

Abstract
The import of acetyl-CoA into the ER lumen by AT-1/SLC33A1 is essential for the Nε-lysine acetylation of ER-resident and ER-transiting proteins. A point-mutation (S113R) in AT-1 has been associated with a familial form of spastic paraplegia. Here, we report that AT-1S113R is unable to form homodimers in the ER membrane and is devoid of acetyl-CoA transport activity. The reduced influx of acetyl-CoA into the ER lumen results in reduced acetylation of ER proteins and an aberrant form of autophagy. Mice homozygous for the mutation display early developmental arrest. In contrast, heterozygous animals develop to full term, but display neurodegeneration and propensity to infections, inflammation, and cancer. The immune and cancer phenotypes are contingent on the presence of pathogens in the colony, whereas the nervous system phenotype is not. In conclusion, our results reveal a previously unknown aspect of acetyl-CoA metabolism that affects the immune and nervous systems and the risk for malignancies.
Introduction
Acetyl-CoA is an important substrate for a large variety of biochemical reactions occurring in the cell. Cytosolic acetyl-CoA mainly originates from the conversion of citrate by ATP-citrate lyase and the condensation of free acetate and CoA by acetyl-CoA synthetase. One of its metabolic functions is to serve as donor of the acetyl group in the reaction of Nε-lysine acetylation of transiting and resident proteins that occurs in the lumen of the ER (Pehar and Puglielli, 2013). Bioavailability of acetyl-CoA into the ER lumen is achieved through active import by the ER membrane transporter AT-1 (also known as SLC33A1 and ACATN; Jonas et al., 2010). Specific ER-based acetyl-CoA:lysine acetyltransferases are then responsible for the acetylation of a long list of ER-based protein substrates (Choudhary et al., 2009; Ko and Puglielli, 2009; Pehar et al., 2012b; for review, see Pehar and Puglielli, 2013).
The influx of acetyl-CoA into the ER lumen appears to regulate the induction of ER Associated Degradation type II, ERAD(II), downstream of the unfolded protein response (UPR; Pehar et al., 2012a). This process is essential for autophagy-mediated degradation of large protein aggregates that accumulate in the ER. When dysfunctional, cells can either overaccumulate misfolded/unfolded protein aggregates [i.e., hypoactive ERAD(II)] or undergo autophagic cell death [i.e., hyperactive/uncontrolled ERAD(II); Klionsky, 2006; Bergmann, 2007; Pehar and Puglielli, 2013]. A dysfunctional autophagy/ERAD(II) machinery has been linked to a variety of human diseases (Mizushima et al., 2008; Buchberger et al., 2010).
Although proteomic studies assessing the ER acetylome predict wide biological impact (Choudhary et al., 2009; Pehar et al., 2012b), the contribution of the ER-based acetylation to human physiology and pathology has not been evaluated. Lin et al. (2008) reported that the gene encoding AT-1 is mutated in patients affected by autosomal dominant spastic paraplegia-42 (SPG42). The mutation, S113R, manifests itself in heterozygous individuals, displays incomplete penetrance, and affects adult individuals. Mutations in AT-1 have also been identified in children affected by severe developmental delay and premature death (Huppke et al., 2012). In contrast to SPG-42 patients, they were all homozygous for the mutation (for review, see Pehar and Puglielli, 2013).
Here, we provide insights into how the S113R mutation may lead to disease. Specifically, we show that AT-1S113R is unable to form homodimers in the plane of the ER membrane and is devoid of acetyl-CoA transport activity. Mice homozygous for the mutation display early developmental arrest. In contrast, heterozygous animals are born with Mendelian ratio and no developmental defect. Adult animals develop defects of both the immune and nervous systems. The defects of the immune system result in increased propensity to infections, aberrant ongoing inflammation, and increased propensity to malignancies. The defects of the nervous system result into motor and sensory deficits as well as degenerative features of the CNS and PNS. At the cellular level, the animals display a deregulated form of autophagy that can be rescued by a dominant “gain-of-acetylation” mutant form of the ER-associated autophagy protein 9A (Atg9A).
Materials and Methods
Generation of knock-in animals
Site-directed mutagenesis was used to introduce a serine to arginine substitution at amino acid 113 in the mouse Slc33a1 gene. Because the modified Slc33a1 gene remains under the control of its endogenous promoter, any aberration in level, timing, or tissue specificity of expression, problems commonly encountered when using a conventional transgenic approach, will be avoided. A Murine SV/129 BAC clone generated from AB2.2 ES DNA and containing the Slc33a1 gene and flanking regions were obtained from Geneservice. A portion of this BAC clone was used to construct a knock-in (KI) targeting vector in which serine (S) residue at position 113 was replaced by an arginine (R).
Recombineering (Liu et al., 2003) was used to introduce an 8.2 kb fragment of mouse strain 129/SV genomic DNA containing a portion of the Slc33a1 gene into the HSV-TK cassette containing vector pL253. The resulting plasmid, Slc33a1-pL253, was used as the backbone for constructing the Slc33a1-S113R targeting vector. A mini-targeting vector was constructed by cloning three PCR fragments, the 418 bp 5′ mini-homology unit, the 529 bp internal homology unit, and the 728 bp 3′ homology unit that contains the S113R substitution and a translationally silent HindIII site into the recombineering vector PL452 containing a loxP flanked pGK promoter/EM7 promoter-Neo-pGHpA cassette. The mini-targeting cassette was excised from PL452 vector sequence. This mini-homology unit fragment was electroporated into recombination-competent DY380 bacteria cells previously transformed with the Slc33a1-pL253 plasmid. Homologous recombination between the mini-targeting fragment and Slc33a1-pL253 plasmid resulted in a plasmid that integrated the Slc33a1-S113R mutation, the pGK promoter/EM7 promoter-Neo-pGHpA selection cassette, and the HSV-TK negative selection cassette. Restriction mapping and DNA sequencing were used to confirm the homologous integration of the Slc33a1-S113R mini-targeting cassette into the Slc33a1-pL253 plasmid and the completion of the Slc33a1-S113R targeting vector.
The targeting vector was linearized by digestion with NotI and introduced by electroporation into murine Sv129 AB2,2 embryonic stem (ES) cells (Nagy et al., 1993). ES cells that integrated the targeting vector either by homologous or random integration were selected by growth on G418. Gancyclovir (GANC) selected against clones that contained the HSV-TK cassette, thus enriching for clones that integrated the Neo cassette by homologous recombination. The resulting Neor, GANCr colonies were replicated and expanded. DNA was isolated from replica ES clones, digested with BamH1, electrophoresed on agarose gels, transferred to charged nylon membranes, and hybridized to a unique radiolabeled 5′ probe. Correctly targeted clones reveal an 11.2 kb band representing a gene modified by homologous recombination and a 25.9 kb band representing the unmodified gene. DNA from replicas of clones that appeared to be correctly targeted on the 5′ side were digested with BamHI electrophoresed on agarose gels, transferred to charged nylon membranes, and hybridized to unique radiolabeled 3′ probe. Correctly targeted clones reveal a 14.2 kb band representing the gene modified by homologous recombination and a 29.5 kb band representing the unmodified gene. Clones that appeared to be correctly targeted were sequenced to confirm the integrity of the loxP sites and the presence of the Slc33a1-S113R mutation. Six correctly targeted clones were expanded. These expanded cloned were regenotyped by Southern blotting and DNA sequencing. All six clones were karyotyped. Two clones with normal karyotypes were injected into C57BL/6 blastocysts to produce chimeric founders at the University of Michigan's Transgenic Animal Model Core (Ann Arbor, MI). Chimeric founder animals were produced from two ES clones: clone 4F12: 3 males 80% agouti; clone 4E2: 3 males 90% agouti, 5 males 80% agouti, 2 males 70% agouti. Highly chimeric founders were mated to C57BL/6 partners to obtain F1 animals. The floxed neocassette was removed in vivo by crossing mice carrying the Slc33a1-S113R mutation to transgenic mice expressing cre-recombinase in the male germline.
Genotyping was performed by PCR using the following specific primers: forward: 5′-CATGCAATTCTGTGGGCCAAAC-3′ and reverse: 5′-AAGCAGGACTGTGTCTCAAGCTGG-3′. Amplicons were analyzed on a 2% agarose gel. Amplicons size was as follows: 636 bp for wild-type (WT) AT-1 and 670 bp for mutant S113R-AT-1.
Animals were maintained in accordance with guidelines for the ethical care and treatment of animals from the Institutional Animal Care and Use Committee of the University of Wisconsin–Madison. The day of the experiment, mice were euthanized and tissues were immediately processed for analysis. Euthanasia was performed according to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
Animal testing
Inverted cage.
Mice were placed on a cover grid. The grid was slowly rotated to inverted position and held at ∼15–20 cm above the surface. The latency to fall was noted. Animals were tested three consecutive times, each time after appropriate rest in the home cage. The average latency of the three tests was calculated and used for statistical analysis. All animals were tested on the same day.
Hot plate.
Mice received one trial on a hot plate set to 55°C. Latency to first pain response (licking forepaws or back paws or other pain response) was noted. Mice were then removed from the hotplate and placed back in their home cage. Mice were tested for nociception on the same day.
Cell cultures
Human neuroglioma cells.
The human neuroglioma 4 (H4) cell line was maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin/glutamine solution (Mediatech). Stable transfection was performed using Lipofectamine 2000 (Invitrogen/Life Technologies) and the culture medium was supplemented with 350 μg/ml G418 sulfate (Mediatech) and/or 200 μg/ml zeocin (Invitrogen/Life Technologies). Cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
Mouse embryonic fibroblasts.
Embryos from timed pregnant females were collected at embryonic day 12.5 (E12.5) to E13.5. Embryos (without heads or visceral organs) were minced in cell culture grade trypsin-ethylenediaminetetraacetic acid (EDTA; 0.25%; Mediatech) and incubated in a 37°C CO2 incubator for 30–45 min. Trypsin was quenched by adding complete mouse embryonic fibroblast (MEF) media (DMEM-high glucose, 10% FBS, penicillin/streptomycin/glutamine mixture, fungizone) and tissue was further broken by gentle pipetting. Cells were collected into a centrifuge tube, spun at 1000 rpm for 5 min, and the medium was eliminated. Cells were washed again in MEF culture media and plated at postnatal day 0 (P0). Confluent cells were passaged at 1:4 dilutions by using trypsin-EDTA. For storage, P1 cells were frozen in regular MEF freezing medium. Cultures were used at P1–P2. For the Atg9AGln rescue, MEFs were transfected with Nucleofector II using the Basic Fibroblast Nucleofector Kit (Lonza). Cells were harvested 48 h later for Western blot.
