

Abstract
Apigenin (4′,5,7,-trihydroxyflavone), an anticancer agent, selectively toxic to cancer cells induces cell cycle arrest and apoptosis through mechanisms that have not been fully elucidated. Our studies indicate that apigenin-mediated growth inhibitory responses are due to inhibition of class I histone deacetylases (HDACs) in prostate cancer cells. Treatment of PC-3 and 22Rv1 cells with apigenin (20–40μM) resulted in the inhibition of HDAC enzyme activity, specifically HDAC1 and HDAC3 at the protein and message level. Apigenin-mediated HDAC inhibition resulted in global histone H3 and H4 acetylation, as well as localized hyperacetylation of histone H3 on the p21/waf1 promoter. A corresponding increase was observed in p21/waf1 and bax protein and mRNA expression after apigenin exposure, consistent with the use of HDAC inhibitor, trichostatin A. The downstream events demonstrated cell cycle arrest and induction of apoptosis in both cancer cells. Studies of PC-3 xenografts in athymic nude mice further demonstrated that oral intake of apigenin at doses of 20 and 50μg/mouse/day over an 8-week period resulted in a marked reduction in tumor growth, HDAC activity, and HDAC1 and HDAC3 protein expression at both doses of apigenin. An increase in p21/waf1 expression was observed in apigenin-fed mice, compared to the control group. Furthermore, apigenin intake caused a significant decrease in bcl2 expression with concomitant increase in bax, shifting the bax/bcl2 ratio in favor of apoptosis. Our findings confirm for the first time that apigenin inhibits class I HDACs, particularly HDAC1 and HDAC3 and its exposure results in reversal of aberrant epigenetic events that promote malignancy.
Keywords: prostate cancer, apigenin, epigenetics, histone modification, chromatin remodeling
Introduction
Epigenetic modifications play an important role in the deregulation of gene expression during cancer development (1–3). The deregulation of genes has been associated with acetylation of histone proteins by histone deacetylases (HDACs) and histone acetyltransferases (HATs), the two opposing group of enzymes involved in chromatin remodeling. HATs catalyze histone acetylation on the ε-amino groups of lysine residues in the N-terminal tails of core histones, neutralizing the positive charge and facilitating the binding of transcription factors to nucleosomal DNA, thereby enhancing its transcription (4, 5). In contrast, HDACs catalyze deacetylation by cleavage of acetyl groups, typically producing a compact chromatin configuration that restricts transcription factor access to DNA and repressing gene expression. Studies of HDAC inhibitors indicate that they facilitate re-expression of epigenetically silenced genes, a property that suggests they may be useful in the field of cancer chemoprevention (6).
HDACs are sub-categorized into 4 classes, based on their sequence homology, as follows: class I (HDACs1-3 and 8), class II (HDACs 4–7 and HDACs 9–10), class III (Sirt1-Sirt7) and class IV (HDAC11). Class I contain a deacetylase domain and are the homologs of yeast RPD3, whereas class II HDACs are homologs of yeast Hda1 (7, 8). Class III (Sirt1-Sirt7) HDACs are homologs of yeast Sir2 (silent mating type information regulation 2) and form a structurally distinct class of NAD-dependent enzymes, whereas class IV HDAC consists of HDAC11 having properties of both class I and class II HDACs (7, 8). Class I HDACs are frequently over-expressed in various human cancers (9). Studies have shown that class I HDACs are highly expressed in prostate cancers compared to high-grade prostatic intraepithelial neoplasia (HGPIN) and adjacent normal prostate tissue (10, 11). HDAC1 and HDAC3 are highly expressed in prostate cancer and HDAC2 has been shown to be associated with shorter prostate-specific antigen (PSA) relapse time (11). In addition, class I HDACs are expressed at higher levels in several human prostate cancer cell lines, compared to benign prostatic hyperplasia cells (12, 13). Over-expression of HDAC1 in prostate cancer PC-3 cells causes increased cell proliferation and reduction in cell differentiation markers (13, 14). Importantly, inhibitors of HDAC such as trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), valproic acid, depsipeptide and sodium butyrate are effective in inhibiting cell proliferation in experimental models of cancer by reactivation of p21/waf1 resulting in cell cycle arrest (15, 16).
