

Abstract
Exaggerated intracellular Ca2+ signaling is a robust proximal phenotype observed in cells expressing familial Alzheimer's disease (FAD)-causing mutant presenilins (PSs). The mechanisms that underlie this phenotype are controversial and their in vivo relevance for AD pathogenesis is unknown. Here, we used a genetic approach to identify the mechanisms involved and to evaluate their role in the etiology of AD in two FAD mouse models. Genetic reduction of the type 1 inositol trisphosphate receptor (InsP3R1) by 50% normalized exaggerated Ca2+ signaling observed in cortical and hippocampal neurons in both animal models. In PS1M146V knock-in mice, reduced InsP3R1 expression restored normal ryanodine receptor and cAMP response element-binding protein (CREB)-dependent gene expression and rescued aberrant hippocampal long-term potentiation (LTP). In 3xTg mice, reduced InsP3R1 expression profoundly attenuated amyloid β accumulation and tau hyperphosphorylation and rescued hippocampal LTP and memory deficits. These results indicate that exaggerated Ca2+ signaling, which is associated with FAD PS, is mediated by InsP3R and contributes to disease pathogenesis in vivo. Targeting the InsP3 signaling pathway could be considered a potential therapeutic strategy for patients harboring mutations in PS linked to AD.
Keywords: Mouse model, Alzheimer's disease, amyloid, calcium, ion channel, memory
Introduction
Mutations in presenilin 1 (PS1) or PS2 or amyloid precursor protein (APP) are linked to dominantly inherited familial Alzheimer's disease (FAD). The discovery that PSs form the catalytic core of the γ-secretase responsible for APP cleavage and Aβ release provides support for the amyloid cascade hypothesis (Hardy and Selkoe, 2002). Nevertheless, clinical trials targeting Aβ directly or the secretases involved in Aβ production have failed to show therapeutic benefit in even mild-to-moderate AD patients (Mullane and Williams, 2013). This could indicate that interventions were initiated too late in the disease or it may reflect an impasse in AD therapeutic development that underscores a critical need to better understand the etiology of AD.
The etiologies of sporadic Alzheimer's disease (SAD) and FAD are widely assumed to be similar, with FAD simply presenting more rapidly and aggressively. Nevertheless, demonstration that this is the case is lacking. The causes of SAD are unknown, but may be heterogeneous (Bettens et al., 2013). Similarly, it is unknown whether the etiology of FAD pathogenesis is similar in patients with mutations in PS versus APP. Therefore, AD may be a spectrum of diseases with similar end-stage pathology, but with distinct etiologies that could provide targets for personalized therapy.
A clue to the etiology of mutant PS-associated FAD is the repeated observations of altered intracellular Ca2+ ([Ca2+]i) signaling as a robust, proximal phenotype (LaFerla, 2002). Ca2+ signaling regulates many cellular and neuronal processes (Berridge, 1998; Woods and Padmanabhan, 2012). Exaggerated [Ca2+]i signaling is observed in non-neuronal cells from presymptomatic FAD patients (Etcheberrigaray et al., 1998) and at early ages in neurons in FAD mouse models (Stutzmann et al., 2004). Although the mechanisms are controversial (Honarnejad and Herms, 2012), these observations indicate that exaggerated Ca2+ release caused by FAD PS is an early phenotype and could accordingly contribute to disease pathogenesis.
WT and FAD PSs interact with the endoplasmic-reticulum-localized inositol-1,4,5-trisphosphate receptor (InsP3R) Ca2+ release channel and FAD mutations in PS provide a gain-of-function enhancement of InsP3R channel gating and sensitivity to its ligand, InsP3 (Cheung et al., 2008). However, the consequence of this enhancement for exaggerated Ca2+ release in vivo is not clear, nor is the contribution of exaggerated Ca2+ release to PS-associated FAD pathogenesis known. Here, for the first time, we used a genetic approach to test the hypothesis that exaggerated InsP3R-mediated [Ca2+]i signaling contributes to the development of AD pathogenesis in vivo. We found that decreasing InsP3R protein expression by ∼50% normalized FAD PS-associated exaggerated neuronal [Ca2+]i signaling. Importantly, this normalization rescued AD-like biochemical, electrophysiological, and behavioral phenotypes observed in two PS FAD mouse models. These observations suggest that exaggerated [Ca2+]i signaling in PS FAD is InsP3R dependent and that it contributes to disease pathogenesis.
Materials and Methods
Transgenic mice.
M146V knock-in (Guo et al., 1999), 3xTg (Oddo et al., 2003) and InsP3R1Opt+/− [Opisthotonos (Opt); Street et al., 1997] mice were generated and characterized previously and kindly provided by Dr. M. Mattson (National Institutes of Health), Dr. H. Wei (University of Pennsylvania), and Dr. J. Chen (University of San Diego), respectively. Mice were housed in a pathogen-free, temperature- and humidity-controlled facility with a 12 h light/dark cycle. Mice were fed a standard laboratory chow diet and double-distilled water ad libitum. All procedures involving mice were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania in accordance with the National Institutes of Health's Guidelines for the Care and Use of Experimental Animals.
M146V/Opt, 3xTg/Opt, and control lines were generated by crossing M146V+/+ (C57BL/6 background) or 3xTg mice (C57BL/6/129S6 background) to Opt mice (C57BL/6 background). First-generation M146V+/−/Opt mice were crossed to M146V+/−/InsP3R1Opt−/− littermates to generate the M146V/Opt line and control lines. First-generation 3xTg+/−/Opt were backcrossed to parental 3xTg mice to restore M146V to homozygosity and the copy numbers of the APPSWE and tauP301L transgenes or crossed to littermates to generate control lines. PS1 and InsP3R1 genotyping was conducted as described previously (Street et al., 1997; Guo et al., 1999). Real-time PCR (RT-PCR) of genomic DNA verified restoration of APPSWE and tauP301L transgene copy numbers in the 3xTg/Opt line using a 7300 Real Time PCR System (Applied Biosystems), SYBR green PCR Master Mix (Applied Biosystems), 3 μm primers, and cycling of 2 min at 50°C, 10 min at 95°C, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Primer specificity was validated by the presence of a single PCR product for each primer set after agarose gel analysis. In addition, a dissociation phase was used at the end of each RT-PCR assay, which yielded only a single peak for each primer set. Two sets of primers for each transgene were used. Oligonucleotide primers were synthesized by Integrated DNA Technologies: APP set 1 forward (F): 5′-GGACCAAAACCTGCATTGAT-3′, reverse (R): 5′-CTGGTTGGTTGGCTTCTACC-3′; APP set 2 (F): 5′-CACCAGGAGAGGATGGATGT-3′, (R): 5′-CTACCCCTCGGAACTTGTCA-3′; tau set 1 (F): 5′-GGGGGACAGGAAAGATCAG-3′, (R): 5′-GTGACCAGCAGCTTCGTCTT-3′; tau set 2 (F): 5′-AAGACGAAGCTGCTGGTCAC-3′, (R): 5′-GGCGATCTTCGTTTTACCAT-3′. Two sets of primers for actin (set 1 (F): 5′-ACTGGGACGACATGGAGAAG-3′, (R): 5′-CTTTTCACGGTTGGCCTTAG-3′; set 2 (F): 5′-TACAGCTTCACCACCACAGC-3′, reverse (R): 5′-TCTCCAGGGAGGAAGAGGAT-3′) were used to control for [DNA]. The comparative cycle threshold (ΔΔCt) method was used to analyze amplification data. Six backcrosses to parental 3xTg mice were required to restore transgene copy number. Four sets of crosses were used to produce the eight genotypes used; M146V+/+ with M146V/Opt, Opt (C57BL/6) with WT (C57BL/6), 3xTg with 3xTg/Opt, and Opt (C57BL/6/129S6) with WT (C57BL/6/129S6). Only male mice were used for experiments. Mice were assigned to groups based on birth order and randomly matched with littermate controls for analyses.
RT-PCR.
Immediately after euthanasia, brains were quickly removed and washed in ice-cold PBS. Hippocampal and cortical tissues were isolated and stored in RNAlater (QIAGEN) following the manufacturer's protocols. Samples were stored at −80°C until processed. RNA was isolated using an RNeasy Mini Kit (QIAGEN) and the manufacturer's recommended protocols and cDNA was synthesized using a M-MLV Reverse Transcriptase kit (Invitrogen), Oligo(dT)12–18 primers (Invitrogen), and manufacturer's recommended protocols. A volume of cDNA, corresponding to 10 ng of starting RNA, was evaluated using a 7300 Real Time PCR System as described in the preceding paragraph. PS1 (F): 5′-GTCTGAGGACAGCCACTCCA-3′, (R): 5′-TGGCTCAGGGTTGTCAAGTC-3′, human tau primer sets described above. The geometric mean of three reference genes, actin ((F): 5′-CCAACCGTGAAAAGATGACC-3′ (R): 5′-ACCAGAGGCATACAGGGACA-3′)), β2 microglobin ((F): 5′-CTGACCGGCCTGTATGCTAT-3′ (R): 5′-TATGTTCGGCTTCCCATTCT-3′), and β-glucuronidase ((F): 5′-GGTTTCGAGCAGCAATGGTA-3′ (R): 5′-TGCTTCTTGGGTGATGTCATT-3′) was used to control for [cDNA]. The relative mRNA expression was calculated using the ΔCt method, with 2(Ct(ref) − Ct(exp)) as the reported value.
Isolation of primary cell lines.
Primary cortical neuron (PCN) cultures were established from single E14–E16 mouse embryos as described previously (Meberg and Miller, 2003). Cultures were maintained in neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen), l-glutamine (Mediatech), and antibiotics and antimycotics (Invitrogen) at 37°C with 5% CO2. Half of the medium was replaced every third day; 1 μm cytosine β-d-arabinofuranoside was added to the culture medium 3 d after plating. Experiments were performed on 9-d-old cultures. Genotyping of embryos was conducted on noncortical brain tissue from each embryo.
Ca2+ measurements.