Neurons.
Cortical neurons were prepared as described previously (Costantini et al., 2005; Costantini et al., 2006) from E16–E18 mice. Cultures were maintained in neurobasal medium containing 2% (v/v) B27 supplement (Invitrogen) and 0.5 mm GlutaMAX (Invitrogen) in the absence of serum or antibiotics. Medium was changed every 3 d. Cultures yielded approximately 99.5% neurons. The autophagy assay with LC3B-GFP imaging was conducted as described previously (Pehar et al., 2012a).
Plasmid constructs and site-directed mutagenesis
Human AT-1 (NM_004733) cDNA (SC117182) was obtained from Origene and cloned into pcDNA3.1AMyc/His (Invitrogen) and pcDNA3.1V5/His/TOPO (Invitrogen; Jonas et al., 2010). The cDNA of human AT-1 with a C-terminal TurboGFP fusion in the expression vector pCMV6-AC-GFP was obtained from Origene (RG204131). Mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) according to manufacturer's protocol. Serine113 was mutated to arginine using the following primers: forward: 5′-CAAGCTTTCTTCAGgTTTGTCTTTTGGCCC-3′ and reverse: 5′-GGGCCAAAAGACAAAcCTGAAGAAAGCTTG-3′. The presence of the mutation and the full-length gene sequences were confirmed by DNA sequencing performed at the DNA Sequencing Facility of the University of Wisconsin–Madison Biotechnology Center.
Antibodies and Western blotting
Protein extracts were prepared in GTIP buffer (10 mm Tris, pH 7.6, 2 mm EDTA, 0.15 m NaCl) supplemented with 1% Triton X-100 (Roche), 0.25% NP40 (Roche), complete protein inhibitors mixture (Roche), and phosphatase inhibitors (mixture set I and set II; Calbiochem). Protein concentration was measured by the bicinchoninic acid method (Pierce). Protein electrophoresis was performed as described previously (Costantini et al., 2006; Jonas et al., 2008; Jonas et al., 2010; Pehar et al., 2010) on a NuPAGE system using 4–12% Bis-Tris gels (Invitrogen). The following primary antibodies were used: anti-AT1/SLC33A1 (1:500; Abnova); anti-c-myc (1:1000; Santa Cruz Biotechnology); anti-V5 (1:300; Abcam); anti-acetylated lysine residue (1:1000; Abcam); anti-calreticulin (1:1000; Abcam); anti-beclin 1 (1:1000; Cell Signaling Technology); and anti-LC3β (1:1000; Cell Signaling Technology). For visualization of primary antibodies, membranes were incubated with goat anti-rabbit Alexa Fluor 680- or goat anti-mouse Alexa Fluor 800-conjugated secondary antibodies (LI-COR). Membranes were then imaged and quantified using the Odyssey Infrared Imaging System (LI-COR) as described previously (Jonas et al., 2008).
Affinity purification of AT-1
For affinity purification of AT-1-myc we used the ProFound c-Myc-Tag IP/Co-IP Kit (Pierce) as described previously (Costantini et al., 2007; Jonas et al., 2008; Ko and Puglielli, 2009; Jonas et al., 2010). Briefly, cell microsomes were generated from stably transfected H4 cells and then solubilized in the presence of 1.5% Triton X-100 as described in Antibodies and Western blotting, above. The protein extract (input) was loaded onto the above anti-myc immobilized column, washed, and eluted by lowering the pH to 2.0. The pH was immediately neutralized by adding 10 μl of neutralizing buffer (1 m Tris, pH 9.5) per 200 μl of elution buffer.
Cell fluorescence microscopy
The cellular localization of AT-1 was assessed in living H4 cells expressing a GFP-tagged version of WT or S113R mutant AT-1. In both cases, the GFP-tag was inserted at the C terminus so as not to disrupt the N-terminal signal anchor that ensures correct membrane insertion of AT-1. Cells were plated at a density of 9000 cells/cm2 in DMEM supplemented as described in Cell cultures, above. Forty-eight hours later, cells were incubated under normal growth conditions for 15 min in Hank's balanced salt solution with calcium and magnesium (HBSS/Ca/Mg; Invitrogen-Invitrogen) containing ER-Tracker Blue-White DPX dye (Invitrogen) at a final concentration of 100 nm. After washing with fresh HBSS/Ca/Mg, cells were imaged on a Zeiss Axiovert 200 inverted fluorescent microscope.
Glycerol gradient
Pooled ER fractions from a subcellular fractionation of AT-1WT and AT-1S113R overexpressing cells were solubilized with 0.5% Triton X-100, diluted to 0.2% Triton X-100, and then separated by analytical ultracentrifugation on a 10 ml of 8–30% glycerol gradient for 48 h at 4°C as described previously (Puglielli et al., 1999a; Puglielli et al., 1999b). Fractions of 0.35 ml were collected from the top to the bottom. Individual fractions were assayed for acetyl-CoA transport activity by in vitro reconstitution as described previously (Puglielli et al., 1999a; Puglielli et al., 1999b). Carbonic anhydrase, bovine serum albumin (BSA), and alcohol dehydrogenase (Sigma) were used as sedimentation controls.
Acetyl-CoA membrane transport
Highly purified ER vesicles were generated as described previously (Costantini et al., 2007; Ko and Puglielli, 2009; Jonas et al., 2010). Acetyl-CoA transport into intact native vesicles was performed as described previously (Costantini et al., 2007; Jonas et al., 2010). The specific activity of [3H]Acetyl-CoA was 656 cpm/pmol and the final concentration was 10 μm.
In vitro reconstitution
C-terminal myc-tagged versions of WT and S113R mutant AT-1 were purified from stable transfected cells with anti-myc immobilized columns (Costantini et al., 2007; Jonas et al., 2010). Affinity-purified AT-1 was reconstituted into artificial liposomes with five cycles of freeze-thaw in an acetone-dry ice bath (Puglielli et al., 1999a; Jonas et al., 2010). The protein to lipid ratio and the lipid composition of the artificial liposomes has been described previously (Jonas et al., 2010). Reconstituted proteoliposomes were assayed for acetyl-CoA transport immediately after the last freeze-thaw cycle. As control, transport was also assayed in liposomes alone.
Immune system characterization
Serum and mononuclear cell isolation.
Blood (100 μl) was collected transcardially from mice with an insulin syringe. For serum, blood was allowed to clot on ice for 15 min, centrifuged at 1000 g for 10 min at 4°C and the supernatant was collected. Single cell suspensions of mononuclear cells were isolated from all tissues as described previously (Zozulya et al., 2009). Briefly, blood was transferred into 10 ml of HBSS containing 10 mm EDTA. Leukocytes were also collected from bone marrow, thymus, spleen, and lymph nodes (cervical, axillary, mesenteric, and popliteal). Single-cell suspensions were generated by gently dissociating tissues with frosted slides (thymus, spleen, and lymph nodes) or passing sample through a 23 gauge needle (bone marrow). To remove contaminating erythrocytes, samples from blood, bone marrow, and spleen were allowed to lyse in ammonium chloride potassium containing red blood cell lysis buffer for 60 s before resuspending in 15 volumes of HBSS.
Flow cytometry.
From single-cell suspensions, 106 leukocytes were resuspended in PBS containing 1% BSA and stained with saturating concentrations of monoclonal antibodies against surface markers for 30 min at 4°C: CD2 (RM2-5), CD3e (17A2), CD4 (RM4-5), CD8a (53.67), CD11a (M17/4), CD11b (M1/70), CD11c (HL3), CD19 (1D3), CD25 (7D4), CD40 (MR1), CD43 (01605B), CD44 (IM7), CD45R(B220; RA3-6B2), Ly6g (GR-1; RB6-8C5), Ly6c (AL-21), and MHC-II (AF6-120.1) (all from BD Biosciences). For intracellular cytokine staining, T cells were activated in vitro with antibodies against CD3 (1452C11; 1 μg/ml) and CD28 (37.51; 2 μg/ml) before surface staining. Cells were incubated for 4 h at 37°C in complete RPMI medium containing 10% fetal calf serum in the presence of GolgiStop (BD Biosciences). After surface staining, these cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences), resuspended in PermWash Buffer (BD Biosciences), and stained with saturating concentrations of monoclonal antibodies against intracellular cytokines for 30 min at 4°C: IL-4 (11B11), IL-10 (JES5-16E3), IL-17 (TC11-18H10), and IFN-gamma (XMG1.2). Cell staining was acquired on a FACSCalibur or LSR II (BD Biosciences) and analyzed with FlowJo (TreeStar) software version 5.4.5.
Serum analytes.
Serum was collected as above and analytes were determined by MyriadRBM.
Diffusion tensor imaging
To obtain the highest resolution images, which is only possible when using extended scanning times (Lerch et al., 2012), animal brains were scanned ex vivo. Because fixation reduces the sensitivity of diffusion tensor imaging (DTI) for detecting pathology (Sun et al., 2009), brains were scanned freshly immediately after euthanasia. Specimens were immersed in free susceptibility-matching fluid (FluorInert FC-77; AMS Materials) and imaged on a Varian 4.7T Small Animal MR scanner. DTI was performed using echoplanar imaging. One non-diffusion-weighted reference image and 81 diffusion-weighted images were acquired using a multiple-shell acquisition technique. The gradient directions were generated from the square (9 × 9 = 81) sparse and optimal acquisition design recently proposed in Koay et al. (2006). The maximum b-value was 7510.15 s/mm2. Fourteen contiguous sections were acquired coronally with a field of view of 24 mm × 24 mm and a slice thickness of 1 mm. Other imaging parameters for this sequence were as follows: TR = 2000 ms TE = 40 ms, NEX = 3, matrix = 256 × 256. To improve field homogeneity, high-order shimming was performed before the DTI scan. Imaging time was as follows: 34 h: 59 m: 16 s. For data processing, the diffusion tensor was estimated using a least-squares estimation approach (Koay et al., 2006). Fractional anisotropy (FA) maps were generated using in-house software written in MATLAB version 7.11.0 (MathWorks) together with tools from the FMRIB Software Library (Smith et al., 2004). For analysis, regions of interest (ROIs) were placed on native space images using MRIcron software (Rorden et al., 2007). ROIs were placed on coronal sections of the FA maps by a trained operator blind to transgenic mutation status. Before ROI placement, maps were enlarged and windowed to an equivalent size and contrast across specimens. ROIs (∼1.3 mm in size) were placed within the corpus callosum at the midline for each animal, as per Paxinos and Franklin (2007).