Epidemiologic studies suggest that intake of plant flavonoids may lower the overall risk of various cancers, including prostate cancer (17–19). Apigenin (4′ 5, 7,-trihydroxyflavone) is a plant flavone abundantly present in common fruits and vegetables such as grapefruit, parsley, chamomile, and wheat sprouts (20). Apigenin has been identified as an effective chemoprotective agent inhibiting multiple signal transduction pathways such as NF-κB, IGF-I axis, PI3K-Akt, HIF-1α, and β-catenin in various experimental models of prostate cancer (20–25). Studies have shown that apigenin-induced prostate cancer cell death is initiated by generation of reactive oxygen species and p53 activation (26). Apigenin has been shown to modulate MAPK, PI3K-Akt and loss of cyclin D1 associated with dephosphorylation of the tumor suppressor, retinoblastoma (27). Apigenin inhibits focal adhesion kinase and Src kinase resulting in decrease in cell motility and cytoskeleton remodeling in highly invasive prostate cancer cells (28). We have previously demonstrated that apigenin treatment of prostate cancer cells caused arrest in the G0-G1 phase of the cell cycle, a function that was associated with increased levels of p21/waf1 and bax proteins (29). Additionally, there was evidence for the loss of bcl-2 expression and increase in multicaspase activity, resulting in apoptosis (30). Based on these findings, we sought to determine whether apigenin has ability to suppress HDAC levels in human prostate cancer cells. Our results indicate that apigenin inhibits class I HDACs, particularly HDAC1 and HDAC3 expression, resulting in increased acetylation of histone H3 and H4, enhancing accessibility of the promoter region of the p21/waf1 gene causing cell cycle arrest in prostate cancer cells, and inhibiting tumor growth of PC-3 xenograft in nude mice. These findings provide evidence that apigenin may exert its chemopreventive effects, at least in part, through inhibition of class I HDACs.
Materials & Methods
Cell lines and treatments
Human prostate cancer cell lines 22Rv1 and PC-3, obtained from American Type Culture Collection (Manassas, VA) were maintained in RPMI 1640 containing glutamine (Lonza Walkersville Inc., Walkersville, MD) with 10% and 5% FBS, respectively, supplemented with penicillin and streptomycin in a humidified incubator at 37°C with an atmosphere of 5% CO2. Cells treatments were provided as follows: 20 μM or 40 μM apigenin (Sigma, St. Louis, MO) and 20 ng/ml or 80 ng/ml trichostatin A (Sigma) for 24 h. The final concentration of the vehicle DMSO (Sigma) did not exceed 0.1% in all the treatments.
HDAC activity assay
HDAC activity was measured with the HDAC Fluorometric Cellular Activity Assay Kit (Biomole, Plymouth Meeting, PA) in fluorometric 96-well plates according to the manufacturer’s protocol with slight modification. Briefly, 5μg of nuclear cell lysate from 22Rv1 and PC-3 treated as mentioned earlier was taken in 96-well plates in triplicates and HDAC assay buffer added to a final volume of 25μl, followed by the addition of 25 μl 1X Fluor de Lys substrate and shaking for 20 min. 50 μl of the Fluor de Lys developer containing 2 μM TSA was then added, shaken for 10 min and the fluorescence was read in a microplate reading fluorimeter at an excitation wavelength of 360 nm and emission wavelength of 460nm.
