

Abstract
Melanin-concentrating hormone (MCH) is a neuropeptide produced in neurons sparsely distributed in the lateral hypothalamic area. Recent studies have reported that MCH neurons are active during rapid eye movement (REM) sleep, but their physiological role in the regulation of sleep/wakefulness is not fully understood. To determine the physiological role of MCH neurons, newly developed transgenic mouse strains that enable manipulation of the activity and fate of MCH neurons in vivo were generated using the recently developed knockin-mediated enhanced gene expression by improved tetracycline-controlled gene induction system. The activity of these cells was controlled by optogenetics by expressing channelrhodopsin2 (E123T/T159C) or archaerhodopsin-T in MCH neurons. Acute optogenetic activation of MCH neurons at 10 Hz induced transitions from non-REM (NREM) to REM sleep and increased REM sleep time in conjunction with decreased NREM sleep. Activation of MCH neurons while mice were in NREM sleep induced REM sleep, but activation during wakefulness was ineffective. Acute optogenetic silencing of MCH neurons using archaerhodopsin-T had no effect on any vigilance states. Temporally controlled ablation of MCH neurons by cell-specific expression of diphtheria toxin A increased wakefulness and decreased NREM sleep duration without affecting REM sleep. Together, these results indicate that acute activation of MCH neurons is sufficient, but not necessary, to trigger the transition from NREM to REM sleep and that MCH neurons also play a role in the initiation and maintenance of NREM sleep.
Keywords: ablation, cell fate, channelrhodopsin2, hypothalamus, optogenetics, REM sleep
Introduction
Melanin-concentrating hormone (MCH)-producing neurons are coexpressed with orexin/hypocretin-producing neurons in the lateral hypothalamic area (LHA) but are more numerous and extend further rostrocaudally (Bittencourt et al., 1992; Peyron et al., 1998; Elias et al., 2008). MCH neurons project widely throughout the brain and densely innervate the cholinergic and monoaminergic arousal centers (Bittencourt et al., 1992). MCH neurons have been reported to be GABAergic (Del Cid-Pellitero and Jones, 2012). While MCH receptor-1 (MCHR1; the only receptor found in rodents) binding activates Gq, Gi, and Go subunits (Hawes et al., 2000), the major effect of MCHR1 binding is to decrease cAMP levels (Chambers et al., 1999; Lembo et al., 1999) and cellular electrophysiological studies have revealed presynaptic and postsynaptic inhibitory effects of MCH (Gao and van den Pol, 2001; Wu et al., 2009). Thus, MCH neurons exert neuroinhibitory effects on downstream targets.
MCH has been implicated in several functions, including feeding, energy balance, and locomotor activity (Shimada et al., 1998; Asakawa et al., 2002; Marsh et al., 2002; Semjonous et al., 2009); its involvement in sleep and wakefulness has also been extensively studied. Intracerebroventricular injections of MCH early in the dark period increased rapid eye movement (REM) sleep in a dose-dependent manner (Verret et al., 2003); some doses also increased non-REM (NREM) sleep. A recent study reported that optogenetic stimulation of MCH neurons increased both NREM and REM sleep (Konadhode et al., 2013). These results are consistent with the observation that intracerebroventricular injections of MCH increase both sleep states, but are at variance with results from functional neuroanatomical, pharmacological (Ahnaou et al., 2008), and optogenetic (Jego et al., 2013) studies that specifically indicate a role for MCH neurons in REM sleep regulation.
To this point, the precise role of MCH neurons in REM sleep regulation remains unclear and the involvement of MCH neurons in NREM sleep is uncertain (Jego and Adamantidis, 2013; Jones and Hassani, 2013; Luppi et al., 2013b; McGinty and Alam, 2013; Pelluru et al., 2013). One potential source of the discrepant results from the optogenetic studies could be the number of MCH neurons transfected with the light-sensitive protein and subsequently stimulated. Therefore, we generated new transgenic mice (Berndt et al., 2011; Han et al., 2011; Tanaka et al., 2012) to control MCH neurons and bred this strain with three other transgenic lines to create novel bigenic strains that enabled cell-specific manipulation of MCH neurons: an E123T/T159C double mutant of channelrhodopsin2 (ChR2) for stimulation (Berndt et al., 2011), archaerhodopsinTP009 (ArchT) for inhibition (Han et al., 2011), and diphtheria toxin A (DTA) for ablation. We find that optogenetic stimulation of MCH neurons induced REM sleep with a short latency and that continuous stimulation resulted in a net increase of REM sleep in conjunction with a decrease of NREM sleep. Although optogenetic inhibition of MCH neurons had no effect on sleep/wakefulness, conditional ablation of MCH neurons resulted in hyposomnia. These results demonstrate a critical role for MCH neurons in REM sleep regulation under acute stimulation and provide unequivocal evidence for this LHA neuronal population in sleep/wake control.
Materials and Methods
Animal usage.
All experimental procedures involving animals were approved by the animal care and use committees at Nagoya University, Hokkaido University, and SRI International and were in accordance with National Institutes of Health guidelines. All efforts were made to minimize animal suffering or discomfort and to reduce the number of animals used.
Generation of MCH-tetracycline-controlled transactivator bacterial artificial chromosome transgenic mice.
The codons of bacterial tetracycline repressor protein and viral VP16 activator domain [tetracycline-controlled transactivator (tTA)] were fully mammalianized (Inamura et al., 2012). Mouse bacterial artificial chromosome (BAC) DNA (clone RP23-348H6) was initially modified by inserting an Rpsl-Zeo cassette (a gift from Dr. Hisashi Mori, Toyama University) into the translation initiation site of the MCH gene followed by the replacement with a cassette containing tTA and the SV40 polyadenylation signal. BAC DNA was linearized by PI-SceI enzyme digestion (New England Biolabs) and injected into fertilized eggs from BDF1 mice. Two founders were obtained and line B was used in this report.
Generation of tetracycline operator ChR2 (E123T/T159C) knock-in mice.
To achieve sufficient opsin expression (ChR2) in MCH neurons to drive photocurrents, we used a knockin-mediated enhanced gene expression by improved tetracycline-controlled gene induction (KENGE-tet) strategy (Tanaka et al., 2012). We used the same housekeeping gene, actb, but the position of gene targeting was 800 bp downstream of the polyA signal, which was different from our initial report (Tanaka et al., 2012).
We constructed a plasmid containing the tetracycline operator (TetO) ChR2 polyA cassette with the Neo selection gene flanked on both sides by flippase recombinase target (FRT) sites in which the following elements were connected in tandem: TetO sequence, rabbit β-globin intron, ChR2(E123T/T159C) enhanced yellow fluorescent protein (EYFP) cDNA, SV40 polyadenylation signal, and FRT-flanked phosphoglycerate kinase-EM7-Neo. To insert the above cassette downstream of the mouse β-actin gene, a 265 bp 5′ homology arm consisting of the sequence from 535 to 799 bp downstream of the mouse β-actin gene polyadenylation signal (AATAAA) was connected to the 5′ end of the TetO sequence, and a 265 bp 3′ homology arm consisting of the sequence from 800 to 1064 bp downstream of the AATAAA sequence was connected to the 3′ end of Neo. To perform BAC recombination, DNA fragments with both homology arms were electroporated into bacteria carrying the BAC (clone RP23-289L7) and the pBADTcTypeG plasmid (a gift from Dr. Manabu Nakayama, Kazusa Institute, Japan; Nakayama and Ohara, 2005). Kanamycin-resistant clones were selected as the modified BAC and the TetO ChR2 polyA cassette was subsequently inserted 800 bp downstream of mouse β-actin gene AATAAA. The targeting vector was isolated from the kanamycin-resistant, modified BAC clone using a retrieval technique involving insertion into the pMCS-DTA plasmid (a gift from Dr. Kosuke Yusa, Osaka University, Japan).
We subsequently obtained a targeting vector comprising a 11 kb 5′ homology arm, TetO ChR2 polyA cassette with Neo, a 1.5 kb 3′ homology arm, and a DTA subunit. EGR-G01 ES cell (established by F1 from 129S2 and C57BL-6-Tg(CAG/Acr-EGFP)) cells were used. We obtained 28 recombinant clones out of 96 G418-resistant clones. Recombination was confirmed by Southern blotting with a 424 bp 3-prime outside probe, which recognized an 8.1 kb fragment of the wild-type allele and a 6 kb fragment of the targeted allele in genomic DNA digested with XbaI. Germline-transmitted offspring were established as TetO ChR2-Neo knock-in mice. TetO ChR2-Neo mice were crossed with ROSA-Flp mice (Farley et al., 2000), and FRT-flanked Neo selection markers were removed. TetO ChR2 knock-in mice were subsequently generated.
Generation of bigenic MCH mouse strains.
To express ChR2, ArchT, or DTA in MCH neurons, MCH-tTA mice were bred with TetO ChR2 mice, TetO ArchT mice (Tsunematsu et al., 2013), or TetO DTA mice (B6.Cg-Tg(tetO-DTA)1Gfi/J, 008468, Jackson Laboratory), respectively.
Brain slice preparation.
Male and female MCH-tTA; TetO ChR2 or MCH-tTA; TetO ArchT bigenic mice (3–6 weeks old) were used for whole-cell recordings. The mice were deeply anesthetized with isoflurane (Abbott Japan) and decapitated. Brains were quickly isolated in ice-cold cutting solution consisting of (in mm) the following: 280 sucrose, 2 KCl, 10 HEPES, 0.5 CaCl2, 10 MgCl2, 10 glucose, pH 7.4 with NaOH, bubbled with 100% O2. Brains were cut coronally into 350 μm slices with a microtome (VTA-1200S, Leica). Slices containing the LHA were transferred to an incubation chamber shielded from light and filled with a physiological solution containing the following (in mm): 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4 with NaOH, bubbled with 100% O2. This was incubated for ≥1 h at room temperature (RT; 24–26°C).
In vitro electrophysiological recordings.