Positron emission tomography imaging
[11C]PBR28 was synthesized following the steps described previously (Wang et al., 2009) with high specific activity ranging from 150 to 630 GBq/μmol. Dynamic positron emission tomography (PET) scans were obtained using the Genisys-4 (Sophie Biosciences) and were initiated with a bolus injection of [11C]PBR28 ranging from 0.59 to 6.55 MBq. Data were acquired for 60 min. PET data were corrected for decay, random coincidence events, attenuation, and dead time. Subject-specific ROIs were hand drawn on transverse slices over anterior, medial, and posterior regions of the brain, each with an average volume of 3.8 mm3, as well as over the whole brain with an average volume of 11.4 mm3. Standard uptake values (SUVs, units Bq/cc/Bqinj*g) were calculated by dividing the amount of radioactivity in the ROI by the injected dose of [11C]PBR28 per total body weight using the data collected between 40 and 60 min. Two-tailed unpaired t tests (α ≤ 0.01) were performed to test for group differences in SUV.
Histology and immunostaining
Tissues were collected immediately after euthanasia, fixed overnight in 10% neutral buffered formalin, and paraffin embedded using standard techniques. Coronal tissue sections were prepared using a microtome. After standard deparaffinization and rehydration, the tissue sections were processed for H&E, Nissl, and Kluver–Barrera staining or for classical immunofluorescence as described previously in detail (Pehar et al., 2010). For toluidine staining, sections were fixed in 2.5% glutaraldehyde in phosphate buffer, pH 7.2, before staining (Oberley et al., 2008). The following primary antibodies were used for immunofluorescence: anti-neurofilament 200 (clone RT97, 1:200; EMD-Millipore), anti-NeuN (clone A60, 1:200) and anti-GFAP (polyclonal, 1:500; Dako); and anti-Iba1 (polyclonal; 1:200; Wako). Nuclei were counterstained with DAPI.
Secondary antibodies were Alexa Fluor 488- and Alexa Fluor 594-conjugated goat anti-rabbit (5 μg/ml; Invitrogen) for GFAP and Iba1. For NeuN and neurofilament immunofluorescence, the secondary antibodies were biotin-labeled goat anti-mouse (5 μg/ml; Invitrogen), followed by Alexa Fluor 488- or Alexa Fluor 594-conjugated streptavidin (5 μg/ml; Invitrogen). Controls were performed by omitting the primary antibody. Processed slides were imaged on a Zeiss Axiovert 200 inverted fluorescent microscope. Computer-assisted quantitation was performed as described previously (Pehar et al., 2010; Ma et al., 2011; Yin et al., 2012). Areas quantified, number of sections, and animals analyzed are reported in the appropriate figure.
Electron microscopy
Transmission electron microscopy was performed at the Electron Microscopy Facility of the University of Wisconsin–Madison and VA Medical Center as described previously (Pehar et al., 2010). Tissue sections were observed with a transmission electron microscope (H-600; Hitachi) operated at 75 kV.
Statistical analysis
Data analysis was performed using GraphPad InStat 3.06 statistical software. Data are expressed as means ± SD. Comparison of the means was performed using a t test or one-way ANOVA followed by Tukey-Kramer multiple-comparisons test. For lifespan assessment, data were analyzed with the Kaplan-Meier lifespan test and log-rank test using GraphPad Prism version 4.0. Differences were declared statistically significant if p ≤ 0.05.
Results
SPG42-associated mutant AT-1 is unable to form homodimers in the ER membrane and is deficient in acetyl-CoA transport activity
To determine whether mutant AT-1S113R retained transport activity, we used the in vitro reconstitution system that we described previously (Jonas et al., 2010). Transgenic myc-tagged versions of the WT (AT-1WT) and mutant (AT-1S113R) AT-1 were purified from stably transfected H4 (human neuroglioma) cells with an immobilized anti-myc column. Affinity-purified AT-1 was then reconstituted into artificial liposomes before biochemical assessment of acetyl-CoA transport activity. In contrast to AT-1WT, AT-1S113R was completely devoid of transport activity (Fig. 1A). Figure 1B shows that similar levels of AT-1WT and AT-1S113R were reconstituted for functional analysis.
Figure 1.

AT-1S113R is unable to form homodimers in the membrane and is deficient of acetyl-CoA transport activity. A, Functional reconstitution of affinity-purified AT-1 into artificial liposomes. AT-1S113R is devoid of transport activity. Values are the mean (n = 3) ± SD. B, Immunoblot showing levels of AT-1WT and AT-1S113R in cell lysates, after affinity purification (output), and after reconstitution. Lanes in the output are from the same membrane. Asterisk (*) indicates a background band visible in both transfected and nontransfected (-) cells. C, Endogenous AT-1 migrates as a homodimer on analytical ultracentrifugation. Values are the mean (n = 4) ± SD. Bars on top show the sedimentation of molecular standards (Mol. St.): carbonic anhydrase (CA; 29 kDa), BSA (66 kDa) and alcohol dehydrogenase (AlchD; 150 kDa). D, Immunoblots showing co-IP of myc- and V5-tagged versions of AT-1WT and AT-1S113R. AT-1WT forms homodimers while AT-1S113R does not. A schematic view of the experiment is shown in the bottom.
Nucleotide sugar transporters and ATP membrane transporters function as homodimers in the membrane (Hirschberg et al., 1998). When solubilized in the presence of 0.2–0.5% Triton X-100, they maintain their functional assembly state and migrate as dimers on analytical ultracentrifugation (Mandon et al., 1994; Puglielli et al., 1999a). To determine whether AT-1 also behaved as a dimer, we solubilized active ER fractions of nontransfected H4 cells and loaded them on top of an 8–30% glycerol gradient. After centrifugation, the individual fractions were reconstituted into artificial liposomes and assayed for endogenous acetyl-CoA transport activity. The migration of protein standards was also assessed in parallel. The transport activity was found in fractions that corresponded to the ∼120 kDa area, suggesting that the functional acetyl-CoA transport unit has a molecular mass of ∼120 kDa (Fig. 1C), which is twice that of AT-1 as assessed by reducing gel electrophoresis. To confirm that AT-1 is a homodimer and to determine whether the S113R mutation interferes with the dimerization status of the transporter, we coexpressed myc- and V5-tagged versions of the WT and mutant AT-1 in the same cell and determined whether the immunoprecipitation of the myc-tagged version was able to pull down the V5-tagged version of the transporter. This was indeed the case for AT-1WT, but not for AT-1S113R (Fig. 1D, left, lanes 1 and 3). Figure 1D (right) shows that the anti-myc antibody was able to immunoprecipitate similar levels of AT-1WT and AT-1S113R. The ability of mutant AT-1 to form dimers was partially restored by the coexpression of WT AT-1 (Fig. 1D, left, lane 2).
To determine whether the mutation also affected the cellular distribution profile of AT-1, we studied H4 cells expressing a GFP-tagged version of AT-1WT or AT-1S113R. In both cases, the GFP-tag was inserted at the C terminus so as not to disrupt the N-terminal signal anchor that ensures membrane insertion of AT-1. Both the WT and S113R mutant versions of the transporter were found in the ER (Fig. 2A). No mislocalization was caused by the mutation. The ER localization of WT AT-1 has already been confirmed by both immunolabeling (Kanamori et al., 1997) and subcellular fractionation (Jonas et al., 2010).
Figure 2.
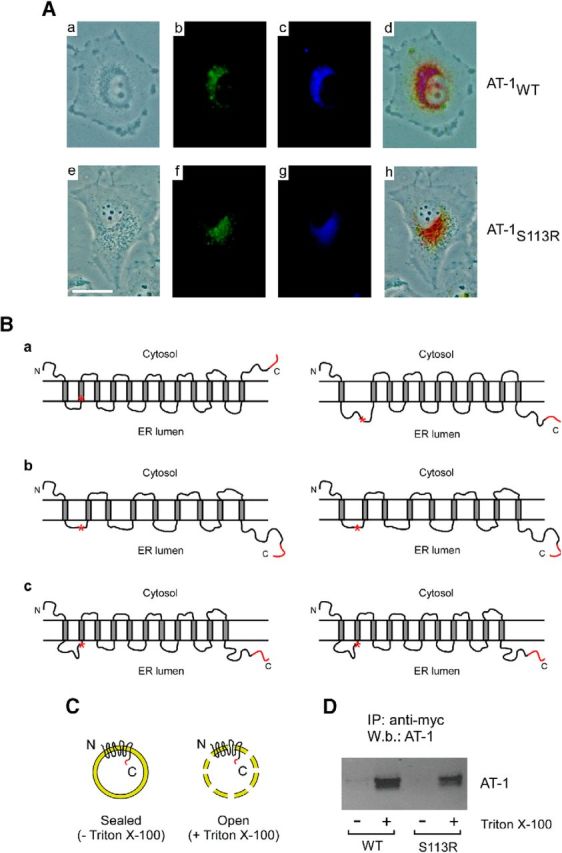
The S113R mutation does not affect the subcellular localization or the orientation of the C terminus of AT-1. A, Subcellular localization of AT-1 assessed in living H4 cells overexpressing a GFP-tagged version of WT (AT-1WT) or mutant (AT-1S113R) AT-1. In both cases, the GFP-tag was inserted at the C terminus so as not to disrupt the N-terminal signal anchor that ensures membrane insertion of AT-1. a, Phase-contrast; b, GFP fluorescence; c, ER tracker; d, merge of a, b, and c; e, phase-contrast; f, GFP fluorescence; g, ER tracker; and h, merge of e, f, and g. B, Predicted topology of WT (left) and SPG42-associated S113R mutant (right) AT-1. The following prediction models were used: a, SOSUI; b, PredictProtein; and c, UniProtKB. C, Schematic view of the experiment reported in D. The myc tag is shown in red. D, ER vesicles from AT-1WT and AT-1S113R expressing cells were incubated with an anti-myc antibody covalently attached to aldehyde-activated agarose beads for immunoprecipitation. After extensive washing, bound proteins were eluted by lowering the pH and analyzed by SDS-PAGE and immunoblotting. Vesicles were used under the sealed (no detergent) or open (with detergent) condition.