Cell cycle analysis
Asynchronized (70–80%) confluent cells were treated with 20 and 40 μM apigenin for 24 h. After treatment cells were collected, washed twice with chilled PBS and spun in a cold centrifuge at 600×g for 10 min. The pellet was fixed and resuspended in 50 μl PBS and 450 μl chilled methanol for 1 h at 4°C. The cells were washed twice with PBS at 600×g for 5 min and again suspended in 500 μl PBS and incubated with 5 ml RNase (20 μg/ml final concentration) for 30 min at 37°C. The cells were chilled over ice for 10 min and stained with propidium iodide (50 μg/ml final concentration) for 1 h and analyzed by flow cytometry and evaluated using Cell Quest & ModFit cell cycle analysis software.
Detection of apoptosis
Apoptosis was assayed in control and treated 22Rv1 and PC-3 cells by staining with Annexin V-FITC using the staining protocol provided by the manufacturer and analyzed on EPICS-XL MCL flow cytometer.
Isolation of RNA, RT-PCR and q-PCR reactions
Total RNA was extracted from 22Rv1 and PC3 cells (untreated and cells treated with 20 μM, 40 μM apigenin, 20 ng/ml TSA for 24 h) with the RNAqueous-4PCR Kit (Applied Biosystems, Foster City, CA) as per the manufacturers’ protocol and quantitated on Nanodrop (Thermo Scientific, Wilmington, DE). 500 ng RNA was amplified with random primers in the Superscript III First-Strand Synthesis Supermix (Invitrogen, Carlsbad CA) as per the manufacturers’ protocol. p21/waf1 was amplified using the forward primer 5′-GTTCCTTGTGGAGCCGGAGC-3′ and the reverse primer 5′-GGTACAAGACAGTGACAGGTC-3′; bax forward primer 5′-GGCCCACCAGCTCTGAGCAGA-3′ and the reverse primers 5′-GCCA CGTGGGCGTCCCAAAGT-3′, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward primer 5′-CAACGGATTTGGTCGTATTGG-3′; reverse primer 5′-GCAACAATATCCACTTTACCAGA GTTAA-3′) using Px2 thermal cycler (Thermo Electron Corp.).
Levels of HDAC1 and HDAC3 mRNA were quantified by real-time PCR and normalized to GAPDH after validating that the efficiency for both HDAC1 and HDAC3 and the endogenous control GAPDH was −1.1. Taqman gene expression assay kits for HDAC1 (Hs00606262_g1), HDAC3 (Hs00187320_m1) and GAPDH (4333764T) were used as per the manufacturer’s protocol (Applied Biosystems, Foster City, CA) in 20μL reactions. All reactions were performed in triplicate and standard deviation calculated using the Comparative Ct method (ΔΔCt Method, ABI PRISM 7500 Real-Time PCR Software). The mean expression levels are represented as the ratio between the HDAC1/GAPDH and HDAC3/GAPDH expression.
Western blot analysis
Cells from treated and control groups were lysed in RIPA buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% SDS in PBS) to prepare whole cell lysate and protein was estimated by Bradford Protein Assay Reagent (Bio-Rad Laboratories, Hercules, CA) using the manufacturer’s protocol. 40 μg of cell lysate was resolved in 4–20% Tris-glycine polyacrylamide gel, transferred onto nitrocellulose membrane, blocked in 5% nonfat dry milk and probed using appropriate primary antibodies for anti-HDAC1 (sc-7872), anti-HDAC3 (sc-11417), anti-bax (sc-493), anti-p53 (sc-126), anti-bcl2 (sc-7382), anti-β-actin (sc-1616) from Santa Cruz Biotechnology, Santa Cruz, CA, and p21/waf1 (MS-891-P1) from Neomarkers Inc. Fremont, CA, in the blocking buffer. The membrane was then incubated with appropriate secondary antibody conjugated with horseradish peroxidase followed by detection using an enhanced chemiluminescence kit (Amersham Life Sciences Inc.).