At RT, the slices were transferred to a recording chamber (RC-27L, Warner Instrument) on a fluorescence microscope stage (BX51WI, Olympus). Neurons having EGFP and EYFP fluorescence were identified as MCH neurons and were subjected to electrophysiological recordings. The fluorescence microscope was equipped with an infrared camera (C2741-79, Hamamatsu Photonics) for infrared differential interference contrast imaging and a cooled charge-coupled device (CCD) camera (Cascade 650, Roper Scientific) for fluorescent imaging. Each image was displayed separately on a monitor and saved on a computer. Recordings were performed with an Axopatch 200B amplifier (Molecular Devices) using a borosilicate pipette (GC150-10, Harvard Apparatus) prepared by a micropipette puller (P-97, Sutter Instruments), and filled with intracellular solution (4–6 MΩ) consisting of (in mm) the following: 138 K-gluconate, 10 HEPES, 8 NaCl, 0.2 EGTA-Na3, 2 MgATP, 0.5 Na2GTP, pH 7.3 with KOH. The osmolality of the solution was checked by a vapor pressure osmometer (model 5520, Wescor). The osmolality of the internal and external solutions was 280–290 and 320–330 mOsm/l, respectively. The liquid junction potential of the patch pipette and perfused extracellular solution was estimated to be 16 mV and was corrected in the data. Recording pipettes were under positive pressure while advancing toward individual cells in the slice, and tight seals on the order of 1.0–1.5 GΩ were made by negative pressure. The membrane patch was then ruptured by suction. The series resistance during recording was 10–25 MΩ. The reference electrode was an Ag-AgCl pellet immersed in bath solution. During recordings at RT, cells were superfused with extracellular solution at a rate of 1.6 ml/min using a peristaltic pump (Dynamax, Rainin). Blue (475 ± 17.5 nm, 2.5 mW) and green light (549 ± 7.5 nm, 0.9 mW) was generated by a SPECTRA light engine (Lumencor).
The output signal was low-pass filtered at 5 kHz and digitized at 10 kHz. Data were recorded on a computer through a Digidata 1322A analog-to-digital converter using pClamp software version 10.2 (Molecular Devices).
Drugs.
Tetrodotoxin (TTX; Wako) was dissolved in extracellular solution at a concentration of 1 μm. During experiments, TTX was applied by bath application.
Immunohistochemistry.
Male and female MCH-tTA; TetO ChR2 and MCH-tTA; TetO ArchT bigenic mice (6 weeks old) were deeply anesthetized with isoflurane and perfused sequentially with 20 ml of chilled saline and 20 ml of chilled 10% formalin solution (Wako). The brains were removed and immersed in the above fixative solution for 24 h at 4°C and then immersed in a 30% sucrose solution for ≥2 d. The brains were quickly frozen in embedding solution (Sakura Finetechnical). For MCH and GFP double staining, coronal sections (40 μm) of MCH-tTA; TetO ChR2 and MCH-tTA; TetO ArchT bigenic mice brains were incubated with mouse anti-GFP antiserum (1:1000; Wako) for 24 h at 4°C. These sections were incubated with Alexa 488-labeled donkey anti-mouse IgG (1:1000; Biotium) for 1 h at RT. The sections were then incubated with rabbit anti-MCH antiserum (1:2000; Sigma-Aldrich) for 24 h at 4°C and incubated with Alexa 594-labeled donkey anti-rabbit IgG (1:1000; Biotium) for 1 h at RT. To confirm the specificity of antibodies, incubations without primary antibody were conducted as a negative control in each experiment and, in each case, no signal was observed.
To determine the number of MCH and orexin neurons, coronal sections (40 μm) were stained by the avidin-biotin-peroxidase method. Brain sections were incubated for 40 min in phosphate buffer containing 0.3% H2O2 to inactivate endogenous peroxidase. Sections were transferred into PBS containing 0.25% Triton X-100 and 1% bovine serum albumin fraction V (PBS-BX) for 30 min and then incubated with rabbit anti-MCH antiserum (Sigma-Aldrich) diluted 1:4000 in PBS-BX, goat anti-orexin IgG antibody (Santa Cruz Biotechnology) diluted 1:2000 in PBS-BX overnight at 4°C. Sections were then incubated with biotin-labeled goat anti-rabbit IgG antibody (1:1000; Vector Laboratories) or biotin-labeled horse anti-goat IgG antibody (1:1000; Vector Laboratories) for 1 h at RT followed by incubation with avidin and biotinylated peroxidase complex solution for 1 h at RT. Bound peroxidase was visualized by DAB-buffer tablet (Merck) with 0.0015% H2O2, resulting in a golden-brown reaction product. Approximately 10 slices from a one-in-four series were selected for counting of MCH and orexin neurons. Sections were mounted and examined with a fluorescence microscope (BZ-9000, Keyence) or a confocal microscope (LSM710, Zeiss). Cell counts were made bilaterally. The number of MCH neurons in the brain is presented as the sum of the number of MCH neurons counted in every fourth brain slice at a thickness of 40 μm.
In vivo photo illumination using freely moving mice.
Male MCH-tTA; TetO ChR2 and MCH-tTA; TetO ArchT bigenic mice, 12 weeks of age or older, were housed under controlled lighting (12 h light/dark cycle; lights on from 8:00 to 20:00) and temperature (22 ± 3°C) conditions. Food and water were available ad libitum. Mice were anesthetized with isoflurane using a vaporizer for small animals (Bio Research Center) and positioned in a stereotaxic frame (David Kopf Instruments). Plastic fiber optics (0.5 mm in diameter; Eska, Mitsubishi Rayon) were bilaterally implanted into the hypothalamus ∼1 mm above the LHA (1.2 mm posterior, 1 mm lateral from bregma, 3.5 mm depth from brain surface). Electrodes for EEG and EMG were implanted on the skull and neck muscles, respectively. For the head-fixed mice, a U-shaped plastic plate was attached to the skull to enable fixation of the mouse's head to the stereotaxic frame during recordings. These were attached to the skull using dental cement (GC). The mice were then housed separately for a recovery period of ≥7 d.
Continuous EEG and EMG recordings were performed through a slip ring (Air Precision, Le Pressis Robinson) designed so that the movement of the mouse was unrestricted. EEG and EMG signals were amplified (AB-610J, Nihon Koden), filtered (EEG, 1.5–30 Hz; EMG, 15–300 Hz), digitized at a sampling rate of 128 Hz, and recorded using SleepSign software version 3 (Kissei Comtec). Blue (475 ± 17.5 nm) and green light (542 ± 13.5 nm) was generated by the SPECTRA light engine and applied through plastic optical fibers bilaterally inserted 1 mm above the LHA. An optical swivel (COME2, Lucir) was used for unrestricted in vivo photo illumination. The power intensities of blue and green light at the tip of the plastic fiber optics (0.5 mm diameter) were 25.8 and 33.4 mW/mm2, respectively, as measured by a power meter (VEGA, Ophir Optronics). Each animal's behavior was monitored through a CCD video camera and recorded on a computer synchronized with EEG and EMG recordings using the SleepSign video option system (Kissei Comtec). The infrared activity monitor was a sensor mounted on top of the cage to measure locomotor activity. The sensor's output signals (representing the magnitude of each animal's movement) were digitally converted and transferred to a computer.
MCH-tTA; TetO DTA bigenic mouse surgery. MCH-tTA; TetO DTA bigenic mice were housed under controlled lighting (12 h light/dark cycle; lights on from 7:00 to 19:00) and temperature (22°C) conditions. Food and water were available ad libitum. Doxycycline-containing chow (Dox chow) was made by adding 10% doxycycline (Dox) powder (Kyoritsu Seiyaku) to normal chow (Labo MR Stock, Nosan) at a final concentration of 100 mg/kg. Labo MR stock was provided during the Dox(−) period (from 10 weeks of age). Mating pairs of MCH-tTA mice and TetO DTA mice were fed with Dox-containing chow [Dox(+) condition] from the day of mating. During the prenatal and early postnatal periods, Dox was supplied via maternal circulation or lactation, respectively. After weaning, MCH-tTA; TetO DTA mice were fed with Dox(+) chow until the day of the experiment.
MCH-tTA; TetO DTA bigenic mice (8 weeks of age) were anesthetized with an intraperitoneal injection of a drug mixture containing ketamine (75 mg/kg) and medetomidine (1 mg/kg), placed in a stereotaxic device, and chronically implanted with EEG and EMG electrodes for polysomnographic recording of sleep and wakefulness states. Two stainless-steel screws (diameter, 1.0 mm) were implanted into the skull over the left cerebral hemisphere (frontal: 1.5 mm lateral to midline, 1.0 mm anterior to bregma; parietal: 1.5 mm lateral to midline, 1.0 mm anterior to lambda) according to the atlas of Franklin and Paxinos (1997). A third screw was placed over the frontal bone to serve as a ground electrode. EMG activity was monitored using stainless-steel Teflon-coated wires inserted bilaterally into the neck muscles. All electrodes were attached to a microconnector, and the entire assembly was fixed to the skull with dental cement. Mice were injected with indomethacin (1 mg/kg, i.p.) and penicillin G (200 U/kg, i.m.) immediately after surgery. All procedures were performed on a heating pad, and mice were allowed to recover for 10–14 d before being transferred to sleep recording chambers.
Vigilance state determination.
Polysomnographic recordings were automatically scored offline as wakefulness, NREM sleep, or REM sleep by SleepSign, in 4 s epochs (optogenetics experiments) or 10 s epochs (ablation experiments), according to standard criteria (Tobler et al., 1997; Yamanaka et al., 2002). All vigilance state classifications assigned by SleepSign were examined visually and corrected if necessary. The same individual, blinded to genotype and experimental condition, scored all EEG/EMG recordings. Spectral analysis of the EEG was performed by fast Fourier transform (FFT; sampled at 128 Hz). This analysis yielded a power spectral profile over a 0–25 Hz window with a 1 Hz resolution divided into delta (1–5 Hz), theta (6–10 Hz), alpha (10–13 Hz), and beta (13–25 Hz) waves.
Statistical analysis.
Data were analyzed by paired t test, unpaired t test, one-way ANOVA, two-way ANOVA, or nonparametric Kruskal–Wallis test (as appropriate for the parameters examined) using KaleidaGraph 4.0 software (Hulinks). When appropriate, ANOVA tests were followed by post hoc analysis of significance using Fisher's protected least significant difference test or Bonferroni's test. p values <0.05 were considered statistically significant.
Results
Specific activation of MCH neurons using ChR2
MCH neurons in the brains of MCH-tTA; TetO ChR2 bigenic mice specifically expressed ChR2 (Fig. 1A) as confirmed by double-labeled immunohistochemistry. An anti-GFP antibody was used to detect ChR2-EYFP fusion proteins. Merged pictures show that ChR2-EYFP was exclusively observed in MCH neurons in bigenic MCH-tTA; TetO ChR2 mice (Fig. 1B,C). Ectopic expression of ChR2 outside of MCH neurons was not observed. Confocal microscopic observation revealed that ChR2 was located in the plasma membrane of somata and dendrites of MCH neurons. MCH neurons expressing ChR2 did not show blebbing or other features indicative of inappropriate trafficking (Fig. 1C). The number and morphology of MCH neurons in MCH-tTA; TetO ChR2 mice were indistinguishable from those of monogenic littermate mice (MCH-tTA or TetO ChR2 mice, data not shown), suggesting that ChR2 expression is not toxic to MCH neurons. The ChR2 expression rate (YFP-immunoreactive/MCH-immunoreactive × 100%) in MCH-tTA; TetO ChR2 mice was 88.0 ± 1.3% (n = 9).