Next, we investigated whether the mutation affected the topology of the transporter across the ER membrane. In fact, model prediction with the SOSUI engine suggests a membrane topology with 12 transmembrane (TM) domains and a C-terminal tail facing the cytosol. According to this model, the S113R mutation would cause the transporter to flip across the membrane. Specifically, it would dislodge the second TM domain and result in an 11 TM protein with the C-terminal tail in the ER lumen (Fig. 2B). However, two different prediction models (PredictProtein and UniProtKB) do not support this conclusion. Although they differ in the number of potential TM domains, they both predict the C-terminal tail in the lumen of the ER and discard any effect on the topology of the protein as a result of the S113R mutation (Fig. 2B). To address this issue experimentally, we took advantage of the fact that both the WT and the S113R mutant versions of AT-1 used for the in vitro reconstitution (Fig. 1A) had a myc-tag at the C terminus. Specifically, we prepared highly purified ER vesicles from AT-1-overexpressing H4 cells and then analyzed the distribution of the tag across the membrane by using anti-myc antibodies covalently attached to aldehyde-activated agarose beads. A similar strategy was previously used to assess the topology of ER-based acetyl-CoA:lysine acetyltransferases (Ko and Puglielli, 2009). In fact, when prepared, ER vesicles are sealed and of the same topographical orientation as in vivo (Carey and Hirschberg, 1981; Puglielli et al., 1999a; Puglielli et al., 1999b). Vesicles were incubated with the immobilized anti-myc column in the presence (Fig. 2C, open) or absence (Fig. 2C, sealed) of mild concentrations (0.2–0.5%) of Triton X-100. The results show that AT-1 could only be immunoprecipitated under the “open” condition, which allowed the anti-myc antibody to have access to and interact with the C-terminal myc tag (Fig. 2D). These findings support the conclusion that the C-terminal tail of AT-1 faces the lumen of the ER. Similar results were also obtained with AT-1S113R, indicating that the S113R mutation does not cause the transporter to flip across the ER membrane as predicted by the SOSUI program (Fig. 2D).
In conclusion, these experiments indicate that S113R mutation only affects the ability of the transporter to form homodimers in the ER membrane. It is conceivable that changes in the folding and structural rigidity of the first intraluminal loop caused by the S113R mutation might be responsible for the inability of the two monomers to interact. Interestingly, currently identified mutations either block translation of the protein or are confined to the first ER luminal loop of the transporter, suggesting possible common mechanistic events (Lin et al., 2008; Huppke et al., 2012).
To confirm that AT-1S113R is devoid of acetyl-CoA transport activity, we analyzed transport under native conditions. Specifically, we generated mice expressing the S113R mutation (Fig. 3A,B) and determined transport in highly purified ER vesicles prepared from the liver of nontransgenic WT and KI (AT-1S113R/+) mice. The liver was chosen because it is uniquely suited for high-quality and high-mass isolation of ER. Due to the lethality of the mutation (see the following section), we were not able to test homozygous animals. Therefore, only heterozygous mice (AT-1S113R/+) were studied (Fig. 3C). As predicted by the in vitro reconstitution system (Fig. 1A), AT-1S113R/+ animals displayed an ∼50% decrease of acetyl-CoA transport across the ER membrane (Fig. 3C), supporting the conclusion that the animals are functionally haploinsufficient. The reduced influx of acetyl-CoA import into the ER lumen was also accompanied by reduced acetylation of ER proteins (Fig. 3D).
Figure 3.

AT-1S113R/+ mice are functionally haploinsufficient. A, Generation of AT-1S113R/+ mice. A, SLC33a1-mini targeting vector fragment. B, SLC33a1 retrieval vector. C, SLC33a1 targeting vector. D, Murine chromosome 3. E, Targeted chromosome 3. F, Neo excised chromosome 3. B, SLC33a1 retrieval vector. C, SLC33a1 targeting vector. D, Murine chromosome 3. E, Targeted chromosome 3. F, Neo-excised chromosome 3. B, AT-1S113R/+ mice are heterozygous for the mutation. C, Native ER vesicles from AT-1S113R/+ mice display reduced acetyl-CoA influx. Two independent animals for each genotype were analyzed. Values are the mean ± SD (n = 3). D, Immunoblot showing reduced Nε-lysine acetylation of ER proteins in AT-1S113R/+ mice. Calreticulin was used as an ER loading marker.
AT-1S113R/+ mice develop defects of the immune system and increased propensity to cancer when exposed to pathogens
Animals homozygous for the mutation suffer from severe retardation early in their development. Based on the stage of the neural tube closure, we estimated that, in most cases, the arrest occurs at E8–E8.5 (Fig. 4A). In contrast, heterozygous animals are born with Mendelian ratio and without any apparent defect. To assess the phenotype of the animals, AT-1S113R/+ mice were housed in a mouse specific pathogen free (SPF) facility, as well as in an “open” facility, where they are exposed to normal mouse pathogens. When housed in an open facility, AT-1S113R/+ mice developed signs of active infections, such as severe conjunctivitis (Fig. 4B, left), dermatitis (Fig. 4B), and disseminated abscesses that resulted positive for common pathogens. At death, several organs/tissues displayed moderate to severe inflammatory infiltration (Fig. 4D, Table 1). Typically, the histology revealed neutrophils, mononuclear leukocytes, and multinucleate giant cells intermixed to morphologically altered cells. The animals also displayed increased propensity to malignancies that were either the immediate cause of death/euthanasia or discovered while conducting the necropsy (Fig. 4E, Table 1). These included sarcoma, lymphoma, carcinoma, and carcinomatosis. The increased propensity to infections and malignancies resulted in reduced survival of AT-1S113R/+ mice compared with WT littermates housed in the same facility (Fig. 4C, top). Importantly, when housed in the SPF facility AT-1S113R/+ mice displayed no infection, tissue inflammation, or propensity to cancer (Table 1) and had a normal lifespan (Fig. 4C, bottom).
Figure 4.

AT-1S113R/+ mice display increased propensity to infections, disseminated tissue inflammation, and cancer. A, Mice homozygous for the S113R mutation display developmental arrest. Embryos in the figure were collected at E10.5. B, Adult AT-1S113R/+ mouse with conjunctivitis and severe dermatitis. C, Lifespan of WT and AT-1S113R/+ mice housed in an open (top; n = 30) and pathogen-free (bottom; n = 50) facility. AT-1S113R/+ mice display reduced survival in the open facility. D, Postmortem autopsy of the animal shown in B revealed inflammatory infiltration of different tissues/organs: a, skin (ulcerative dermatitis); b, stomach mucosa (gastritis); c, prostate parenchyma (prostatitis); d, bladder mucosa (cystitis); e, salivary gland (sialadenitis); and f, preputial gland (adenitis). E, Postmortem autopsy of the animal shown in B revealing different malignant foci: a, spindle cell sarcoma of the spine; b, spindle cell sarcoma invading the pleura; c, focal pulmonary adenoma; and d, sarcoma invading the bone structure of the knee.
Table 1.
Postmortem frequency of inflammation/infections and cancer
| Open (n = 30) Dead | Inflammation/infections | Cancer | Inflammation/infections | Cancer | SPF (n = 50) Dead | |||
|---|---|---|---|---|---|---|---|---|
| WT | 7 | 2 (28%) | 0 (0%) | 8 | 1 (12.5%) | 0 (0%) | ||
| AT-1S113R/+ | 18** | 18 (100%)** | 10 (55.5%)** | 10 | 1 (10%) | 0 (0%) | ||
Table shows postmortem frequency of inflammation/infections and cancer among animals included in Figure 4C.
**p < 0.005.
The increased propensity to inflammation and malignancies displayed by AT-1S113R/+ mice in the open facility suggested that the mutant animals might have aberrancies in their immune system, which could lead to either immune deficiency or hyperresponsiveness. When we compared immune cell development in healthy mice housed in an SPF facility, we found that B-cell precursor frequency in bone marrow (Fig. 5A) and T-cell precursor frequencies in thymus (Fig. 5C) were similar in WT and AT-1S113R/+ animals. Moreover, we observed no difference in the frequency of T-cell and B-cell populations in peripheral lymphoid tissues of healthy WT and AT-1S113R/+ mice (data not shown). Similarly, WT and AT-1 mutant mice had comparable levels of CD11b+ myeloid cells and Ly6c-hi monocytes in the bone marrow and spleen (data not shown). Together, these data suggest that there was no intrinsic aberration in the immune system of AT-1S113R/+ mice and that, in the absence of pathogen challenge, major components of both the innate and adaptive immune system develop normally in AT-1S113R/+ mice compared with WT littermates.
Figure 5.

Phenotypic characterization of the immune system. A, Dot plots showing frequency of B220+ B-cell precursors in bone marrow including: pre-pro B cells (CD43+CD19−), pro-B cells (CD43+CD19+), pre-B cells (CD43−CD19+CD2−B220int), and mature B cells (CD43−CD19+CD2+B220high). B, Representative histograms showing the percentage of cells expressing the activation marker LFA-1 on CD4+ and CD8+ cells isolated from lymph nodes (summarized in bar graphs). C, Representative dot plots showing frequency of T-cell precursors in thymus. CD4−CD8− double-negative (DN) cells were subdivided into developmental stages DN1 (CD44+CD25−), DN2 (CD44+CD25+), and DN3 (CD44−CD25+). CD4+ and CD8+ single-positive populations are also shown, along with percentage of cells expressing the T-cell coreceptor CD3 (histogram). D, Frequency of granulocytic CD11bhighLy6g+SSChigh and monocytic CD11b+Ly6g−SSClow myeloid cells in the spleen. E, T-helper lineage-specific intracellular cytokine production among lymphocytes stimulated with anti-CD3 and anti-CD28 for 4–5 h. Representative dot plots show IL-17 (Th17) and IFNγ (Th1); IL-4 (Th2) and IL-10 (Treg) are not depicted. In all panels, values are the mean (n = 3) ± SD. *p < 0.05; **p < 0.005; ***p < 0.0005.
Next, we compared innate and adaptive immune responses in WT and AT-1S113R/+ mice housed in an open facility. As expected, we detected higher expression of the T-cell activation marker LFA-1 on T cells from pooled lymph nodes of AT-1S113R/+ mice compared with WT mice, which is indicative of an ongoing immune response (Fig. 5B). We also observed an increase in the frequency of B cells isolated from peripheral lymph nodes, which corresponded with decreased B-cell frequency in the blood (data not shown). Upon ex vivo T-cell receptor stimulation, these T cells produced higher levels of inflammatory cytokines, including IL-17, IFN-γ, and IL-4 (Fig. 5E). IL-17 is known to promote chemokine production from endothelial cells, promoting the recruitment of monocytes and especially neutrophils. Interestingly, among CD11b+ myeloid cells isolated from the spleens of AT-1S113R/+ mice, there were proportionally more cells with a neutrophilic surface marker expression pattern (Ly6c-intermediate, side-scatter-high, Ly6g+; Fig. 5D), whereas there was no difference in the frequency of neutrophils among bone marrow cells isolated from WT and AT-1S113R/+ mice (data not shown). This increase in neutrophils was accompanied by a decrease in the frequency of MHC-II+ and CD40+ cells among myeloid cells in the spleen (data not shown). We also examined cytokine and chemokine levels in blood plasma from these mice. Compared with WT littermates housed in the same facility, we detected elevated concentrations of several chemokines and cytokines in AT-1S113R/+ mice, including IL-7, IL-18, CXCL6, CCL9 (MIP-3beta), and CCL19 (MIP-1gamma; Table 2). Among these, CXCL6 is notable in that it is highly chemotactic for neutrophils, which were found to be abundant in the spleens of the AT-1S113R/+ animals. In contrast, CCL22, which is chemotactic for dendritic cells, monocytes, and chronically activated T cells, was significantly decreased in serum compared with that of WT mice.