All imaging was conducted on an Eclipse FN1 (Nikon) microscope with a mounted Live Scan Swept Field Confocal head equipped with an Innova 70C Ar/Kr ion laser (Coherent) and operated from NIS Elements software (Nikon). A Cascade 512B EM-CCD camera (Photometrics) was used with continuous exposure through slit mode. ImageJ software was used for image analysis, Microsoft Excel was used for background subtraction, and Igor Pro (WaveMetrics) was used to determine magnitudes and rates of change of Ca2+ indicator fluorescence. The magnitude of the change of the Ca2+ indicator fluorescence is expressed as the ratio of the change in fluorescence (F − F0) relative to the resting fluorescence (F0). To determine rates, curves were filtered using the minimum amount of Gaussian smoothing required to obtain a dominant peak in the plot of the derivative of ΔF/F0 versus time. This value is reported as ∂(ΔF/F0)/∂t.
In vitro PCN imaging.
PCNs were plated onto poly-d-lysine-coated glass coverslips at a density of 50,000/ml. Before loading, PCNs were washed once in the buffer used for loading and imaging, which contained the following (in mm): 120 NaCl, 4 KCl, 20 HEPES, 2 CaCl2, 1 MgSO4, and 15 glucose, and then incubated for 60 min at 37°C and 5% CO2 with 1 μm Oregon Green 488 BAPTA-1 AM (Invitrogen) and 5 μm caged InsP3-AM (c-InsP3; Sirius Fine Chemicals). Because intracellular Ca2+ stores are depleted in PCNs of this age (Smith et al., 2005), the cells were depolarized for 90 s by replacing 50 mm Na+ with K+, followed by a 60 s recovery period before each experiment. Photolysis of c-InsP3 was achieved by whole-field illumination (350–400 nm; ∼40 mW/cm2) derived from a X-Cite 120 PC (Lumen Dynamics) controlled by a shutter (Uniblitz) and a Master-8 pulse generator (A.M.P.I.). Images were captured at 25 Hz using a 16× water-immersion lens [numerical aperture (NA) = 0.8]. Stimulus strength was regulated by pulse duration and neutral density filters. Only one ultraviolet (UV) pulse was applied to each coverslip and experiments were performed on PCNs from at least three different embryos for each genotype per UV illumination duration.
Ex vivo slice imaging.
Brains were quickly removed from 10- to 12-d-old pups and transferred to ice-cold sucrose artificial CSF (saCSF) containing the following (in mm): 87 NaCl, 75 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 CaCl2, 2 MgSO4, and 10 glucose bubbled with 95%O2/5%CO2. After removal of the cerebellum, the tissue was mounted on the stage of a vibratome and 300 μm coronal sections containing hippocampal tissue were obtained. Nonhippocampal tissue was trimmed from sections before loading with 4 mm Oregon Green in saCSF bubbled with 95%O2/5%CO2 at 39°C for 60 min. Slices were then transferred to a holding chamber containing saCSF until used. For imaging, slices were transferred to a recording chamber and perfused at a rate of 1–2 ml/min with aCSF containing the following (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgSO4, and 10 glucose bubbled with 95% O2/5% CO2 for 10 min before the start of each experiment. Images were captured at 10 Hz using a 40× water-immersion lens (NA = 0.8). After obtaining a baseline recording, the solution was switched to aCSF containing 10 μm dihydroxyphenylglycine for the remainder of the recording period. Experiments were conducted on slices from at least three mice of each genotype. When more than one Ca2+ release event was observed, only the first was analyzed.
Protein extraction and immunoblotting.
PCNs or brain tissue were sonicated in lysis buffer containing the following (in mm): 5 EGTA, 50 Tris, pH 7.4, and 100 NaCl with 1% Triton X-100 and protease and phosphatase inhibitors (Roche). Homogenates were spun at 14,000 rpm for 20 min at 4°C. The supernatant was added to 4× loading buffer containing the following (in mm): 275 SDS, 5.7 bromophenol blue, and 250 Tris, pH 6.8, with 40% glycerol and 8% β-mercaptoethanol (Bio-Rad), incubated at 95°C for 2 min, and stored at −80°C until use. Lysates were run by SDS-PAGE or Tris acetate 3–8% gradient gels (for RyR; NuPAGE), transferred onto nitrocellulose membranes (GE Healthcare), blocked with 5% nonfat dried milk, and probed with primary antibodies at 4°C overnight. The antibodies used were as follows: InsP3R1, kindly provided by Dr. R. Neumar (University of Pennsylvania) (1:1000); CaMKIV (1:2000; Cell Signaling Technology); phospho-CaMKIV (Thr-196, 1:1000; Santa Cruz Biotechnology); cAMP response element-binding protein (CREB; 1:2000; Cell Signaling Technology); phospho-CREB (Ser-133, 1:1000; Millipore); c-fos (1:1000; Cell Signaling Technology); neuronal nitric oxide synthase (nNOS; 1:1000; Cell Signaling Technology); brain-derived neurotrophic factor (BDNF; 1:1000; Millipore); GAPDH (1:1000; Millipore); acetyl-histone 3 (H3, 1:1000; Millipore); pan-RyR (MA3–925, 1:2000; Affinity Bioreagents); RyR2 (MA3–916, 1:1000; Affinity Bioreagents); RyR3 (AB9082, 1:1000; Millipore); β-amyloid (clone 6E10, 1:500; Covance); htau (clone HT7, 1:500; Thermo Scientific); phospho-tau (AT8 and AT180, 1:500; Thermo Scientific); and the secondary horseradish peroxidase-conjugated antibodies anti-mouse (1:5000; GE Healthcare) and anti-rabbit (1:5000; GE Healthcare). An Alpha Innotech FlourChem Q imaging system was used to visualize protein bands. Band intensities were normalized to anti-β-tubulin (1:5000; Invitrogen) or anti-heat-shock protein 90 (HSP90, 1:1000; Cell Signaling Technology) using AlphaView software version 3.1.1.0.
Immunofluorescence of fresh-frozen tissue.
Brains were quickly removed and washed in ice-cold PBS, followed by embedding in optimal cutting temperature medium (OCT). Tissue was stored at −80°C until processed. Then, 8 μm sections were cut using a cryostat and stored at −80°C. For staining, slides were transferred to −20°C for 20 min and then immersed for 10 min in ice-cold PBS to remove OCT. Sections were fixed for 10 min in 4% paraformaldehyde in PBS, washed 3 times for 10 min each in cold PBS, then permeabilized for 30 min at room temperature (RT) in PBS containing 5% bovine serum albumin (BSA), pH 7.4 (BSA/PBS), and 0.05% Triton X-100. Sections were blocked in PBS/BSA for 1 h at RT, followed by overnight incubation at 4°C in a moist chamber with anti-RyR2 (1:100; Millipore) in BSA/PBS. On the following day, slides were washed three times for 10 min each in PBS/BSA, incubated for 90 min in anti-rabbit 488 Alexa Fluor (1:500; Invitrogen) in BSA/PBS, and washed three times for 10 min each in PBS/BSA. Slides were mounted using anti-fade medium with DAPI (hardest, Vectashield) and kept in the dark at 4°C until confocal microscope analyses (LSM 710; Zeiss).
Immunfluorescence of paraffin-embedded tissue.
Half of the brain was fixed in paraformaldehyde (4% in PBS) overnight at 4°C and then processed for paraffin embedding by the University of Pennsylvania Cancer Histology Core. Then, 5 μm sections were mounted onto silane-coated slides and dried overnight at 37°C. Slices were deparaffinized by two washes in 100% xylene (Fisher Scientific) for 20 min at 37°C, two washes in 100% ethanol for 10 min, one wash in 90% ethanol for 10 min, one wash in 70% ethanol for 10 min, and two washes in H2O for 10 min. Antigen retrieval was accomplished by placing slices in 96°C Target Retrieval Solution (Dako) for 30 min, followed by a 20 min cool-down incubation at RT. Slides were then washed twice for 10 min in H2O, permeabilized for 30 min in BSA/PBS containing 0.025% Triton X-100, and blocked for 1 h in BSA/PBS at RT. After blocking, slices were incubated in the primary antibodies phospho-CaMKIV (Thr-196, 1:100; Santa Cruz Biotechnology), phospho-CREB (Ser-133, 1:100; Cell Signaling Technology), and InsP3R1 (1:100; provided by Dr. R. Neumar) diluted in BSA/PBS overnight at 4°C in a moist chamber. The slices were washed three times for 10 min each in BSA/PBS at RT and then incubated with secondary Alexa Fluor antibodies (1:500) in BSA/PBS for 1 h at RT, washed three times for 10 min in BSA/PBS at RT, mounted using Vectashield with DAPI (Vector Laboratories), and sealed with nail polish. Slides were stored in the dark at 4°C for up to 10 weeks before confocal imaging (LSM 710; Zeiss). All slices were visualized using the same exposure settings. CA1 fluorescence was quantified using ImageJ software.
Immunohistochemistry.
For immunohistochemistry (IHC), half of the brain was fixed and processed as described for immunofluorescence with the following modifications: 10 μm sections were used and antigen retrieval was accomplished by placing slides into 96°C PT Module Buffer 1 (antibodies AT180, 1:250, and AT8, 1:100; Thermo Scientific) for 30 min, followed by a 30 min cool-down incubation at RT or 70% formic acid for 30 min (antibodies 6E10 and 12F4, both 1:1000). After overnight incubation with primary antibodies, a DAB detection system (Covance) and manufacturer's recommended protocols were used to visualize antigen localization. Slices were counterstained using Hematoxylin Gill 3× (Fisher) for 20 s, rinsed in tap water, and cleared with 95% ethanol for 5 min, two washes for 10 min in 100% ethanol, and two washes for 10 min in xylene. Slides were mounted using Permount (Fisher). All slices were processed in parallel.
ELISA.
Invitrogen ELISA kits were used to determine Aβ content in cortical and hippocampal lysates following the manufacturer's recommended protocols. Briefly, homogenates were prepared from dissected cortical or hippocampal tissue by adding 8 volumes/weight of 5 m guanidine buffered with 50 mm Tris HCl, pH 8.0, in 90 μl aliquots and grinding thoroughly with a hand-held pestle after each addition. Homogenates were then rocked at RT for 3–4 h. All lysates were stored at −80°C until used. All samples of each time point were run on a single ELISA plate. Plates were read on a Spectr max 340 PC (Molecular Devices). Results were normalized to total [protein] in each homogenate as determined by protein assay (Bio-Rad) using an Evolution 60 spectrophotometer (Thermo Scientific).
Hippocampal electrophysiology.