Figure 1.
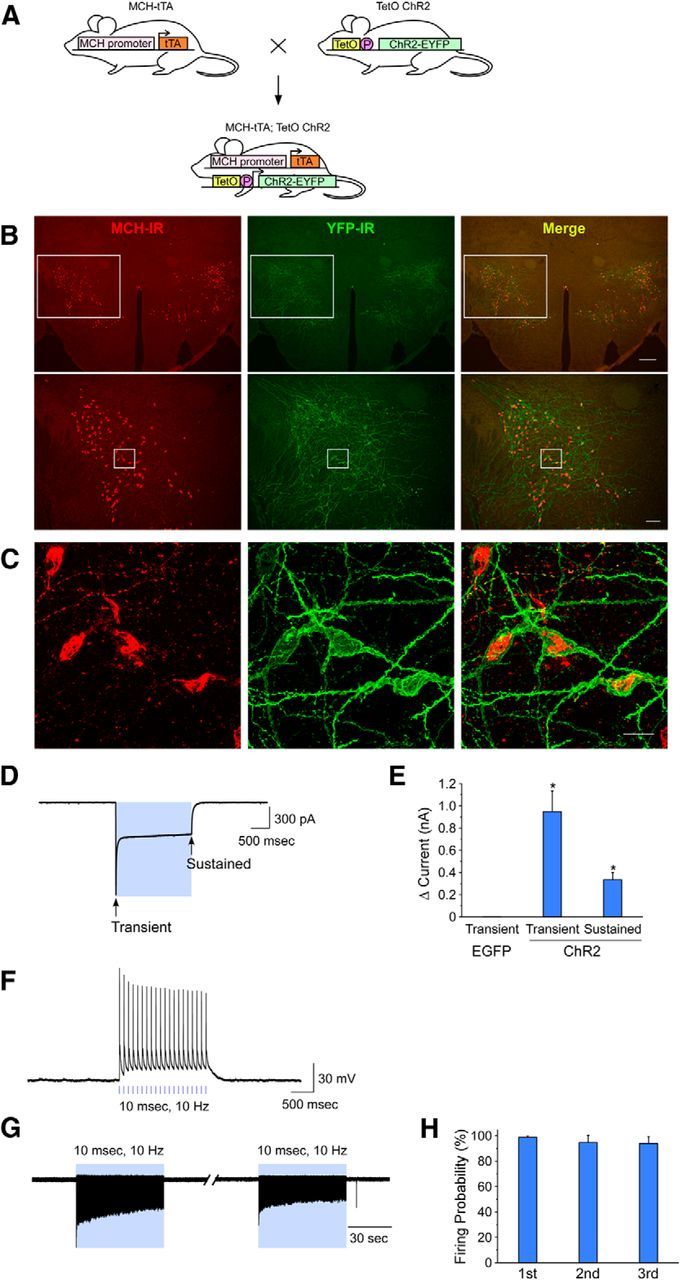
Specific activation of MCH neurons. A, Schematic showing generation of bigenic MCH-tTA; TetO ChR2 mice. P, Minimal promoter. B, C, Immunohistochemical analyses revealed that ChR2 is specifically expressed in MCH neurons in the MCH-tTA; TetO ChR2 bigenic mouse brain. B, MCH-immunoreactive (MCH-IR) neurons (left, Alexa594, red) and ChR2::EYFP-immunoreactive (YFP-IR) neurons (middle, Alexa488, green) located in the LHA and the zona incerta. Right, Merged image shows specific expression of ChR2 in MCH neurons. Bottom row represents higher magnifications of the regions enclosed by the squares in the top row. Scale bars: (in top, right) top row, 200 μm; (in bottom, right) bottom row, 100 μm. C, Confocal microscopic image of the region indicated by the squares in B, bottom row. Scale bar, 20 μm. D–H, Slice patch-clamp recordings from MCH neurons. D, Photocurrent induced by blue light (475 ± 17.5 nm, 2.5 mW, 2 s; Vh = −60 mV, TTX 1 μm). E, Bar graph summarizing the data obtained from D. F, Current-clamp mode recording, blue light pulses (2.5 mW, 10 ms, 10 Hz) generated action potentials in MCH neurons. G, Loose cell-attached recording from MCH neurons. Blue light pulses (2.5 mW, 10 ms, 10 Hz) for 1 min every 5 min generated action potentials. H, Summary data from G. Values represent means ± SEM. *p < 0.05 versus EGFP-expressing MCH neurons.
To confirm the function of ChR2 in MCH neurons, slice patch-clamp analyses were performed. Most MCH neurons did not exhibit spontaneous firing (0.0 ± 0.0 Hz, n = 5). Under whole-cell voltage-clamp mode, blue light (475 ± 17.5 nm, 2.5 mW) was illuminated through the objective lens in the presence of TTX. At a holding potential of −60 mV, blue light illumination for 2 s induced robust inward currents (Fig. 1D). Blue light illumination-induced transient and sustained currents were 948.2 ± 185.5 pA (n = 9, p < 0.001, ANOVA) and 335.0 ± 64.3 pA (n = 9, p = 0.04, ANOVA), respectively (Fig. 1E). Under current-clamp mode, a blue light pulse (10 ms, 10 Hz) was applied. Blue light illumination instantaneously caused depolarization and action potentials were generated in conjunction with light pulses (Fig. 1F). Next, activation of MCH neurons was confirmed using loose cell-attached recordings, which recorded firing frequencies without affecting intracellular conditions. Blue light illumination for 1 min (10 ms, 10 Hz) evoked repetitively induced action potentials with high fidelity (Fig. 1G,H). These results strongly suggested that blue light illumination would activate MCH neurons in vivo.
Activation of MCH neurons increases total time in REM sleep
To study the physiological significance of MCH neuronal activity in the regulation of sleep/wakefulness, MCH neurons were activated in vivo using freely moving MCH-tTA; TetO ChR2 mice. To determine sleep/wakefulness states, mice were chronically implanted with EEG and EMG electrodes and bilateral optical fibers. The tip of the optical fiber was stereotaxically placed 1 mm above MCH neurons to illuminate ChR2-expressing MCH neurons.
First, the vigilance state distribution of MCH-tTA; TetO ChR2 bigenic mice under basal conditions was compared with that of littermate monogenic mice (MCH-tTA mice or TetO ChR2 mice). There were no significant differences between these strains in the total time, mean episode duration, or the number of episodes of wakefulness, REM sleep, or NREM sleep (Table 1), indicating that ChR2 expression in MCH neurons had no effect on sleep/wakefulness patterns.
Table 1.
Vigilance state parameters recorded from MCH-tTA; TetO ChR2 bigenic mice, monogenic littermate mice, and MCH-tTA; TetO ArchT bigenic mice under basal conditions
| Wakefulness |
REM sleep |
NREM sleep |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | MCH-tTA; TetO ChR2 | MCH-tTA; TetO ArchT | Control | MCH-tTA; TetO ChR2 | MCH-tTA; TetO ArchT | Control | MCH-tTA; TetO ChR2 | MCH-tTA; TetO ArchT | |
| 24 h | |||||||||
| Total time (min) | 792.5 ± 40.7 | 734.5 ± 30.3 | 755.9 ± 14.8 | 61.5 ± 6.8 | 81.2 ± 9.3 | 80.8 ± 10.6 | 586.0 ± 10.6 | 624.2 ± 25.1 | 603.2 ± 12.3 |
| Episode duration (s) | 479.1 ± 91.7 | 479.3 ± 81.9 | 546.7 ± 86.9 | 65.8 ± 4.8 | 75.1 ± 1.6 | 71.7 ± 4.7 | 281.6 ± 26.2 | 251.6 ± 12.1 | 251.2 ± 13.2 |
| Number of episodes (counts) | 118.0 ± 4.6 | 108.1 ± 8.1 | 131.4 ± 11.4 | 52.6 ± 3.4 | 48.7 ± 4.6 | 59.4 ± 6.6 | 133.3 ± 5.1 | 127.0 ± 6.8 | 148.2 ± 10.5 |
| Light period | |||||||||
| Total time (min) | 232.4 ± 14.7 | 210.2 ± 9.2 | 204.9 ± 5.9 | 50.5 ± 8.1 | 62.5 ± 5.0 | 68.1 ± 8.1 | 437.1 ± 16.5 | 447.3 ± 12.2 | 446.9 ± 6.2 |
| Episode duration (s) | 212.3 ± 25.9 | 167.3 ± 13.7 | 169.8 ± 30.7 | 71.5 ± 7.6 | 83.7 ± 3.7 | 79.8 ± 3.1 | 344.8 ± 42.8 | 285.7 ± 33.9 | 269.0 ± 15.4 |
| Number of episodes (counts) | 79.4 ± 9.0 | 80.9 ± 10.4 | 95.2 ± 5.7 | 39.5 ± 4.3 | 41.3 ± 3.8 | 48.2 ± 5.1 | 90.4 ± 9.8 | 97.7 ± 8.9 | 108.6 ± 6.1 |
| Dark period | |||||||||
| Total time (min) | 551.0 ± 43.9 | 524.4 ± 37.6 | 551.0 ± 16.8 | 12.7 ± 2.9 | 18.7 ± 5.7 | 12.7 ± 3.4 | 156.4 ± 41.3 | 176.9 ± 32.2 | 156.3 ± 14.9 |
| Episode duration (s) | 745.8 ± 204.8 | 791.3 ± 153.4 | 923.6 ± 147.3 | 60.2 ± 3.4 | 66.4 ± 2.9 | 63.6 ± 9.7 | 218.3 ± 12.6 | 217.4 ± 22.4 | 233.4 ± 24.6 |
| Number of episodes (counts) | 38.6 ± 8.2 | 27.3 ± 5.9 | 36.2 ± 7.2 | 13.1 ± 3.0 | 7.4 ± 2.0 | 11.2 ± 1.7 | 42.9 ± 9.4 | 29.3 ± 6.5 | 39.6 ± 6.4 |
Blue light pulses (475 ± 17.5 nm, 25.8 mW/mm2, 10 ms, 10 Hz) were applied during the first half of the light period (from 10:00 to 13:00) and MCH-tTA; TetO ChR2 mice without light illumination were used as a comparison. The total time in REM sleep was significantly increased in MCH-tTA; TetO ChR2 mice during activation of MCH neurons (Fig. 2A,B). During this 3 h period, the total time spent in wakefulness, REM sleep, and NREM sleep under basal conditions and after activation of MCH neurons was 51.8 ± 2.9 min (n = 6) and 56.3 ± 4.2 min [n = 6, p = 0.25, not significantly different (NS) paired t test]; 15.1 ± 1.2 min (n = 6) and 46.3 ± 2.7 min (n = 6, p < 0.001, paired t test); and 113.1 ± 3.0 min (n = 6) and 77.4 ± 3.6 min (NREM, n = 6, p < 0.001, paired t test), respectively. These results indicated that activation of MCH neuronal activity resulted in increased time spent in REM sleep accompanied by a decrease in time spent in NREM sleep, but without affecting the time spent in wakefulness. The mean episode duration of NREM sleep was significantly decreased from 5.3 ± 0.4 min (n = 6) to 2.0 ± 0.2 min (n = 6, p < 0.001, paired t test) by activation of MCH neurons (Fig. 2C). Meanwhile, the number of episodes of wakefulness, REM sleep, and NREM sleep was significantly increased (Fig. 2D). These results suggest that activation of MCH neurons increased the probability of transitions from NREM to REM sleep, resulting in increased REM and decreased NREM sleep over the 3 h period. Since the duration of REM episodes was unchanged, however, an overall fragmentation of sleep/wakefulness resulted, as indicated by the increased number of episodes of all states. After stimulation of MCH neurons ceased, NREM sleep was significantly increased with decreases in wakefulness and REM sleep (Fig. 2E), suggesting a compensatory rebound in NREM sleep.