Table 2.
Plasma inflammatory molecules that are altered in the two groups
| WT | AT-1S113R/+ | |||
|---|---|---|---|---|
| IgA (μg/ml) | 171 ± 97 | 608 ± 387 | ▴ 3.5-fold | p < 0.05 |
| IL-7 (ng/ml) | <0.17 | 0.57 ± 0.39 | ▴ 3.5-fold | p < 0.005 |
| IL-18 (ng/ml) | 9.67 ± 4.13 | 14.2 ± 3.87 | ▴ 46% | p < 0.05 |
| VCAM-1 (ng/ml) | 1644 ± 99 | 2067 ± 358 | ▴ 25% | p < 0.05 |
| CXCL-6 (ng/ml) | 4.83 ± 1.56 | 16.7 ± 5.5 | ▴ 3.5-fold | p < 0.005 |
| CCL-22 (pg/ml) | 2608 ± 291 | 1932 ± 231 | ▾ 26% | p < 0.005 |
| CCL-9 (ng/ml) | 13.7 ± 4.7 | 19.5 ± 4.2 | ▴ 42% | p < 0.05 |
| CCL-19 (ng/ml) | 2.57 ± 0.48 | 3.48 ± 0.88 | ▴ 35% | p < 0.05 |
Plasma analytes that were significantly affected out of a total of 60 assessed.
Collectively, these data indicate that, when housed in an open facility and exposed to common mouse pathogens, AT-1S113R/+ mice display signs of ongoing inflammation and increased propensity to cancer. Importantly, no infections, abnormal innate immune response activation, or cancer propensity was observed in the AT-1S113R/+ mice housed in an SPF facility, indicating that an initial noxa (infection) was necessary to activate the immune response and cause this phenotype. Upon infection, AT-1S113R/+ mice are prone to an IL-17 neutrophil-dominated response that resulted in the pathologies observed.
AT-1S113R/+ mice develop defects of the PNS and CNS
At birth, AT-1S113R/+ mice appear normal and do not display any PNS or CNS phenotype. However, around the age of 10–12 months, they develop defective motor functions. Specifically, they display intermittent hind-leg clasping and abnormal body rotation, as assessed by the hind-leg clasping reflex (Fig. 6A–C), and reduced grip strength on the inverted cage lid (Fig. 6D). No histological defects of the muscles were observed. Finally, when evaluated with the hot-plate test, AT-1S113R/+ mice displayed increased latency to pain response, suggesting deficits in sensory processing (Fig. 6E). No significant differences were observed when the animals were evaluated with open-field or rotarod tests (data not shown), suggesting that they do not suffer from widespread motor function deficits. Lack of motor function defects, as assessed by the rotarod test, has also been reported in other related mouse models (Xia et al., 2003; Hadano et al., 2006; Kasher et al., 2009). Severe spasticity and/or paraplegia were visible in <5% of the animals.
Figure 6.
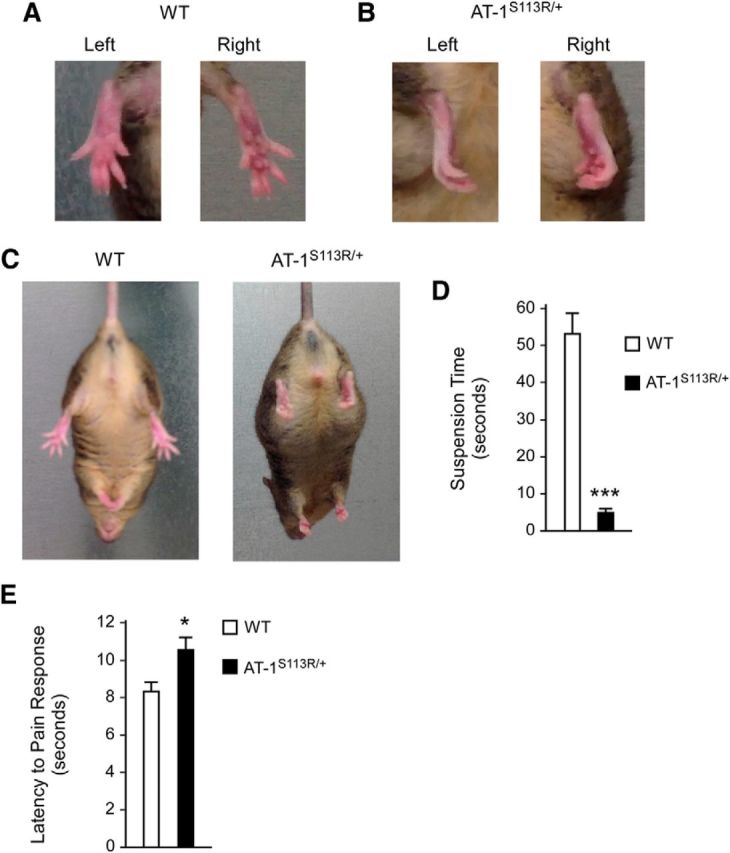
AT-1S113R/+ mice display defective hind-leg clasping, reduced grip strength, and reduced sensitivity to pain. A, Normal clasping in WT mice. B, Pathological clasping in AT-1S113R/+ mice. C, Pathological hind-leg clasping reflex in AT-1S113R/+ animals. D, AT-1S113R/+ mice have a reduced latency to fall on the inverted cage lid test. Values are the mean ± SD (n = 10). ***p < 0.0005. E, AT-1S113R/+ mice display reduced sensitivity to pain. Values are the mean ± SD (n = 5). *p < 0.05. Animals were 10–12 months old when studied.
Next, we analyzed both proximal and distal sections of the sciatic nerve and observed a significant loss of axonal fibers (Fig. 7A–D). Fibers were visualized by both toluidine staining and immunolabeling with an antibody targeting axonal neurofilament protein. Both strategies yielded similar results (Fig. 7A–D). Remaining fibers displayed several abnormalities that typically characterize progressing axonopathies (Fig. 7A,E). Specifically, EM showed features that are consistent with axonal degeneration and myelinopathy (Fig. 7E). Analysis of the optic nerve did not reveal significant morphological changes (data not shown), indicating that the mutation affects axonal fibers differently.
Figure 7.
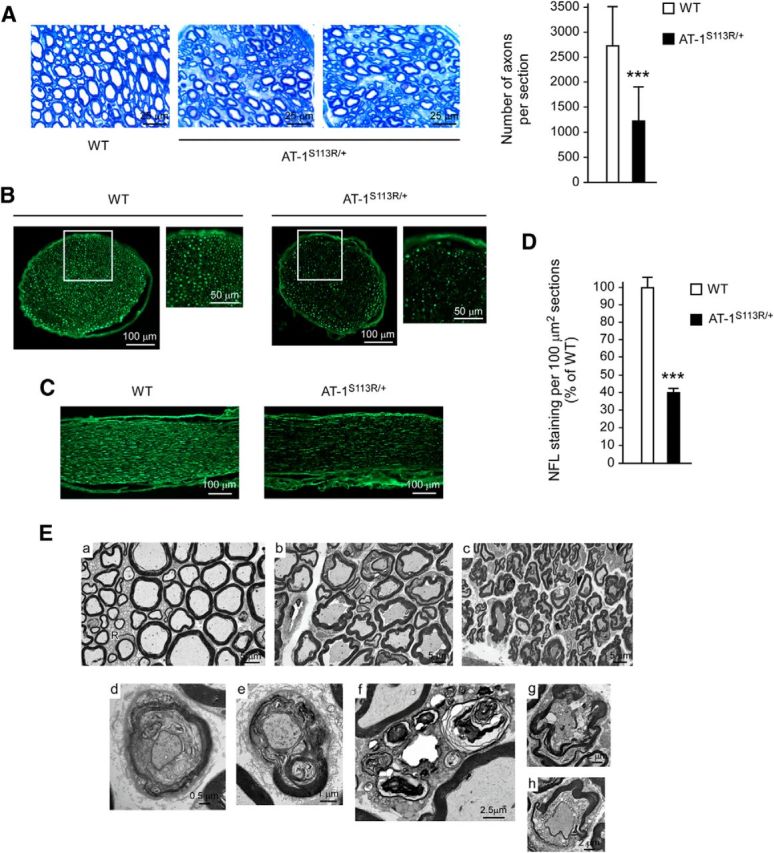
Loss of axonal fibers in the sciatic nerve of AT-1S113R/+ mice. A, Toluidine staining. Left, Representative images. Right, Computer-assisted quantitation (n = 3 animals; 7 nerve sections per animal). Computer-assisted quantitation was done with entire/intact serial sections. Error bars indicate SD. ***p < 0.0005. B, C, Immunolabeling with anti-neurofilament (NFL) antibody. Both transverse (B) and longitudinal (C) sections are shown. In B, high-magnification of indicated areas is also shown. D, Computer-assisted quantitation of NFL staining per 100 μm2 section-areas (n = 3 animals; 5 nerve sections per animal). Error bars indicate SD. ***p < 0.0005. E, Electron microscopy of sciatic nerve. a, WT; b and c, AT-1S113R/+ (two different animals are shown). AT-1S113R/+ mice show several features of myelin and axonal degeneration. Selected degenerating features are shown in d–h. They include “onion bulbs” with degenerating and regenerating myelin, myelin outfolds, and axonal degeneration. R, Remak bundle. Animals were 10–12 months old when studied.
To further characterize the disease phenotype, we also analyzed different regions of the spinal cord. Immunolabeling with the neuronal marker NeuN and Nissl staining showed a reduced number of neurons (Fig. 8A–C). The neuronal loss appeared to affect both dorsal and ventral horns, but was more evident in lumbar sections of the spinal cord. Immunolabeling with GFAP for the visualization of astrocytes and with Iba1 for the visualization of microglia revealed the presence of reactive astrocytes (Fig. 8D) and activated microglia (Fig. 8E) in AT-1S113R/+ mice. Both findings underline the ongoing neurodegenerative disease affecting the spinal cord. Finally, EM assessment of the white matter revealed several features of axonal degeneration and myelinopathy (Fig. 8F).
Figure 8.