Mice were killed by cervical dislocation and hippocampi were quickly collected in ice-cold oxygenated saCSF bubbled with 95% O2/5% CO2. Transverse, 400 μm hippocampal slices were obtained using a Mcllwain tissue chopper, transferred to an interface recording chamber, and perfused (1–2 ml/min) with oxygenated aCSF containing the following (in mm): 124 NaCl, 4.4 KCl, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, 2.5 CaCl2, and 10 d-glucose at 28°C for at least 2 h before recording. A bipolar 0.5 mm nichrome stimulating electrode (AM Systems) was positioned in the Shaffer collateral pathway and a glass micropipette (2–5 mΩ; AM Systems) filled with oxygenated aCSF was placed adjacent to the stimulating electrode in the CA1 hippocampal region. Data were acquired using Clampex 9.2 and a Digidata 1322 A/D converter (Molecular Devices) at 20 kHz and low-pass filtered at 2 kHz. Slices were used only if they provided a maximum response ≥5 mV. Basal synaptic transmission was measured by monitoring responses over a range of voltages applied to the stimulating electrode from 30 V decreasing until the presynaptic fiber volley (PFV) was undetectable. LTP experiments were conducted by applying a stimulus strength that elicited 40% of the maximum evoked-field EPSP (fEPSP) amplitude. fEPSP slopes were monitored every 60 s. The first 20 min baseline values were averaged and used to normalize the initial fEPSP slopes. Tetani were applied at 100 Hz for 1 s.
Behavioral experiments.
All behavioral experiments were conducted in the Neurobehavioral Testing Core at the University of Pennsylvania. Mice were singly housed for 1 week, followed by 5 d of handling before testing. Handling was done in the experimental room for 90 s per day per mouse. Spatial object recognition (SOR) experiments were conducted as described previously (Wimmer et al., 2012). Briefly, mice were placed in the training arena four times for 10 min each. Between trials, mice were returned to their home cage. The first session was a context habituation period without objects in the arena; in the next three sessions, mice were placed in the arena with two distinct objects. The objects used were a glass bottle, a metal rectangular tower, and a plastic cylinder tower. Objects and the training arena were cleaned with 70% ethanol before each session. Twenty-four hours after training, mice were placed back into the arena for a 10 min trial with one of the objects displaced to a new location [displaced object (DO)]; the other object was not moved. All sessions were recorded using a digital camera and subsequently scored for time spent exploring each object, blinded to the mouse genotype. Object exploration was defined as the amount of time a mouse was oriented toward the object with its nose within 0.5 cm of the object. Object preference was calculated as the time spent exploring the DO relative to the total time spent exploring both objects.
Open-field testing was conducted to assess locomotion. Mice were brought to the testing room and allowed to acclimate for 30 min. They were then placed in a Plexiglas arena (14 in2) with a white floor and clear walls. Activity data were collected as beam breaks during a 10 min trial for each mouse using a Photobeam Activity System (San Diego Instruments).
Fear-conditioning tests were conducted by placing mice in a conditioning chamber (Med Associates) for 2 min before the onset of a 2800 Hz, 85 dB tone for 30 s. During the last 2 s of the tone, a 0.7 mA continuous foot shock was applied. Mice remained in the chamber for 30 s after the foot shock and were then returned to their home cage. Twenty-four hours later, mice were tested for time spent frozen (motionless except for respiratory movements) in the chamber during a 3 min period. Mice were also placed in a novel chamber with unique context (smooth flat floor, posters on the walls, novel odorant). After a 2 min habituation period, they were tested for time spent frozen in response application of a 2800 Hz, 85 dB tone during a 3 min period. Scoring of time spent frozen was automated using FreezeScan software.
Materials.
Photoactivatable membrane-permeant InsP3 was dissolved in dimethyl sulfoxide (DMSO) with 10% Pluronic F-127 to a stock concentration of 2.5 mm, aliquoted, and stored at −80°C until needed. Each aliquot was thawed only once. Oregon Green was dissolved in DMSO with 10% Pluronic F-127 to a stock concentration of 4 mm and used fresh daily. Unless otherwise stated, all chemicals were purchased from Sigma and were of the highest purity.
Statistical analyses.
To determine whether the AD mice were abnormal, the null hypothesis that AD mice = WT was tested by performing unpaired two-tailed t tests using STATA software. If the null hypothesis was rejected, two additional unpaired two-tailed t tests were performed to determine whether the Opt allele attenuated or rescued the abnormality; null hypotheses: AD-mice/Opt = WT and AD-mice/Opt = AD-mice. Multiple testing adjustments were not necessary because all t tests performed were hypothesis driven. For LTP data, STATISTICA software was used to conduct repeated-measures ANOVA. Normality of data was checked using normal quantile plots for each dataset. Comparisons were made using Student's t tests or unequal variance t tests when groups were found to have unequal variance. Sample size was chosen by power analysis to ensure adequate power to detect differences between groups. Data from all animals were included in the final analyses unless they were found to be an outlier using a Grubbs' test. Statistical significance was set at a threshold of p ≤ 0.05. Quantitative data represent mean ± SE.
Results
Expression of FAD PS1M146V causes exaggerated [Ca2+]i signaling in mouse cortical and hippocampal neurons
We used two AD mouse models that both express the targeted PS1M146V (M146V) FAD mutation in the endogenous PS1 locus (Guo et al., 1999; Oddo et al., 2003). We chose M146V mice because they do not develop canonical AD pathology, providing a model with which to examine the contribution of the InsP3R to exaggerated [Ca2+]i signaling and the effects of altered [Ca2+]i signaling in vivo uncomplicated by the effects of elevated Aβ and hyperphosphorylated tau (P-tau; LaFerla, 2002). To evaluate the contribution of altered [Ca2+]i signaling to development of canonical AD-like pathology and hippocampal synaptic deficits, we used the 3xTg mouse (Oddo et al., 2003), which contains human APPSWE and human tauP301L transgenes in addition to the PS1M146V knock-in allele.
PCN cultures were established, aged 9 d in vitro, and loaded with a Ca2+ indicator and membrane-permeable caged InsP3. After obtaining a baseline recording, varying durations (∼1–500 ms) of UV light pulses generated a range of cytosolic free [InsP3] (Fig. 1A–C). Both the magnitudes (ΔF/F0) and maximum rates (∂(ΔF/F0)/∂t) of Ca2+ release were greater in M146V and 3xTg than WT PCNs over a wide range of pulse durations (unpaired two-tailed t test, n = 10–186 responding neurons from at least three embryos each per UV flash duration, p < 0.05; Fig. 1D–G). Notably, M146V and 3xTg PCNs responded to the minimum InsP3 release, whereas WT PCNs did not.
Figure 1.
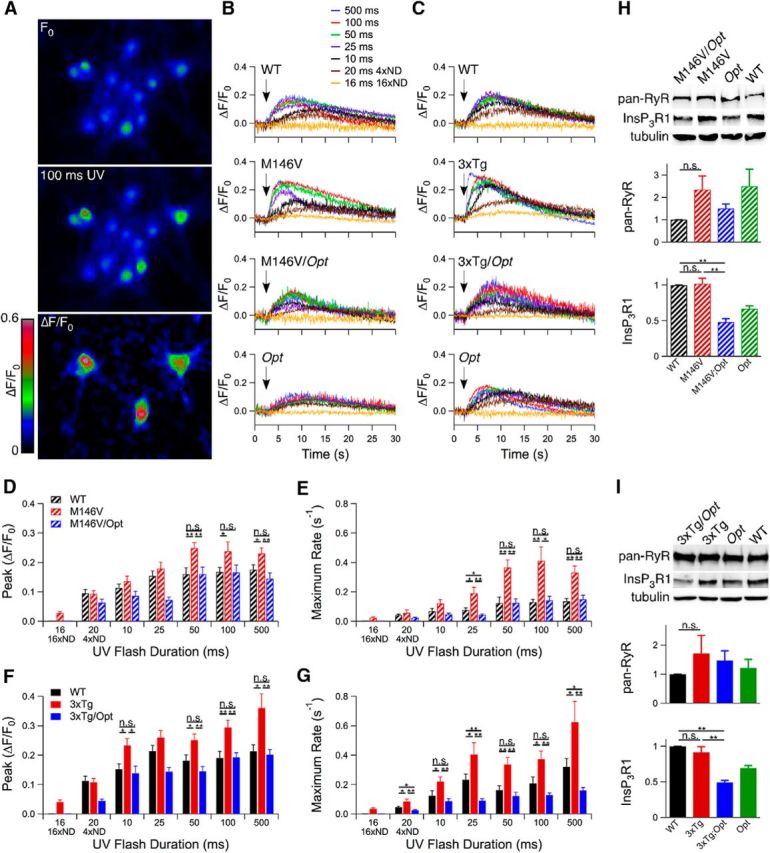
Oregon Green Ca2+ imaging in PCNs loaded with caged InsP3. A, After obtaining a baseline recording (F0), a 100 ms UV pulse was applied. B, C, Representative single cell traces of each genotype after UV illuminations (indicated by arrows) ranging from ∼1–500 ms. Stimulus strength was regulated by pulse duration and use of neutral density (ND) filters. D–G, Magnitudes (D, F) and rates (E, G) of Oregon Green fluorescence change after UV illumination. Unpaired two-tailed t test, n = 10–186 responding neurons from three embryos each per flash duration, *p < 0.05, **p < 0.005. H, I, InsP3R1 and RyR protein levels in PCNs from M146V (n = 4 mice each; H) and 3xTg (n ≥ 5 mice each; I) lines, and tubulin as a loading control. Unpaired two-tailed t test, p < 0.005. Error bars show mean and SEM.
Hippocampal slices from P10–P12 mice were loaded with the Ca2+ indicator Oregon Green and dentate gyrus granular cell layer neurons were imaged. After obtaining a baseline recording, the group 1 metabotropic glutamate receptor agonist dihydroxyphenylglycine was added to the perfusate to induce InsP3R-mediated Ca2+ release (Fig. 2A–C) independently of neuronal depolarization (Fig. 2D,E). Neurons in slices from M146V and 3xTg mice displayed enhanced magnitudes and rates of Ca2+ release compared with WT (unpaired two-tailed t test, n = 72–207 responding neurons from at least three mice each, p ≤ 0.005; Fig. 2F–I). These results demonstrate that the expression of FAD PS1 is associated with exaggerated Ca2+ release in cortical and hippocampal neurons.
Figure 2.

