

Abstract
Defects in p21-activated kinase (PAK) are suspected to play a role in cognitive symptoms of Alzheimer's disease (AD). Dysfunction in PAK leads to cofilin activation, drebrin displacement from its actin-binding site, actin depolymerization/severing, and, ultimately, defects in spine dynamics and cognitive impairment in mice. To determine the role of PAK in AD, we first quantified PAK by immunoblotting in homogenates from the parietal neocortex of subjects with a clinical diagnosis of no cognitive impairment (n = 12), mild cognitive impairment (n = 12), or AD (n = 12). A loss of total PAK, detected in the cortex of AD patients (−39% versus controls), was correlated with cognitive impairment (r2 = 0.148, p = 0.027) and deposition of total and phosphorylated tau (r2 = 0.235 and r2 = 0.206, respectively), but not with Aβ42 (r2 = 0.056). Accordingly, we found a decrease of total PAK in the cortex of 12- and 20-month-old 3xTg-AD mice, an animal model of AD-like Aβ and tau neuropathologies. To determine whether PAK dysfunction aggravates AD phenotype, 3xTg-AD mice were crossed with dominant-negative PAK mice. PAK inactivation led to obliteration of social recognition in old 3xTg-AD mice, which was associated with a decrease in cortical drebrin (−25%), but without enhancement of Aβ/tau pathology or any clear electrophysiological signature. Overall, our data suggest that PAK decrease is a consequence of AD neuropathology and that therapeutic activation of PAK may exert symptomatic benefits on high brain function.
Introduction
Synaptic dysfunction occurs early in Alzheimer's disease (AD) and is thought to underlie cognitive impairment before widespread neuronal loss (Selkoe, 2001; Calon et al., 2004; Moolman et al., 2004; Masliah et al., 2006; Scheff and Price, 2006; Rodríguez and Verkhratsky, 2011; Selkoe, 2011; Huang and Mucke, 2012). It has been postulated that p21-activated kinase (PAK)-related molecular pathways are essential to synaptic health and therefore are potential therapeutic targets in AD (Zhao et al., 2006; Ma et al., 2008; Huang et al., 2011; Dubos et al., 2012). PAKs are serine/threonine protein kinases (Bokoch, 2003; Boda et al., 2006; Kreis and Barnier, 2009; Huang et al., 2011) that play critical roles in regulating the formation and maintenance of postsynaptic dendritic spines through actin remodeling (Rousseau et al., 2003; Salminen et al., 2008; Kreis and Barnier, 2009; Szczepanowska, 2009). PAK inhibition causes cognitive impairment in dominant-negative PAK-transgenic mice (dnPAK; Hayashi et al., 2004), in adult mice after intracerebral infusion of inhibitors (Zhao et al., 2006), and in double PAK1/3 knock-out mice (Huang et al., 2011).
Decreases in cytosolic PAK concentrations have been found in the cortex and hippocampus of persons who died with advanced AD (Heredia et al., 2006; Zhao et al., 2006; Ma et al., 2008; Nguyen et al., 2008). Moreover, immunohistochemical studies showed a cofilin-positive cluster labeling of active PAK in AD patients (Zhao et al., 2006). PAK dysfunction was proposed to lead to cofilin activation, drebrin displacement from its actin-binding site, actin depolymerization/severing, and, ultimately, defects in spine dynamics (Minamide et al., 2000; Bokoch, 2003; Zhao et al., 2006; Kreis and Barnier, 2009; Dubos et al., 2012). This pathogenic hypothesis is supported by data in cultured hippocampal cells and in rodents, in which intracerebral injections of PAK inhibitors induced drebrin translocation from membrane to cytosol (Zhao et al., 2006; Ma et al., 2008). Accordingly, reduction of PAK catalytic activity in vivo induces a decrease in the spine density within cortical neurons and shifts the overall spine distribution toward spines of shorter length with larger postsynaptic densities (Hayashi et al., 2004; Hayashi et al., 2007). Converging data indicate that PAK activation is essential for the maturation and maintenance of dendritic spines and thus for the proper functioning of the brain. PAK activation therefore represents a promising therapeutic target in AD and other dementias.
To determine the significance of a decrease of PAK activity in AD pathophysiology, we: (1) investigated the relationship of PAK, cognitive deficits, and Aβ/tau neuropathologies in subjects with no cognitive deficits, mild cognitive impairment, or AD; (2) characterized age-related PAK alterations in an animal model of AD, the 3xTg-AD mouse; (3) investigated whether PAK inactivation potentiates an AD-relevant behavioral and molecular phenotype in vivo by crossing 3xTg-AD mice with dnPAK mice (Hayashi et al., 2004); and (4) sought to determine whether the electrophysiological parameters known to be impaired in AD mice models (Harris et al., 2011; Verret et al., 2012), and more specifically in 3xTg-AD mice (Arsenault et al., 2011), were corrected or aggravated by PAK defects.
Materials and Methods
Patients and handling of brain tissue
Samples from the parietal cortex of 36 individuals from the Religious Orders Study were divided into three groups based on clinical diagnoses: 12 with mild cognitive impairment (MCI), 12 with AD, and 12 with no cognitive impairment, as described previously (Bennett et al., 2002; Bennett, 2006; Tremblay et al., 2007; Julien et al., 2009). MCI defines a group of otherwise healthy elderly subjects with a markedly elevated risk of developing AD and a more rapid rate of decline in cognitive functions (Boyle et al., 2006). In addition, MCI is considered as a possible early clinical manifestation of AD (Bennett et al., 2005). Each participant underwent a uniform structured baseline clinical evaluation, details of which have been described previously (Bennett et al., 2002; Bennett et al., 2005; Bennett, 2006). Briefly, the composite index of global cognition combined 19 separate tests including measures of episodic, semantic, and working memory, as well as perceptual speed and visuospatial ability (Wilson et al., 2002). At death, each case was assigned a Braak score based on neurofibrillary tau pathology, a senile plaque score based on modified Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria, and a diagnosis based on the National Institute on Aging–Reagan criteria by a neuropathologist shielded to all clinical data (Bennett et al., 2005). To further investigate the link between PAK and the CERAD-based neuropathological diagnosis, we added parietal cortex samples of 19 AD patients and 22 controls obtained from the Douglas Hospital Research Centre (Quebec) brain bank, which have been fully described in previous studies (Fig. 1C; Julien et al., 2008; Julien et al., 2009). In Julien et al. (2009), the neuropathological diagnosis of AD was made only at neuropathological examination and was based on CERAD scores. In all subjects, cerebellar pH was measured to assess the degree of preservation of the tissue (Kingsbury et al., 1995; Calon et al., 2003). Concentrations of Aβ and tau were assessed using ELISA and Western immunoblotting, respectively, in the inferior parietal cortex of all volunteers, as described previously (Tremblay et al., 2007; Julien et al., 2008). Relevant information regarding all subjects involved in this study is available in a previous publication (Julien et al., 2009).
Figure 1.

PAK loss in AD correlates with disease severity in human. A, Actin-normalized PAK was decreased in AD patients compared with controls, based on clinical diagnoses. B, C, Decreases in actin-normalized PAK (B) and pPAK (C) were observed in individuals with definitive neuropathological diagnosis of AD (CERAD 1) compared with those with a “probable” diagnosis of AD (CERAD 2) or low AD probability (possible or none, CERAD 3 or 4). D–G, Linear regression analyses showed that actin-normalized PAK (in the soluble fraction) was correlated negatively with the ratio of insoluble tau fraction on the soluble tau fraction (D) and insoluble PHFtau (E), but not with the accumulation of insoluble Aβ42 (F), but was positively associated with global antemortem cognitive score (G). H, Examples of Western blot results obtained for PAK and pPAK in control, MCI, and AD patients. All samples were run simultaneously on the same SDS-PAGE gels. Statistical analyses: one-way ANOVA/Dunnett's multiple-comparison test (A), Kruskal-Wallis/Dunn's multiple-comparison test (B,C), and linear regression (D–G).