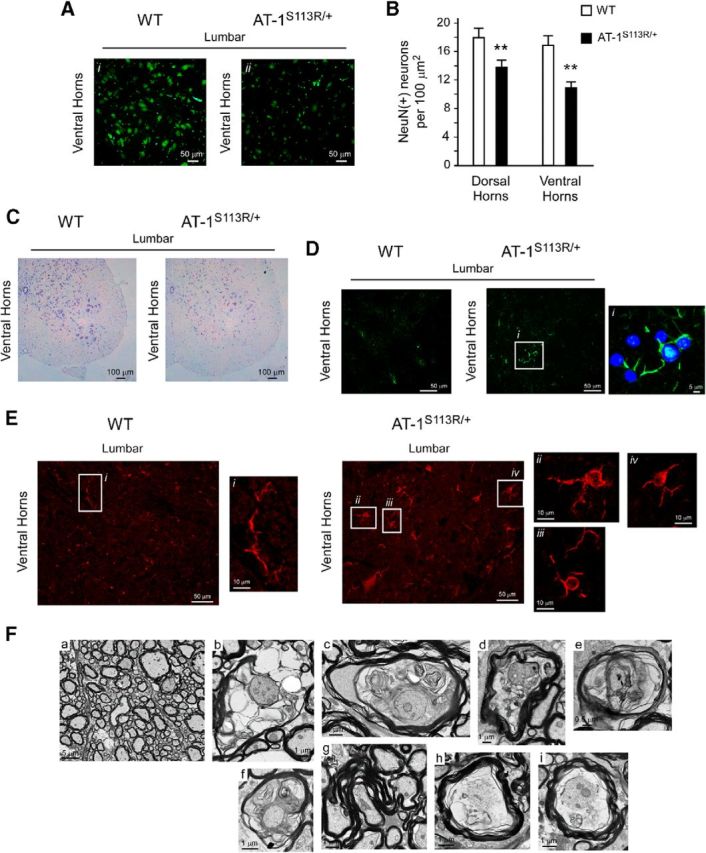
Neuronal loss and microglia activation in the spinal cords of AT-1S113R/+ mice. A, NeuN staining in the ventral horn of the spinal cord showing neuronal loss in AT-1S113R/+ mice. Representative images of the ventral horns of the lumbar section are shown. B, Computer-assisted quantification of NeuN(+) neurons per 100 μm2 section areas (n = 3 animals; 7 sections per animal). Error bars indicate SD. **p < 0.005. C, Nissl staining showing neuronal loss in AT-1S113R/+ mice. D, GFAP staining showing reactive astrocytes in AT-1S113R/+ mice. E, Iba1 staining showing microglia activation in AT-1S113R/+ mice. High-magnification insets show typical features of resting microglia in WT (left) and activated microglia in AT-1S113R/+ (right) mice. F, Electron microscopy showing myelin and axonal degeneration in the white matter of AT-1S113R/+ mice. Representative images of the lateral columns (lumbar section) are shown. a, WT; b–i, AT-1S113R/+. Selected degenerating features are shown in b–i, including vacuolar myelinopathy, myelin outfolds, degenerating and regenerating myelin, and axonal degeneration. Animals were 10–12 months old when studied.
To determine whether the brain was also affected by the mutation, we initially used DTI, a magnetic resonance technique that is sensitive to the structural organization of white matter (Basser and Pierpaoli, 1996). In the present study, DTI was used to measure white matter anisotropy, specifically FA, an index of fiber coherence (Beaulieu, 2002). The analysis revealed that AT-1S113R/+ mice display lower FA of the corpus callosum compared with WT littermates (Fig. 9A,B). Lower FA reflects abnormalities in the underlying cellular microstructure. A similar conclusion (albeit more evident) was reached with Kluver–Barrera (luxol fast blue-cresyl violet) staining of consecutive coronal sections (bregma 1–7) of the brain (Fig. 9C).
Figure 9.
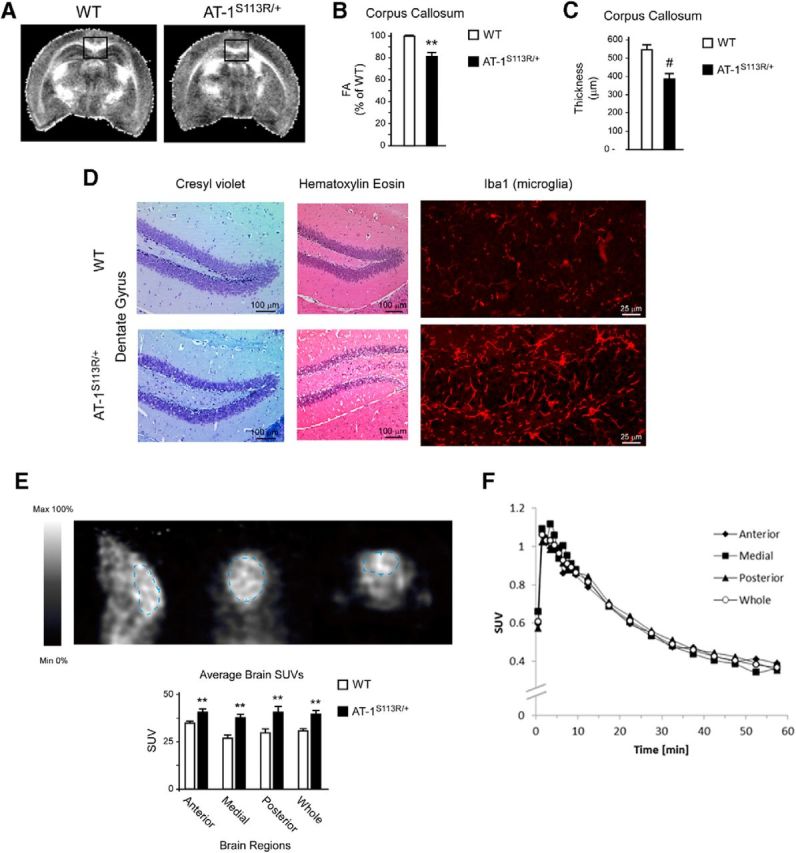
Loss of association fibers, gliosis, and microglia activation in the brains of AT-1S113R/+ mice. A, B, MRI imaging showing reduced FA of the corpus callosum in AT-1S113R/+ mice. Representative images are shown in A and FA changes are shown in B. Results are expressed as percentage of WT ± SD (n = 3 animals; 14 contiguous sections per animal). **p < 0.005. C, Thickness of the corpus callosum determined after Kluver–Barrera staining of consecutive coronal sections. Results are the average ± SD (n = 5 animals; 7 contiguous sections per animal). #p < 0.0005. D, Cresyl violet, H&E, and Iba1 staining showing gliosis and microglia activation in the dentate gyrus of AT-1S113R/+ mice. E, [11C]PBR28 PET imaging showing widespread microglia activation and inflammation of the brain in AT-1S113R/+ mice. Values are the mean ± SD (n = 4). **p < 0.005. F, No overall difference can be seen in the clearance of [11C]PBR28 from different brain regions. Animals were 10–12 months old when studied.
Histological assessment of the brain with H&E or Kluver–Barrera staining revealed possible gliosis in both the cortex and the hippocampal formation (Fig. 9D; only the dentate gyrus is shown). The gliosis was confirmed by the presence of activated microglia in the same areas of the brain (Fig. 9D; only the dentate gyrus is shown). Microglial cells respond to neuronal damage; therefore, the presence of activated microglia in the brain normally reflects the inflammatory response to the damage (Kettenmann et al., 2011). To confirm the microglia activation in vivo, we used PET imaging with [11C]PBR28, which is congenial for global and unbiased analysis of the inflammatory state of the brain (Venneti et al., 2006, 2013). The analysis revealed a significant retention of [11C]PBR28 in AT-1S113R/+ mice (Fig. 9E). The different brain areas behaved very similarly (Fig. 9E,F), indicating that the microglia activation and consequent inflammation was widespread. No histological defects were observed in the cerebellum (data not shown).
Collectively, the results indicate that AT-1S113R/+ mice suffer from a neurodegenerative disease that affects the PNS and the CNS. Importantly, these defects were observed with mice housed in both SPF and open facilities, indicating that the neuronal phenotype is independent of the presence of pathogens in the environment.
AT-1S113R/+ mice display abnormal induction of autophagy
We reported previously that downregulation of AT-1 in cultured cells leads to aberrant induction of the autophagy machinery and consequent autophagic cell death downstream of the inositol-requiring protein-1 (IRE1)/X-box-binding-1 protein (XBP1) branch of the UPR (Jonas et al., 2010; Pehar et al., 2012a). To determine whether AT-1S113R/+ suffered from excessive/aberrant autophagy, we initially used EM. AT-1S113R/+ neurons displayed large autophagosomes and autolysosomes, together with features that are consistent with excessive or aberrant induction of autophagy (Fig. 10A–C). These features were observed in the spinal cord and brain and in isolated neurons. Next, we determined whether neurons in vivo were positive for microtubule-associated light chain 3B (LC3β) puncta, which are normally used as an indication of ongoing autophagy (Mizushima et al., 2010). Specifically, we studied the spinal cord, where a higher level of neuronal loss was observed (Fig. 8). The analysis revealed the presence of LC3β puncta-positive neurons in AT-1S113R/+, but not in WT mice (Fig. 10D). Finally, we generated neuronal cultures from WT and AT-1S113R/+ mice and assessed the induction of autophagy after transduction with a GFP-tagged version of LC3β. As expected, WT neurons displayed a diffuse cytoplasmic distribution of GFP-LC3β, whereas AT-1S113R/+ neurons displayed clear puncta corresponding to the autophagosomes (Fig. 10E). At this stage, the gross morphology of the neurons did not appear to be altered.
Figure 10.

AT-1S113R/+ mice display abnormal activation of autophagy-neuronal studies. A, EM of spinal cord neurons shows alterations that are consistent with abnormal autophagy. Asterisks indicate vacuolar material resembling lipofuscin; arrowhead indicates a large structure resembling a complex autophagosome. N, Nucleus. B, C, Similar EM alterations were observed in the cortex (B) and in culture (C). D, LC3β-positive neurons can be observed in the spinal cord of AT-1S113R/+ mice. Arrowheads in the NeuN staining indicate two different neurons: one displays LC3β puncta and one does not. No LC3β-positive neurons were observed in WT mice. E, LC3β puncta in AT-1S113R/+ cultured neurons.