Ex vivo Oregon Green Ca2+ imaging in hippocampal dentate gyrus. A, Hippocampal slices were obtained from 10- to 12-d-old animals, loaded with Oregon Green, and dentate gyrus granular cell layer neurons were imaged by confocal microscopy. After obtaining a baseline recording (F0), the solution was switched to aCSF containing 10 μm dihydroxyphenylglycine (DHPG) for the remainder of the recording period. B, C, Representative traces for M146V lines (B) and 3xTg lines (C). D, E, Magnitudes (D) and rates (E) of Oregon Green fluorescence change in dentate gyrus granular cell layer neurons in WT mice (C57BL/6 background) with and without tetrodotoxin present. Unpaired two-tailed t test, n = 102–125 responding neurons from two mice, no significant differences observed. F, H, Magnitudes of Oregon Green fluorescence change after DHPG perfusion for M146V lines (F) and 3xTg lines (H). Unpaired two-tailed t test, n = 72–207 responding neurons from ≥ 3 mice each, p < 0.005. G, I, Rates of Oregon Green fluorescence change after DHPG perfusion for M146V lines (G) and 3xTg lines (I). Unpaired two-tailed t test, n = 72–207 responding neurons from ≥ 3 mice each, p < 0.005. J, K, Western blot analyses of hippocampal lysates from P10–P12 mice from M146V (n = 6 mice each; J) and 3xTg (n ≥ 6 mice each; K) lines using a pan-RyR antibody or antibodies specific to RyR2, RyR3, or InsP3R1, and tubulin as a loading control. Unpaired two-tailed t test, *p < 0.05, **p < 0.005. Error bars show mean and SEM.
Generation of AD mice with only one functional InsP3R1 allele
Based on previous in vitro studies (Cheung et al., 2008; Cheung et al., 2010), we reasoned that elimination of one allele of the gene encoding the InsP3R1, the major InsP3R isoform expressed in the CNS (Furuichi et al., 1993), would reduce exaggerated [Ca2+]i signaling. We used the Opisthotonos (Opt) mouse, which harbors a spontaneous in-frame deletion that removes 107 aa from the InsP3R1 (Street et al., 1997). The Opt allele acts as a null-like allele in vivo, resulting in InsP3R1 protein levels ∼50% of WT (p < 0.05; Figs. 1H,I; 2J,K; 3A; 6A,B; 7A; and 8A). Heterozygote Opt mice were crossed with M146V or 3xTg mice to create M146V/Opt and 3xTg/Opt mice (Fig. 3C–F). There was no effect of the Opt allele on PS expression (Fig. 3B). Total ryanodine receptor (RyR) protein levels in PCNs were similar to WT across all genotypes (Fig. 1H,I). Total RyR and RyR3 protein levels were similar to WT levels in hippocampi from P10–P12 mice across all genotypes (Fig. 2J,K). RyR2 protein levels were reduced in P10–P12 M146V/Opt hippocampi (unpaired two-tailed t test, n = 6 mice each, p < 0.05) compared with WT, whereas they were similar to WT in all other genotypes (Fig. 2J,K). Therefore, genetic deletion of one InsP3R1 allele results in ∼50% of InsP3R1 expression with no compensatory changes in RyR expression, validating the use of the AD/Opt mice as models to test for the role of the InsP3R1 in FAD PS1 associated phenotypes.
Figure 3.

Creation of PS1M146V-KIN, InsP3R1Opt+/− (M146V/Opt) and 3xTg, InsP3R1Opt+/− (3xTg/Opt) mice. A, Immunofluorescence analysis of hippocampal slices from 5-week-old mice using an InsP3R1-specific antibody. B, Reverse transcriptase RT-PCR on hippocampal RNA from 3-month-old animals for PS1 expression. Unpaired two-tailed t test, n = 3 mice each, no significant differences observed. C, D, A C57BL/6 mouse carrying the Opt allele was crossed to a C57BL/6 mouse homozygote for the PS1M146V-KIN mutation. First-generation M146V+/−/Opt+/− mice were crossed to M146V+/− littermates without the Opt allele to isolate the Opt allele on the WT background and to generate the M146V/Opt line. E, F, A C57BL/6 mouse carrying the Opt allele was crossed to a C57BL/6/129S6 3xTg mouse (containing the PS1M146V-KIN mutation and the human APPSWE and tauP301L transgenes). E, First-generation 3xTg+/−/Opt+/− mice were backcrossed to parental 3xTg mice to restore the PS1M146V-KIN mutation to homozygosity and the APPSWE and tauP301L transgene copy number. Bar graph presents data from RT-PCR conducted on genomic DNA for APPSWE and tauP301L transgenes after six backcrosses to the 3xTg line. F, First-generation 3xTg+/−/Opt+/− mice were crossed to littermates without the Opt allele to generate the control lines. Error bars show mean and SEM.
Figure 6.

The Opt allele rescues aberrant activation of the CaMKIV-CREB transcriptional pathway in M146V mice. A–D, Western blot analyses of hippocampal lysates from 5-week-old (n = 5 mice each; A, C) and 3-month-old mice (n = 6 mice each; B, D) for CaMKIV-CREB pathway activation, protein levels of CREB-dependent genes (nNOS, BDNF, and c-fos), a CREB-independent gene (GAPDH), and genomic accessibility [acetyl-H3, P-CaMKIV (Thr-196), and P-CREB (Ser-133)]. Unpaired two-tailed t test, *p < 0.05, **p < 0.005. Error bars show mean and SEM. E, F, Immunofluorescence in hippocampal slices from 5-week-old mice for P-CaMKIV (E) and P-CREB (F). Hippocampal region CA1 is shown. G, H, Quantification of immunofluorescence in region CA1 for P-CaMKIV (G) and P-CREB (H). Unpaired two-tailed t test, *p < 0.05. Error bars show mean and SEM.
Figure 7.
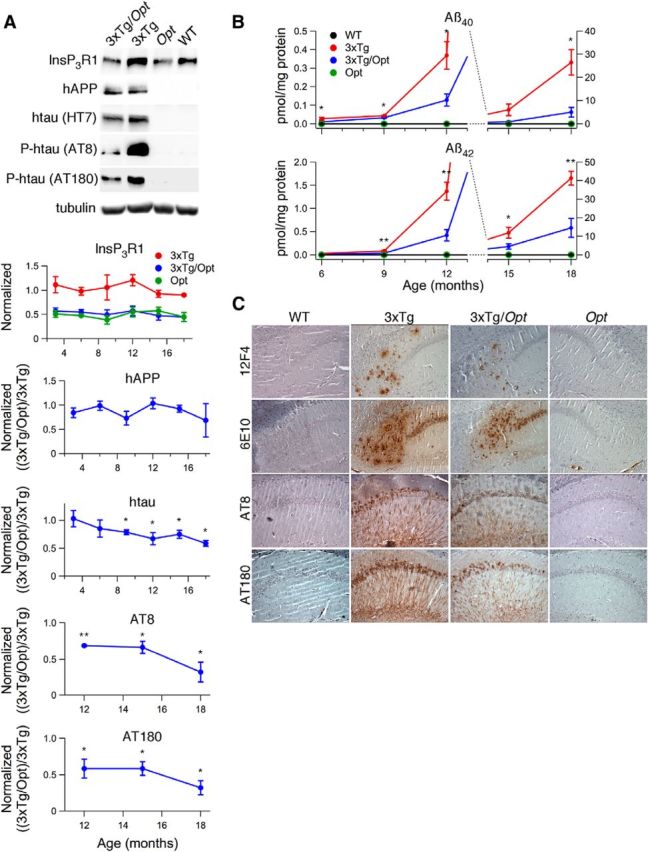
The Opt allele attenuates hippocampal Aβ and P-tau accumulation in 3xTg mice. A, Western blot analyses of hippocampal lysates from 3- to 18-month-old mice using antibodies specific for InsP3R1 (normalized to WT), hAPP (6E10), htau (HT7), htau phosphorylated at Ser-202 (AT8), or Thr-231 (AT180) with tubulin as a loading control. Representative blots are from experiments conducted on 18-month-old mice. Unpaired two-tailed t tests, n ≥ 3 mice each per age, *p < 0.05, **p < 0.005. B, ELISA of hippocampal homogenates for Aβ40 and Aβ42 in mice ranging in age from 6 to 18 months. Aβ content in homogenates from WT and Opt animals were always below the detection limit of the ELISA. Unpaired 2-tailed t test, n ≥ 5 mice each per age, *p ≤ 0.05, **p ≤ 0.005. Error bars show mean and SEM. C, Immunohistochemistry on hippocampal slices from 18-month-old mice for Aβ and P-tau. 6E10 intracellular staining is most likely full-length APP (Winton et al., 2011).
Figure 8.

The Opt allele attenuates cortical AD-like pathology in 3xTg mice. A, Western blot analyses of cortical lysates from 3- to 18-month-old mice using antibodies specific for InsP3R1 (normalized to WT), hAPP (6E10), htau (HT7), and tubulin as loading control. Representative blots from experiments conducted on 18-month-old mice. Unpaired two-tailed t test, n ≥ 3 mice each per age, p ≤ 0.005. B, ELISA of cortical homogenates for Aβ40 and Aβ42 in mice ranging in age from 6 to 18 months. Aβ content in homogenates from WT and Opt animals were always below the ELISA detection limit. Unpaired two-tailed t test, n ≥ 4 mice each per age, *p ≤ 0.05, **p ≤ 0.005. C, Reverse transcriptase RT-PCR for htau expression using cortical RNA isolated from 6-month-old mice. Unpaired two-tailed t test, n = 3 mice each, p = 0.06. Error bars show mean and SEM.
Opt allele rescues FAD PS1M146V-associated exaggerated [Ca2+]i signaling
The presence of a single Opt allele completely normalized exaggerated magnitudes and rates of Ca2+ release observed in M146V and 3xTg PCNs to WT values or less (Fig. 1B–G). Similarly, the Opt allele rescued enhanced [Ca2+]i signaling in hippocampal neurons in M146V/Opt and 3xTg/Opt slices to WT values (Fig. 2B,C, F–I). These results indicate that a single Opt allele rescues FAD PS1-associated exaggerated [Ca2+]i signaling in PCNs and acute hippocampal slices in both AD mouse models. Accordingly, these mice provide models with which to investigate the role of exaggerated [Ca2+]i signaling in vivo on FAD pathogenesis.