Transgenic models
All procedures were approved by the Laval University animal ethics committee (protocol 10-037) and were performed according to the guidelines of the Canadian Council on Animal Care. The 3xTg-AD mouse model has been described previously (Oddo et al., 2003a; Oddo et al., 2003b; Mastrangelo and Bowers, 2008; Phivilay et al., 2009; Julien et al., 2010; Arsenault et al., 2011). In brief, 3xTg-AD mice express the mutated gene PS1M146V (knock-in) and mutated human genes APPSwe and tauP301L in the same locus, both under the control of the mouse Thy1.2 regulatory element (Oddo et al., 2003a; Oddo et al., 2003b; Mastrangelo and Bowers, 2008). These transgenic mice develop an age-related progressive neuropathological phenotype that includes both plaques and tangles distributed along a regional pattern similar to AD (Oddo et al., 2003a; Ackl et al., 2005; Mastrangelo and Bowers, 2008; Martinez-Coria et al., 2010). Finally, this AD mouse model presents behavioral and cognitive changes that are correlated with the development of the Aβ and tau pathology (Sterniczuk et al., 2010). Nontransgenic (NonTg) mice were derived from the original mouse line and were of the same genetic background.
PAK inactivation in 3xTg-AD mice was generated by crossing homozygous 3xTg-AD mice with heterozygous mice expressing the dnPAK transgene, which codes for a PAK autoinhibitory domain binding to PAK catalytic site (Hayashi et al., 2004). Expression of this autoinhibitory domain therefore decreases PAK1/2/3 kinase activity (Zhao et al., 1998; Zenke et al., 1999; Hayashi et al., 2004). dnPAK transgene expression is under control of the CaMKII promoter, leading to an expression restricted to the postnatal forebrain (Hayashi et al., 2004). In addition, dnPAK was marked with a myc-tag sequence fused in-frame to the N terminus of the autoinhibitory domain-PAK sequence (Hayashi et al., 2004). This crossing produced heterozygous 3xTg-AD mice expressing the dnPAK transgene (3xTg-AD+/− × dnPAK+/−) or not (3xTg-AD+/− × dnPAK−/−). Animals were killed at either 12 or 20 months and the parietotemporal cortex was used to quantify proteins and neuropathological markers. Both females and males were used in this study.
Preparation of tissue samples
Tissue extracts (50 mg of mouse tissue or 100 mg of human tissue) were homogenized in 4 (human) or 8 (mouse) volumes of TBS containing a complete protease inhibitor mixture (Roche), 10 μg/ml pepstatin A, 0.1 mm EDTA, and phosphatase inhibitors (1 mm each of sodium vanadate and sodium pyrophosphate, 50 mm sodium fluoride). The frozen samples were sonicated briefly (3 × 10 s) and centrifuged at 100,000 × g for 20 min at +4°C to generate a TBS-soluble fraction (soluble or cytosolic fraction). The TBS-insoluble pellets were sonicated in 8 (mice) or 4 (human) volumes of lysis buffer (150 mm NaCl, 10 mm NaH2PO4, 1% Triton X-100, 0.5% SDS, and 0.5% deoxycholate) containing the same protease and phosphatase inhibitor mixture. The resulting homogenate was centrifuged at 100,000 × g for 20 min at +4°C to produce a lysis buffer-soluble fraction (detergent-soluble or membrane fraction). The pellet (detergent-insoluble fraction) was homogenized in 175 μl of 90% formic acid followed by sonication (3 × 10 s). The mouse supernatant was divided in two portions and dried out with a SpeedVac (Thermo Savant). One portion was solubilized in guanidine-HCl (5 m guanidine in Tris-HCl 0.05 m) and then sonicated shortly for solubilization to be used for ELISA; the other portion was solubilized in Laemmli's buffer for Western immunoblotting. For the human tissue, the resultant suspension was centrifuged (13,000 × g at +4°C for 20 min) and 20 μl of each supernatant was neutralized with 1:30 dilution of Tris-base 1 m, pH 10, to be used for ELISA (see below). The rest of the supernatants were dried out with a SpeedVac, solubilized in Laemmli's buffer, and processed for Western immunoblotting (insoluble fraction; see below).
ELISA
Aβ40 and Aβ42 from human brain samples were measured with the hAmyloid β40/β42 ELISA HS kits (The Genetics Company), as described previously (Tremblay et al., 2007). Amyloid β40/β42 ELISA kits (Biosource) were used to analyze mouse tissue. Both ELISAs were performed in the soluble and insoluble protein fraction according to the manufacturers' recommendations and the plates were read at 450 nm using a Synergy HT multidetection microplate reader (Biotek).
Western immunoblotting
Protein concentration was determined using bicinchoninic acid assays (Pierce). For Western immunoblotting, equal amounts of protein per sample (15 μg) were added to Laemmli's loading buffer, heated to +95°C for 5 min before loading, and subjected to SDS-PAGE (8%). Proteins were electroblotted onto PVDF membranes (Millipore) before blocking in 5% nonfat dry milk and 1% BSA in PBS-Tween 20 for 1 h. Membranes were immunoblotted with appropriate primary and secondary antibodies followed by chemiluminescence reagents (Lumiglo Reserve; KPL). Band intensities were quantified using a Kodak Image Station 4000 MM Digital Imaging System (Molecular Imaging Software version 4.0.5f7; Carestream Health). The following antibodies were used in this study: anti-actin (ABM), anti-cofilin (Cell Signaling Technology), anti-drebrin (MBL International), anti-synaptophysin clone SVP-38 (Millipore), anti-PAK1 (Invitrogen), anti-PAK3 and anti-total PAK (Cell Signaling Technology), anti-phospho-PAK1/2/3 (phosphorylated at serine 141; Invitrogen), anti-total tau clone tau13 (Covance), and anti-phospho-tau clones CP13 (phosphorylated at serine 202/threonine 205; gift from Dr Peter Davies, Albert Einstein College of Medicine, New York), and AD2 (phosphorylated at serines 396 and 404; Bio-Rad).
Immunohistofluorescence staining
Hemi-brains were postfixed with 4% PFA (4°C overnight), cryoprotected with 20% sucrose-PBS, snap frozen at −80°C, and microtome sectioned into coronal (25 μm) sections. Washes in 0.1 m PBS, pH 7.4, were performed between each step of the immunohistofluorescence protocols. Free-floating brain sections from 3xTg-AD mice with or without dnPAK transgene were blocked for 1 h in a PBS solution containing 5% horse serum (Invitrogen) and 0.4% Triton X-100. Sections were then incubated overnight at 4°C with primary antibodies in the blocking solution: rabbit anti-Myc-Tag (1:100; Cell Signaling Technology), rabbit anti-pPAKs141 (1:100; Invitrogen Corporation), and mouse anti-neuronal nuclei (NeuN, 1:1000; Millipore). After incubation with primary antibodies, slices were exposed to AF-conjugated donkey anti-rabbit and anti-mouse secondary antibodies (1:1000; Invitrogen). Finally, slices were counterstained with DAPI (Invitrogen), mounted on SuperFrost Plus slides (Thermo Fisher Scientific), treated with 0.5% Sudan black (in 70% methanol) for 5 min, and placed under coverslips with Mowiol mounting medium. Sections were transferred onto SuperFrost Plus slides and placed under coverslips with Mowiol mounting medium.
Slice preparation for electrophysiology recordings
Brain slices were prepared as described previously (Zhang and Arsenault, 2005; Arsenault and Zhang, 2006). Briefly, mice were deeply anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and decapitated. The brain was removed quickly (<60 s) and placed in an ice-cold solution containing the following (in mm): 210 sucrose, 3.0 KCl, 1.0 CaCl2, 3.0 MgSO4, 1.0 NaH2PO4, 26 NaHCO3, and 10 glucose saturated with 95% O2/5% CO2. Horizontal slices of 300 μm were cut from inferior to superior brain with a vibrating tissue slicer (VT 1000s; Leica) and kept at room temperature in ACSF containing the following (in mm): 124 NaCl, 3.0 KCl, 1.5 CaCl2, 1.3 MgCl2, 1.0 NaH2PO4, 26 NaHCO3, and 20 glucose saturated with 95% O2/5% CO2. Slices were allowed to recover for at least 1 h before recording (n = 9–10).
Patch-clamp recording
For recording, a slice was transferred to a submerge-type chamber and continuously exposed to ACSF heated to 30–32°C saturated with 95% O2/5% CO2 and flowing at a rate of 2.0 ± 0.2 ml/min. The slices were viewed first with a 4× objective and the deep layer of the entorhinal cortex was located beside the hippocampus. For most animals, two to three slices were recorded per hemisphere. Large deep layer neurons in the entorhinal cortex were then viewed under near-infrared illumination with a 40× water-immersion objective (Fluor, 40×, 0.80W; Nikon) and a charge-coupled device camera (IR-1000; Dage MTI).