To further assess whether AT-1S113R/+ mice suffered from excessive/aberrant autophagy, we generated MEFs. Consistent with the results displayed in Figure 10, we detected enlarged ER and aberrant autophagic structures by EM (Fig. 11A). These findings were supported by the increased levels of beclin and LC3βII in the mutant mice (Fig. 11B,F, lanes 1–2). Both beclin and LC3βII are commonly used markers of autophagy. The induction of autophagy can be accompanied by reduced levels of p62, an autophagosome-associated protein that is degraded as a result of the autophagic process itself (Mizushima et al., 2010). As expected, we detected reduced levels of p62 in AT-1S113R/+ MEFs (Fig. 11C), thus supporting the results obtained with beclin and LC3β. Finally, we also observed an increased number of cells containing LC3β-GFP puncta among AT-1S113R/+ MEFs compared with WT MEFs (Fig. 11E). Therefore, different experimental strategies in tissue, isolated neurons, and cultured MEFs support the conclusion that AT-1S113R/+ mice suffer from excessive and/or aberrant induction of autophagy. AT-1 is an ER-resident protein; therefore, it is likely that the deficient influx of acetyl-CoA affects early rather than late stages of the autophagic process. However, to confirm this, we also monitored the autophagic flux after BPE treatment to impair lysosomal acidification. As expected, accumulation of LC3β was observed in both WT and AT-1S113R/+ MEFs; however, the changes were more evident in cells from AT-1S113R/+ animals (Fig. 11D). These results rule out a block in the later stages of the autophagic process and support the conclusion that AT-1S113R/+ cells suffer from excessive/aberrant induction of autophagy.
Figure 11.

AT-1S113R/+ mice display abnormal activation of autophagy-MEF studies. A, EM of MEFs. a, MEFs from WT animals. b–f, MEFs from AT-1S113R/+ mice. Different features that are consistent with abnormal activation of autophagy are evident in AT-1S113R/+ MEF cells. A large autolysosome is evident in d, whereas cells undergoing autophagic (type II) cell death are shown in e and f. B, Immunoblots showing increased levels of autophagy markers beclin-1 and LC3β. Error bars indicate SD of n = 3 MEF lines. *p < 0.05. C, Immunoblots showing decreased levels of the autophagy “cargo” protein p62. Error bars indicate SD of n = 3 MEF lines. *p < 0.05. D, Immunoblots showing the autophagy flux in WT and AT-1S113R/+ MEFs. BPE, bafilomycin (500 nm), pepstatin A (10 μg/ml), and E64 (10 μg/ml). E, LC3β-GFP activation and redistribution in AT-1S113R/+ MEF cells. The increased induction of autophagy observed in AT-1S113R/+ MEFs is rescued by expressing the dominant Atg9AGln mutant. Error bars indicate SD of n = 3 MEF lines. ***p < 0.0005. F, Immunoblots showing rescue of the autophagy phenotype after expression of Atg9AGln mutant. Lane 1, WT; lane 2, AT-1S113R/+; and lane 3, AT-1S113R/+ + Atg9AGln. G, Colocalization of Atg9A and LC3β in AT-1S113R/+ MEFs.
We showed previously that the influx of acetyl-CoA into the ER lumen controls the induction of autophagy by regulating the acetylation status of Atg9A, the only membrane-bound autophagy protein (Pehar et al., 2012a). Indeed, a mutant form of Atg9A that resembles a “gain-of-acetylation” version (Atg9AGln) of the protein was able to prevent the induction of autophagy and the autophagic cell death after XBP1 and AT-1 downregulation (Pehar et al., 2012a). In the Atg9A mutant, both lysine residues that normally undergo acetylation (Lys359 and Lys363) were mutated into glutamine, which removes a positive charge and mimics a bona fide form of lysine acetylation (Masumoto et al., 2005; Costantini et al., 2007). To determine whether we could rescue the excessive/aberrant autophagy observed in AT-1S113R/+ MEF cells, we made use of the dominant mutant. Expression of Atg9AGln was able to rescue the induction of autophagy as assessed by direct quantification of LC3β-GFP puncta (Fig. 11E) and protein levels of beclin and LC3βII (Fig. 11F). Atg9A is known to translocate out of the ER and Golgi apparatus and associate with LC3β during the progression of autophagy (Young et al., 2006; Mizushima et al., 2010; Ohashi and Munro, 2010). To determine whether AT-1S113R/+ MEFs displayed increased colocalization of Atg9A and LC3β, we transfected WT and AT-1S113R/+ MEFs with a GFP-tagged version of Atg9A. The results show a marked colocalization with endogenous LC3β, which was not observed in WT cells (Fig. 11G).
In conclusion, at the cellular level, AT-1 haploinsufficiency results in decreased influx of acetyl-CoA into the ER lumen, reduced Nε-lysine acetylation of ER-resident and ER-transiting proteins, and aberrant autophagy.
Discussion
The ER-based Nε-lysine acetylation machinery was discovered while studying the posttranslational regulation of a type I membrane protein (Costantini et al., 2007). Subsequent studies showed that this modification affects both ER-transiting and ER-resident proteins (Choudhary et al., 2009; Pehar et al., 2012b). The intraluminal acetylation of ER proteins requires one membrane transporter and at least two acetyltransferases (Pehar and Puglielli, 2013). The transporter AT-1 ensures continuous supply of acetyl-CoA, the donor of the acetyl group for the reaction of acetylation.
Here, we show that a mutant version of AT-1 (AT-1S113R) associated with a familial form of spastic paraplegia is unable to make homodimers in the plane of the ER membrane and is devoid of acetyl-CoA transport activity. Animals homozygous for the mutation display early developmental arrest. In contrast, heterozygous animals develop defects of both the immune and nervous systems during adulthood. The defects of the immune system result in increased propensity to infections, aberrant inflammatory response, and increased propensity to malignancies. The defects of the nervous system result into degenerative features of the PNS and CNS. Overall, the nervous system phenotype of AT-1S113R/+ mice recapitulates the clinical features of hereditary spastic paraplegias (Salinas et al., 2008) and confirms the disease association of the S113R mutation. The phenotype also indicates that the ER-based acetylation machinery plays an important role in human physiology and pathology.
The molecular basis of the nervous system phenotype involves an excessive and/or aberrant induction of autophagy leading to progressive neurodegeneration. Autophagy/ERAD(II) is essential for the degradation of large protein aggregates that accumulate in the ER. When “hypoactive,” cells accumulate protein aggregates in the ER lumen, whereas when they are “hyperactive,” they undergo autophagic cell death (Klionsky, 2006; Bergmann, 2007). Therefore, the autophagic process must be constantly maintained under strict control to ensure the elimination of toxic protein aggregates and to prevent excessive autophagy (Kroemer et al., 2010). AT-1 regulates the induction of autophagy/ERAD(II) downstream of XBP1 by ensuring constant influx of acetyl-CoA into the ER lumen (Pehar et al., 2012a). The intraluminal levels of acetyl-CoA appear to be “sensed” by Atg9A: nonacetylated Atg9A stimulates, whereas acetylated Atg9A reduces the induction of autophagy at the ER (Pehar et al., 2012a). Therefore, reduced influx of acetyl-CoA into the ER lumen would alter the above equilibrium (Fig. 12). The conclusion that the nervous system phenotype is linked to an aberrant form of autophagy is also supported by the fact that altered induction and/or progression of autophagy is invariably associated with neurodegeneration (Mizushima et al., 2008; Buchberger et al., 2010).
Figure 12.

Schematic interpretation of our results. WT AT-1 (AT-1WT) is a homodimer in the ER membrane, where it ensures constant influx of acetyl-CoA into the lumen of the organelle. SPG-associated AT-1 (AT-1S113R) is unable to form homodimers and, as a result, is devoid of transport activity. Animals heterozygous for the mutation have reduced influx of acetyl-CoA into the ER lumen, which results in deacetylation (or nonacetylation) of Atg9A and aberrant induction of autophagy. The aberrant levels of autophagy lead to propensity to infections, diffuse inflammation, propensity to cancer, and neurodegeneration of both the PNS and CNS. Animals homozygous for the mutation suffer from early developmental arrest.
Although the molecular basis of the nervous system phenotype seems to be logically connected to the aberrant autophagy, the increased propensity to infections, inflammation and cancer remains to be fully understood. We initially thought that AT-1S113R/+ mice had a defect in the maturation of B cells. In fact, the differentiation of B cells into Ig-secreting plasma cells is accompanied by upregulation of AT-1 downstream of XBP1 (Wiest et al., 1990; Shaffer et al., 2004). However, the development of B cells did not appear to be perturbed in the animals. Although we recognize that this does not fully rule out an intrinsic defect of plasma cells, it is likely that a more generalized defect of the immune system is responsible for the phenotype. It is worth stressing that autophagy, specifically its induction at the ER level, regulates antigen presentation and influences major histocompatibility complex-dependent events (Gagnon et al., 2002; Menéndez-Benito and Neefjes, 2007; Schmid et al., 2007). This process is crucial when targeting self-antigens and pathogens and has important implications for autoimmunity, tumor immunity, and intracellular pathogen immunity (Huang and Klionsky, 2007; Crotzer and Blum, 2009). Prolonged activation of autophagy appears to present intracellular proteins preferentially (Muntasell et al., 2002; Dengjel et al., 2005), thus altering the fidelity of the antigen-presentation process and leading to autoimmunity and aberrant inflammatory response (Lleo et al., 2007; Crotzer and Blum, 2009). This, in turn, is likely to be the immediate cause of the increased propensity to malignancies as well (Aggarwal et al., 2006). Indeed, compelling evidence supports a causative link between ongoing inflammation and several steps involved with tumorigenesis (Aggarwal et al., 2006). In many instances, a paracrine or autocrine effect of interleukins, such as IL-17, which was abnormally elevated in the AT-1S113R/+ mice, has also been revealed (Ogura et al., 2008; Chae et al., 2010; Zou and Restifo, 2010; Wilke et al., 2011). Particularly, the paracrine effects of IL-17 appear to stimulate spontaneous tumorigenesis in the mouse (Chae et al., 2010). Tumorigenic properties have also been reported for several CC- and CXC-motif chemokines (Balkwill, 2004; Aggarwal et al., 2006).
In conclusion, our studies have discovered a novel aspect of acetyl-CoA metabolism that affects the nervous system, the immune system, and the propensity for cancer. It is worth stressing that a large number of genes found mutated in hereditary SPGs encode proteins that reside in the ER or ERGIC, regulate ER morphology/organization, or control post-ER membrane trafficking (for review, see Pehar and Puglielli, 2013). Finally, defects in the CoA biosynthetic pathway have been associated with different forms of neurodegenerative diseases both in humans and model animals (for review, see Pehar and Puglielli, 2013). Further studies will now be necessary to determine whether changes in AT-1 activity in the general population are associated with increased propensity to neurological diseases, infections, and/or cancer.
