduction events including those stimulated by Ang II [6]. Caveolin 1 (Cav1) is the major structural protein of caveolae in the vasculature and is expressed both in vascular smooth muscle cells (VSMC) and endothelial cells (EC) [7]. Recent studies including those with Cav1−/− mice further suggest a strong link between caveolae and Cav1 with cardiovascular diseases and their diverse pathophysiology including atherosclerosis, dyslipidemia, cardiac fibrosis, insulin resistance, inflammation and oxidative stress [8]. However, whether Cav1 and the caveolae-compartmentalized signaling have any role in AAA development has never been investigated.
In VSMC, the metalloprotease ADAM17/tissue necrosis factor-α (TNF-α) converting enzyme is primarily responsible for the epidermal growth factor receptor (EGFR) trans-activation induced by Ang II [9]. This process relies on ADAM17 compartmentalization in caveolae and is crucial for vascular remodeling [10, 11]. In human AAA as well as in a mouse model where AAA is induced by aortic CaCl2 application, significant up-regulation of ADAM17 has been reported [12]. Moreover, temporal and systemic deletion of ADAM17 gene in mice prevented CaCl2-induced AAA formation that was associated with attenuations of oxidative stress, inflammation and extracellular matrix disruption [13]. In addition, genome-wide transcriptional profiling identified AT1, ADAM17, EGFR and a lysyl oxidase, Lox, as central genes among highly ranked gene sub-networks associated with AAA induced by Ang II [14].
Lox crosslinks collagen and elastin fibers to create insoluble proteins resistant to proteolytic degradation, thus stabilizing the vessel wall [15]. Lox secretion and maturation seem to require its caveolae localization in VSMC [16] and Lox gene transfer prevented CaCl2-induced AAA in mice [17]. Although genetically hyperlipidemic mice with Ang II infusion is an established model for AAA [18], mechanistic dissection of this AAA model may be hampered by associated metabolic pathophysiology including atherosclerosis, obesity, hyperlipidemia and type II diabetes. A new mouse model of AAA has recently been created in normolipidemic mice with co-infusion of Ang II and a Lox inhibitor, β-aminopropionitrile (BAPN) [19]. While genetic ablation of Lox in mice causes aortic aneurysm and rupture [20], inhibition of Lox by BAPN is insufficient to promote AAA in mice and requires co-infusion of Ang II [19].
Given that the caveolae-compartmentalized specific signaling process may be the key feature in AAA, we hypothesized that Cav1 in the vasculature plays a critical role for development of AAA at least in part via its function in regulating caveolae based signaling events. This hypothesis was tested using Cav1−/− mice with co-infusion of Ang II and BAPN.
MATERIALS AND METHODS
Animal protocol
All animal procedures were performed with the prior approval of the Temple University Institutional Animal Care and Use Committee and in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals. 8 weeks old male Cav1−/− mice (B6.Cg-Cav1tm1Mls/J) and control Cav1+/+ mice (C57BL/6J) were obtained from the Jackson Laboratory and housed under barrier conditions. Standard sterilized laboratory diet and water were available ad libitum. The mice were co-infused with Ang II (1 μg/kg/min) for 4 weeks and BAPN (150 mg/kg/day) for the first 2 weeks or infused with control saline through osmotic-pump (Alzet, Durect Corp) [19], which was subcutaneously implanted under anesthesia (Ketamine 80 mg/kg and xylazine 4 mg/kg, i.p.). Adequacy of anesthesia was monitored by withdrawal response to foot pinch. This mouse model reproducibly produces AAA associated with hypertension with morphological and histological characteristics similar to human AAA, but without atherosclerosis seen in other Ang II-dependent AAA models in hyperlipidemic mice [19]. Blood pressure and heart rate were evaluated in all animals in the conscious state at day 28 by telemetry (DSI equipped with AD Instrument 6 software) via carotid catheter (PA-C10 transmitter). Mice were euthanized by exsanguination via carotid artery under anesthesia (Ketamine 120 mg/kg and xylazine 6 mg/kg, i.p.). To prepare samples for histological analysis, mice were perfused with saline followed by 10% paraformaldehyde at 100 mmHg. Aortas were dissected, cleaned of extraneous tissue and then photographed. To prepare samples for mRNA analysis, mice were euthanized, aortas dissected free, extraneous tissue removed and photographed. Tissue was flash-frozen for mRNA isolation. AAA was defined as a localized dilation of the abdominal aortic wall with maximal outside diameter greater than 50% of the outside diameter of the infra-renal aorta measured at the midpoint between the renal artery and bifurcation of the iliac artery. Compared to saline controls, Ang II plus BAPN did not induce AAA or aortic dilation at infra-renal aorta in any of the experimental animals. In regards to other segments of the aorta, only one wild type mouse receiving co-infusion demonstrated a class I thoracic aortic aneurysm (TAA). As such, all maximum aortic diameters were measured at supra-renal aortas after excision. In total, 24 wild type mice and 16 Cav1−/− mice were used for the Ang II plus BAPN infusion experiments. Among the 16 Cav1−/− AngII plus BAPN infusion mice, a total of 12 were included in the survival study (4 Cav1−/− mice with Ang II plus BAPN infusion died by telemetry failure/bleeding death). Of these mice, 10 were analyzed for aortic diameter and 2 were collected without the diameter evaluation for mRNA analysis. In wild type mice with Ang II plus BAPN infusion, aortic diameters were analyzed in all 10 surviving mice. For saline control experiments, 10 wild type and 9 Cav1−/− mice were used for analysis. No deaths were observed in the saline infusion groups and all the aortic diameters were analyzed. In the saline control group, successful telemetry recordings were obtained in 8 wild type and 8 Cav1−/− mice while telemetry data was collected in 6 wild type and 9 Cav1−/− mice infused with Ang II plus BAPN. The severity of AAA was classified based on the gross appearance of the aorta as previously described [21]. Serial cross-sections (5 μm thick) from abdominal aortas were mounted on microscope slides. To avoid sample selection bias, aortas from the first 4 animals in each infusion group were examined by histology and immunohistochemistry while aorta’s from the last 4 animals were processed for qPCR.