Patch pipettes were pulled from thick-walled borosilicate glass (1.5/0.84 mm; WPI) on a horizontal puller (P-97; Sutter Instruments). The pipette solution contained the following (in mm): 100 KMeSO4, 15 KCl, 4 ATP-Mg, 10 creatine phosphate, 10 HEPES, 0.5 EGTA, pH 7.2, adjusted with KOH, 275–280 mOsm. Electrodes had resistances between 4 and 6 MΩ. The seal resistance was >2 GΩ. Whole-cell recordings were made at the soma with a Multiclamp 700A amplifier (Molecular Devices). The access resistance, usually between 10 and 30 MΩ, was monitored throughout each experiment and only recordings with stable access were used. Experiments were conducted using the Axograph 4.9 program (Molecular Devices) or pClamp 9.2 (Molecular Devices). Data were digitized at 8 or 16 kHz and were either not filtered or filtered at 1 kHz, depending on the recording protocol.
Data analysis for electrophysiology experiments
This study included two sets of electrophysiological experiments. The first investigated association between PAK and selected physiologic parameters (passive properties and spontaneous EPSC) known to change in 3xTg-AD mice (Arsenault et al., 2011). This investigation was performed in homozygous mice and the mean of three to five recorded neurons was used to compare with molecular data. The second set of experiments were performed to determine physiologic parameters modulated by PAK activity. These experiments were conducted in heterozygous mice and comparison was performed for each neuron.
Electrophysiological recordings were analyzed with Clampfit 9.2 (Molecular Devices). Passive properties (input resistance, cell capacitance [CC], resting potential) were studied in current-clamp mode given the stronger accuracy for this recording configuration than voltage-clamp mode (Golowasch et al., 2009). The input resistance was estimated from the slope of the graph of voltage variation versus hyperpolarized current injection using the equation V = IR * I, where IR is input resistance, V is voltage variation, and I is the injected current. The injected current duration was 400 ms and current intensities were 50, 100, 150, and 200 pA. CC was estimated from the linear slope of the plot of I * T = CC * V (i.e., for a first-order resistance-capacitance circuit), where T is the time constant of voltage variation measured by fitting a single exponential function for a voltage decay over time, V = V∞ (1 − e −T/(IR*CC)), and V∞ is the asymptote, so that T = RC (i.e., V = 0.632 V∞) using a graphical method. CC corresponds to the linear slope of the graph displaying the relationship between I * T versus V. sEPSCs were quantified in voltage-clamp mode and neurons were held at −60 mV (near the reversal potential for GABAAR-mediated currents). The sEPSCs were automatically detected using the event detection package of Clampfit 9.2. This package uses multiple preestablished templates to optimize the detection of synaptic events.
Behavioral analysis
Social recognition.
This test was based on the propensity of an adult mouse to interact less with a known mouse than with an unknown mouse of the same sex and age (Kaidanovich-Beilin et al., 2011; Winslow, 2001). In this paradigm, two mice were exposed to each other for a 20 min period in a transparent plastic arena (40 cm × 22 cm × 18 cm) and the time that animal spent exploring its dyad partner (sniffing, following, grooming the partner, crawling over or under) was measured. Then, the same mice were reexposed 24 h later and the reduction of exploratory activity for the known partner was used as an indicator of social memory. Mice were tested at 20 months of age in dyads of one 3xTg-AD+/− × dnPAK−/− and one 3xTg-AD+/− × dnPAK+/− mice.
Anxiety-like behavior.
Anxiety was assessed using light/dark box test, which is based on the innate aversion of rodents to brightly illuminated areas and on the spontaneous exploratory behavior of rodents in response to the mild stressors novel environment and light. The test apparatus consists of a small dark “safe” compartment and a large illuminated “aversive” compartment (Bourin and Hascoët, 2003). Animals were positioned in the dark compartment and the time spent in the illuminated compartment was measured for 10 min. In addition, the number of alternances between each compartment was analyzed.
Vertical/motor activity.
To determine whether motor impairments could affect the social and exploratory behaviors in mice, a 10 min vertical activity test was also performed in a transparent plastic arena (20 cm × 40 cm × 30 cm) in parallel with black and white box testing.
Exploratory behavior.
Exploratory behavior of mice was assessed in a squared arena (1 m × 1 m) with holes in each corner. Hole board exploration is a behavioral paradigm that is used frequently in mice and requires no training (Crawley, 1985). The time spent in active exploration for the 4 holes was measured during a single 5 min daily session for 3 consecutive days.
Statistical analysis
Values are expressed as mean ± SEM. Statistical comparisons between groups were performed depending on the normality of distribution and variances equivalence between groups. For analysis in human samples, in cases of equal variance and normal distribution, ANOVA followed by post hoc pairwise comparisons (e.g., Dunnett) were used when appropriate. Groups of data that failed tests for normality and equal variance were analyzed by the nonparametric Kruskal–Wallis test followed by Dunn's test. In the animal studies, statistical comparisons were performed using a two-way ANOVA for the study of two variables simultaneously. When variable interaction was detected, statistical comparisons were performed using a one-way ANOVA followed by the Tukey-Kramer post hoc test. When only two groups were compared, unpaired Student's t test was performed, except for unequal variance, for which a Welch's t test was used. Finally, for both human and animal studies, the coefficient of determination (r2) and the significance of the degree of linear relationship between various parameters were determined with a simple regression model. Statistical analyses were performed using JMP statistical analysis software (version 8.0.2).
Results
Loss of PAK in AD is associated with symptoms and neuropathology
We first quantified PAK in the TBS-soluble protein fractions from the parietal cortex of individuals clinically determined as MCI or AD and compared them with controls. We found 39% lower PAK concentrations in subjects with a diagnosis of AD, but no significant differences in those with MCI (Welch ANOVA: p = 0.012, Dunnett's multiple-comparison test: p < 0.01 for AD vs controls; Fig. 1A). To further link actin-normalized PAK level with the neuropathological diagnosis, we added parietal cortex samples from the Douglas Hospital cohort to the analysis. All subjects were classified according to CERAD-based neuropathology diagnosis. We detected a 30% lower PAK concentration in individuals with a definite neuropathologic diagnosis of AD compared with individuals with a lower probability of AD (Kruskal–Wallis: p = 0.0095, Dunn's multiple-comparison test: p < 0.01 for definite AD versus probable/possible/no AD; Fig. 1B). We also observed a significant decrease in phospho-PAK in persons with a CERAD score of 1 (definitive AD; Kruskal-Wallis: p = 0.0036, Dunn's multiple-comparison test: p < 0.01 for definite AD vs possible/no AD and, p < 0.05 for definite AD vs “probable” AD; Fig. 1C). Next, we sought to establish the relationship between PAK levels and other common neuropathological markers of AD. Correlative analyses showed that actin-normalized PAK was inversely correlated with the insoluble/soluble tau ratio (Fig. 1D) and the accumulation of insoluble phospho-tau (Fig. 1E). However, no relationship was detected with the levels of insoluble Aβ42 (Fig. 1F). Finally, a positive correlation was observed between postmortem cortical PAK levels and the global cognitive score measured antemortem (Fig. 1G). The strongest associations were established with episodic memory (r2 = 0.16; p = 0.0212) and perceptual speed (r2 = 0.23; p = 0.0047). Partial correlation analyses showed that, after adjusting for insoluble phospho-tau, the significance of the relationship between PAK and cognitive scores was lost (r2 = 0.08; p = 0.39), whereas the correlation between phospho-tau and cognitive scores was not affected by PAK levels (r2 = 0.26; p = 0.0018). Such data may suggest that the association between PAK and cognition is mediated by tau, which is consistent with PAK loss being a consequence of AD neuropathology. Overall, these data confirm that the clinical and neuropathological progression of AD is associated with a loss of PAK.
Decrease of PAK is replicated in the 3xTg-AD mouse
To further probe whether brain expression of tau and Aβ neuropathologies can cause a reduction in PAK in vivo, we quantified PAK levels in the parietotemporal cortex from the 3xTg-AD mouse model at different ages (Fig. 2). We found a decrease in cortical concentrations of total PAK in 12- and 20-month-old 3xTg-AD mice compared with NonTg controls, but not at 4 months of age, which is consistent with an age-related progressive loss of PAK (Fig. 2A). In contrast, the concentration of phospho-PAK remained unchanged (Fig. 2B). In addition, the levels of the two main cerebral isoforms of PAK, PAK1 and PAK3, were both decreased in the parietotemporal cortex of 12-month-old (−26%, p < 0.01, and −9%, p < 0.05, respectively) and 20-month-old (−11% and −18%, respectively, p < 0.05 for both) 3xTg-AD mice (Fig. 2C). These results indicate that the 3xTg-AD mouse replicates the decrease in PAK observed in AD patients.