Figure 2.

In vivo optogenetic activation of MCH neurons increased time spent in REM sleep. A, F, Bar graphs illustrating the time in each vigilance state during the light period (9:00–16:00) in (A) MCH-tTA; TetO ChR2 bigenic mice and (F) MCH-tTA monogenic mice and TetO ChR2 monogenic mice. Blue light pulses (475 ± 17.5 nm, 25.8 mW/mm2, 10 ms, 10 Hz for 3 h from 10:00 to 13:00) were applied. Blue background indicates the period of illumination. B, G, Bar graphs summarizing the data from A and F. The time spent in each vigilance state during illumination for 3 h is summarized. Black bars, No light illumination; blue bars, blue light pulse illumination. C, H, Mean episode duration. D, I, Number of episodes. E, J, Bar graph summarizing the time in each vigilance state for 1 h after cessation of illumination (from 13:00 to 14:00) in MCH-tTA; TetO ChR2 bigenic mice (E) and MCH-tTA monogenic mice and TetO ChR2 monogenic mice (J). W, Wakefulness; R, REM sleep; NR, NREM sleep. Values are represented as means ± SEM. *p < 0.05 versus basal (no illumination).
Blue light illumination (475 ± 17.5 nm, 25.8 mW/mm2, 10 ms, 10 Hz) had no effect on sleep/wakefulness in monogenic MCH-tTA or TetO ChR2 mice (Fig. 2F). Neither the total time (Fig. 2G), nor mean episode duration (Fig. 2H), nor the number of episodes (Fig. 2I) during illumination for 3 h, nor the total time after illumination (Fig. 2J) of each vigilance state were significantly affected by blue light pulse.
Does activation of MCH neurons induce physiological REM sleep?
Although activation of MCH neurons increased the time spent in REM sleep as determined from EEG and EMG recordings in freely moving mice, it was unclear whether this state met criteria for physiological REM sleep other than those based on EEG and EMG recordings. To evaluate this further, eye and whisker movements during activation of MCH neurons were visually analyzed. To monitor eye and whisker movements during MCH neuron stimulation, mice were affixed to a stereotaxic frame via a plastic U-shape frame fixed onto the skull. After a week adaptation and training to these conditions, mice spontaneously cycle between wakefulness and sleep. While a mouse was in spontaneous NREM sleep, MCH neurons were activated and the EEG and EMG indicated signs of REM sleep shortly after initiation of stimulation (475 ± 17.5 nm, 25.8 mW/mm2, 10 ms, 10 Hz). During MCH neuron activation-induced REM sleep, the eyes and whiskers moved intermittently, similar to that observed during spontaneously occurring REM sleep. These results suggest that MCH neuron activation-induced REM sleep is indistinguishable from physiologically occurring REM sleep in terms of EEG, EMG, and eye and whisker movement indicators.
Activation of MCH neurons during NREM sleep but not during wakefulness induces REM sleep
The results presented in Figure 2 clearly demonstrate that REM sleep can be induced by MCH neuron stimulation during NREM sleep. To determine whether activation of MCH neurons during wakefulness also induces a transition to REM sleep, MCH neurons were intermittently activated for 1 min (475 ± 17.5 nm, 25.8 mW/mm2, 10 ms, 10 Hz) every 5 min from 20:00 (the onset time of the active dark period) to 16:46 (the second half of the inactive light period). MCH-tTA; TetO ChR2 mice without light illumination were used as controls. Figure 3A shows a representative hypnogram from MCH-tTA; TetO ChR2 mice under basal conditions (without light illumination). In comparison, the hypnogram of MCH-tTA; TetO ChR2 mice with blue light illumination (Fig. 3B) shows frequent transitions to REM sleep in conjunction with light illumination. However, transitions to REM sleep were restricted to epochs during which mice were in NREM sleep. Activation of MCH neurons in NREM sleep immediately decreased EEG delta power and increased theta power, suggesting a transition to REM sleep (Fig. 3C,D). Mean REM sleep latency calculated from the initiation of blue light illumination was 12.4 ± 1.2 s (n = 129). REM sleep was often terminated when blue light illumination ended. In contrast, activation of MCH neurons did not induce a transition to REM sleep when mice were awake (Fig. 3E,F). EEG and EMG were indistinguishable from before MCH neuron activation.
Figure 3.
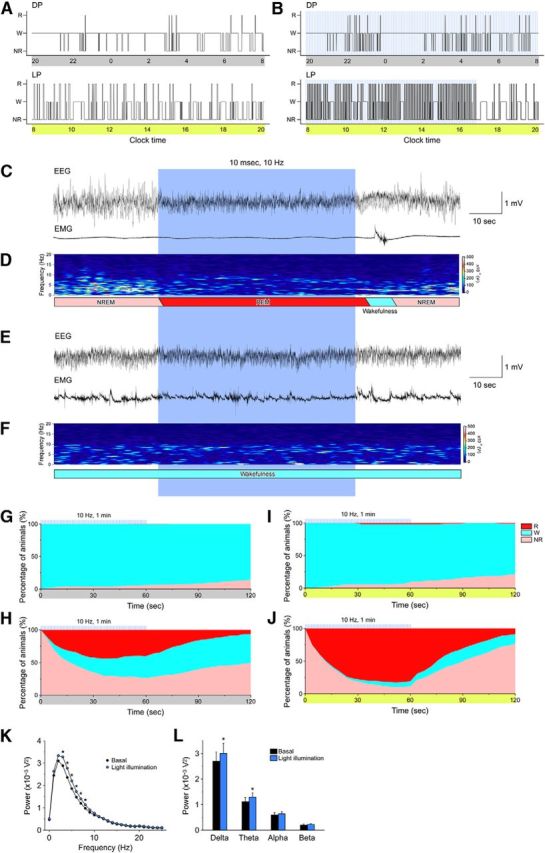
Acute activation of MCH neurons induces transitions from NREM sleep to REM sleep. A, B, Representative hypnograms of (A) no illumination and (B) blue light illumination for 1 min every 5 min (25.8 mW/mm2, 10 ms, 10 Hz) from 20:00 to 16:46. The upper panel is the dark period (20:00–8:00); lower panel is the light period (8:00–20:00); blue bars indicate 1 min periods of blue illumination. C, E, Representative traces for EEG (upper trace) and EMG (lower trace). Blue pulses were applied during NREM sleep (C) or wakefulness (E). D, F, EEG power spectra corresponding to C and E. G–J, Graphs show percentage of animals in each vigilance state during blue pulse illumination. G, H, Dark period. I, J, Light period. Blue light illumination occurred during a waking period in G and I and during NREM sleep in H and J. The blue lines above each graph indicate 1 min blue light illumination pulses. K, Power spectral analysis of EEG in the light period (8:00–16:46) without illumination (black) and with illumination (blue). L, The average EEG power densities in the delta, theta, alpha, and beta wave bands in the light period (8:00–16:46). W, Wakefulness; R, REM sleep; NR, NREM sleep; LP, light period; DP, dark period. Values are represented as means ± SEM. *p < 0.05 versus no illumination.
Figure 3G–J presents the percentage of transitions during and after 1 min MCH neuron activation during the dark period (Fig. 3G,H) and in the light period (Fig. 3I,J). Activation of MCH neurons was initiated either when the mice were awake (Fig. 3G,I) or in NREM sleep (Fig. 3H,J). Activation of MCH neurons while the mice were awake during either the dark or light period did not affect vigilance states; wakefulness was maintained both during and after activation of MCH neurons (Fig. 3G,I). In contrast, activation of MCH neurons during NREM sleep greatly increased the probability of transition to REM sleep (Fig. 3H,J). REM sleep duration was ∼1 min, probably due to the duration of light illumination. MCH neuron activation during NREM sleep in the light period was particularly effective in facilitating the transition from NREM to REM sleep (Fig. 3J): the proportion of mice transitioning from NREM sleep to REM sleep within 1 min of MCH neuron activation was 89.7 ± 1.2% (n = 7, p < 0.001, unpaired t test). To exclude the effect of blue light illumination alone on the transition to REM sleep, littermate monogenic MCH-tTA or TetO ChR2 mice were subjected to blue light illumination. In these mice, the transition probability from NREM sleep to REM sleep after 1 min blue light MCH neuron illumination was only 10.4 ± 1.6% (n = 8). The EEG power spectral analysis by FFT showed that EEG power of 3–8 Hz was significantly increased during light illumination in the light period (8:00 to 16:46; Fig. 3K). The average EEG power density in the delta (1–5 Hz) and theta (6–10 Hz) wave bands was significantly increased as well during light illumination (Fig. 3L), indicating an increase in sleep intensity during activation of MCH neurons.
Acute inhibition of MCH neurons has no effect on sleep/wake states
Next, the effect of acute inhibition of MCH neurons on vigilance states was determined. To strongly inhibit MCH neurons over a long period using optogenetics, TetO ArchT mice (Tsunematsu et al., 2013) were bred with MCH-tTA mice to generate bigenic MCH-tTA; TetO ArchT mice (Fig. 4A).