Immunohistochemistry
Sections from abdominal aortas were deparaffinized and blocked in 5% goat serum and 1% BSA for 1h at room temperature, incubated with primary antibody in PBS containing 1% BSA and 0.1% Tween 20 for 18 h at 4 °C, followed by biotinylated secondary antibody for 90 min at room temperature. Slides were incubated with avidin–biotin peroxidase complex for 30 min at room temperature and staining was visualized with the substrate diaminobenzidine (Vector) producing a brown color and counterstained with haematoxylin. An equal concentration of control IgG was used side-by-side with each antibody to ensure staining specificity. Quantification of the antibody staining was performed as reported previously with subtraction of IgG background staining [22]. All images were visualized on a Photometrics CoolSNAP HQ digital camera and acquired with SPOT 4.7 Basic software using the same exposure time. Images were loaded into the ImageJ program (http://rsb.info.nih.gov/ij) for analysis. A region of interest was drawn around the entire aorta with the freehand selection tool. Adventitia was excluded from the quantification since the adventitia areas were quite limited in aortas except those with AAA. All images were set to the same hue, saturation, and brightness. The area and intensity (integrated density) in the region of interest were then measured and analyzed. Data were obtained from three to four non-overlapping fields per aortic cross-section for each antibody (n=4–3 aortas per treatment or genotype). Results are presented as fold increase over control, which was set at 1.
Quantitative real-time PCR
Abdominal aorta was homogenized by Biomasher and total RNA was extracted using TRIzol reagent (Invitrogen). cDNA was synthesized RevertAid First Strand cDNA Synthesis Kit (Thermo). Quantitative real-time PCR (qPCR) was performed with SYBR Green qPCR Master Mix (Fermentas) as described previously [23]. mRNA abundance was calculated by normalization to ribosome 18S. The primers used are ADAM17: Forward GGC GCG GGA GGG AGA AGT TT, Reverse CGC CGC CTC ATG TTC CCG TC, Ribosome 18S: Forward AGT TCC AGC ACA TTT TGC GAG, Reverse TCA TCC TCC GTG AGT TCT CCA.
Cell culture
VSMC were prepared from thoracic aorta of male Sprague-Dawley rats (~350 g) by the explant method as described previously [24]. Rats were euthanized by exsanguination under anesthesia (Ketamine 100 mg/kg and xylazine 5 mg/kg, i.p.). VSMC were subcultured in DMEM containing 10% fetal bovine serum, penicillin and streptomycin. Cells from passage 3 to 10 at 80~90% confluence in culture wells were made quiescent by incubation with serum-free medium for 2–3 days. To avoid any potential phenotypic alteration, VSMC were renewed every 2–3 months and VSMC from frozen stock were not used in this study. The results were confirmed in at least 2 distinct cell lines.
RNA interference by recombinant adenovirus
Replication-incompetent adenoviruses expressing engineered miRNA encoding murine miR-155 stem loop and embedded siRNAs were constructed using the BLOCK-iT™ Adenoviral RNAi Expression System (Invitrogen) according to the manufacturer’s instructions [25]. In this system, virally encoded engineered miRNA is processed by the endogenous cellular machinery to produce siRNA specifically to the target [26, 27]. A 21mer siRNA sequence (siR Cav1-226: 5′- GTG GTC AAG ATT GAC TTT GAA -3′) perfectly complementary to target coding regions of rat Cav1 (Accession: NM_031556) was designed using the Invitrogen BLOCK-iT™ RNAi online designer program and was subsequently cloned into the pcDNA™ 6.2-GW/EmGFP-miR vector. The pcDNA™6.2-GW/EmGFP-miR control plasmid with a 21mer sequence, which is predicted not to target any known mammalian gene was used as a scramble control (referred to as miR control). Adenoviruses encoding the EmGFP-miRNA cassette from these constructs were generated using the ViraPower™ Adenoviral Expression System (Invitrogen) to produce crude adenoviral stocks. For convenience, we abbreviated the miRNA-embedded Cav1 siRNA as miR Cav1. Viral titers were calculated as previously described [28] and are expressed in units of multiplicity of infection (MOI). VSMC were infected with adenovirus as described with modification to include 3% FuGENE6 to enhance infection efficiency [29].
Immunoblotting
Immunoblotting was performed as previously described [24]. Quiescent VSMCs grown on 6-well plates were stimulated for specified durations. The reaction was terminated by the replacement of medium with 100 μL of 1xSDS sample buffer. 40 μL of the cell lysates were subjected to SDS-PAGE gel electrophoresis and electrophoretically transferred to a nitrocellulose membrane. The membranes were then exposed to primary antibodies overnight at 4 °C. After incubation with the peroxidase linked secondary antibody for 1 h at room temperature, immunoreactive proteins were visualized using a chemiluminescence reaction kit.
Heparin binding EGF-like growth factor (HB-EGF) shedding assay
Ang II-mediated HB-EGF shedding was quantified in VSMC using an alkaline-phosphatase-tagged HB-EGF encoding adenovirus (HB-EGF-ALP) as previously described [10]. Following serum starvation, VSMC were co-infected with adenoviral vectors encoding HB-EGF-ALP and control or Cav1 miRNA. 3 days after infection, cells were stimulated with 100 nmol/LAng II for 60 min. Secreted HB-EGF was measured using a colorimetric alkaline phosphatase activity assay as previously described [10].
Promoter assay
pADAM17-Luc vectors encoding murine ADAM17 promoter (−2340 to −1 bp), its 5′ deletion constructs or HRE mutants located at H4 site were constructed as reported previously [30]. pSEAP2-control vector (Clontech) was used to normalize transfection efficiency. VSMC (2×106 cells) were transfected by Amaxa Nucleofector program U-25 in basic SMC Nucleofector™ solution (Lonza) containing 2 μg pADAM17-Luc and 1 μg pSEAP2-control vector together with 3 μg pcDNA™ 6.2-GW/EmGFP-miR Cav1 or the control vector. 24 h after the transfection, cells were incubated with serum-free DMEM for 24 h. The cells were then stimulated with 100 nmol/L Ang ll for 24 h. Cellular luciferase activity was measured by Luciferase Reporter Gene Assay kit (Roche) and secreted alkaline phosphatase activity in the medium was measured as described.