Figure 2.

Characterization of PAK, membrane cofilin, and drebrin impairments in homozygous 3xTg-AD mice according to age. A, Soluble PAK1/2/3 was decreased in 12- and 20-month-old 3xTg-AD mice compared with NonTg mice of the same age. No statistical differences were found between the two genotypes in young animals (4 months). B, When comparing both genotypes at different ages, we observed no alteration of soluble phospho-PAK in 3xTg-AD mice at 4, 12, or 20 months. C, Loss of soluble PAK1/2/3 was observed in both isoforms 1 and 3 at all ages. Despite an age effect for drebrin level, we did not found any change of membrane cofilin (D) or drebrin (E) levels in 3xTg-AD compared with NonTg mice. Values are expressed as mean ± SEM. Statistical comparisons were performed using a one-way ANOVA followed by a Tukey-Kramer post hoc test (age comparison, A–C) and an unpaired t test (D,E). *p < 0.5, **p < 0.01, ***p < 0.001.
Based on previously described cellular pathways, inactivation of PAK should lead to downstream cofilin activation and removal of the spine actin-regulatory protein drebrin from the membrane compartment (Zhao et al., 2006). However, despite an age-induced decrease of drebrin levels, the concentration of cofilin and drebrin in the detergent-soluble membrane fraction remained unaltered in 3xTg-AD mice compared with NonTg mice (Fig. 2D,E, respectively), which is consistent with our previous results (Julien et al., 2008; Phivilay et al., 2009).
Transgenic PAK inactivation leads to a decrease of the postsynaptic protein drebrin
So far, the results gathered from human samples and in the 3xTg-AD mice strongly suggested a tight association between PAK and AD-like neuropathology, clinical symptoms, and electrophysiological defects, but did not infer a causal role of PAK. To discriminate whether alterations found in 3xTg-AD mice are dependent of PAK activity, we crossed the 3xTg-AD mice with dnPAK mice to generate heterozygous 3xTg-AD mice with (3xTg-AD+/− × dnPAK+/−) or without (3xTg-AD+/− × dnPAK−/−) PAK activity deficiencies.
Because the product of dnPAK transgene is labeled with a myc-tag sequence (Hayashi et al., 2004), we first performed immunofluorescence analysis using an antibody raised against anti myc-tag. As expected, we observed a strong staining of the dnPAK transgene in 3xTg-AD mice with genetically PAK inactivation, but no staining in 3xTg-AD mice (Fig. 3A). This confirms that dnPAK expression was restricted to the cortex of 3xTg-AD × dnPAK mice in our study. Moreover, we observed a strong colocalization with the neuronal nuclei (NeuN) protein, indicating that the dnPAK transgene was mostly expressed in neuronal cells (Fig. 3A). We next conducted immunohistofluorescence experiments using an antibody directed against pPAK (ser141, activated PAK). An apparent decrease of pPAK cortical staining was observed in 3xTg-AD × dnPAK mice compared with 3xTg-AD mice (Fig. 3A). Colocalization with NeuN labeling was also confirmed (Fig. 3A). Using Western blotting analysis, we found a decrease of phospho-PAK (ser141) in 3xTg-AD+/− × dnPAK+/− at 12 and 20 months of age (−27% and −33%, respectively, p < 0.01), confirming the effectiveness of the dnPAK transgene in reducing PAK activation (Fig. 3B). A small compensatory upregulation of PAK1 (+14%, p < 0.05), but not PAK3, was observed in 3xTg-AD+/− × dnPAK+/− mice compared with 3xTg-AD+/− × dnPAK−/− mice at 12 months, but not at 20 months (Fig. 3C). dnPAK transgene expression did not induce any effect on detergent-soluble cofilin levels in 3xTg-AD mice regardless of their age (Fig. 3D). Despite a reduction with age (two-way ANOVA, p = 0.014), no significant difference in phospho-cofilin levels was detected between 3xTg-AD and 3xTg-AD × dnPAK mice. Nevertheless, PAK inactivation induced a 25% decrease of detergent-soluble drebrin level, a postsynaptic protein (p < 0.05; Fig. 3F), in 20-month-old 3xTg-AD+/− × dnPAK+/− mice. This result was consistent with studies indicating that the membrane localization of drebrin is dependent on PAK activation (Zhao et al., 2006; Ma et al., 2008). Separate Western blot analyses showed that dnPAK expression in NonTg mice led to a nonsignificant trend toward a decrease of drebrin (2.68 ± 0.62 in NonTg mice vs 1.35 ± 0.26 in dnPAK mice, n = 6 per group, p = 0.0776). Although these data must be interpreted carefully due to low statistical power, they suggest that drebrin loss requires both PAK inactivation and AD neuropathology. Detergent-soluble levels of the presynaptic protein synaptophysin were also analyzed, but no significant change was observed whatever the age of 3xTg-AD mice (Fig. 3G).
Figure 3.
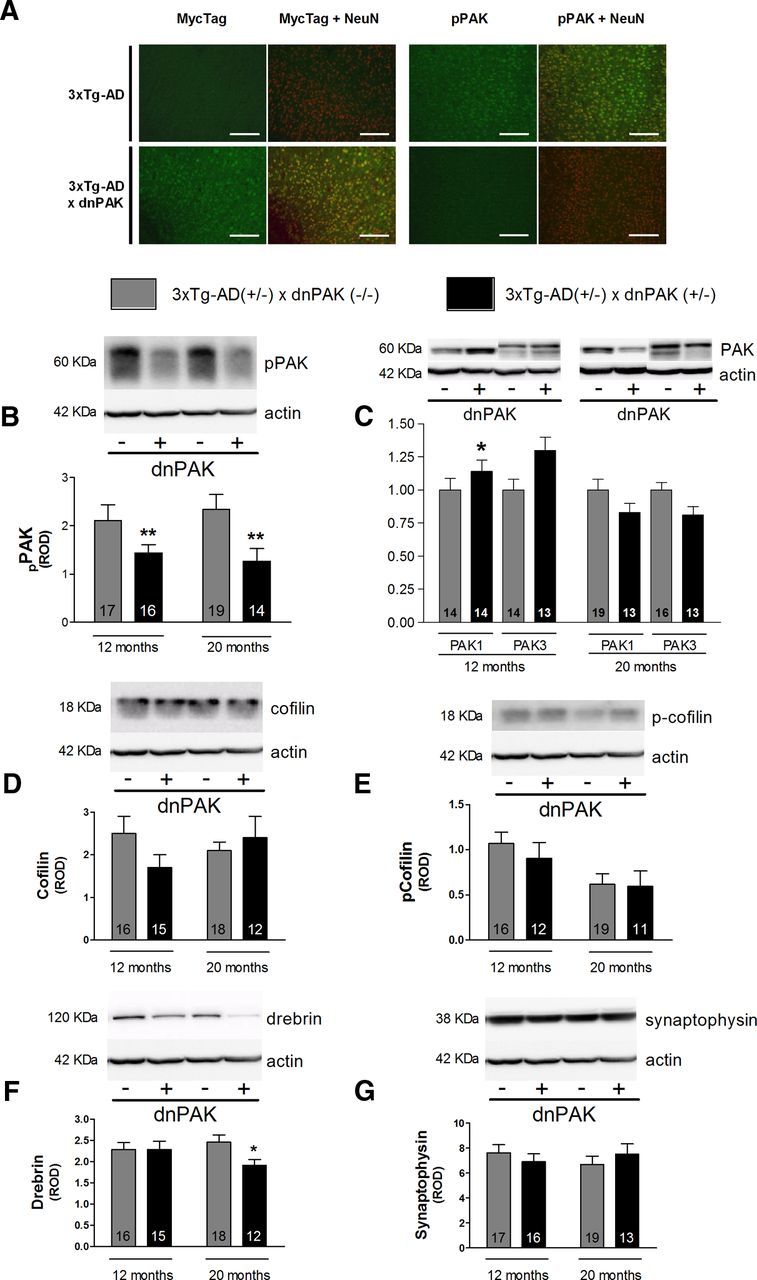
Consequences of PAK inactivation in 12- or 20-month-old heterozygous 3xTg-AD mice. A, Myc-Tag immunofluoresence (green) in the cortex confirmed that the dnPAK transgene was only expressed in neurons (NeuN in red) from 3xTg-AD × dnPAK mice compared with 3xTg-AD mice. pPAK immunostaining (green) in the cortex highlighted a neuronal localization (NeuN in red) of pPAK (s141) and an apparent decrease in 20-month-old 3xTg-AD mice with genetically PAK inactivation compared with 3xTg-AD mice (n = 3 per group). Western blot analyses revealed that dnPAK transgene expression reduced pPAK (B) and increased the level of PAK1 in 12-month-old mice, but was inefficient to modulate PAK3 at both ages (C) in the soluble fraction. dnPAK transgene expression did not induce any effect on cofilin (D) or phospho-cofilin (E) levels in the cortex of 3xTg-AD mice. Interestingly, drebrin level was reduced in 20-month-old mice in the membrane fraction (F). Finally, no significant effect was observed on detergent-soluble synaptophysin level whatever the age of mice (G). Square bar equals 100 μm. Values are expressed as mean ± SEM. Statistical analyses were performed using an unpaired t test. For PAK3 analysis (C), only the upper band was used. *p < 0.05, **p < 0.01.