Figure 4.
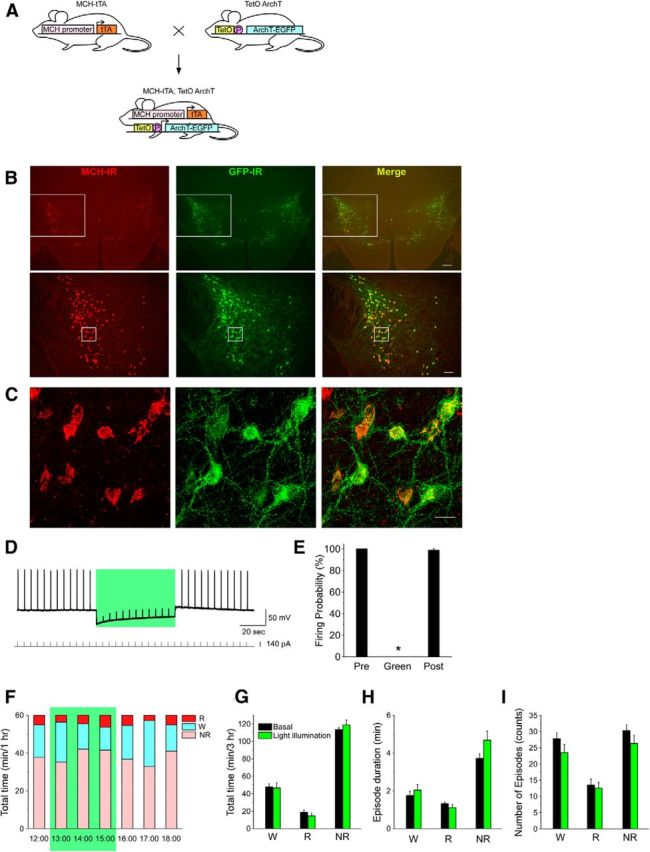
In vivo optogenetic inhibition of MCH neurons has no effect on vigilance states. A, Schematic shows generation of bigenic MCH-tTA; TetO ArchT mice. P, Minimal promoter. B, C, Immunohistochemical analyses revealed that ArchT is specifically expressed in MCH neurons in the MCH-tTA; TetO ArchT bigenic mouse brain. B, MCH-immunoreactive (MCH-IR) neurons (left, red; Alexa594) and ArchT::GFP-immunoreactive (GFP-IR) neurons (middle, green; Alexa488) located in the LHA and the zona incerta. Right, Merged image shows specific expression of ArchT in MCH neurons. Bottom row are higher magnifications of the regions enclosed by the squares in the top row. Scale bars: (in top, right) top row, 200 μm; (in bottom, right) bottom row, 100 μm. C, Confocal microscopic image of the region indicated by the squares in B, bottom row. Scale bar, 20 μm. D, E, Slice patch-clamp recordings from MCH neurons. D, Upper trace, Current-clamp recording from MCH neuron. Lower trace, Injected current through the recording electrode (0.2 Hz, 140 pA, 100 ms) to generate action potentials. Green light illumination (549 ± 7.5 nm, 0.9 mW, 1 min) inhibited firing elicited by current injection. E, Bar graph indicating the firing probability calculated from the data in D (n = 7). Firing probability is normalized to the 1 min before light illumination (Pre). Green light was applied for the duration indicated by the green bars. Values represent means ± SEM. *p < 0.05 versus Pre. F, Green light illumination (542 ± 13.5 nm, 33.4 mW/mm2, 3 h from 13:00 to 16:00) in the light period. G–I, Bar graphs indicating the total time in each vigilance state (G), episode duration (H), and number of episodes (I) during green light illumination. W, Wakefulness; R, REM sleep; NR, NREM sleep. Values are represented as means ± SEM. *p < 0.05 versus basal (no illumination).
The specific expression of ArchT in MCH neurons was confirmed by a double-label immunohistochemical study. An anti-GFP antiserum was used to detect ArchT-EGFP fusion proteins. A merged picture (GFP-immunoreactive and MCH-immunoreactive) revealed that ArchT-EGFP was exclusively expressed in MCH neurons (Fig. 4B,C). ArchT expression was restricted to MCH neurons; ectopic expression of ArchT outside of MCH neurons was not observed. Confocal microscopic observation revealed that ArchT was present in the soma and dendrites of MCH neurons. The number and morphology of MCH-immunoreactive neurons in MCH-tTA; TetO ArchT mice were indistinguishable from those of monogenic littermate mice (MCH-tTA or TetO ArchT mice; data not shown), suggesting that ArchT expression is not toxic to MCH neurons. ArchT was expressed in 97.3 ± 2.0% (n = 6, GFP-immunoreactive/MCH-immunoreactive × 100%) of MCH-immunoreactive neurons.
Slice patch-clamp recording from MCH neurons confirmed the function of ArchT in MCH neurons. Under whole-cell current-clamp mode, a rectangular depolarizing current (0.2 Hz, 100 ms) was injected through the recording electrode to generate artificial action potentials since most MCH neurons were silent. Green light illumination (549 ± 7.5 nm, 0.9 mW) for 1 min completely inhibited current injection-generated action potentials (Fig. 4D). Green light illumination completely inhibited generation of action potentials. The firing probability during green light illumination was 0.0 ± 0.0% (n = 7, p < 0.001; Fig. 4E). After termination of green light illumination, neuronal discharge rate recovered to 98.8 ± 1.3% of the preillumination firing rate (n = 7, p = 0.24, NS).
During the light (inactive) period, green light (542 ± 13.5 nm, 33.4 mW/mm2) was illuminated through fiber optics onto the hypothalamus of MCH-tTA; TetO ArchT mice for 3 h from 13:00 to 16:00 using the same methods used for activation of MCH neurons. MCH-tTA; TetO ArchT mice without illumination were used as controls. Optogenetic inhibition of MCH neurons had no significant effect on time spent in any vigilance state (Fig. 4F,G). In the control condition, the time spent in wakefulness, REM sleep, and NREM sleep over the 3 h period was 47.7 ± 3.5 min (n = 6), 18.6 ± 2.9 min (n = 6), and 113.6 ± 2.1 min (n = 6), respectively. The time spent in wakefulness, REM sleep, and NREM sleep during green light illumination was 46.6 ± 5.9 min (n = 6, p = 0.88, NS, paired t test), 14.5 ± 3.3 min (n = 6, p = 0.054, NS, paired t test), and 118.9 ± 5.5 min (n = 6, p = 0.41, NS, paired t test), respectively. Inhibition of MCH neurons also had no effect on episode duration (Fig. 4H) or the number of episodes (Fig. 4I). As indicated in Table 1, ArchT expression in MCH neurons had no effect on basal sleep/wakefulness patterns (total time and episode duration of each state). These results suggest that silencing of MCH neurons has little effect on the initiation and maintenance of REM sleep.
Ablation of MCH neurons decreases time spent in NREM sleep
Inhibition of MCH neurons using optogenetics had little effect on sleep/wakefulness. However, due to the technical limitations of optogenetic inhibition, particularly for long periods of inhibition, we wanted to induce a chronic loss of function to further evaluate the role of MCH neurons in sleep/wakefulness regulation. Thus, we used the tet-off system mice to specifically ablate MCH neurons. MCH-tTA mice were bred with TetO DTA mice to generate MCH-tTA; TetO DTA bigenic mice (Fig. 5A). In these mice, DTA expression is restricted to MCH neurons and DTA induces cell death by inhibiting protein synthesis. The timing of DTA expression is controlled by the presence or absence of Dox in the chow. In the presence of DTA, tTA loses its ability to bind the TetO sequence (Fig. 5B). However, in the absence of Dox, tTA induces DTA expression. Here, MCH-tTA; TetO DTA mice were fed with chow containing Dox (100 mg/kg) until 10 weeks of age. Then, Dox(+) chow was replaced with Dox(−) chow, the timing of which was defined as Dox(−) 0 week (Fig. 5C).
Figure 5.