Reagents
Ang II was purchased from Sigma-Aldrich and Bachem. BAPN was purchased from TCI. Antibodies for immunoblotting against ADAM17 (sc-13973) and for immunohistochemistry against interleukin (IL)-1β (sc-7884) and TNF-α (sc-52746) were purchased from Santa Cruz Biotechnology. Antibodies for immunohistochemistry against ADAM17, matrix metalloprotease-2 (MMP2) and MMP9 were purchased from Abcam (ab39163, ab37150 and ab38898), respectively. Antibody against IL-6 (bs-0782R) was purchased from Bioss. Antibody against Tyr1068-phosphorylated EGFR (2234) was purchased from Cell Signaling. Antibody against GAPDH (MRB374) and nitro-tyrosine (06–284) were purchased from Millipore. Antibody against KDEL (ADI-SPA-827) was from Enzo Life Sciences. Antibodies against Cav1 (610060) and Nox2/gp91 Phox (611414) were purchased from BD Biosciences.
Statistical analysis
Kaplan-Meier survival curves were constructed and analyzed using log-rank (Mantel-Cox) test. Fisher’s exact test was used to analyze categorical data. Differences between multiple groups were analyzed by 2-way ANOVA, followed by the Tukey-Kramer post hoc test. Data were presented as mean±SEM. Statistical significance was taken at p<0.05.
RESULTS
Cav1 deficiency attenuated Ang II-dependent AAA development
There was a significant difference in survival rates between Cav1+/+ and Cav1−/− mice during 28 days of AngII plus BAPN co-infusion. Among 24 control Cav1+/+ mice that received Ang II plus BAPN, a total of 14 mice died before 4 weeks (Figure 1A). We were able to perform necropsy for 9 mice and ruptured aortae were recognized in all of these mice (6 at thoracic level and 3 at abdominal level) suggesting that the major cause of death in these mice was rupture (Figure 1B). No deaths were observed in the group of Cav1−/− mice that received Ang II plus BAPN or in the saline infusion groups.
Figure 1. Increased mortality and fatal aortic rupture were prevented in Cav1 deficient mice co-infused with Ang II and BAPN.
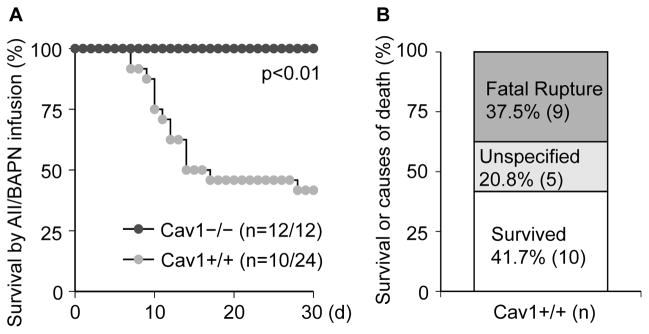
8 week old Cav1−/− mice and the control Cav1+/+ (C57Bl/6) mice were infused with Ang II (1 μg/kg/min for 4 weeks) and BAPN (150 mg/kg/day for the first 2 weeks). Percentage survival curve with black and grey circles represent Cav1−/− and Cav1+/+ mice, respectively (n=survived number/total number) (A). Summary data showing percentages of animals with fatal rupture or unspecified death following treatment with Ang II plus BAPN (B).
After 4 weeks of either saline or Ang II plus BAPN infusion, surviving mice were characterized. There was no significant difference in body weights or heart rates among the groups. Blood pressure was significantly elevated in both strains receiving Ang II plus BAPN (Table 1). In agreement with a previous report [31], Cav1−/− mice under saline control condition have slightly elevated diastolic blood pressure.
Table 1.
Characteristics of the mice infused with saline or Ang II + BAPN
| Infusion | Cav1 | BW g (n) | SBP/MBP/DBP mmHg (n) | HR b/m (n) |
|---|---|---|---|---|
| Saline | +/+ | 22.7±0.4 (10) | 125±4/101±4/79±4 (8) | 604±20 (8) |
| −/− | 23.4±0.6 (9) | 118±3/104±3/93±3† (8) | 543±44 (8) | |
| Ang II+BAPN | +/+ | 23.8±0.8 (10) | 195±6*/151±5*/116±7* (6) | 570±41 (6) |
| −/− | 24.3±1.3 (10) | 194±7*/161±7*/135±9* (9) | 526±22 (9) |
Data are means ± SEM. BW, body weight; SBP, systolic blood pressure; MBP, mean blood pressure; DBP, diastolic blood pressure; HR, heart rate; b/m, beats/min. p<0.01, significantly different from saline infusion within strains (*). p<0.05, significantly different from Cav1+/+ within infusions (†).
All surviving control Cav1+/+ mice with AngII and BAPN co-infusion exhibited AAA (Figures 2A and 2B) and most of the AAA (80%) were at advanced stage (class III or IV) (Supplementary Table 1). In contrast, only one mouse among 10 Cav1−/− mice receiving the co-infusion had class I AAA. Ang II plus BAPN infusion increased abdominal aortic diameter in Cav1+/+ mice but not in Cav1−/− mice (Figure 2C). Thoracic aortic aneurysm was observed in one Cav1+/+ mouse with the co-infusion but not in the Cav1−/− group with the infusion.
Figure 2. Prevention of Ang II-dependent AAA development in Cav1 deficient mice.