PAK modulation can influence social recognition in 3xTg-AD mice
A battery of behavioral tests was performed to determine the cognitive impact of dnPAK expression in 3xAD-Tg mice. First, the general motor activity was found to be decreased in 3xTg-AD mice, but unaltered by PAK inactivation (Fig. 4A, p < 0.05). Second, in the black and white box paradigm, animals from the three different groups spent similar time in the illuminated box (Fig. 4B). However, NonTg mice showed a higher number of alternances between compartments compared with 3xTg-AD and 3xTg-AD × dnPAK mice (Fig. 4C, p < 0.001). Therefore, our results are suggestive of a higher anxiety level in 3xTg-AD mice, which, however, remained unaffected by the expression of dnPAK. Spontaneous exploration in a new environment was also evaluated. All animals showed a normal decrease of their exploratory behavior in the second and the third day compared with the first (Fig. 4D). The number of exploratory events recorded was lower in both 3xTg-AD and 3xTg-AD × dnPAK mice compared with NonTg mice (Fig. 4D). Therefore, as described previously (Arsenault et al., 2011; Bories et al., 2012), 3xTg-AD transgenes decreased the motivation to explore a new environment (Fig. 4D), a behavior that remained unaltered by the expression of dnPAK.
Figure 4.

PAK inactivation impaired social recognition in 20-month-old 3xTg-AD mice. (A) NonTg mice showed higher general activity than 3xTg-AD mice, as reflected in a vertical activity test over a 10 min observation span. B, Time spent in the illuminated compartment of the black and white box was not significantly different in NonTg, 3xTg-AD, and 3xTg-AD × dnPAK mice. C, 3xTg-AD and 3xTg-AD × dnPAK mice showed a similar number of alternances, but both were lower than NonTg, suggesting that the level of anxiety was not influenced by PAK inhibition. D, Exploratory behavior was similarly decreased in 3xTg-AD and 3xTg-AD × dnPAK mice compared with NonTg animals. E, Social events in which a mouse interacts with its dyad partner were recorded (sniffing, following, grooming the partner, crawling over or under) at day 1 and at day 2. F, 3xTg-AD mice with normal PAK activity had a 40% reduction of interaction time with their dyad mate at day 2 compared with day 1. In contrast, PAK-deficient 3xTg-AD mice displayed the same interaction time with their partner at day 1 and day 2, consistent with a complete absence of social recognition. Values are expressed as mean ± SEM. Statistical comparisons were performed using an unpaired Student's t test. *p < 0.05, ***p < 0.001.
To determine whether PAK inactivation translated into social behavior defects, the capacity of groups of animals to recognize their peers was documented. To assess sociability in animal models, several behavioral tests have been developed (for review, see Silverman et al., 2010). The social recognition test is based on the propensity of an adult mouse to spend more time interacting with an unknown mouse than with a known mouse (Winslow, 2001; Kaidanovich-Beilin et al., 2011; Fig. 4E) and is frequently used to show memory impairments in rodents (Winslow, 2001; Bilkei-Gorzo et al., 2005; Zhao et al., 2006). Our data revealed a significant increase of the social recognition ratio in 3xTg-AD+/− × dnPAK+/− mice compared with 3xTg-AD+/− × dnPAK−/− mice at 20 months of age (+56%, p < 0.001; Fig. 4F), which was not explained by changes in motor performances, motivation, or anxiety levels (Fig. 4A–D). In other words, social interactions with a known partner were not reduced in 3xTg-AD+/− × dnPAK+/− mice on day 2 compared with day 1, whereas a clear decrease in social interactions was noted in 3xTg-AD+/− × dnPAK−/− mice. Therefore, disrupted PAK activity led to a complete loss of social recognition in 3xTg-AD+/− × dnPAK+/− mice.
Transgenic PAK inactivation leads to a transient decrease of Aβ and tau deposition
To determine whether PAK plays a causal or amplifying role in the development of neuropathological markers of AD, we quantified Aβ and tau in the soluble and insoluble fractions of parietotemporal cortex of 12- or 20-month-old 3xTg-AD mice with or without concomitant dnPAK transgene expression. We first found that PAK inactivation did not affect the soluble levels of Aβ40 and Aβ42 in 3xTg-AD mice (Fig. 5A,B). However, PAK dysfunction significantly reduced the amount of both Aβ40 and Aβ42 in the insoluble fraction in 3xTg-AD+/− × dnPAK+/− mice at 12 months of age (−35% for both, p < 0.05), but not at 20 months of age (Fig. 5C,D). In addition, we observed a 58% decrease of insoluble/soluble tau ratio in 3xTg-AD+/− × dnPAK+/− mice compared with 3xTg-AD+/− × dnPAK−/− mice at the age of 12 months (p < 0.01; Fig. 5E). Again, this decrease was ultimately lost at 20 months of age (Fig. 5E). No significant changes were observed in the soluble phospho-tau/soluble tau ratio or the concentration of insoluble phospho-tau (Fig. 5F,G). These experiments suggest that a reduction in brain PAK activity can exert a beneficial effect on Aβ and tau deposition, at least transiently, and that the disruptive effect of PAK inactivation on social recognition does not require an enhancement of Aβ and tau deposition in the brains of 3xTg-AD mice.
Figure 5.

PAK inactivation reduced Aβ and tau depositions in 12- and 20-month-old heterozygous 3xTg-AD mice. PAK inactivation did not alter concentrations of sAβ40 (A) and sAβ42 (B) in soluble fractions at any age, but decreased iAβ40 (C) and iAβ42 (D) levels in insoluble fractions from 12-month-old 3xTg-AD mice. Whereas the inactivation of PAK induced a decrease of the insoluble/soluble tau ratio in 12-month-old mice (E), it did not induced any effect on the soluble phospho-tau/total tau ratio in mice whatever their age (F). Finally, no effect was observed on phospho-tau levels in the insoluble fraction (G). Values are expressed as mean ± SEM. Statistical comparisons were performed using an unpaired t test. ROD indicates relative optical density. Comparison between genotype: *p < 0.05, **p < 0.01; comparison between age: #p < 0.05, ##p < 0.01, ###p < 0.001.
Loss of CC and hyperactivity of excitatory synapses in 3xTg-AD mice were not influenced by PAK activity
As reported previously (Arsenault et al., 2011), entorhinal cortex deep layer neurons of 12-month-old homozygous 3xTg-AD mice present normal resting potential (Fig. 6A) and decreased CC (Fig. 6B). Interestingly, here we found a strong positive correlation between soluble total PAK and the CC (Fig. 6C). In addition, the frequency of sEPSC recordings was higher in 3xTg-AD neurons (Fig. 6D,F), whereas their amplitudes were lower (Fig. 6E,F). This suggests that homozygous 3xTg-AD cortical neurons display abnormalities of intrinsic or synaptic properties that are associated with PAK concentrations.
Figure 6.