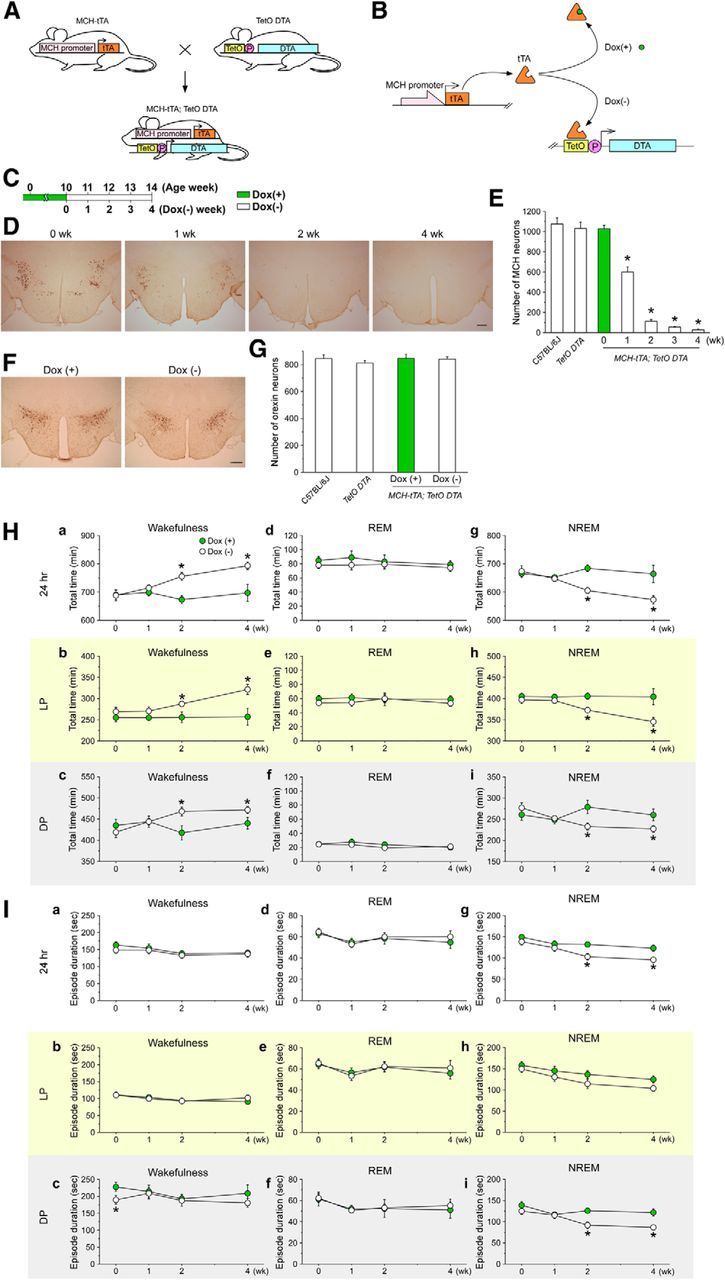
Ablation of MCH neurons increases wakefulness and decreases NREM sleep in MCH-tTA; TetO DTA bigenic mice. A, Schematic showing generation of bigenic MCH-tTA; TetO DTA mice. P, Minimal promoter. B, Schematic illustration of the tetracycline-controlled gene expression system and tTA-induced DTA expression in MCH neurons. In the presence of Dox, Dox binds tTA protein and DTA expression is repressed. C, Schematic shows the experimental protocol. In the presence of Dox, DTA expression is suppressed and MCH neurons are intact. Green bar, Chow contained Dox [Dox(+)]. Open bar, Chow without Dox [Dox(−)]. D, Immunohistochemistry indicates MCH neuron-specific ablation in Dox(−); 0, 1, 2, and 4 weeks (wk) indicates time after Dox(−). Scale bar, 300 μm. E, Bar graph shows the number of MCH-immunoreactive neurons (n = 3–4). F, Orexin neurons were stained with anti-orexin antibody to produce a golden-brown color (scale bar, 300 μm). G, Bar graph summarizing the number of orexin-immunoreactive cell bodies in the LHA. H, Line graphs indicate the total time of each vigilance state. Ha, Hd, Hg, Total time in wakefulness, REM sleep, and NREM sleep, respectively. Hb, He, Hh, Total time for 24 h during the light period in wakefulness, REM sleep, and NREM sleep. Hc, Hf, Hi, Total time during the dark period in wakefulness, REM sleep, and NREM sleep. I, Line graphs indicate the mean duration of each vigilance state. Ia, Id, Ig, Mean duration over 24 h in wakefulness, REM sleep, and NREM sleep, respectively. Ib, Ie, Ih, Mean duration during the light period in wakefulness, REM sleep, and NREM sleep, respectively. Ic, If, Ii, Mean duration during the dark period in wakefulness, REM sleep, and NREM sleep. Green circles, Dox(+) condition (MCH neurons are intact); open circles Dox(−) (MCH neurons are ablated). Values are represented as means ± SEM. *p < 0.05 versus Dox(+). LP, Light period; DP, dark period.
MCH neuron-specific and temporally controlled ablation was confirmed by immunohistochemical studies using an anti-MCH antibody. The number of MCH-immunoreactive neurons was counted in C57BL/6J wild-type mice, in TetO DTA monogenic mice at 10 weeks of age, and in MCH-tTA; TetO DTA bigenic mice at DOX(−) 0, 1, 2, 3, and 4 weeks. At Dox(−) 0 week, the number of MCH neurons was 1028 ± 33 (n = 3). This was comparable to the number of MCH neurons in the C57BL/6J wild-type mice (1076 ± 61, n = 4, p = 1, Kruskal–Wallis) and in the TetO DTA monogenic mice (1031 ± 65, n = 3, p = 1, Kruskal–Wallis). Meanwhile, the number of MCH neurons dramatically decreased after Dox removal (Fig. 5D,E). The number of MCH neurons at Dox(−) 1, 2, 3, and 4 weeks was 600 ± 52 (58.3%, n = 3, p < 0.001, Kruskal–Wallis), 112 ± 18 (10.9%, n = 4, p < 0.001, Kruskal–Wallis), 55 ± 4 (5.4%, n = 3, p < 0.001, Kruskal–Wallis), and 28 ± 9 (2.7%, n = 4, p < 0.001, Kruskal–Wallis), respectively. To confirm whether DTA-induced cell death induction was restricted to MCH neurons, orexin/hypocretin neurons were stained and counted (Fig. 5F,G). Orexin neurons are distributed in the lateral hypothalamic area but do not colocalize with MCH (Broberger et al., 1998; Elias et al., 1998; Peyron et al., 1998; Bayer et al., 2002). The number of orexin neurons was comparable to that of C57BL/6J wild-type mice and that of TetO DTA monogenic mice (Fig. 5G). In addition, the number of orexin neurons was unaffected by Dox removal for 4 weeks in MCH-tTA; TetO DTA mice (Fig. 5G). No signs of unhealthiness, shape, or size of orexin neurons were found even though orexin neurons are located near MCH neurons. These results confirm that the tet-off system functioned correctly in the transgenic mice and that MCH neurons were specifically ablated after removing Dox from the chow.
Next, the effect of ablation of MCH neurons on sleep/wakefulness regulation was analyzed. EEG and EMG electrodes were implanted in MCH-tTA; TetO DTA mice at 8 weeks of age and EEG and EMG recordings were started after 2 weeks recovery at 10 weeks of age.
MCH-tTA; TetO DTA mice, fed with Dox(+) chow [the Dox(+) group], were used as controls. The total time in wakefulness, REM sleep, and NREM sleep was unaffected in the Dox(+) group during recordings [from Dox(+) 0 week to Dox(+) 4 weeks]. In contrast, the time spent in wakefulness was significantly increased and the time in NREM sleep decreased in the Dox(−) group (Fig. 5H). At Dox(−) 4 weeks, total time in wakefulness in the Dox(+) group and the Dox(−) group was 697.1 ± 29.6 min (n = 5) and 793.1 ± 13.5 min (n = 8, p < 0.001, unpaired t test), respectively. At Dox(−) 4 weeks, the total time in NREM sleep in the Dox(+) group and the Dox(−) group was 664.2 ± 30.5 min (n = 5) and 572.7 ± 14.8 min (n = 8, p = 0.01, unpaired t test), respectively. Although increases in wakefulness were observed during both the light and dark periods, the increase was more pronounced in the light period. Interestingly, however, the time spent in REM sleep was unaffected in either the light or dark period (Fig. 5He,Hf).
The mean episode duration of NREM sleep across the 24 h period and during the dark period significantly decreased from Dox(−) 2 weeks (Fig. 5Ig,Ii). The mean episode durations of wakefulness and REM sleep were not significantly affected (Fig. 5Ia–If). These results provide evidence that MCH neurons are involved in the regulation of NREM sleep in addition to their role in REM sleep demonstrated above.
To confirm that the ablation of MCH neurons did not affect normal cortical activity, cortical EEG power spectral analyses were compared between Dox(−) 0 week and Dox(−) 4 weeks in MCH-tTA; TetO DTA mice (Fig. 6). The power spectra in both REM and NREM sleep of Dox(−) 4 weeks mice were indistinguishable from those of Dox(−) 0 week mice, suggesting that ablation of MCH neurons had no effect on basal cortical activity.
Figure 6.
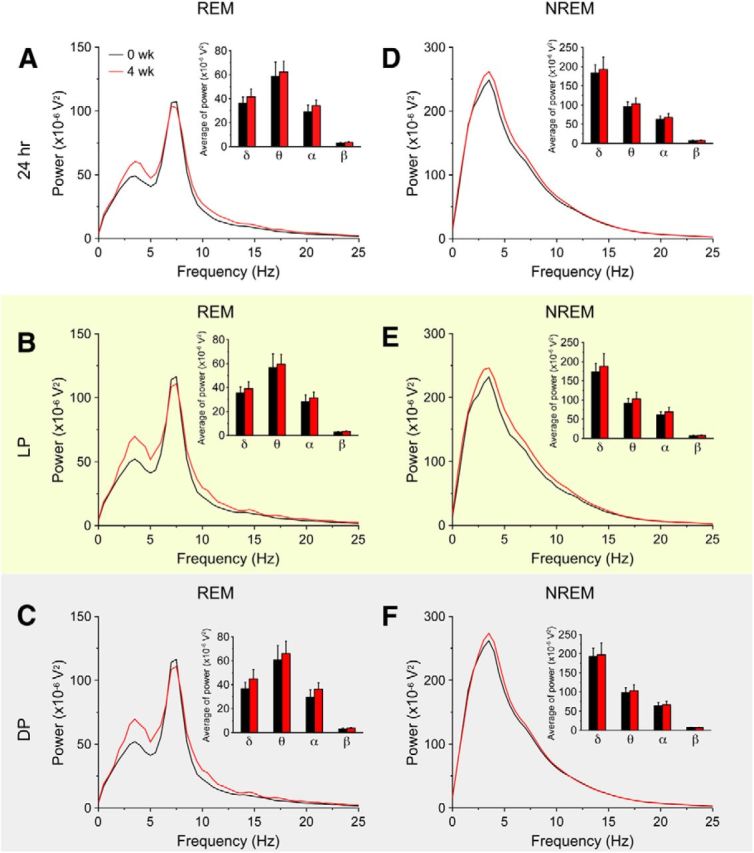
Ablation of MCH neurons had no effect on EEG power spectra during REM sleep and NREM sleep. A–F, Power spectral analysis of EEG recorded from MCH-tTA; TetO DTA bigenic mice. Black lines indicate the presence of Dox (MCH neurons are intact); Red lines show Dox removal for 4 weeks (MCH neurons are ablated). Inset summarizing the average EEG power density in the delta (1–5 Hz), theta (6–10 Hz), alpha (10–13 Hz), and beta (13–25 Hz) bands during REM sleep (A–C) and NREM sleep (D–F). Values are represented as means ± SEM. *p < 0.05 versus 0 week.
Discussion
In the present study, newly developed transgenic mouse strains enabled manipulation of the activity and fate of MCH neurons in vivo. The results obtained support a role for MCH neurons in the regulation of both REM and NREM sleep.
Acute and reversible manipulation of MCH neuronal activity using optogenetics
A role for MCH neurons in sleep/wake regulation has previously been suggested based on studies using gene knock-out mice and pharmacological studies. Prepro-MCH gene knock-out mice exhibited longer wake bouts during the dark period but REM sleep was unchanged (Willie et al., 2008). MCHR1 gene knock-out mice increased wakefulness and decreased NREM sleep (Ahnaou et al., 2011). MCHR1 antagonists decreased deep NREM and REM sleep quantities primarily by reducing the mean episode duration (Ahnaou et al., 2008). MCH neurons are silent during waking and discharge during sleep and are maximally active during REM sleep (Hassani et al., 2009). Despite these reports, however, the specific role of MCH neurons in sleep/wakefulness regulation remains uncertain.