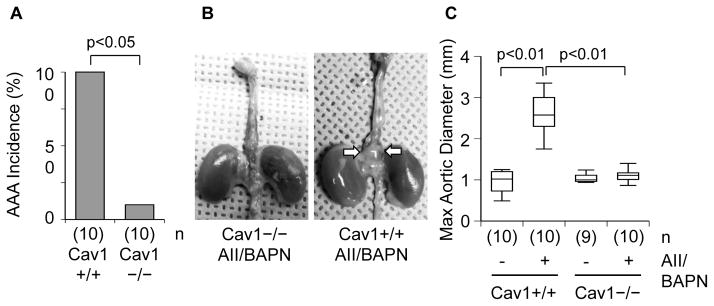
8 week old Cav1−/− mice and the control Cav1+/+ (C57Bl/6) mice were infused with Ang II (1 μg/kg/min for 4 weeks) and BAPN (150 mg/kg/day for the first 2 weeks) or saline for 4 weeks. Incidence of AAA in surviving mice after Ang II plus BAPN infusion for 4 weeks (A). Representative aorta after Ang II plus BAPN infusion, arrows indicate AAA (B). Measurements of maximal external width of abdominal aortas after the 4 week infusion (C).
Histological characterization of abdominal aortas with Masson’s trichrome staining and hematoxylin eosin staining demonstrated marked thickening of the vascular wall, degeneration of elastic lamina, presence of intramural thrombus, and thickened, collagen-rich adventitia in Cav1+/+ mice with the co-infusion (Figure 3). In contrast, Ang II plus BAPN infusion only slightly enhanced medial layer and adventitia thickness and elastic lamina remained intact in Cav1−/− mice. Taken together, systemic Cav1 ablation appears to protect mice from AAA development and rupture induced by Ang II plus BAPN co-infusion.
Figure 3. Histological assessment of abdominal aortas.

8 week old Cav1−/− mice and the control Cav1+/+ (C57Bl/6) mice were infused with Ang II (1 μg/kg/min 4 weeks) and BAPN (150 mg/kg/day 2 weeks) or saline for 4 weeks. Cross sections of abdominal aortas were stained with Masson’s trichrome (MT) or hematoxylin and eosin (HE) protocol. Representative staining was shown from n=4 for each.
Requirement of Cav1 for induction and activation of ADAM17 by Ang II
Immunohistochemical analysis was performed in abdominal aortae in order to evaluate the status of caveolae-localized signaling proteins associated with AAA formation (Supplementary Figure A). ADAM17 was markedly up-regulated in medial layers and adventitia of AAA induced by the co-infusion compared with normal abdominal aorta with saline infusion. The ADAM17 induction was associated with EGFR activation as assessed with an auto-phosphorylation site antibody. The induction of ADAM17 and EGFR phosphorylation induced by the co-infusion were markedly suppressed in aorta of Cav1−/− mice. In addition, quantitative real-time PCR analysis of abdominal aortas demonstrated induction of ADAM17 mRNA by Ang II plus BAPN infusion in wild-type mice that was attenuated in Cav1−/− mice (Figure 4A).
Figure 4. ADAM17 induction was prevented by Cav1 silencing.
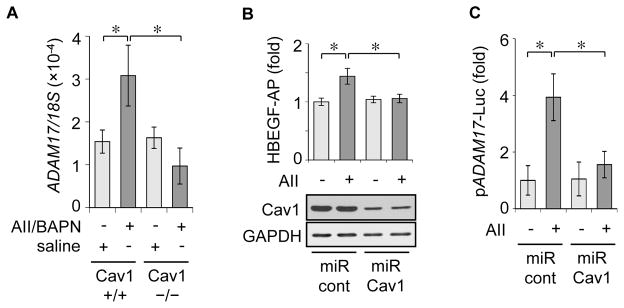
8 week old Cav1−/− mice and the control Cav1+/+ (C57Bl/6) mice were infused with Ang II+BAPN or saline for 4 weeks as in Fig 1. qPCR analysis of ADAM17 mRNA expression in abdominal aorta (n=4) (A). Rat VSMC were infected with adenoviruses (50 moi) encoding HB-EGF-AP and miRNA targeting rat Cav1 or non-targeting control miRNA (100 moi) for 72 h and HB-EGF-AP shedding assay was performed with or without 100 nM Ang II stimulation (means ± SEM from three separate experiments). The combined cell lysates from each experimental condition were analyzed by immunoblotting as indicated (B). Rat VSMC cotransfected with ADAM17 promoter-Luc construct and miR ADAM17 or the control miR construct were stimulated with or without 100 nM Ang II for 24 hours (means ± SEM from three separate experiments) (C). *p<0.05
The role of Cav1 in regulating ADAM17 induction and activity was further studied with cultured VSMC in vitro. Infection of adenovirus vector encoding engineered miRNA-embedded Cav1 targeting siRNA [25] markedly reduced VSMC Cav1 expression (Supplementary Figure B) and inhibited Ang II-induced ADAM17 activation as assessed by HB-EGF shedding (Figure 4B). Cav1 silencing also prevented ADAM17 promoter activation by Ang II (Figure 4C).
Cav1 deficiency attenuated ER stress, oxidative stress and inflammatory responses associated with AAA
Both oxidative stress and endoplasmic reticulum (ER) stress have been implicated in a variety of cardiovascular diseases [32, 33] and each can be induced through Ang II signaling mechanisms [34, 35]. While oxidative stress seems critical for AAA development [5], the role of ER stress in AAA remains obscure. Enhanced staining of an ER stress marker, KDEL, an oxidative stress marker, nitro-tyrosine, and NADPH oxidase subunit, Nox2, was observed in aorta with AAA, whereas all staining was markedly prevented in Cav1−/− mice with co-infusion (Supplementary Figure C).
The effects of Cav1 deficiency on AAA-associated cytokine and MMP expression were further analyzed. AAA induced by Ang II and BAPN co-infusion was associated with enhanced expression of TNF-α, IL-6 and MMP-2 but not IL-1β or MMP-9 in medial layers. These AAA associated responses were attenuated in Cav1−/− mice with the co-infusion. Enhanced expression of TNF-α, MMP-2 and MMP-9 was also observed in adventitia of AAA (Supplementary Figure D).