Intrinsic properties of entorhinal cortex neurons in 12-month-old homozygous 3xTg-AD mice and in 20-month-old 3xTg-AD mice with genetically induced PAK inactivation. A–F, Expression of transgenes in homozygous 3xTg-AD mice did not change resting potential (A) but decreased CC (B) of entorhinal cortex deep layer neurons. Interestingly, we found a positive correlation between soluble PAK1/2/3 and the CC (C). Events of sEPSC occurred more frequently in 3xTg-AD neurons (D), but event amplitudes were lower (E). F, Examples of spontaneous EPSC recordings made in NonTg and 3xTg-AD neurons. NonTg mice are represented in white and 3xTg-AD mice are represented by stripes in graphs. G–K, To investigate the physiological function of PAK in AD, we studied effects of PAK inactivation in intrinsic and synaptic properties of deep-layer lateral entorhinal cortex neurons of NonTg (white), NonTg with a deficiency in PAK activity (white with black stripes), heterozygous 3xTg-AD (gray), and heterozygous 3xTg-AD mice with a deficiency of PAK activity (black). Genetically, PAK inactivation in NonTg or 3xTg-AD mice did not influence impairments of passive properties of entorhinal cortex observed in 3xTg-AD mice, as reflected by resting potential (G) and CC (H) analyses. Similarly, frequency (I) and amplitude (J) of sEPSCs observed in 3xTg-AD mice were not influenced by PAK activity. K, Examples of sEPSC recordings made in NonTg, 3xTg-AD, and PAK-deficient 3xTg-AD neurons. Values are expressed as mean ± SEM of the numbers of cells analyzed. Statistical analyses were performed using unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001.
To further investigate the electrophysiological function of PAK in AD, we studied the effect of PAK inactivation on the same intrinsic and synaptic properties of deep-layer lateral entorhinal cortex neurons of NonTg mice (3xTg-AD−/− × dnPAK−/−), heterozygous 3xTg-AD mice (3xTg-AD+/− × dnPAK−/−), and heterozygous 3xTg-AD mice with a deficiency of PAK activity (3xTg-AD+/− × dnPAK+/−). We first confirmed that dnPAK expression in NonTg mice had no effect on each intrinsic parameter under study (Fig. 6) and then showed that neurons from heterozygous 3xTg-AD+/− × dnPAK−/− mice displayed decreased CC and higher sEPSC frequencies compared with NonTg mice (Fig. 6H,I, respectively), as shown previously in homozygous 3xTg-AD mice, but without any change in their resting potential (Fig. 6G). However, we found that CC and sEPSC frequencies of entorhinal cortex neurons in 3xTg-AD mice remained unchanged with PAK inactivation (Fig. 6H,I, respectively). Finally, we did not find any difference in the mean amplitude of sEPSCs between groups (Fig. 6J). These results suggest that the effects of PAK inactivity on social recognition are not related to the electrophysiological parameters altered in 3xTg-AD animals evaluated here.
Discussion
This study shows that the postmortem PAK concentrations in the cortex of AD patients are strongly associated with the clinical and neuropathological expression of the disease (Fig. 7). The decrease in PAK can be replicated in animal models transgenically designed to develop Aβ and tau pathologies. Expression of the dnPAK transgene in 3xTg-AD mice led to reduced PAK activation, as observed in AD patients. However, this did not aggravate Aβ or tau pathologies, suggesting that the loss of PAK lies downstream of these canonical AD markers. More strikingly, despite no direct deteriorating effects of PAK dysfunction on CC or synaptic activity in 3xTg-AD mice, a loss of PAK activity obliterated their social recognition capacity, a behavior defect reminiscent of a common symptom of AD (Larson et al., 1992). Therefore, although our study does not pinpoint PAK dysfunction as an early causal mechanistic event in AD, it suggests that PAK activity is essential for the maintenance of social recognition functions that are severely compromised in AD.
Figure 7.

Counterbalancing the loss of PAK might be of therapeutic value in AD. Late during AD progression, and not in individuals with mild cognitive impairment, the levels of PAK decrease in the brain in close relationship with accumulation of tau neuropathology and declining cognitive performance. In the 3xTg-AD mouse model, expression of transgenes leading to Aβ and tau pathologies induced a global decrease of PAK. Conversely, genetic deactivation of PAK in the 3xTg-AD mouse led to a transient decrease in insoluble Aβ and tau. This suggests that PAK decrease is a consequence rather than a cause of Aβ and tau neuropathologies. However, inactivating PAK in the brains of 3xTg-AD mice generated an animal model closer to AD by decreasing drebrin levels and social recognition capacities. Overall, our study supports the hypothesis that a therapeutic intervention aimed at increasing PAK activity may improve AD symptoms without correcting Aβ and tau pathologies.
PAK alteration in AD
In the present study, the diagnosis of AD was associated with lower PAK and phospho-PAK concentrations in the parietal cortex. This is in agreement with previous Western blot and immunostaining analyses performed in the cortex and hippocampus of AD patients (Zhao et al., 2006; Ma et al., 2008; Nguyen et al., 2008). However, no significant difference in PAK was detected in individuals with mild cognitive impairment, suggesting that the loss of PAK is a relatively late event. Therefore, our results indicate that a decrease in PAK concentration is closely associated with both clinical symptoms and Aβ/tau neuropathologies and argue in favor of an important role of PAK in the disease progression.
Impairments of PAK observed in AD patients are partly replicated in 3xTg-AD mice
The 3xTg-AD mouse simultaneously expresses tau and Aβ pathologies and is therefore used to model AD (Oddo et al., 2003a). We observed a loss of PAK in 3xTg-AD mice at 12 and 20 months of age, but not at 4 months, indicating that PAK is affected by age, a key factor in the development of AD. This result also demonstrates that the loss of soluble PAK in 3xTg-AD mice is not the result of an interaction between transgene expression and developmental processes, because the loss of the protein occurs in adult mice. Interestingly, the loss of PAK coincides with the age when the animal presents abundant deposition of insoluble proteins (i.e., 12 months; Oddo et al., 2003a; Julien et al., 2008). Our results indicate that the 3xTg-AD model replicates PAK abnormalities found in AD, but, consistent with the human data, these observations in animals strongly suggest that the loss of PAK is a consequence rather than a cause of AD neuropathology.
Downstream alterations of drebrin and cognitive impairment in AD: a consequence of PAK dysfunction?
PAK is essential to the maturation and maintenance of dendritic spines and its dysfunction leads to cognitive impairments through a disruption of actin scaffold within dendrites (Zhao et al., 2006). PAK dysfunction induces downstream cofilin activation, actin filament depolymerization, drebrin displacement, and, ultimately, defects in spine dynamics (Minamide et al., 2000; Bokoch, 2003; Zhao et al., 2006; Kreis and Barnier, 2009). This hypothesis is supported by data in cultured hippocampal cells and in rodents, in which intracerebral injections of PAK inhibitors induced the translocation of drebrin from the membrane to the cytosol (Zhao et al., 2006; Ma et al., 2008). Incidentally, a loss of drebrin in the membrane compartment of the brain of AD patients has been consistently observed by various groups using different methodologies (Shim and Lubec, 2002; Calon et al., 2004; Counts et al., 2006; Aoki et al., 2007; Lacor et al., 2007; Julien et al., 2008). A decrease in drebrin in the membrane fractions can also be detected in the brain of AD transgenic mouse models, particularly after exposure to dietary risk factors (Calon et al., 2004; Julien et al., 2010). Therefore, the specific loss of detergent-soluble drebrin observed here after chronic PAK inactivation is consistent with drebrin being a downstream element of PAK signalization. In contrast, consistent with previous studies performed in AD patients and mouse models (Zhao et al., 2006; Kang et al., 2011), no changes in cofilin and phospho-cofilin concentrations were observed in our study.
Because PAK is essential for cognitive performance in animals (Ramakers, 2002; Meng et al., 2005; Zhao et al., 2006; Nekrasova et al., 2008) and humans (Boda et al., 2004; Zhao et al., 2006; Morrow et al., 2008; Nikolić, 2008), one could assume that a loss of PAK activity is detrimental. The loss of drebrin observed here, as in AD, is consistent with such a hypothesis (Zhao et al., 2006; Kojima and Shirao, 2007; Kojima et al., 2010; VanGuilder et al., 2012). Social interaction is a fundamental and adaptive component of the biology in numerous species. In mice, recognition of conspecifics may be important for maintaining social hierarchy and for mate choice (Kaidanovich-Beilin et al., 2011). Two unfamiliar mice placed in a neutral arena will usually display important interactions (Kwon et al., 2006). Our results showed a profound social-cognitive impairment in the 3xTg-AD mice with PAK dysfunction, suggesting that PAK activity is critical to the maintenance of these higher social functions. Previous work showed that dnPAK mice exhibit specific deficits in the consolidation phase of hippocampus-dependent memory (Hayashi et al., 2004), suggesting that the loss of social recognition is a consequence of memory impairment. Finally, the impaired social recognition functions and drebrin loss observed suggest that the 3xTg-AD+/− × dnPAK+/− mouse may mimic more closely the human disease.