To help clarify the role of MCH neurons in sleep/wake control, we determined the effects of acute optogenetic activation of MCH neurons. To activate these cells, we used a ChR2 (E123T/T159C) double mutant that is suitable for reliable and fast optical stimulation (Berndt et al., 2011). We found that acute activation of MCH neurons significantly increased the time spent in REM sleep in association with decreased NREM sleep. These results were in good agreement with those of a previous study in which ChR2 was expressed in MCH neurons using a Cre-dependent viral vector in prepro-MCH-cre transgenic mice (Jego et al., 2013). Interestingly, NREM rebound was observed after stimulation of MCH neurons. This might be caused simply by NREM suppression during activation of MCH neurons. Alternatively, this might suggest that homeostatic pressure for NREM sleep accumulates not only in wakefulness, but also in REM sleep.
On the other hand, optogenetic activation of MCH neurons has also been reported to increase both NREM and REM sleep (Konadhode et al., 2013). This discrepancy may be due to differential ChR2 expression among MCH neurons. The present study and that of Jego et al. (2013) report that >80% of MCH neurons expressed ChR2, whereas Konadhode et al. targeted MCH neurons that were dorsal to the fornix (53%) with relatively little expression (∼20%) in the lateral and ventral hypothalamic populations (Konadhode et al., 2013). Activation of a subset of MCH neurons might have distinct effects on NREM sleep. Jego et al. (2013) expressed ChR2, eNpHR3.0, or ArchT in MCH neurons by viral injections into Cre transgenic mice. However, the expression rate among individual mice could differ since this technique is dependent upon interanimal replicability of each virus injection. In contrast, the bigenic mouse strains used in the present study do not require viral injections and we have found interanimal expression to be reliable and reproducible.
We further demonstrated that acute activation of MCH neurons during NREM sleep reliably triggered transitions to REM sleep. MCH neuron activation-induced REM sleep following NREM sleep suggests a similarity to physiological REM sleep control. In contrast, optogenetic silencing of MCH neurons had no effect on either total REM time or the duration of REM sleep episodes. These results demonstrate that MCH neuronal activity is sufficient to induce REM sleep, but is not necessary for REM sleep occurrence. This conclusion is consistent with the observation that REM sleep occurs even in pontine cats in which all structures rostral to the brainstem have been removed (Jouvet, 1962), implicating neurons in the brainstem as responsible for the generation of REM sleep.
MCH neurons may cooperatively interact with other brain regions to regulate REM sleep. Recent studies using cFos immunostaining with retrograde tracing have revealed an interaction between MCH neurons and pontine sublaterodorsal tegmental nucleus (SLD) neurons (Clément et al., 2012; Luppi et al., 2013a). Other studies indicate that REM sleep is generated by glutamatergic neurons in the SLD that are specifically active during REM sleep (Sakai and Koyama, 1996; Boissard et al., 2002; Lu et al., 2006; Clément et al., 2011). These studies also suggest that MCH neurons inhibit GABAergic REM-off neurons located in the ventrolateral periaqueductal gray region during REM sleep and that these GABAergic REM-off neurons gate the activation of the REM-on glutamatergic neurons in the SLD. MCH neurons might function to open this gate to initiate REM sleep during NREM sleep. This hypothesis is supported by our results following intermittent activation of MCH neurons. Although activation of MCH neurons during NREM sleep induced REM, activation during wakefulness had no effect on either REM or NREM sleep.
Direct transitions from wakefulness to REM sleep do not occur under normal conditions, but it occurs in narcoleptics in whom orexin signaling pathways are interrupted. This observation indicates that, in contrast to MCH, orexin inhibits the initiation of REM sleep. Indeed, local administration of orexin into the locus ceruleus or the laterodorsal tegmental nucleus dramatically suppresses REM sleep without affecting NREM sleep quantity (Bourgin et al., 2000; Xi et al., 2001). Orexin neurons and MCH neurons are coextensive in the LHA, which leads us to imagine a functional interaction between these neurons in the regulation of sleep/wakefulness. Our observation that MCH neuron activation during NREM sleep induces REM sleep with higher probability in the light period compared with the dark period might reflect this interaction. Orexin concentrations in the CSF are higher during the dark period compared with the light period (Yoshida et al., 2001), which might interfere with the transition from NREM to REM sleep when MCH neurons are activated.
Chronic and irreversible ablation of MCH neurons
To further elucidate the role of MCH neurons in sleep/wakefulness, MCH neuron-specific ablation was performed. Near-complete MCH neuron ablation increased the total time in wakefulness with decreased NREM sleep. However, the total time in REM sleep was not affected. These observations indicate that MCH neurons also play a crucial role in the regulation of NREM sleep. The result from this chronic disruption of MCH neurotransmission conflicted with our results demonstrating that acute activation of MCH neurons increases total time in REM sleep. How could such different results between chronic ablation and acute activation of MCH neurons arise? One possible explanation is suggested by a slice patch-clamp study that revealed that MCH exhibited an inhibitory effect on orexin neurons (Rao et al., 2008). The orexin and MCH systems often display opposing effects on physiological functions. For example, MCH knock-out mice were hyperactive during fasting (Willie et al., 2008), whereas orexin neuron-ablated mice were hypoactive but did not increase locomotor activity during fasting (Hara et al., 2001; Yamanaka et al., 2003). Orexin neurons are thought to be active during wakefulness and silent during REM sleep (Lee et al., 2005; Mileykovskiy et al., 2005; Hassani et al., 2009), whereas MCH neurons are active in REM sleep and inactive during wakefulness (Hassani et al., 2009). Putative somasomatic, axosomatic, and axodendritic contacts between orexin and MCH neurons have been observed (Bayer et al., 2002; Guan et al., 2002). Additionally, MCHR1 knock-out mice showed an increase in glutamatergic transmission onto orexin neurons. These facts support the concept that MCH neurons have a role in the inhibition of orexin neuronal activity. Therefore, ablation of MCH neurons might result in a chronic activation of orexin neurons, which might cause an increase in the total wake time and decrease in NREM sleep in MCH neuron-ablated mice. However, acute inhibition of MCH neurons affected neither the time in wakefulness nor NREM sleep duration. Optogenetic inhibition for 3 h might not be enough to affect orexin neurons. Alternatively, after ablation of MCH neurons, neural network reorganization might occur, resulting in chronic activation of orexin neurons.
MCH neurons also project to forebrain areas, such as the hippocampus and cortex (Bittencourt et al., 1992; Saito et al., 1999; Hervieu et al., 2000). High levels of MCHR1 mRNA and MCHR1 immunoreactivity have been found in these areas (Bittencourt et al., 1992; Saito et al., 1999; Hervieu et al., 2000). Therefore, in addition to its role in sleep/wakefulness regulation, the release of MCH onto cortical and hippocampal cells during REM sleep might modulate long-term plasticity and contribute to the processing of learning and memory. Similar to the result of Konadhode et al., the increase of delta and theta power during activation of MCH neurons might suggest the role of MCH on the cortex (Konadhode et al., 2013). Additional studies are also required to determine the role of MCH release during REM sleep in these brain areas.
Together, the present results show that MCH neurons in the hypothalamus play a significant role in the regulation of both NREM and REM sleep. Further experiments are necessary to identify the specific neural pathways that underlie the differential roles that MCH neurons play in the regulation of NREM versus REM sleep. The creation of MCH-tTA; TetO DTA mice, which enable partial lesions of the MCH neuron population to be produced by varying the duration of Dox removal from the diet, may be a useful tool in this regard.
Notes
Supplemental material for this article is available at https://dl.dropboxusercontent.com/u/32320004/MCH-REM.mov. This material has not been peer reviewed.
Footnotes
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Mesoscopic Neurocircuitry” (23115103), a Grant-in-Aid for Scientific Research (B) (23300142; A.Y.), a Grant-in-Aid for Scientific Research (C) (22500830; A.T.), and a Grant-in-Aid for Young Scientist (A) (23680042; K.T.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; by the Prescursor Research for Embryonic Science and Technology (PRESTO) program from Japan Science and Technology Agency (A.Y.); by Takeda Science Foundation and Kanae Foundation by a Japan Society for Promotion of Science postdoctoral fellowship (T.T. and S.T.); and by National Institutes of Health Grant R01 NS077408 (T.K.). We thank Dr. Y. Ootsuka and Dr. K. Matsui for assisting in data analyses. We thank C. Saito, K. Nishimura, Y. Esaki (Nonprofit Organization Biotechnology Research and Development), and S. Nishioka (Nonprofit Organization Biotechnology Research and Development) for technical assistance.
The authors declare no competing financial interests.