DISCUSSION
By using a novel rodent model of AAA induced by Ang II plus BAPN co-infusion [19], the present study demonstrated a remarkable prevention of AAA development and rupture in Cav1−/− mice compared with the wild-type control C57Bl/6 mice. However, hypertension associated with the co-infusion was unaltered in Cav1−/− mice. Our findings are consistent with a past publication reporting that Cav1−/− mice were protected from cardiac hypertrophy but not hypertension induced by co-treatment with Ang II and an endothelial nitric oxide synthase (eNOS) inhibitor [36]. Although hypertension is a frequently cited risk factor for human AAA, its contribution to AAA development appears rather weak or negative in human studies [37]. Past studies with several distinct pharmacological [4, 38–40] or genetic manipulations [41–43] together with our current observations further support the dispensable role of hypertension in Ang II-dependent AAA formation.
While the induction of AAA in this model requires co-infusion of BAPN (a Lox inhibitor), the AAA analyses on this model may provide a unique alternative to other established AAA mouse models. We, and others, have observed higher incidence of AAA formation and rupture in the Ang II/BAPN co-infusion models than the hyperlipidemic mice infused with Ang II alone [19, 44], whereas incidence of thoracic aortic aneurysm seems to be varied among the studies. The rupture or mortality rate we observed seems comparable to previously reported rupture-prone AAA models including C57Bl/6 with Ang II plus TGF-β antibody co-infusion [45] and Tukuba hypertensive mice with 1% salt loading [46]. The Ang II BAPN co-infusion appears to be a useful model to study potential prevention of AAA development and rupture in a genetically engineered mouse without the need for hybrid double knockouts such as those bred with apoE−/− mice.
Our data demonstrating suppression of ADAM17 induction in Cav1−/− mice with the co-infusion and in VSMC stimulated with Ang II suggest one potential mechanism by which Cav1 silencing protects from AAA formation. A recent study showed that CaCl2-induced AAA did not develop in mice with inducible ADAM17 silencing [13]. Two major ADAM17 substrates likely mediating vascular remodeling are HB-EGF and TNF-α [11]. Our data demonstrated that EGFR activation is associated with AAA. We also observed induction of ADAM17 and activation of EGFR in AAA in LDLr−/− mice infused with Ang II (unpublished observation). Cav1 silencing in VSMC in vitro also prevented HB-EGF shedding in VSMC stimulated with Ang II. ADAM17 appears to be one of the central genes forming a gene cluster involving AT1 receptor and EGFR in early stage AAA observed in apoE−/− mice infused with Ang II [14]. It is intriguing to note that both Cav1 and ADAM17 are up-regulated through hypoxia inducible factor a [30, 47], which is another Ang II inducible factor in VSMC [48]. In addition, both the Cav1 silencing presented here and also its over-expression [10] blocked EGFR transactivation suggesting a tight regulation of the ADAM17 EGFR signaling axis via Cav1 and caveolae. However, further studies are required to support the involvement of the VSMC ADAM17/HB-EGF/EGFR signaling axis in AAA development via Cav1 such as with VSMC selective rescue and or silencing of each component.
In apoE−/− mice, Cav1 expression was abundant in EC and VSMC, whereas no expression was evident in infiltrating macrophage [49]. A study with EC-selective Cav1 rescue in athero-protective Cav1/apoE double knockout mice has demonstrated a critical role of EC Cav1 to promote cell adhesion molecule expression and macrophage infiltration [50]. In addition, Cav1−/− mice have been reported to show eNOS hyper-reactivity [50]. EC function has a negative relationship with AAA development in human [51] and eNOS/apoE double knockout mouse shows AAA development and rupture without Ang II infusion [52]. Cav1 upregulation has been implicated in endothelial dysfunction of apoE−/− mice upon Ang II infusion [53]. Therefore, whether EC Cav1 also participates in AAA development and rupture by inhibiting eNOS remains to be investigated.
Protecting against oxidative stress either pharmacologically [4] or genetically [5] appears effective for AAA prevention. Our data suggest that Cav1 silencing protects from AAA via suppression of oxidative stress and ER stress. Oxidative stress, particularly in VSMC, seems critical for AAA development [54, 55]. While little is known regarding the role of ER stress in AAA formation or rupture, this stress seems to be a key player in development of atherosclerosis and diabetes and is a major contributor to accelerated oxidative stress in these diseases [33]. The conceptual importance of ER stress in CVD is further supported by recent reports demonstrating the effectiveness of pharmacological ER stress suppression of hypertensive end organ damage induced by Ang II [35].
Clinical as well as experimental evidence suggests that inflammatory cytokines such as TNF-α [56], IL-1β [57], IL-6 [58] and MMPs [59] are critical for AAA development. Induction of TNF-α has also been implicated in target organ damage induced by Ang II [60]. In macrophages, Cav1 was reported to exert anti-inflammatory effects with strong reductions in TNF-α [61], which may involve transcriptional repression of NFκB [62]. While we did not evaluate MMP-2/9 activity in the present study, induction of MMP-2/9 mRNAs and proteins is associated with greater MMP-2/9 activities in experimental AAA [63]. Ang II has been shown to induce MMP-2 and MMP-9 in a NAD(P)H oxidase- and NFκB-dependent manner in VSMC, respectively [64, 65]. Moreover, induction of TNF-α and activation of MMP-2/9 associated with AAA were prevented in ADAM17 silenced mice [13]. Therefore, it is intriguing to imagine that Cav1 deficiency prevents AAA through suppressions of TNF-α and MMP-2/9 via its anti-inflammatory properties. However, the contribution of TNF-α receptor to AAA appears to be debatable [66].
IL-6 was induced in VSMC stimulated by Ang II via a transcriptional mechanism involving NFκB [67] and cAMP-responsive element binding protein (CREB) [68]. The CREB activation appears down-stream of EGFR transactivation in VSMC [69]. Moreover, Ang II-induced vascular inflammation and aortic dissection were prevented in IL-6 deficient mice [70]. Our findings coordinate well with these past findings. By contrast, the lack of IL-1β induction in this AAA model remains unclear at present.