PAK inactivation does not promote the formation of insoluble depots of Aβ and tau
To determine whether PAK is a cause or a consequence of AD neuropathology, we investigated the effect of a genetically induced PAK dysfunction on Aβ and tau concentrations in the 3xTg-AD mouse model. Potentiation of AD-like neuropathology in 3xTg-AD+/− × dnPAK+/− mice would have been consistent with a benefic action of PAK. However, in our study, PAK inactivation rather led to a diminution of insoluble Aβ and tau in 12-month-old animals, an effect shown to be transient because it was lost at 20 months of age. It is important to underscore that the causal role of both Aβ and tau neuropathologies in AD remains unclear and relies mostly on correlative clinicopathology (Steinerman et al., 2008; Nelson et al., 2012) or genetic studies (Götz et al., 2008; O'Brien and Wong, 2011). The present data indicate that PAK may play a role in the formation of insoluble Aβ or tau deposits, and thus deserve further investigation. For example, PAK targeting could be used to modulate the aggregation of proteins into their insoluble form. These data chiefly indicate that the disruptive effect of PAK inactivation on social recognition does not require any clear enhancement of Aβ and tau deposition in the 3xTg-AD mouse model.
Electrophysiological abnormalities in 3xTg-AD are not modulated by PAK
Electrophysiological abnormalities such as a loss of CC and increased frequency of sEPSCs were described recently in 3xTg-AD mice (Arsenault et al., 2011). These observations are consistent with a study showing that Aβ peptide could enhance sEPSCs (Abramov et al., 2009), leading to a vicious cycle because synaptic activity is known to stimulate the production of Aβ peptides (Kamenetz et al., 2003). CC is a well accepted indication of the membrane surface area of the patched cell (Golowasch et al., 2009; Arsenault et al., 2011). The decrease in CC observed in 3xTg-AD animals may reflect neuronal atrophy (Golowasch et al., 2009), as reported previously in 3xTg-AD mice (Arsenault et al., 2011). The strong relationship between PAK levels and the decrease of CC in entorhinal cortical neurons prompted us to investigate the effect of PAK inactivation on this neuronal population.
Our patch-clamp experiments in 20-month-old heterozygous 3xTg-AD+/− × dnPAK+/− mice confirmed what has been found earlier in homozygous 3xTg-AD mice: a decrease in CC and an increase of sEPSCs. However, PAK dysfunction did not correct or aggravate CC or synaptic current changes in the 3xTg-AD mouse, suggesting that PAK impairments are either downstream of or occur in parallel with these electrophysiological abnormalities. In summary, our results suggest that the social recognition deficit induced by PAK inactivation in 3xTg-AD mice is more likely to be related to defect in PAK-drebrin signaling rather than to changes in basic neuron physiology.
Overall, the human and animal data show that a loss of PAK is associated with AD and is therefore likely a consequence rather than a cause of Aβ and tau neuropathology. Transgenic deactivation of PAK in the brain led to AD-like obliteration of social recognition in 3xTg-AD mice without either enhancing brain Aβ/tau pathologies or leaving a clear electrophysiological signature in neurons. Although the PAK loss does not appear to play a causal role in AD pathogenesis, our results suggest that increasing the activation of the PAK molecular cascade may prove beneficial against cognitive or neuropsychiatric symptoms of AD.
Footnotes
We thank Atsou Dzini Memelio for technical assistance and Dr. Zhong-Wei Zhang for instrumentation support.
The authors declare no competing financial interests.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Ackl N, Ising M, Schreiber YA, Atiya M, Sonntag A, Auer DP. Hippocampal metabolic abnormalities in mild cognitive impairment and Alzheimer's disease. Neurosci Lett. 2005;384:23–28. doi: 10.1016/j.neulet.2005.04.035. [DOI] [PubMed] [Google Scholar]
- Aoki C, Mahadomrongkul V, Fujisawa S, Habersat R, Shirao T. Chemical and morphological alterations of spines within the hippocampus and entorhinal cortex precede the onset of Alzheimer's disease pathology in double knock-in mice. J Comp Neurol. 2007;505:352–362. doi: 10.1002/cne.21485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenault D, Zhang ZW. Developmental remodelling of the lemniscal synapse in the ventral basal thalamus of the mouse. J Physiol. 2006;573:121–132. doi: 10.1113/jphysiol.2006.106542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenault D, Julien C, Tremblay C, Calon F. DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PLoS One. 2011;6:e17397. doi: 10.1371/journal.pone.0017397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA. Postmortem indices linking risk factors to cognition: results from the Religious Order Study and the Memory and Aging Project. Alzheimer Dis Assoc Disord. 2006;20:S63–68. doi: 10.1097/00002093-200607001-00009. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/WNL.59.2.198. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–841. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- Bilkei-Gorzo A, Racz I, Valverde O, Otto M, Michel K, Sarstre M, Zimmer A. Early age-related cognitive impairment in mice lacking cannabinoid CB1 receptors. Proc Natl Acad Sci U S A. 2005;102:15670–15675. doi: 10.1073/pnas.0504640102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boda B, Alberi S, Nikonenko I, Node-Langlois R, Jourdain P, Moosmayer M, Parisi-Jourdain L, Muller D. The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J Neurosci. 2004;24:10816–10825. doi: 10.1523/JNEUROSCI.2931-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boda B, Nikonenko I, Alberi S, Muller D. Central nervous system functions of PAK protein family: from spine morphogenesis to mental retardation. Mol Neurobiol. 2006;34:67–80. doi: 10.1385/MN:34:1:67. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Bories C, Guitton MJ, Julien C, Tremblay C, Vandal M, Msaid M, De Koninck Y, Calon F. Sex-dependent alterations in social behaviour and cortical synaptic activity coincide at different ages in a model of Alzheimer's disease. PLoS One. 2012;7:e46111. doi: 10.1371/journal.pone.0046111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourin M, Hascoët M. The mouse light/dark box test. Eur J Pharmacol. 2003;463:55–65. doi: 10.1016/S0014-2999(03)01274-3. [DOI] [PubMed] [Google Scholar]
- Boyle PA, Wilson RS, Aggarwal NT, Tang Y, Bennett DA. Mild cognitive impairment: risk of Alzheimer disease and rate of cognitive decline. Neurology. 2006;67:441–445. doi: 10.1212/01.wnl.0000228244.10416.20. [DOI] [PubMed] [Google Scholar]
- Calon F, Morissette M, Rajput AH, Hornykiewicz O, Bédard PJ, Di Paolo T. Changes of GABA receptors and dopamine turnover in the postmortem brains of parkinsonians with levodopa-induced motor complications. Mov Disord. 2003;18:241–253. doi: 10.1002/mds.10343. [DOI] [PubMed] [Google Scholar]
- Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N, Jr, Ashe KH, Frautschy SA, Cole GM. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron. 2004;43:633–645. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Exploratory behavior models of anxiety in mice. Neurosci Biobehav Rev. 1985;9:37–44. doi: 10.1016/0149-7634(85)90030-2. [DOI] [PubMed] [Google Scholar]
- Dubos A, Combeau G, Bernardinelli Y, Barnier JV, Hartley O, Gaertner H, Boda B, Muller D. Alteration of Synaptic Network Dynamics by the Intellectual Disability Protein PAK3. J Neurosci. 2012;32:519–527. doi: 10.1523/JNEUROSCI.3252-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golowasch J, Thomas G, Taylor AL, Patel A, Pineda A, Khalil C, Nadim F. Membrane capacitance measurements revisited: dependence of capacitance value on measurement method in nonisopotential neurons. J Neurophysiol. 2009;102:2161–2175. doi: 10.1152/jn.00160.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Ittner LM, Schonrock N, Cappai R. An update on the toxicity of Abeta in Alzheimer's disease. Neuropsychiatr Dis Treat. 2008;4:1033–1042. doi: 10.2147/NDT.S3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L. Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2011;68:428–441. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain-specific dominant-negative PAK transgenic mice. Neuron. 2004;42:773–787. doi: 10.1016/j.neuron.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci U S A. 2007;104:11489–11494. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia L, Helguera P, de Olmos S, Kedikian G, Solá Vigo F, LaFerla F, Staufenbiel M, de Olmos J, Busciglio J, Cáceres A, Lorenzo A. Phosphorylation of Actin-Depolymerizing Factor/Cofilin by LIM-Kinase Mediates Amyloid beta-Induced Degeneration: A Potential Mechanism of Neuronal Dystrophy in Alzheimer's Disease. J Neurosci. 