Opt allele rescues mild cognitive impairment-like phenotypes in M146V mice
M146V mice display enhanced RyR2 expression (Stutzmann et al., 2006) and hippocampal synaptic plasticity (Auffret et al., 2010), abnormalities mirroring those observed in presymptomatic FAD PS carriers and patients suffering from mild cognitive impairment (MCI; Dickerson et al., 2005; Bruno et al., 2012; Reiman et al., 2012), a condition that commonly precedes AD. Total hippocampal RyR protein levels were elevated in 5-week-old M146V animals compared with WT (unpaired two-tailed t test, n = 6 mice each, p < 0.05; Fig. 4A). This was due to increased RyR2 (unpaired two-tailed t test, n = 4 mice each, p < 0.05) without changes in RyR3 levels (unpaired two-tailed t test, n = 5 mice each). The Opt allele restored M146V RyR protein expression to WT levels or less (Fig. 4A). Hippocampal RyR2 immunofluorescence was enhanced in M146V mice compared with WT, particularly in the CA1 region, which was normalized in M146V/Opt mice (qualitatively, n = 2 mice each; Fig. 4B). Therefore, the Opt allele rescued elevated RyR2 protein levels in M146V mice.
Figure 4.
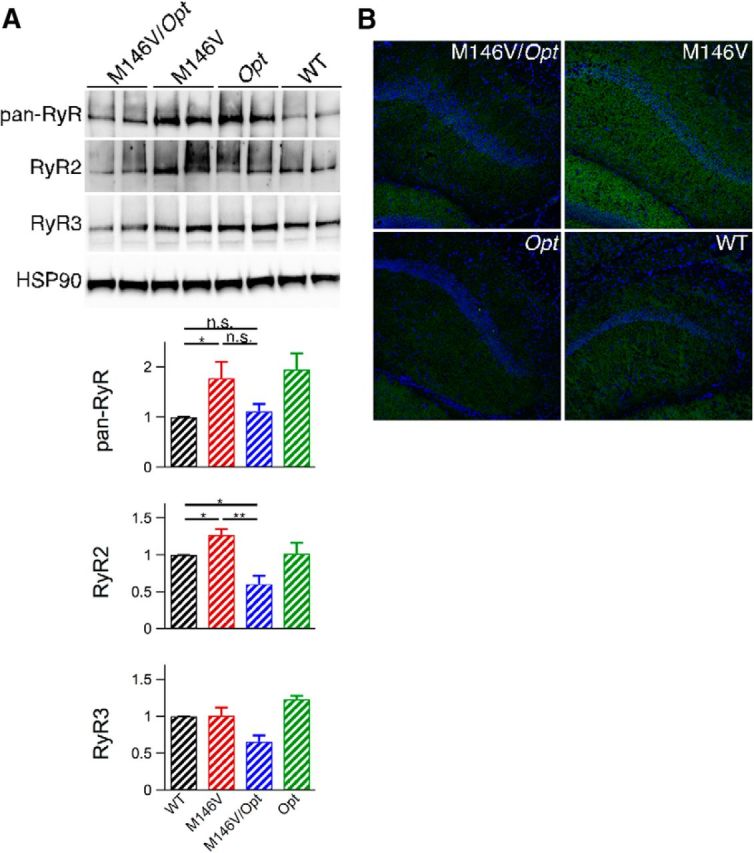
The Opt allele rescues elevated hippocampal RyR protein levels in M146V mice. A, Western blot analyses of hippocampal lysates from 5-week-old animals using a pan-RyR antibody (n = 6 mice each) or antibodies specific to RyR2 (n = 4 mice each) or RyR3 (n = 5 mice each) and heat-shock protein 90 as a loading control. Unpaired two-tailed t test, *p ≤ 0.05, **p ≤ 0.005. Error bars show mean and SEM. B, Immunofluorescence in brain slices from 5-week-old animals using an antibody specific to RyR2. Hippocampal region CA1 is shown.
Enhanced hippocampal activation during associative memory tasks is observed in presymptomatic FAD PS and MCI patients (Dickerson et al., 2005; Reiman et al., 2012). FAD PS expressing mice have been reported to display enhanced hippocampal synaptic potentiation (Auffret et al., 2010) that may be a correlate of the patient phenotypes. Although the molecular basis for the patient phenotypes is unknown, enhanced synaptic potentiation in M146V mice has been suggested to involve altered [Ca2+]i signaling (Auffret et al., 2010). Hippocampal LTP induced by a single tetanus was monitored in CA1 of 5-week-old M146V animals by measuring fEPSP elicited by stimulation of the Schaeffer collateral pathway. There were no differences in basal synaptic transmission among 5-week-old WT, M146V, and M146V/Opt mice (unpaired 2-tailed t test, n ≥ 5 mice each; Fig. 5A,B). Enhanced posttetanic potentiation (PTP) over the first 10 min after tetanus and enhanced LTP over the last 20 min of recording were observed in 5-week-old M146V mice compared with WT (repeated-measures ANOVA, n ≥ 5 mice each, p < 0.005; Fig. 5C). Notably, potentiation in M146V/Opt slices was indistinguishable from that observed in WT (Fig. 5C). Similar results were obtained in adult (3-month-old) mice (Fig. 5D–F). Consistent findings in 5-week-old and 3-month-old M146V mice indicate that developmental differences do not underlie FAD PS-associated enhanced synaptic potentiation. Therefore, enhanced RyR2 expression and hippocampal synaptic potentiation, abnormalities reminiscent of those reported in presymptomatic FAD PS carriers and patients suffering from MCI, are features present in M146V mice. Importantly, both phenotypes are corrected by a single Opt allele.
Figure 5.

The Opt allele rescues enhanced hippocampal potentiation in M146V mice. A, B, Basal synaptic transmission in hippocampal region CA1 as measured by slope of the input-output curve, the ratio of fEPSP slope to PFV amplitude at each applied voltage, and the maximum evoked fEPSP slope for slices from 5-week-old mice. Unpaired two-tailed t test, n ≥ 6 mice each, no significant differences observed. C, Hippocampal LTP resulting from a single 100 Hz, 1 s tetanus applied at t = 0 in slices from 5-week-old mice. The average of the baseline fEPSPs (black) and those recorded during the last 20 min (red) is presented. Comparisons with WT were made over the first 10 min after tetani and over the last 20 min of the recording. Repeated-measures ANOVA, n ≥ 5 mice each, p < 0.005. D, E, Basal synaptic transmission in hippocampal region CA1 as measured by slope of the input-output curve, the ratio of fEPSP slope to PFV amplitude at each applied voltage, and the maximum evoked fEPSP slope for slices from 3-month-old mice. Unpaired two-tailed t test, n ≥ 6 mice each, no significant differences observed. F, Hippocampal LTP resulting from a single 100 Hz, 1 s tetanus applied at t = 0 in slices from 3-month-old mice. The average of the baseline fEPSPs (black) and those recorded during the last 20 min (red) is presented. Comparisons with WT were made over the first 10 min after tetani and over the last 20 min of the recording. Repeated-measures ANOVA, n = 5 mice each, *p < 0.05, **p < 0.005. Error bars show mean and SEM.
Enhanced kinase activities and hippocampal transcription factor activation in M146V mice is attenuated by the Opt allele
We previously observed constitutive activation of Ca2+-dependent CaM kinase IV (CaMKIV) and CREB and elevated protein levels of CREB-dependent genes in whole-brain lysates from M146V mice (Müller et al., 2011). CREB-dependent gene expression is important for the conversion of transient to long-lasting potentiation (Yin and Tully, 1996), suggesting that its constitutive activation may underlie the observed enhancement of hippocampal synaptic potentiation in M146V mice. The proportion of total CaMKIV phosphorylated at Thr-196 (P-CaMKIV), a Ca2+-dependent autophosphorylation that activates its kinase activity (Selbert et al., 1995), was elevated in hippocampal lysates from 5-week-old and 3-month-old M146V mice compared with WT (unpaired 2-tailed t test, n ≥ 5 mice each, p < 0.05; Fig. 6A–D). Constitutive CaMKIV phosphorylation was confirmed by immunofluorescence, which showed a strong somal P-CaMKIV signal in CA1 neurons of M146V mice compared with WT (unpaired two-tailed t test, n = 3 mice each, p < 0.05; Fig. 6E,G). An antibody specific to CREB phosphorylated on Ser-133 (P-CREB), a phosphorylation that activates its transcriptional activity (Gonzalez and Montminy, 1989), revealed an increased proportion of phosphorylated CREB at both ages compared with WT (unpaired 2-tailed t test, n ≥ 5 mice each, p < 0.05; Fig. 6A–D). In agreement, immunofluorescence showed strong P-CREB nuclear staining in M146V hippocampal neurons compared with WT (unpaired 2-tailed t test, n = 3 mice each, p < 0.05; Fig. 6F,H). CREB-dependent protein expression was also specifically enhanced in M146V hippocampal lysates; nNOS, BDNF, and c-fos were elevated at both ages compared with WT (unpaired 2-tailed t test, n ≥ 5 mice each, p < 0.05; Fig. 6A–D), whereas levels of glyceraldehyde 3-phosphate dehydrogenase, a CREB-independent gene, were similar, as was genomic accessibility as measured by histone-3 acetylation (Turner, 1998; Fig. 6A–D). Therefore, enhanced hippocampal P-CaMKIV, P-CREB, and CREB-dependent gene expression are specifically activated in the hippocampus of M146V mice. Strikingly, a single Opt allele normalized elevated hippocampal P-CaMKIV and P-CREB to WT values at both ages and reduced the strong immunofluorescence of P-CREB and P-CaMKIV observed in M146V hippocampi to WT levels (Fig. 6A–H). Furthermore, hippocampal CREB-dependent gene expression was normalized to WT levels by a single Opt allele at both ages (Fig. 6A–D). These results indicate that genetic correction of exaggerated [Ca2+]i signaling rescues the constitutive activation of this transcriptional pathway.