As noted the major limitation of our study was not determining the main causal cell type due to unavailability of Cav1 conditional knockout mice. Inclusion of EC-selective rescue on Cav1−/− background is ongoing to partially compensate for this limitation. We also acknowledge significantly fewer Cav1−/− mice were included than the wild type mice in the surviving study, the reproducibility of aortic diameter measurement was not confirmed by an additional evaluator, and there was a potential bias in selecting samples for histology, as minor limitations. In conclusion, Cav1 silencing protected mice from AAA development and rupture but not hypertension induced by Ang II plus BAPN. Furthermore, Cav1 silencing was associated with suppression of ADAM17 activation/induction, oxidative stress, ER stress and inflammatory responses. Cav1 and caveolae localized signal transduction molecules may play critical roles in development of AAA.
Supplementary Material
CLINICAL PERSPECTIVES.
Although contribution of the renin angiotensin system for AAA development has been well acknowledged, the proximal signal transduction by which Ang II initiates AAA remains uncertain. The ADAM17/EGFR signaling axis localized specifically at caveolae/lipid raft is one of the most proximal signal transduction events by which Ang II participates in vascular remodeling. Our data demonstrated that silencing Cav1, a major structural protein of vascular caveolae, prevented Ang II-dependent AAA formation and rupture in normolipidemic mice and was associated with attenuation of ADAM17 induction/activation, ER stress, oxidative stress and inflammatory responses. The membrane localized signal components (Cav1, ADAM17 and EGFR) are therefore potential therapeutic targets for prevention of AAA development and rupture.
Summary statement.
Caveolin-1 deficiency protected mice from abdominal aortic aneurysm development induced by angiotensin II, which is likely due to suppression of ADAM17 activation and subsequent oxidative stress. Caveolin-1 may provide a novel therapeutic target for prevention of abdominal aortic aneurysm.
Acknowledgments
FUNDING
This study was supported in part by the National Institutes of Health (HL076770 to S.E., HL086551 to V.R., NS055876 to T.H.); and the American Heart Association (13GRNT17060036 to S.E.).
Footnotes
AUTHOR CONTRIBUTION
Takehiko Takayanagi and Kevin J. Crawford performed animal experiments, part of the immunohistochemical and cell culture experiments, and contributed to the discussion. Tomonori Kobayashi performed the part of animal studies and immunohistochemical experiments. Katherine J. Elliott performed in vitro experiments and wrote/edited the paper. Takashi Obama and Toshiyuki Tsuji performed the revision experiments. Tomoki Hashimoto provided expertise in the AAA model, contributed to the discussion and edited the paper prior to submission. Satoru Eguchi and Victor Rizzo conceived, designed and coordinated the research plan and wrote/edited the paper.
References
- 1.Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2005;142:203–211. doi: 10.7326/0003-4819-142-3-200502010-00012. [DOI] [PubMed] [Google Scholar]
- 2.Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. 2005;365:1577–1589. doi: 10.1016/S0140-6736(05)66459-8. [DOI] [PubMed] [Google Scholar]
- 3.Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci (Lond) 2012;123:531–543. doi: 10.1042/CS20120097. [DOI] [PubMed] [Google Scholar]
- 4.Gavrila D, Li WG, McCormick ML, Thomas M, Daugherty A, Cassis LA, Miller FJ, Jr, Oberley LW, Dellsperger KC, Weintraub NL. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1671–1677. doi: 10.1161/01.ATV.0000172631.50972.0f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas M, Gavrila D, McCormick ML, Miller FJ, Jr, Daugherty A, Cassis LA, Dellsperger KC, Weintraub NL. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation. 2006;114:404–413. doi: 10.1161/CIRCULATIONAHA.105.607168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ushio-Fukai M, Alexander RW. Caveolin-dependent angiotensin II type 1 receptor signaling in vascular smooth muscle. Hypertension. 2006;48:797–803. doi: 10.1161/01.HYP.0000242907.70697.5d. [DOI] [PubMed] [Google Scholar]
- 7.Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovascular research. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang B, Rizzo V. TNF-{alpha} potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H954–962. doi: 10.1152/ajpheart.00758.2006. [DOI] [PubMed] [Google Scholar]
- 9.Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, Suzuki H, Nakashima H, Eguchi K, Eguchi S. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2006;26:e133–137. doi: 10.1161/01.ATV.0000236203.90331.d0. [DOI] [PubMed] [Google Scholar]
- 10.Takaguri A, Shirai H, Kimura K, Hinoki A, Eguchi K, Carlile-Klusacek M, Yang B, Rizzo V, Eguchi S. Caveolin-1 negatively regulates a metalloprotease-dependent epidermal growth factor receptor transactivation by angiotensin II. J Mol Cell Cardiol. 2011;50:545–551. doi: 10.1016/j.yjmcc.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takaguri A, Kimura K, Hinoki A, Bourne AM, Autieri MV, Eguchi S. A disintegrin and metalloprotease 17 mediates neointimal hyperplasia in vasculature. Hypertension. 2011;57:841–845. doi: 10.1161/HYPERTENSIONAHA.110.166892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satoh H, Nakamura M, Satoh M, Nakajima T, Izumoto H, Maesawa C, Kawazoe K, Masuda T, Hiramori K. Expression and localization of tumour necrosis factor-alpha and its converting enzyme in human abdominal aortic aneurysm. Clin Sci (Lond) 2004;106:301–306. doi: 10.1042/CS20030189. [DOI] [PubMed] [Google Scholar]
- 13.Kaneko H, Anzai T, Horiuchi K, Kohno T, Nagai T, Anzai A, Takahashi T, Sasaki A, Shimoda M, Maekawa Y, Shimizu H, Yoshikawa T, Okada Y, Yozu R, Fukuda K. Tumor necrosis factor-alpha converting enzyme is a key mediator of abdominal aortic aneurysm development. Atherosclerosis. 2011;218:470–478. doi: 10.1016/j.atherosclerosis.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, Dyer JS, Maegdefessel L, Dalman RL, Tsao PS. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiological genomics. 2011;43:993–1003. doi: 10.1152/physiolgenomics.00044.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez C, Martinez-Gonzalez J, Raposo B, Alcudia JF, Guadall A, Badimon L. Regulation of lysyl oxidase in vascular cells: lysyl oxidase as a new player in cardiovascular diseases. Cardiovascular research. 2008;79:7–13. doi: 10.1093/cvr/cvn102. [DOI] [PubMed] [Google Scholar]