References
- Ahnaou A, Drinkenburg WH, Bouwknecht JA, Alcazar J, Steckler T, Dautzenberg FM. Blocking melanin-concentrating hormone MCH1 receptor affects rat sleep-wake architecture. Eur J Pharmacol. 2008;579:177–188. doi: 10.1016/j.ejphar.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Ahnaou A, Dautzenberg FM, Huysmans H, Steckler T, Drinkenburg WH. Contribution of melanin-concentrating hormone (MCH1) receptor to thermoregulation and sleep stabilization: evidence from MCH1 (−/−) mice. Behav Brain Res. 2011;218:42–50. doi: 10.1016/j.bbr.2010.11.019. [DOI] [PubMed] [Google Scholar]
- Asakawa A, Inui A, Goto K, Yuzuriha H, Takimoto Y, Inui T, Katsuura G, Fujino MA, Meguid MM, Kasuga M. Effects of agouti-related protein, orexin and melanin-concentrating hormone on oxygen consumption in mice. Int J Mol Med. 2002;10:523–525. [PubMed] [Google Scholar]
- Bayer L, Mairet-Coello G, Risold PY, Griffond B. Orexin/hypocretin neurons: chemical phenotype and possible interactions with melanin-concentrating hormone neurons. Regul Pept. 2002;104:33–39. doi: 10.1016/s0167-0115(01)00320-2. [DOI] [PubMed] [Google Scholar]
- Berndt A, Schoenenberger P, Mattis J, Tye KM, Deisseroth K, Hegemann P, Oertner TG. High-efficiency channelrhodopsins for fast neuronal stimulation at low light levels. Proc Natl Acad Sci U S A. 2011;108:7595–7600. doi: 10.1073/pnas.1017210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol. 1992;319:218–245. doi: 10.1002/cne.903190204. [DOI] [PubMed] [Google Scholar]
- Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–1973. doi: 10.1046/j.1460-9568.2002.02257.x. [DOI] [PubMed] [Google Scholar]
- Bourgin P, Huitrón-Résendiz S, Spier AD, Fabre V, Morte B, Criado JR, Sutcliffe JG, Henriksen SJ, de Lecea L. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J Neurosci. 2000;20:7760–7765. doi: 10.1523/JNEUROSCI.20-20-07760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberger C, De Lecea L, Sutcliffe JG, Hökfelt T. Hypocretin/orexin- and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems. J Comp Neurol. 1998;402:460–474. doi: 10.1002/(SICI)1096-9861(19981228)402:4<460::AID-CNE3>3.3.CO%3B2-J. [DOI] [PubMed] [Google Scholar]
- Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM. Melanin-concentrating hormone is the cognate ligand for the orphan G-protein-coupled receptor SLC-1. Nature. 1999;400:261–265. doi: 10.1038/22313. [DOI] [PubMed] [Google Scholar]
- Clément O, Sapin E, Bérod A, Fort P, Luppi PH. Evidence that neurons of the sublaterodorsal tegmental nucleus triggering paradoxical (REM) sleep are glutamatergic. Sleep. 2011;34:419–423. doi: 10.1093/sleep/34.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément O, Sapin E, Libourel PA, Arthaud S, Brischoux F, Fort P, Luppi PH. The lateral hypothalamic area controls paradoxical (REM) sleep by means of descending projections to brainstem GABAergic neurons. J Neurosci. 2012;32:16763–16774. doi: 10.1523/JNEUROSCI.1885-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Cid-Pellitero E, Jones BE. Immunohistochemical evidence for synaptic release of GABA from melanin-concentrating hormone containing varicosities in the locus coeruleus. Neuroscience. 2012;223:269–276. doi: 10.1016/j.neuroscience.2012.07.072. [DOI] [PubMed] [Google Scholar]
- Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol. 1998;402:442–459. doi: 10.1002/(SICI)1096-9861(19981228)402:4<442::AID-CNE2>3.3.CO%3B2-I. [DOI] [PubMed] [Google Scholar]
- Elias CF, Sita LV, Zambon BK, Oliveira ER, Vasconcelos LA, Bittencourt JC. Melanin-concentrating hormone projections to areas involved in somatomotor responses. J Chem Neuroanat. 2008;35:188–201. doi: 10.1016/j.jchemneu.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28:106–110. doi: 10.1002/1526-968X(200011/12)28:3/4<106::AID-GENE30>3.3.CO%3B2-K. [DOI] [PubMed] [Google Scholar]
- Franklin KGB, Paxinos G. The mouse brain in stereotaxic coordinates. Academic: San Diego; 1997. [Google Scholar]
- Gao XB, van den Pol AN. Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol. 2001;533:237–252. doi: 10.1111/j.1469-7793.2001.0237b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JL, Uehara K, Lu S, Wang QP, Funahashi H, Sakurai T, Yanagizawa M, Shioda S. Reciprocal synaptic relationships between orexin- and melanin-concentrating hormone-containing neurons in the rat lateral hypothalamus: a novel circuit implicated in feeding regulation. Int J Obes Relat Metab Disord. 2002;26:1523–1532. doi: 10.1038/sj.ijo.0802155. [DOI] [PubMed] [Google Scholar]
- Han X, Chow BY, Zhou H, Klapoetke NC, Chuong A, Rajimehr R, Yang A, Baratta MV, Winkle J, Desimone R, Boyden ES. A high-light sensitivity optical neural silencer: development and application to optogenetic control of non-human primate cortex. Front Syst Neurosci. 2011;5:18. doi: 10.3389/fnsys.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/S0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A. 2009;106:2418–2422. doi: 10.1073/pnas.0811400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes BE, Kil E, Green B, O'Neill K, Fried S, Graziano MP. The melanin-concentrating hormone receptor couples to multiple G proteins to activate diverse intracellular signaling pathways. Endocrinology. 2000;141:4524–4532. doi: 10.1210/endo.141.12.7833. [DOI] [PubMed] [Google Scholar]
- Hervieu GJ, Cluderay JE, Harrison D, Meakin J, Maycox P, Nasir S, Leslie RA. The distribution of the mRNA and protein products of the melanin- concentrating hormone (MCH) receptor gene, slc-1, in the central nervous system of the rat. Eur J Neurosci. 2000;12:1194–1216. doi: 10.1046/j.1460-9568.2000.00008.x. [DOI] [PubMed] [Google Scholar]
- Inamura N, Sugio S, Macklin WB, Tomita K, Tanaka KF, Ikenaka K. Gene induction in mature oligodendrocytes with a PLP-tTA mouse line. Genesis. 2012;50:424–428. doi: 10.1002/dvg.20808. [DOI] [PubMed] [Google Scholar]
- Jego S, Adamantidis A. MCH neurons: vigilant workers in the night. Sleep. 2013;36:1783–1786. doi: 10.5665/sleep.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, Friedman J, Burdakov D, Adamantidis AR. Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci. 2013;16:1637–1643. doi: 10.1038/nn.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE, Hassani OK. The role of Hcrt/Orx and MCH neurons in sleep-wake state regulation. Sleep. 2013;36:1769–1772. doi: 10.5665/sleep.3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvet M. Research on the neural structures and responsible mechanisms in different phases of physiological sleep (in French) Arch Ital Biol. 1962;100:125–206. [PubMed] [Google Scholar]
- Konadhode RR, Pelluru D, Blanco-Centurion C, Zayachkivsky A, Liu M, Uhde T, Glen WB, Jr, van den Pol AN, Mulholland PJ, Shiromani PJ. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci. 2013;33:10257–10263. doi: 10.1523/JNEUROSCI.1225-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo PM, Grazzini E, Cao J, Hubatsch DA, Pelletier M, Hoffert C, St-Onge S, Pou C, Labrecque J, Groblewski T, O'Donnell D, Payza K, Ahmad S, Walker P. The receptor for the orexigenic peptide melanin-concentrating hormone is a G-protein-coupled receptor. Nat Cell Biol. 1999;1:267–271. doi: 10.1038/12978. [DOI] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clément O, Fort P. Paradoxical (REM) sleep genesis by the brainstem is under hypothalamic control. Curr Opin Neurobiol. 2013a;23:786–792. doi: 10.1016/j.conb.2013.02.006. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Peyron C, Fort P. Role of MCH neurons in paradoxical (REM) sleep control. Sleep. 2013b;36:1775–1776. doi: 10.5665/sleep.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A. 2002;99:3240–3245. doi: 10.1073/pnas.052706899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty D, Alam N. MCH neurons: the end of the beginning. Sleep. 2013;36:1773–1774. doi: 10.5665/sleep.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Ohara O. Improvement of recombination efficiency by mutation of red proteins. Biotechniques. 2005;38:917–924. doi: 10.2144/05386RR02. [DOI] [PubMed] [Google Scholar]
- Pelluru D, Konadhode R, Shiromani PJ. MCH neurons are the primary sleep-promoting group. Sleep. 2013;36:1779–1781. doi: 10.5665/sleep.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Lu M, Ge F, Marsh DJ, Qian S, Wang AH, Picciotto MR, Gao XB. Regulation of synaptic efficacy in hypocretin/orexin-containing neurons by melanin concentrating hormone in the lateral hypothalamus. J Neurosci. 2008;28:9101–9110. doi: 10.1523/JNEUROSCI.1766-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Nothacker HP, Wang Z, Lin SH, Leslie F, Civelli O. Molecular characterization of the melanin-concentrating-hormone receptor. Nature. 1999;400:265–269. doi: 10.1038/22321. [DOI] [PubMed] [Google Scholar]
- Sakai K, Koyama Y. Are there cholinergic and non-cholinergic paradoxical sleep-on neurones in the pons? Neuroreport. 1996;7:2449–2453. doi: 10.1097/00001756-199611040-00009. [DOI] [PubMed] [Google Scholar]
- Semjonous NM, Smith KL, Parkinson JR, Gunner DJ, Liu YL, Murphy KG, Ghatei MA, Bloom SR, Small CJ. Coordinated changes in energy intake and expenditure following hypothalamic administration of neuropeptides involved in energy balance. Int J Obes (Lond) 2009;33:775–785. doi: 10.1038/ijo.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- Tanaka KF, Matsui K, Sasaki T, Sano H, Sugio S, Fan K, Hen R, Nakai J, Yanagawa Y, Hasuwa H, Okabe M, Deisseroth K, Ikenaka K, Yamanaka A. Expanding the repertoire of optogenetically targeted cells with an enhanced gene expression system. Cell Rep. 2012;2:397–406. doi: 10.1016/j.celrep.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Tobler I, Deboer T, Fischer M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–1879. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu T, Tabuchi S, Tanaka KF, Boyden ES, Tominaga M, Yamanaka A. Long-lasting silencing of orexin/hypocretin neurons using archaerhodopsin induces slow-wave sleep in mice. Behav Brain Res. 2013;255:64–74. doi: 10.1016/j.bbr.2013.05.021. [DOI] [PubMed] [Google Scholar]
- Verret L, Goutagny R, Fort P, Cagnon L, Salvert D, Léger L, Boissard R, Salin P, Peyron C, Luppi PH. A role of melanin-concentrating hormone producing neurons in the central regulation of paradoxical sleep. BMC Neurosci. 2003;4:19. doi: 10.1186/1471-2202-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie JT, Sinton CM, Maratos-Flier E, Yanagisawa M. Abnormal response of melanin-concentrating hormone deficient mice to fasting: hyperactivity and rapid eye movement sleep suppression. Neuroscience. 2008;156:819–829. doi: 10.1016/j.neuroscience.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Dumalska I, Morozova E, van den Pol A, Alreja M. Melanin-concentrating hormone directly inhibits GnRH neurons and blocks kisspeptin activation, linking energy balance to reproduction. Proc Natl Acad Sci U S A. 2009;106:17217–17222. doi: 10.1073/pnas.0908200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi MC, Morales FR, Chase MH. Effects on sleep and wakefulness of the injection of hypocretin-1 (orexin-A) into the laterodorsal tegmental nucleus of the cat. Brain Res. 2001;901:259–264. doi: 10.1016/S0006-8993(01)02317-4. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Tsujino N, Funahashi H, Honda K, Guan JL, Wang QP, Tominaga M, Goto K, Shioda S, Sakurai T. Orexins activate histaminergic neurons via the orexin 2 receptor. Biochem Biophys Res Commun. 2002;290:1237–1245. doi: 10.1006/bbrc.2001.6318. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/S0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, Mignot E, Nishino S. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep–wake activities. Eur J Neurosci. 2001;14:1075–1081. doi: 10.1046/j.0953-816x.2001.01725.x. [DOI] [PubMed] [Google Scholar]