Reduction of InsP3R1 expression attenuates hippocampal and cortical AD-like pathology in 3xTg AD-mice
The contribution of exaggerated [Ca2+]i signaling to canonical AD pathology was explored in 3- to 18-month-old 3xTg mice. Over this period, hippocampal and cortical human APP (hAPP) protein levels were similar in 3xTg and 3xTg/Opt mice (unpaired 2-tailed t test, n ≥ 3 mice each per age; Figs. 7A, 8A). Strikingly, 3xTg/Opt mice had markedly attenuated levels of hippocampal and cortical Aβ40 and Aβ42 accumulation compared with 3xTg littermates (unpaired 2-tailed t test, n ≥ 4 mice each per age, p ≤ 0.05; Figs. 7B, 8B). To confirm the location of biochemical changes, qualitative IHC analyses were conducted on hippocampal slices from 18-month-old mice using an antibody specific to Aβ42 (12F4)or one that recognizes both Aβ species (6E10). These analyses revealed strong extracellular staining in the 3xTg hippocampus that appeared largely reduced in 3xTg/Opt littermates (Fig. 7C). Antibodies specific to human tau (htau) and P-tau were used to evaluate the effects of the Opt allele on tau pathology. Although htau protein levels were similar in hippocampal and cortical lysates from 3-month-old 3xTg and 3xTg/Opt littermates, they were decreased in older 3xTg/Opt mice (unpaired two-tailed t test, n ≥ 3 mice each per age, p < 0.05; Figs. 7A, 8A). This reduction was not due to differences in transgene transcription, as real-time reverse transcriptase PCR did not identify differences in cortical htau mRNA levels between 6-month-old 3xTg and 3xTg/Opt (unpaired two-tailed t test, n = 3 mice each; Fig. 8C). Western blot of hippocampal lysates using P-tau specific antibodies (AT8: Ser-202; AT180: Thr-231) revealed pathologic P-tau in 3xTg mice 12-month and older, which was strongly attenuated by the Opt allele (unpaired two-tailed t test, n ≥ 3 mice each per age, p < 0.05; Fig. 7A). Pathological tau phosphorylation was not detected in 3xTg cortical lysates at any age. Qualitative IHC analyses of hippocampal slices to visualize the location of these biochemical differences were conducted using antibodies AT8 and AT180, which revealed strong somatic and neuritic immunoreactivity in CA1. This immunoreactivity appeared to be largely reduced in 3xTg/Opt littermates (Fig. 7C). These results indicate that normalization of exaggerated [Ca2+]i signals dramatically attenuates both hippocampal and cortical Aβ accumulation and hippocampal tau pathology in 3xTg mice.
Opt allele delays the onset and attenuates the severity of hippocampal LTP impairments in 3xTg AD-mice
3xTg mice develop impairments in hippocampal CA1 synaptic plasticity by 6-month-of-age, manifested as decreases in PTP and LTP (Oddo et al., 2003). No differences in basal synaptic transmission were noted between 6-month-old WT, 3xTg, and 3xTg/Opt mice (unpaired two-tailed t test, n ≥ 7 mice each; Fig. 9A–D). Following a 20 min baseline recording, four 1 s 100 Hz stimuli with 5 min interstimulus intervals were applied to the Schaeffer collateral pathway. 3xTg mice displayed decreased PTP during the first 20 min posttetani and impaired LTP over the last 20 min of recording compared with WT mice (repeated-measures ANOVA, n ≥ 5 mice each, p < 0.05; Fig. 9E). In contrast, these deficits were not present in 3xTg/Opt mice. To track the duration of the rescue by the Opt allele, similar studies were performed on 9- and 12-month-old animals. No differences in the slopes of the input-output curves were observed between the genotypes at 9 months of age (unpaired 2-tailed t test, n ≥ 5 mice each; Fig. 9F). However, 3xTg slices did display deficits in the ratio of the fEPSP slope to the PFV amplitude at each applied voltage and in the maximum evoked fEPSP slope (unpaired 2-tailed t test, p < 0.05), which were not observed in 3xTg/Opt slices (Fig. 9G,H). After stimulation, slices from 9-month-old 3xTg mice displayed impaired PTP and LTP compared with WT (repeated-measures ANOVA, n ≥ 5 mice each, p < 0.005; Fig. 9J), whereas 3xTg/Opt slices did not. At 12 months of age, no differences in basal synaptic transmission were found between the genotypes (unpaired 2-tailed t test, n ≥ 5 mice each; Fig. 9K–N). Again, impaired PTP and LTP were observed in 3xTg mice compared with WT (repeated-measures ANOVA, n = 5 mice each, p < 0.005; Fig. 9O). Twelve-month-old 3xTg/Opt mice also displayed impaired PTP and LTP compared with WT (repeated-measures ANOVA, n = 5 mice each, p < 0.005). However, these impairments were significantly less (p < 0.005) than those observed in 12-month-old 3xTg slices. These findings indicate that the Opt allele delays the onset and attenuates the severity of hippocampal impairments observed in 3xTg mice.
Figure 9.
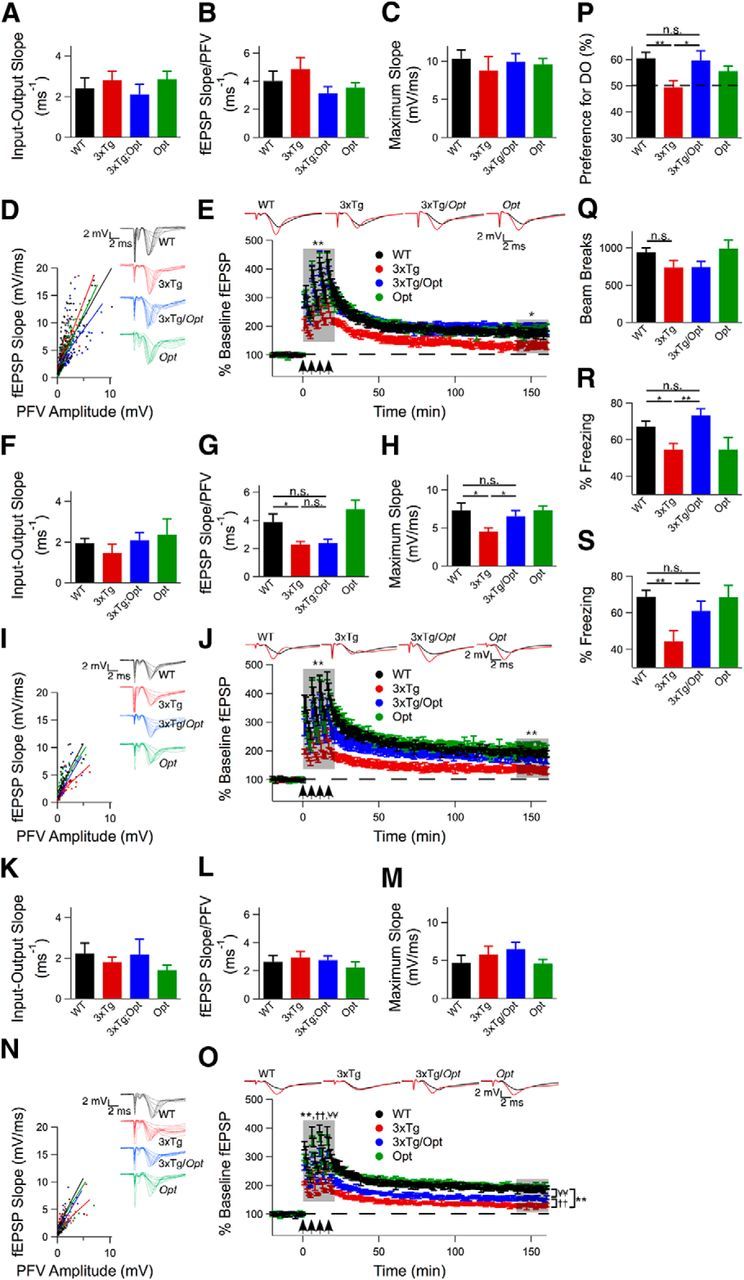
The Opt allele delays the onset and attenuates the severity of hippocampal deficits in 3xTg mice. A–D, Basal synaptic transmission in 6-month-old mice as measured by the slope of the input-output curve, the ratio of the fEPSP slope to the PFV amplitude at each applied voltage, and the maximum evoked fEPSP slope. Unpaired 2-tailed t test, n ≥ 7 mice each, no significant differences observed. E, LTP in hippocampal CA1 of 6-month-old mice induced by 4 100 Hz, 1 s tetani with 5 min interstimulus interval applied at t = 0. The average baseline fEPSPs (black) and those recorded during the last 20 min (red) are presented. Comparisons made over first 20 min after tetani and over the last 20 min of the recording. Repeated-measures ANOVA, n ≥ 5 mice each, *p ≤ 0.05, **p ≤ 0.005. F–I, Basal synaptic transmission in 9-month-old mice as measured by the slope of the input-output curve, the ratio of the fEPSP slope to the PFV amplitude at each applied voltage, and the maximum evoked fEPSP slope. Unpaired 2-tailed t test, n ≥ 5 mice each, p ≤ 0.05. J, LTP in hippocampal CA1 of 9-month-old mice induced by 4 100 Hz, 1 s tetani with 5 min interstimulus interval applied at t = 0. The average baseline fEPSPs (black) and those recorded during the last 20 min (red) are presented. Comparisons made over first 20 min after tetani and over the last 20 min of the recording. Repeated-measures ANOVA, n ≥ 5 mice each, p ≤ 0.005. K–N, Basal synaptic transmission in 12-month old mice as measured by the slope of the input-output curve, the ratio of the fEPSP slope to the PFV amplitude at each applied voltage, and the maximum evoked fEPSP slope. Unpaired two-tailed t test, n ≥ 5 mice each, no significant differences observed. O, LTP in hippocampal CA1 of 12-month-old mice induced by 4 100 Hz, 1 s tetani with 5 min interstimulus interval applied at t = 0. The average baseline fEPSPs (black) and those recorded during the last 20 min (red) are presented. Comparisons made over first 20 min after tetani and over the last 20 min of the recording. Repeated-measures ANOVA,n ≥ 5 mice each, p ≤ 0.005. P, Percentage preference for the DO during spatial object recognition testing of 12- to 13-month-old mice. Dotted line indicates 50% (chance) preference. Unpaired 2-tailed t test, n ≥ 14 mice for WT, 3xTg and 3xTg/Opt, n = 7 mice for Opt, *p ≤ 0.05, **p ≤ 0.005. Q, Ten-minute open-field test conducted on 12- to 13-month-old mice to measure mobility. Unpaired 2-tailed t test, n ≥ 16 mice for WT, 3xTg and 3xTg/Opt, n = 7 mice for Opt, no significant differences observed. R, S, Percentage time spent freezing during a 3 min contextual fear-conditioning probe trial (R) and a cued fear-conditioning probe trial (S). Unpaired 2-tailed t test, n ≥ 13 for WT, 3xTg and 3xTg/Opt, n = 7 for Opt), *p ≤ 0.05, **p ≤ 0.005. Error bars show mean and SEM.