- 16.Ashino T, Sudhahar V, Urao N, Oshikawa J, Chen GF, Wang H, Huo Y, Finney L, Vogt S, McKinney RD, Maryon EB, Kaplan JH, Ushio-Fukai M, Fukai T. Unexpected role of the copper transporter ATP7A in PDGF-induced vascular smooth muscle cell migration. Circulation research. 2010;107:787–799. doi: 10.1161/CIRCRESAHA.110.225334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 18.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 19.Kanematsu Y, Kanematsu M, Kurihara C, Tsou TL, Nuki Y, Liang EI, Makino H, Hashimoto T. Pharmacologically induced thoracic and abdominal aortic aneurysms in mice. Hypertension. 2010;55:1267–1274. doi: 10.1161/HYPERTENSIONAHA.109.140558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maki JM, Rasanen J, Tikkanen H, Sormunen R, Makikallio K, Kivirikko KI, Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 21.Daugherty A, Manning MW, Cassis LA. Antagonism of AT2 receptors augments angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol. 2001;134:865–870. doi: 10.1038/sj.bjp.0704331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou HF, Yan H, Bertram P, Hu Y, Springer LE, Thompson RW, Curci JA, Hourcade DE, Pham CT. Fibrinogen-specific antibody induces abdominal aortic aneurysm in mice through complement lectin pathway activation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4335–4344. doi: 10.1073/pnas.1315512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takayanagi T, Bourne AM, Kimura K, Takaguri A, Elliott KJ, Eguchi K, Eguchi S. Constitutive stimulation of vascular smooth muscle cells by angiotensin II derived from an adenovirus encoding a furin-cleavable fusion protein. Am J Hypertens. 2012;25:280–283. doi: 10.1038/ajh.2011.221. [DOI] [PubMed] [Google Scholar]
- 24.Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, Motley ED, Kawakatsu H, Owada KM, Hirata Y, Marumo F, Inagami T. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J Biol Chem. 1998;273:8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- 25.Elliott KJ, Bourne AM, Takayanagi T, Takaguri A, Kobayashi T, Eguchi K, Eguchi S. ADAM17 silencing by adenovirus encoding miRNA-embedded siRNA revealed essential signal transduction by angiotensin II in vascular smooth muscle cells. J Mol Cell Cardiol. 2013;62C:1–7. doi: 10.1016/j.yjmcc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du G, Yonekubo J, Zeng Y, Osisami M, Frohman MA. Design of expression vectors for RNA interference based on miRNAs and RNA splicing. The FEBS journal. 2006;273:5421–5427. doi: 10.1111/j.1742-4658.2006.05534.x. [DOI] [PubMed] [Google Scholar]
- 27.Holzel M, Rohrmoser M, Orban M, Homig C, Harasim T, Malamoussi A, Gruber-Eber A, Heissmeyer V, Bornkamm G, Eick D. Rapid conditional knock-down-knock-in system for mammalian cells. Nucleic acids research. 2007;35:e17. doi: 10.1093/nar/gkl1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki H, Kimura K, Shirai H, Eguchi K, Higuchi S, Hinoki A, Ishimaru K, Brailoiu E, Dhanasekaran DN, Stemmle LN, Fields TA, Frank GD, Autieri MV, Eguchi S. Endothelial nitric oxide synthase inhibits G12/13 and rho-kinase activated by the angiotensin II type-1 receptor: implication in vascular migration. Arterioscler Thromb Vasc Biol. 2009;29:217–224. doi: 10.1161/ATVBAHA.108.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takayanagi T, Eguchi A, Takaguri A, Hinoki A, Bourne AM, Elliott KJ, Hurlin PJ, Eguchi S. A repressor protein, Mnt, is a novel negative regulator of vascular smooth muscle cell hypertrophy by angiotensin II and neointimal hyperplasia by arterial injury. Atherosclerosis. 2013 doi: 10.1016/j.atherosclerosis.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J Biol Chem. 2007;282:33714–33724. doi: 10.1074/jbc.M704041200. [DOI] [PubMed] [Google Scholar]
- 31.Desjardins F, Lobysheva I, Pelat M, Gallez B, Feron O, Dessy C, Balligand JL. Control of blood pressure variability in caveolin-1-deficient mice: role of nitric oxide identified in vivo through spectral analysis. Cardiovascular research. 2008;79:527–536. doi: 10.1093/cvr/cvn080. [DOI] [PubMed] [Google Scholar]
- 32.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circulation research. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circulation research. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 35.Kassan M, Galan M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler Thromb Vasc Biol. 2012;32:1652–1661. doi: 10.1161/ATVBAHA.112.249318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pojoga LH, Romero JR, Yao TM, Loutraris P, Ricchiuti V, Coutinho P, Guo C, Lapointe N, Stone JR, Adler GK, Williams GH. Caveolin-1 ablation reduces the adverse cardiovascular effects of N-omega-nitro-L-arginine methyl ester and angiotensin II. Endocrinology. 2010;151:1236–1246. doi: 10.1210/en.2009-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nature reviews. Cardiology. 2011;8:92–102. doi: 10.1038/nrcardio.2010.180. [DOI] [PubMed] [Google Scholar]
- 38.Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2003;23:483–488. doi: 10.1161/01.ATV.0000058404.92759.32. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Naggar JC, Welzig CM, Beasley D, Moulton KS, Park HJ, Galper JB. Simvastatin inhibits angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-knockout mice: possible role of ERK. Arterioscler Thromb Vasc Biol. 2009;29:1764–1771. doi: 10.1161/ATVBAHA.109.192609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–1665. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S, Fishbein MC, Blaschke F, Kintscher U, Graf K, Law RE, Hsueh WA. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. The Journal of clinical investigation. 2003;112:1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, Berk BC. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15:649–656. doi: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng GG, Martin-McNulty B, Sukovich DA, Freay A, Halks-Miller M, Thinnes T, Loskutoff DJ, Carmeliet P, Dole WP, Wang YX. Urokinase-type plasminogen activator plays a critical role in angiotensin II-induced abdominal aortic aneurysm. Circulation research. 2003;92:510–517. doi: 10.1161/01.RES.0000061571.49375.E1. [DOI] [PubMed] [Google Scholar]