Footnotes
This work was supported by the National Institutes of Health (L.P. and M.S.), the Department of Veterans Affairs (L.P.), and the Wisconsin Alzheimer's Disease Research Center (B.T.C.). We thank Carlos Hirschberg and Nansi Jo Colley for critical reading of an early version of this manuscript; Patricia Powers and the UW-Madison Transgenic Animal Facility for the generation of AT-1S113R/+ mice; Ruth Sullivan and Shahriar Salamat for help with the pathology; Joan M. Sempf and the VAH-EM Facility for morphology; Beth Meyerand, Beth Rauch, and Guan Koay for help with MRI; and Mohammed Farhoud and Sofie Biosciences for use of PET scanner.
The authors declare no competing financial interests.
References
- Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol. 2006;72:1605–1621. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- Beaulieu C. The basis of anisotropic water diffusion in the nervous system-a technical review. NMR Biomed. 2002;15:435–455. doi: 10.1002/nbm.782. [DOI] [PubMed] [Google Scholar]
- Bergmann A. Autophagy and cell death: no longer at odds. Cell. 2007;131:1032–1034. doi: 10.1016/j.cell.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Carey DJ, Hirschberg CB. Topography of sialoglycoproteins and sialyltransferases in mouse and rat liver Golgi. J Biol Chem. 1981;256:989–993. [PubMed] [Google Scholar]
- Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:5540–5544. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Costantini C, Weindruch R, Della Valle G, Puglielli L. A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem J. 2005;391:59–67. doi: 10.1042/BJ20050700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA to p75(NTR) receptor switch and amyloid beta-peptide generation. EMBO J. 2006;25:1997–2006. doi: 10.1038/sj.emboj.7601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 2007;407:383–395. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol. 2009;182:3335–3341. doi: 10.4049/jimmunol.0803458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, Brock R, Driessen C, Rammensee HG, Stevanovic S. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102:7922–7927. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, Paiement J, Bergeron JJ, Desjardins M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110:119–131. doi: 10.1016/S0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- Hadano S, Benn SC, Kakuta S, Otomo A, Sudo K, Kunita R, Suzuki-Utsunomiya K, Mizumura H, Shefner JM, Cox GA, Iwakura Y, Brown RH, Jr, Ikeda JE. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum Mol Genet. 2006;15:233–250. doi: 10.1093/hmg/ddi440. [DOI] [PubMed] [Google Scholar]
- Hirschberg CB, Robbins PW, Abeijon C. Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu Rev Biochem. 1998;67:49–69. doi: 10.1146/annurev.biochem.67.1.49. [DOI] [PubMed] [Google Scholar]
- Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837–1849. doi: 10.4161/cc.6.15.4511. [DOI] [PubMed] [Google Scholar]
- Huppke P, Brendel C, Kalscheuer V, Korenke GC, Marquardt I, Freisinger P, Christodoulou J, Hillebrand M, Pitelet G, Wilson C, Gruber-Sedlmayr U, Ullmann R, Haas S, Elpeleg O, Nürnberg G, Nürnberg P, Dad S, Møller LB, Kaler SG, Gärtner J. Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am J Hum Genet. 2012;90:61–68. doi: 10.1016/j.ajhg.2011.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 2008;9:916–922. doi: 10.1038/embor.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas MC, Pehar M, Puglielli L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J Cell Sci. 2010;123:3378–3388. doi: 10.1242/jcs.068841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori A, Nakayama J, Fukuda MN, Stallcup WB, Sasaki K, Fukuda M, Hirabayashi Y. Expression cloning and characterization of a cDNA encoding a novel membrane protein required for the formation of O-acetylated ganglioside: a putative acetyl-CoA transporter. Proc Natl Acad Sci U S A. 1997;94:2897–2902. doi: 10.1073/pnas.94.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasher PR, De Vos KJ, Wharton SB, Manser C, Bennett EJ, Bingley M, Wood JD, Milner R, McDermott CJ, Miller CC, Shaw PJ, Grierson AJ. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J Neurochem. 2009;110:34–44. doi: 10.1111/j.1471-4159.2009.06104.x. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ. Neurodegeneration: good riddance to bad rubbish. Nature. 2006;441:819–820. doi: 10.1038/441819a. [DOI] [PubMed] [Google Scholar]
- Ko MH, Puglielli L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J Biol Chem. 2009;284:2482–2492. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koay CG, Chang LC, Carew JD, Pierpaoli C, Basser PJ. A unifying theoretical and algorithmic framework for least squares methods of estimation in diffusion tensor imaging. J Magn Reson. 2006;182:115–125. doi: 10.1016/j.jmr.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch JP, Gazdzinski L, Germann J, Sled JG, Henkelman RM, Nieman BJ. Wanted dead or alive? The tradeoff between in-vivo versus ex-vivo MR brain imaging in the mouse. Front Neuroinform. 2012;6:6. doi: 10.3389/fninf.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Li J, Liu Q, Mao F, Li J, Qiu R, Hu H, Song Y, Yang Y, Gao G, Yan C, Yang W, Shao C, Gong Y. A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42) Am J Hum Genet. 2008;83:752–759. doi: 10.1016/j.ajhg.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleo A, Invernizzi P, Selmi C, Coppel RL, Alpini G, Podda M, Mackay IR, Gershwin ME. Autophagy: highlighting a novel player in the autoimmunity scenario. J Autoimmun. 2007;29:61–68. doi: 10.1016/j.jaut.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CH, Omura T, Cobos EJ, Latrémolière A, Ghasemlou N, Brenner GJ, van Veen E, Barrett L, Sawada T, Gao F, Coppola G, Gertler F, Costigan M, Geschwind D, Woolf CJ. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest. 2011;121:4332–4347. doi: 10.1172/JCI58675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandon EC, Milla ME, Kempner E, Hirschberg CB. Purification of the Golgi adenosine 3′-phosphate 5′-phosphosulfate transporter, a homodimer within the membrane. Proc Natl Acad Sci U S A. 1994;91:10707–10711. doi: 10.1073/pnas.91.22.10707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- Menéndez-Benito V, Neefjes J. Autophagy in MHC class II presentation: sampling from within. Immunity. 2007;26:1–3. doi: 10.1016/j.immuni.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntasell A, Carrascal M, Serradell L, Veelen Pv Pv, Verreck F, Koning F, Raposo G, Abián J, Jaraquemada D. HLA-DR4 molecules in neuroendocrine epithelial cells associate to a heterogeneous repertoire of cytoplasmic and surface self peptides. J Immunol. 2002;169:5052–5060. doi: 10.4049/jimmunol.169.9.5052. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberley TD, Swanlund JM, Zhang HJ, Kregel KC. Aging results in increased autophagy of mitochondria and protein nitration in rat hepatocytes following heat stress. J Histochem Cytochem. 2008;56:615–627. doi: 10.1369/jhc.2008.950873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Ohashi Y, Munro S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol Biol Cell. 2010;21:3998–4008. doi: 10.1091/mbc.E10-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates, third edition. New York: Academic; 2007. [Google Scholar]
- Pehar M, Puglielli L. Lysine acetylation in the lumen of the ER: A novel and essential function under the control of the UPR. Biochim Biophys Acta. 2013;1833:686–697. doi: 10.1016/j.bbamcr.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, O'Riordan KJ, Burns-Cusato M, Andrzejewski ME, del Alcazar CG, Burger C, Scrable H, Puglielli L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell. 2010;9:174–190. doi: 10.1111/j.1474-9726.2010.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, Jonas MC, Hare TM, Puglielli L. SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J Biol Chem. 2012a;287:29921–29930. doi: 10.1074/jbc.M112.363911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, Lehnus M, Karst A, Puglielli L. Proteomic assessment shows that many endoplasmic reticulum (ER)-resident proteins are targeted by N{epsilon}-lysine acetylation in the lumen of the organelle and predicts broad biological impact. J Biol Chem. 2012b;287:22436–22440. doi: 10.1074/jbc.C112.362871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli L, Mandon EC, Hirschberg CB. Identification, purification, and characterization of the rat liver golgi membrane ATP transporter. J Biol Chem. 1999a;274:12665–12669. doi: 10.1074/jbc.274.18.12665. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Mandon EC, Rancour DM, Menon AK, Hirschberg CB. Identification and purification of the rat liver Golgi membrane UDP-N-acetylgalactosamine transporter. J Biol Chem. 1999b;274:4474–4479. doi: 10.1074/jbc.274.7.4474. [DOI] [PubMed] [Google Scholar]
- Rorden C, Karnath HO, Bonilha L. Improving lesion-symptom mapping. J Cogn Neurosci. 2007;19:1081–1088. doi: 10.1162/jocn.2007.19.7.1081. [DOI] [PubMed] [Google Scholar]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23:S208–S219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Sun SW, Liang HF, Xie M, Oyoyo U, Lee A. Fixation, not death, reduces sensitivity of DTI in detecting optic nerve damage. Neuroimage. 2009;44:611–619. doi: 10.1016/j.neuroimage.2008.10.032. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (translocator protein 18kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006;80:308–322. doi: 10.1016/j.pneurobio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wiley CA. Molecular imaging of microglia/macrophages in the brain. Glia. 2013;61:10–23. doi: 10.1002/glia.22357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yoder KK, Gao M, Mock BH, Xu XM, Saykin AJ, Hutchins GD, Zheng QH. Fully automated synthesis and initial PET evaluation of [11C]PBR28. Bioorg Med Chem Lett. 2009;19:5636–5639. doi: 10.1016/j.bmcl.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest DL, Burkhardt JK, Hester S, Hortsch M, Meyer DI, Argon Y. Membrane biogenesis during B cell differentiation: most endoplasmic reticulum proteins are expressed coordinately. J Cell Biol. 1990;110:1501–1511. doi: 10.1083/jcb.110.5.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke CM, Kryczek I, Wei S, Zhao E, Wu K, Wang G, Zou W. Th17 cells in cancer: help or hindrance? Carcinogenesis. 2011;32:643–649. doi: 10.1093/carcin/bgr019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LS. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003;161:55–66. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HZ, Nalbandian A, Hsu CI, Li S, Llewellyn KJ, Mozaffar T, Kimonis VE, Weiss JH. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 2012;3:e374. doi: 10.1038/cddis.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AR, Chan EY, Hu XW, Köchl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–256. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zozulya AL, Ortler S, Lee J, Weidenfeller C, Sandor M, Wiendl H, Fabry Z. Intracerebral dendritic cells critically modulate encephalitogenic versus regulatory immune responses in the CNS. J Neurosci. 2009;29:140–152. doi: 10.1523/JNEUROSCI.2199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]