2006;26:6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Zhou Z, Asrar S, Henkelman M, Xie W, Jia Z. p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol Cell Biol. 2011;31:388–403. doi: 10.1128/MCB.00969-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer Mechanisms and Therapeutic Strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Bendjelloul F, Phivilay A, Coulombe MA, Emond V, Calon F. Decreased drebrin mRNA expression in Alzheimer disease: correlation with tau pathology. J Neurosci Res. 2008;86:2292–2302. doi: 10.1002/jnr.21667. [DOI] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Jr, Bennett DA, Calon F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J, Woodgett J. Assessment of social interaction behaviors. J Vis. Exp. 2011:48. doi: 10.3791/2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kang DE, Roh SE, Woo JA, Liu T, Bu JH, Jung AR, Lim Y. The interface between cytoskeletal aberrations and mitochondrial dysfunction in Alzheimer's disease and related disorders. Exp Neurobiol. 2011;20:67–80. doi: 10.5607/en.2011.20.2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury AE, Foster OJ, Nisbet AP, Cairns N, Bray L, Eve DJ, Lees AJ, Marsden CD. Tissue pH as an indicator of mRNA preservation in human post-mortem brain. Brain Res Mol Brain Res. 1995;28:311–318. doi: 10.1016/0169-328X(94)00219-5. [DOI] [PubMed] [Google Scholar]
- Kojima N, Shirao T. Synaptic dysfunction and disruption of postsynaptic drebrin-actin complex: a study of neurological disorders accompanied by cognitive deficits. Neurosci Res. 2007;58:1–5. doi: 10.1016/j.neures.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Kojima N, Hanamura K, Yamazaki H, Ikeda T, Itohara S, Shirao T. Genetic disruption of the alternative splicing of drebrin gene impairs context-dependent fear learning in adulthood. Neuroscience. 2010;165:138–150. doi: 10.1016/j.neuroscience.2009.10.016. [DOI] [PubMed] [Google Scholar]
- Kreis P, Barnier JV. PAK signalling in neuronal physiology. Cell Signal. 2009;21:384–393. doi: 10.1016/j.cellsig.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten Regulates Neuronal Arborization and Social Interaction in Mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer's Disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EB, Kukull WA, Katzman RL. Cognitive Impairment: Dementia and Alzheimer's Disease. Annual Review of Public Health. 1992;13:431–449. doi: 10.1146/annurev.pu.13.050192.002243. [DOI] [PubMed] [Google Scholar]
- Ma QL, Yang F, Calon F, Ubeda OJ, Hansen JE, Weisbart RH, Beech W, Frautschy SA, Cole GM. p21-activated kinase aberrant activation and translocation in Alzheimer's disease pathogenesis. J Biol Chem. 2008 doi: 10.1074/jbc.M708034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, Parsons CG, Gupta S, Banerjee P, LaFerla FM. Memantine improves cognition and reduces Alzheimer's-like neuropathology in transgenic mice. Am J Pathol. 2010;176:870–880. doi: 10.2353/ajpath.2010.090452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Crews L, Hansen L. Synaptic remodeling during aging and in Alzheimer's disease. J Alzheimers Dis. 2006;9:91–99. doi: 10.3233/jad-2006-9s311. [DOI] [PubMed] [Google Scholar]
- Mastrangelo MA, Bowers WJ. Detailed immunohistochemical characterization of temporal and spatial progression of Alzheimer's disease-related pathologies in male triple-transgenic mice. BMC Neurosci. 2008;9:81. doi: 10.1186/1471-2202-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Meng Y, Hanna A, Janus C, Jia Z. Abnormal Long-Lasting Synaptic Plasticity and Cognition in Mice Lacking the Mental Retardation Gene Pak3. J Neurosci. 2005;25:6641–6650. doi: 10.1523/JNEUROSCI.0028-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Kane A, Goff DC, Walsh CA. Sequence analysis of P21-activated kinase 3 (PAK3) in chronic schizophrenia with cognitive impairment. Schizophrenia research. 2008;106:265–267. doi: 10.1016/j.schres.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasova T, Jobes ML, Ting JH, Wagner GC, Minden A. Targeted disruption of the Pak5 and Pak6 genes in mice leads to deficits in learning and locomotion. Developmental Biology. 2008;322:95–108. doi: 10.1016/j.ydbio.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kövari E, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Galvan V, Huang W, Banwait S, Tang H, Zhang J, Bredesen DE. Signal transduction in Alzheimer disease: p21-activated kinase signaling requires C-terminal cleavage of APP at Asp664. J Neurochem. 2008;104:1065–1080. doi: 10.1111/j.1471-4159.2007.05031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolić M. The Pak1 kinase: an important regulator of neuronal morphology and function in the developing forebrain. Molecular Neurobiology. 2008;37:187–202. doi: 10.1007/s12035-008-8032-1. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003a;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-Transgenic Model of Alzheimer's Disease with Plaques and Tangles: Intracellular A[beta] and Synaptic Dysfunction. Neuron. 2003b;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Phivilay A, Julien C, Tremblay C, Berthiaume L, Julien P, Giguère Y, Calon F. High dietary consumption of trans fatty acids decreases brain docosahexaenoic acid but does not alter amyloid-beta and tau pathologies in the 3xTg-AD model of Alzheimer's disease. Neuroscience. 2009;159:296–307. doi: 10.1016/j.neuroscience.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–199. doi: 10.1016/S0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Rodríguez JJ, Verkhratsky A. Neurogenesis in Alzheimer's disease. J Anat. 2011;219:78–89. doi: 10.1111/j.1469-7580.2011.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau V, Goupille O, Morin N, Barnier JV. A New Constitutively Active Brain PAK3 Isoform Displays Modified Specificities toward Rac and Cdc42 GTPases. J Biol Chem. 2003;278:3912–3920. doi: 10.1074/jbc.M207251200. [DOI] [PubMed] [Google Scholar]
- Salminen A, Suuronen T, Kaarniranta K. ROCK, PAK, and Toll of synapses in Alzheimer's disease. Biochem Biophys Res Commun. 2008;371:587–590. doi: 10.1016/j.bbrc.2008.04.148. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Alzheimer's disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–115. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shim KS, Lubec G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer's disease and Down syndrome. Neurosci Lett. 2002;324:209–212. doi: 10.1016/S0304-3940(02)00210-0. [DOI] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinerman JR, Irizarry M, Scarmeas N, Raju S, Brandt J, Albert M, Blacker D, Hyman B, Stern Y. Distinct pools of beta-amyloid in Alzheimer disease-affected brain: a clinicopathologic study. Arch Neurol. 2008;65:906–912. doi: 10.1001/archneur.65.7.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterniczuk R, Antle MC, Laferla FM, Dyck RH. Characterization of the 3xTg-AD mouse model of Alzheimer's disease: part 2. Behavioral and cognitive changes. Brain Res. 2010;1348:149–155. doi: 10.1016/j.brainres.2010.06.011. [DOI] [PubMed] [Google Scholar]
- Szczepanowska J. Involvement of Rac /Cdc42/PAK pathway in cytoskeletal rearrangements. Acta Biochim Pol. 2009 [PubMed] [Google Scholar]
- Tremblay C, Pilote M, Phivilay A, Emond V, Bennett DA, Calon F. Biochemical characterization of Abeta and tau pathologies in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2007;12:377–390. doi: 10.3233/jad-2007-12411. [DOI] [PubMed] [Google Scholar]
- VanGuilder HD, Farley JA, Yan H, Van Kirk CA, Mitschelen M, Sonntag WE, Freeman WM. Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline. Neurobiol Dis. 2012;43:201–212. doi: 10.1016/j.nbd.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Beckett LA, Barnes LL, Schneider JA, Bach J, Evans DA, Bennett DA. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging. 2002;17:179–193. doi: 10.1037/0882-7974.17.2.179. [DOI] [PubMed] [Google Scholar]
- Winslow JT. Current protocols in neuroscience. New York: Wiley; 2001. Mouse social recognition and preference. [DOI] [PubMed] [Google Scholar]
- Zenke FT, King CC, Bohl BP, Bokoch GM. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J Biol Chem. 1999;274:32565–32573. doi: 10.1074/jbc.274.46.32565. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Arsenault D. Gain modulation by serotonin in pyramidal neurones of the rat prefrontal cortex. J Physiol. 2005;566:379–394. doi: 10.1113/jphysiol.2005.086066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- Zhao Z-S, Manser E, Chen XQ, Chong C, Leung T, Lim L. A conserved negative regulatory region in alphaPAK: inhibition of PAK kinases reveals their morphological roles downstream of Cdc42 and Rac1. Mol Cell Biol. 1998;18:2153–2163. doi: 10.1128/mcb.18.4.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]