- 44.Remus EW, O’Donnell RE, Jr, Rafferty K, Weiss D, Joseph G, Csiszar K, Fong SF, Taylor WR. The role of lysyl oxidase family members in the stabilization of abdominal aortic aneurysms. Am J Physiol Heart Circ Physiol. 2012;303:H1067–1075. doi: 10.1152/ajpheart.00217.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadiere C, Renia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. The Journal of clinical investigation. 2010;120:422–432. doi: 10.1172/JCI38136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishijo N, Sugiyama F, Kimoto K, Taniguchi K, Murakami K, Suzuki S, Fukamizu A, Yagami K. Salt-sensitive aortic aneurysm and rupture in hypertensive transgenic mice that overproduce angiotensin II. Laboratory investigation; a journal of technical methods and pathology. 1998;78:1059–1066. [PubMed] [Google Scholar]
- 47.Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, Roos FC, Chen Y, Finak G, Milosevic M, Wilson BC, Teh BT, Park M, Irwin MS, Ohh M. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4892–4897. doi: 10.1073/pnas.1112129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem. 2000;275:26765–26771. doi: 10.1074/jbc.M003325200. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez-Hernando C, Yu J, Suarez Y, Rahner C, Davalos A, Lasuncion MA, Sessa WC. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell metabolism. 2009;10:48–54. doi: 10.1016/j.cmet.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, Sessa WC. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. The Journal of experimental medicine. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medina F, de Haro J, Florez A, Acin F. Relationship between endothelial dependent vasodilation and size of abdominal aortic aneurysms. Annals of vascular surgery. 2010;24:752–757. doi: 10.1016/j.avsg.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 52.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 53.Seto SW, Krishna SM, Yu H, Liu D, Khosla S, Golledge J. Impaired Acetylcholine-Induced Endothelium-Dependent Aortic Relaxation by Caveolin-1 in Angiotensin II-Infused Apolipoprotein-E (ApoE(−/−)) Knockout Mice. PloS one. 2013;8:e58481. doi: 10.1371/journal.pone.0058481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maiellaro-Rafferty K, Weiss D, Joseph G, Wan W, Gleason RL, Taylor WR. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am J Physiol Heart Circ Physiol. 2011;301:H355–362. doi: 10.1152/ajpheart.00040.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parastatidis I, Weiss D, Joseph G, Taylor WR. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2013;33:2389–2396. doi: 10.1161/ATVBAHA.113.302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiong W, MacTaggart J, Knispel R, Worth J, Persidsky Y, Baxter BT. Blocking TNF-alpha attenuates aneurysm formation in a murine model. J Immunol. 2009;183:2741–2746. doi: 10.4049/jimmunol.0803164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, Owens GK, Upchurch GR, Jr, Ailawadi G. Genetic and pharmacologic disruption of interleukin-1beta signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2013;33:294–304. doi: 10.1161/ATVBAHA.112.300432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones KG, Brull DJ, Brown LC, Sian M, Greenhalgh RM, Humphries SE, Powell JT. Interleukin-6 (IL-6) and the prognosis of abdominal aortic aneurysms. Circulation. 2001;103:2260–2265. doi: 10.1161/01.cir.103.18.2260. [DOI] [PubMed] [Google Scholar]
- 59.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. The Journal of clinical investigation. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang XM, Kim HP, Song R, Choi AM. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. American journal of respiratory cell and molecular biology. 2006;34:434–442. doi: 10.1165/rcmb.2005-0376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang XM, Nadeau PE, Lin S, Abbott JR, Mergia A. Caveolin 1 inhibits HIV replication by transcriptional repression mediated through NF-kappaB. Journal of virology. 2011;85:5483–5493. doi: 10.1128/JVI.00254-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, Viollet B, Xia L, Zhang Y, Zou MH. Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012;18:902–910. doi: 10.1038/nm.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luchtefeld M, Grote K, Grothusen C, Bley S, Bandlow N, Selle T, Struber M, Haverich A, Bavendiek U, Drexler H, Schieffer B. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochemical and biophysical research communications. 2005;328:183–188. doi: 10.1016/j.bbrc.2004.12.152. [DOI] [PubMed] [Google Scholar]
- 65.Guo RW, Yang LX, Wang H, Liu B, Wang L. Angiotensin II induces matrix metalloproteinase-9 expression via a nuclear factor-kappaB-dependent pathway in vascular smooth muscle cells. Regulatory peptides. 2008;147:37–44. doi: 10.1016/j.regpep.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 66.Xanthoulea S, Thelen M, Pottgens C, Gijbels MJ, Lutgens E, de Winther MP. Absence of p55 TNF receptor reduces atherosclerosis, but has no major effect on angiotensin II induced aneurysms in LDL receptor deficient mice. PloS one. 2009;4:e6113. doi: 10.1371/journal.pone.0006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han Y, Runge MS, Brasier AR. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-kappa B transcription factors. Circulation research. 1999;84:695–703. doi: 10.1161/01.res.84.6.695. [DOI] [PubMed] [Google Scholar]
- 68.Funakoshi Y, Ichiki T, Ito K, Takeshita A. Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension. 1999;34:118–125. doi: 10.1161/01.hyp.34.1.118. [DOI] [PubMed] [Google Scholar]
- 69.Funakoshi Y, Ichiki T, Takeda K, Tokuno T, Iino N, Takeshita A. Critical role of cAMP-response element-binding protein for angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem. 2002;277:18710–18717. doi: 10.1074/jbc.M110430200. [DOI] [PubMed] [Google Scholar]
- 70.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. The Journal of clinical investigation. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.