Memory impairments in 3xTg mice are rescued by the Opt allele
Behavioral experiments were conducted on 12- to 13-month-old mice to determine the effects of the Opt allele on memory deficits present in 3xTg mice (Clinton et al., 2007). The SOR test was used to assay hippocampal-dependent spatial memory (Oliveira et al., 2010). Compared with other spatial navigation tests, SOR does not require aversive stimuli to motivate performance, but rather takes advantage of the animal's innate curiosity (Wimmer et al., 2012). Twenty-four hours after training in an arena containing two distinct objects, mice were returned to the arena, but one object was displaced. Preference for the DO was calculated as percentage of exploration time dedicated to it. WT animals demonstrated a preference for the DO (61 ± 2%, n = 16 mice), whereas 3xTg mice did not (49 ± 3%, n = 15 mice), spending less time investigating the DO compared with WT (unpaired two-tailed t test, p < 0.005; Fig. 9P). Diminished preference for the DO was not due to locomotor differences, as determined by open-field testing (unpaired two-tailed t test, n ≥ 16 mice each; Fig. 9Q). Importantly, 3xTg/Opt animals showed a preference for the DO (60 ± 4%, n = 14 mice) that was not different from WT mice (unpaired two-tailed t test; Fig. 9P).
Hippocampal-dependent memory in 3xTg mice was further evaluated by contextual fear-conditioning tests. Twenty-four hours after training, mice were tested for their freezing response during a 3 min period in the conditioning chamber. 3xTg mice spent less time frozen compared with WT (unpaired two-tailed t test, n ≥ 13 mice each, p < 0.05; Fig. 9R), whereas 3xTg/Opt mice did not (unpaired two-tailed t test, n ≥ 16 mice each; Fig. 9R).
Significant AD-like pathology is observed in the amygdala of 3xTg mice (Oddo et al., 2003). To evaluate amygdala-dependent associative memory, mice were subjected to contextual fear-conditioning testing. During a 3 min application of the conditioning tone in the context of a novel chamber, 3xTg mice spent less time frozen compared with WT (unpaired two-tailed t test, n ≥ 16 mice each, p < 0.005; Fig. 9S), whereas 3xTg/Opt mice did not (unpaired two-tailed t test, n ≥ 15 mice each; Fig. 9S).
Together, these experiments strongly suggest that 3xTg mice have hippocampal- and amygdala-dependent memory impairments that are rescued by decreasing InsP3R1 protein levels.
Discussion
The identification of proximal events that predict or drive AD development has remained elusive. Studies of animal models or human patients that express FAD mutant genes may provide the best opportunities to gain insights into the etiologies of the disease. Compelling evidence suggests that FAD PS mutations result in changes in [Ca2+]i signaling in the absence or preceding the appearance of Aβ plaques, indicating that it is an early event that, accordingly, could be involved in disease pathogenesis. Changes in [Ca2+]i signaling can alter transcriptional pathways (Müller et al., 2011), synaptic plasticity (Raymond and Redman, 2006), APP metabolism (Cheung et al., 2008), and activate kinases responsible for tau phosphorylation (Hartigan and Johnson, 1999). Here, we investigated the in vivo mechanism and contribution of enhanced [Ca2+]i signaling to the development of mutant PS-associated FAD. Our results indicate that InsP3R1 contributes to FAD PS-associated exaggerated [Ca2+]i signaling as a proximal event occurring in PCNs and hippocampal neurons from P10–P12 mice months before the appearance of canonical AD phenotypes. Our ex vivo measurements of InsP3R-mediated [Ca2+]i signals were performed in the hippocampal dentate gyrus due to the indicator dye's favorable signal-to-noise ratio in these cells (Namiki et al., 2009). Notably, these measurements were consistent with observations in E14–E16 PCNs (Smith et al., 2005) and hippocampal and cortical neurons in slices from aged animals (Stutzmann et al., 2006; Stutzmann et al., 2007). Our studies demonstrate that reducing InsP3R expression rescues exaggerated [Ca2+]i signaling and its normalization was associated with strongly attenuated AD pathogenesis in two PS FAD mouse models. Our results suggest that enhanced InsP3R-mediated Ca2+ signaling defines a signal transduction pathway that could provide novel therapeutic targets in PS-associated FAD patients.
In vitro studies revealed that FAD PSs modulate gating of the InsP3R receptor by enhancing its open probability (Cheung et al., 2008; Cheung et al., 2010). However, multiple mechanisms have been hypothesized to underlie FAD PS-associated abnormal [Ca2+]i signaling, with contribution of any of the mechanisms to the phenotype in vivo remaining speculative. Here, we demonstrate that reducing InsP3R1 protein levels by ∼50% in M146V and 3xTg mice, both of which harbor the PS1M146V-KIN mutation, normalized exaggerated Ca2+ signaling observed in cortical neurons in vitro and in hippocampal neurons ex vivo. These observations support the hypothesis that exaggerated [Ca2+]i signaling is InsP3R mediated.
The M146V mouse demonstrates phenotypes reminiscent of features observed in presymptomatic FAD PS carriers and MCI patients, including enhanced RyR2 expression (Stutzmann et al., 2006; Bruno et al., 2012). RyR2 has also been proposed to play a role in dysregulated [Ca2+]i homeostasis as a compensatory mechanism in FAD PS mice (Chakroborty et al., 2012). The Opt allele rescued enhanced RyR2 protein levels observed in M146V animals, suggesting that this abnormality is downstream of InsP3R-mediated exaggerated [Ca2+]i signaling. Normalization of RyR levels may have contributed to the rescue of the AD-like phenotypes. Further experimentation is required to determine the specific contributions of each of these pathways.
Enhanced hippocampal activation during the performance of memory tasks, a feature of MCI (Dickerson et al., 2005), is observed in presymptomatic FAD patients (Reiman et al., 2012). Levetiracetam, an anticonvulsant that inhibits intracellular Ca2+ release (Angehagen et al., 2003), reduces hippocampal activation in AD mice (Sanchez et al., 2012) and MCI patients (Bakker et al., 2012) and improves memory performance in both, suggesting that hippocampal hyperactivation is deleterious rather than compensatory. Ca2+ release from intracellular stores plays an important role in hippocampal plasticity (Raymond and Redman, 2006). Enhanced hippocampal synaptic potentiation is observed in M146V mice (Auffret et al., 2010) and several FAD PS transgenic mice (Schneider et al., 2001). This enhancement, which is due to a reduced threshold for potentiation without changes in the maximum potentiation achievable (Schneider et al., 2001), is postulated to be caused by changes in [Ca2+]i signaling (Auffret et al., 2010). The Opt allele rescues M146V-associated enhanced synaptic potentiation, supporting such a hypothesis. Nevertheless, this effect could be indirect, mediated by Ca2+-dependent regulation of downstream signaling pathways, including transcriptional activation (Müller et al., 2011). Indeed, we observed constitutive activation of hippocampal CaMKIV and CREB in M146V mice. CaMKIV overexpression enhances hippocampal synaptic potentiation (Fukushima et al., 2008). CREB is a target of CaMKIV (Matthews et al., 1994) and its activation is important for the conversion of transient to long-lasting synaptic potentiation (Yin and Tully, 1996). Mice harboring constitutively active CREB demonstrate LTP abnormalities similar to those of M146V mice (Barco et al., 2002). The Opt allele rescued CaMKIV and CREB constitutive activation in the M146V mice and reduced elevated protein expression of CREB-dependent genes, including BDNF and nNOS, to WT levels. Both BDNF (Lu et al., 2008) and nNOS (Steinert et al., 2010) have important roles in synaptic plasticity. These findings suggest that enhanced hippocampal potentiation in M146V mice may be due to constitutive activation of the CREB-CaMKIV transcriptional pathway, which is driven by enhanced [Ca2+]i signaling, and identify a molecular mechanism that may underlie enhanced hippocampal activation in presymptomatic FAD PS and MCI patients.
The Opt allele may have effects independent of its normalization of exaggerated [Ca2+]i signaling. InsP3R1 is implicated in modulation of LTP at the CA3-CA1 synapse (Nagase et al., 2003; Taufiq et al., 2005). However, we did not observe effects of the Opt allele alone on LTP induction, possibly because it decreases InsP3R1 levels by ∼50%, whereas previous studies used InsP3R1 KO mice (Nagase et al., 2003) or acute treatment with InsP3R1 antagonists (Taufiq et al., 2005). Although our studies do not rule out the possibility that the Opt allele rescues diminished LTP in 3xTg mice by alleviating LTP suppression, this mechanism does not account for the Opt allele's rescue of enhanced LTP in the M146V mouse. Interestingly, the Opt allele alone was associated with some biochemical changes, notably on RyR expression level, likely as a compensatory response.
The Opt allele strongly attenuated the development of AD-like pathological phenotypes in the 3xTg mouse. The accumulation of Aβ in the hippocampus and cortex from 6- to 18-month-old mice was profoundly reduced by the Opt allele, suggesting that enhanced [Ca2+]i signaling influences APP metabolism in vivo, consistent with previous in vitro studies (Cheung et al., 2008). Because Aβ levels are influenced directly by synaptic activity (Cirrito et al., 2005), early increases in neuronal excitability resulting from exaggerated [Ca2+]i signaling could lead to increased Aβ generation, raising the possibility that life-long [Ca2+]i dysregulation in FAD PS patients may enhance Aβ production and accumulation by promoting APP processing. In addition to reducing the Aβ burden, the Opt allele also attenuated hippocampal P-tau accumulation. The mechanisms by which exaggerated [Ca2+]i signaling impinges on P-tau accumulation remain to be determined, but could involve Ca2+-dependent tau kinases (Hartigan and Johnson, 1999; Noble et al., 2003). In addition, the Opt allele was associated with reduced total htau protein in 3xTg mice. Proteosomal tau degradation is inhibited by Aβ (Tseng et al., 2008). Reduction of Aβ levels by the Opt allele may relieve this inhibition, accounting for the decreased tau levels observed. Notably, reduction of AD-like pathology in 3xTg mice by the Opt allele was correlated with an attenuation of hippocampal LTP and hippocampal- and amygdala-dependent memory impairments. LTP was defective in 6-month-old 3xTg mice, as was observed previously (Oddo et al., 2003). We detected low levels of Aβ in 3xTg hippocampi at this age, whereas P-tau was not detected, consistent with the hypothesis that low [Aβ]-induced synaptic dysfunction is an early manifestation in AD (Oddo et al., 2003). Exaggerated Ca2+ signaling may also contribute to impaired LTP independent of effects on APP metabolism, as suggested by the observation that 9-month-old 3xTg/Opt mice lacked synaptic dysfunction despite having similar hippocampal Aβ loads as 6-month-old 3xTg mice. Ca2+ enhances formation of Aβ oligomers (Itkin et al., 2011), the Aβ species thought to be responsible for impaired LTP maintenance in vivo (Walsh et al., 2002).
We have shown that WT levels of InsP3R1 protein are necessary for exaggerated [Ca2+]i signaling linked to the PS1M146V mutation in two FAD mouse models. Importantly, we have implicated enhanced Ca2+ release as a proximal event in vivo that contributes to MCI-like phenotypes in M146V mice and AD-like phenotypes in 3xTg mice. It is possible that rescue by the Opt allele of AD-like phenotypes observed in both mouse models may be caused by additional factors associated with the expression of only one InsP3R allele. Nevertheless, these results suggest that targeting the InsP3 signaling pathway could be considered a potential therapeutic strategy for patients harboring mutations in PS linked to AD.
Do these results have implications for mutant APP-associated FAD or for SAD? The etiologies of these diseases are unknown and likely quite variable. Altered Ca2+ signaling has been observed in mutant APP models that do not express FAD PS (Oulès et al., 2012). Intracellular oligomeric Aβ activates phospholipase C, triggering InsP3R-mediated Ca2+ release (Demuro and Parker, 2013). Therefore, it is possible that environmental influences and genetic susceptibilities that alter [Ca2+]i signaling play a role in the development of mutant APP-associated FAD and SAD.
Footnotes
This work was supported by the National Institutes of Health (Grant MH059937 to J.K.F. and National Research Service Award Grant AG038240 to D.S.) We thank M. Mattson for PS1M146V-KIN mice; H. Wei for 3xTg mice; J. Chen for Opt mice; R. Neumar for anti-InsP3R1 antibody; the University of Pennsylvania Neurobehavioral Testing Core for assistance; and M. Bridi, A. Park, S. Baratono, K. Cheung, E. Goldberg, M. Wimmer, W. O'Brien, A. Siebert, K. Kopil, and V. Lee.
The authors declare no competing financial interests.
References
- Angehagen M, Margineanu DG, Ben-Menachem E, Rönnbäck L, Hansson E, Klitgaard H. Levetiracetam reduces caffeine-induced Ca2+ transients and epileptiform potentials in hippocampal neurons. Neuroreport. 2003;14:471–475. doi: 10.1097/00001756-200303030-00035. [DOI] [PubMed] [Google Scholar]
- Auffret A, Gautheron V, Mattson MP, Mariani J, Rovira C. Progressive age-related impairment of the late long-term potentiation in Alzheimer's disease presenilin-1 mutant knock-in mice. J Alzheimers Dis. 2010;19:1021–1033. doi: 10.3233/JAD-2010-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/S0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/S0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer's disease. Lancet Neurol. 2013;12:92–104. doi: 10.1016/S1474-4422(12)70259-4. [DOI] [PubMed] [Google Scholar]
- Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2012;33:1001.e1–6. doi: 10.1016/j.neurobiolaging.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, Stutzmann GE. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer's disease. PLoS One. 2012;7:e52056. doi: 10.1371/journal.pone.0052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, McGaugh JL, LaFerla FM. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 2007;28:76–82. doi: 10.1016/j.nbd.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I. Cytotoxicity of intracellular abeta42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J Neurosci. 2013;33:3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etcheberrigaray R, Hirashima N, Nee L, Prince J, Govoni S, Racchi M, Tanzi RE, Alkon DL. Calcium responses in fibroblasts from asymptomatic members of Alzheimer's disease families. Neurobiol Dis. 1998;5:37–45. doi: 10.1006/nbdi.1998.0176. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Maeda R, Suzuki R, Suzuki A, Nomoto M, Toyoda H, Wu LJ, Xu H, Zhao MG, Ueda K, Kitamoto A, Mamiya N, Yoshida T, Homma S, Masushige S, Zhuo M, Kida S. Upregulation of calcium/calmodulin-dependent protein kinase IV improves memory formation and rescues memory loss with aging. J Neurosci. 2008;28:9910–9919. doi: 10.1523/JNEUROSCI.2625-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi T, Simon-Chazottes D, Fujino I, Yamada N, Hasegawa M, Miyawaki A, Yoshikawa S, Guénet JL, Mikoshiba K. Widespread expression of inositol 1,4,5-trisphosphate receptor type 1 gene (Insp3r1) in the mouse central nervous system. Receptors Channels. 1993;1:11–24. [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Johnson GV. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase 3beta-dependent pathway. J Biol Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- Honarnejad K, Herms J. Presenilins: role in calcium homeostasis. Int J Biochem Cell Biol. 2012;44:1983–1986. doi: 10.1016/j.biocel.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Itkin A, Dupres V, Dufrêne YF, Bechinger B, Ruysschaert JM, Raussens V. Calcium ions promote formation of amyloid beta-peptide (1–40) oligomers causally implicated in neuronal toxicity of Alzheimer's disease. PLoS One. 2011;6:e18250. doi: 10.1371/journal.pone.0018250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89:312–323. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107–6116. doi: 10.1128/MCB.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meberg PJ, Miller MW. Culturing hippocampal and cortical neurons. Methods in Cell Biol. 2003;71:111–127. doi: 10.1016/S0091-679X(03)01007-0. [DOI] [PubMed] [Google Scholar]
- Mullane K, Williams M. Alzheimer's therapeutics: continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem Pharmacol. 2013;85:289–305. doi: 10.1016/j.bcp.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Müller M, Cárdenas C, Mei L, Cheung KH, Foskett JK. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer's disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci U S A. 2011;108:13293–13298. doi: 10.1073/pnas.1109297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase T, Ito KI, Kato K, Kaneko K, Kohda K, Matsumoto M, Hoshino A, Inoue T, Fujii S, Kato H, Mikoshiba K. Long-term potentiation and long-term depression in hippocampal CA1 neurons of mice lacking the IP(3) type 1 receptor. Neuroscience. 2003;117:821–830. doi: 10.1016/S0306-4522(02)00803-5. [DOI] [PubMed] [Google Scholar]
- Namiki S, Sasaki T, Matsuki N, Ikegaya Y. Regional difference in stainability with calcium-sensitive acetoxymethyl-ester probes in mouse brain slices. Int J Neurosci. 2009;119:214–226. doi: 10.1080/00207450802330819. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/S0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oliveira AM, Hawk JD, Abel T, Havekes R. Post-training reversible inactivation of the hippocampus enhances novel object recognition memory. Learn Mem. 2010;17:155–160. doi: 10.1101/lm.1625310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Bréchot P, Trebak M, Checler F, Benfenati F, Chami M. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012;32:11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol. 2006;570:97–111. doi: 10.1113/jphysiol.2005.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, Langbaum JB, Kosik KS, Tariot PN, Lopera F. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider I, Reverse D, Dewachter I, Ris L, Caluwaerts N, Kuiperi C, Gilis M, Geerts H, Kretzschmar H, Godaux E, Moechars D, Van Leuven F, Herms J. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem. 2001;276:11539–11544. doi: 10.1074/jbc.M010977200. [DOI] [PubMed] [Google Scholar]
- Selbert MA, Anderson KA, Huang QH, Goldstein EG, Means AR, Edelman AM. Phosphorylation and activation of Ca2+-calmodulin-dependent protein kinase IV by Ca2+-calmodulin-dependent protein kinase Ia kinase. Phosphorylation of threonine 196 is essential for activation. J Biol Chem. 1995;270:17616–17621. doi: 10.1074/jbc.270.29.17616. [DOI] [PubMed] [Google Scholar]
- Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer's disease. J Neurochem. 2005;94:1711–1718. doi: 10.1111/j.1471-4159.2005.03332.x. [DOI] [PubMed] [Google Scholar]
- Steinert JR, Chernova T, Forsythe ID. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist. 2010;16:435–452. doi: 10.1177/1073858410366481. [DOI] [PubMed] [Google Scholar]
- Street VA, Bosma MM, Demas VP, Regan MR, Lin DD, Robinson LC, Agnew WS, Tempel BL. The type 1 inositol 1,4,5-trisphosphate receptor gene is altered in the opisthotonos mouse. J Neurosci. 1997;17:635–645. doi: 10.1523/JNEUROSCI.17-02-00635.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer's mouse models. Ann N Y Acad Sci. 2007;1097:265–277. doi: 10.1196/annals.1379.025. [DOI] [PubMed] [Google Scholar]
- Taufiq AM, Fujii S, Yamazaki Y, Sasaki H, Kaneko K, Li J, Kato H, Mikoshiba K. Involvement of IP3 receptors in LTP and LTD induction in guinea pig hippocampal CA1 neurons. Learn Mem. 2005;12:594–600. doi: 10.1101/lm.17405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. Histone acetylation as an epigenetic determinant of long-term transcriptional competence. Cell Mol Life Sci. 1998;54:21–31. doi: 10.1007/s000180050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wimmer ME, Hernandez PJ, Blackwell J, Abel T. Aging impairs hippocampus-dependent long-term memory for object location in mice. Neurobiol Aging. 2012;33:2220–2224. doi: 10.1016/j.neurobiolaging.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Abeta peptides in 3xTg-AD mice: implications for tau versus Abeta-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Woods NK, Padmanabhan J. Neuronal calcium signaling and Alzheimer's disease. Adv Exp Med Biol. 2012;740:1193–1217. doi: 10.1007/978-94-007-2888-2_54. [DOI] [PubMed] [Google Scholar]
- Yin JC, Tully T. CREB and the formation of long-term memory. Curr Opin Neurobiol. 1996;6:264–268. doi: 10.1016/S0959-4388(96)80082-1. [DOI] [PubMed] [Google Scholar]