Acid extraction and expression of acetylated histone proteins
Histone proteins were extracted as per the protocol provided by Upstate-Millipore (Temecula, CA). Briefly 70% confluent, treated and control 22Rv1 and PC3 cells were scraped, collected by centrifugation at 200×g for 10 min followed by a 10 ml PBS wash, centrifuged at 200×g for 10 min and suspended in lysis buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1.5 mM PMSF). Hydrochloric acid was added to a final concentration of 0.2 M and lysates incubated on ice for 30 min followed by centrifugation at 11,000×g for 10 min at 4°C. The supernatant fraction was collected and either stored at −80°C or 100 μl of the fraction dialyzed on MF-Membrane filters (Millipore, Temecula, CA ) twice against 0.1M (0.1N) acetic acid for 30 min each, followed by three dialysis against H2O for 30 min each. The histone extract was stored at −80°C and the protein content quantified by Bradford assay (BioRad) prior to use. 40 μg lysate was immunoblotted and probed with anti-acetyl histone H3 (07-593); anti-acetyl histone H4 (06-598) and total anti-histone H3 (05-928) and H4 (07-108) antibodies purchased from Upstate-Millipore (Temecula, CA).
Chromatin immunoprecipitation assay
Cells were grown to 70–80% confluency in 150 mm dishes and fixed in 1% formaldehyde for 10 min at room temperature and the reaction was quenched by 0.125 M glycine. Fixed cells were washed twice with PBS at 4°C and lysed for 10 min in SDS Lysis Buffer (1%SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0, supplemented with protease inhibitors (1 mM PMSF, 1 mM benzamidine, 1μg/ml pepstatin, 1μg/ml aprotonin, 1μg/ml leupeptin) at 4°C. Chromatin was sheared by sonication on ice for 12 min at the maximum settings on the Bioruptor 200 sonicator (Diagenode, Liège, Belgium) with 1 min pulses followed by 2 min break, followed by centrifugation for 15 min at 15,000 × g. Diluted supernatants were pre-cleared for 2 h at 4°C with 80 μl 50% (v/v) pre-blocked Protein A agarose (Millipore, Temecula, CA) in Immunoprecipitation (IP) Buffer (16.7 mM Tris-Cl pH 8.0, 1.2 mM EDTA, 167 mM NaCl, 1.1% Triton X-100, supplemented with protease inhibitors (1 mM PMSF, 1 mM benzamidine, 1μg/ml pepstatin, 1μg/ml aprotonin, 1 μg/ml leupeptin) and the sonicated chromatin-DNA complex precipitated overnight with anti-acetyl histone H4 (Upstate-Millipore), followed by the addition of 50 μl of 50% v/v pre-blocked Protein A agarose beads for 2 h after which Protein A bound complexes were serially washed twice each with Low–salt Buffer (20 mM Tris-Cl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 1%SDS), High-salt Buffer (20 mM Tris-Cl pH 8.0, 500 mM NaCl, 2 mM EDTA, Triton X-100, 0.1%SDS), LiCl Wash Buffer (10 mM Tris.Cl pH 8.0, 1 mM EDTA 0.25 M LiCl, 1% NP-40, 1% deoxycholic acid) and TE Buffer, pH 8.0. Bound DNA was eluted by incubating the beads in 250 μl Elution Buffer (0.1 M NaHCO3 and 1% SDS) and reverse cross-linked overnight at 65°C. DNA was purified after phenol-chloroform extraction and ethanol precipitation and purified immunoprecipitated DNA was amplified using primers viz. forward primers, 5′-GTGGCTCTGATTGGCTTTCTG-3′, reverse primers 5′-GTGAAAAC AGGCAGCCCAAG-3′flanking the p21/waf1 promoter.
Tumor xenograft studies
PC-3 tumors were grown subcutaneously in athymic nude mice. Approximately, 1×106 PC-3 cells suspended in 0.05 ml of medium and mixed with 0.05 ml of matrigel were subcutaneously injected into the left and right flank of each mouse to initiate tumor growth. The animals were equally divided into three groups. The first group received only 0.2 ml of vehicle material by gavage daily and served as a control group. The second and third groups of animals received 20 and 50 μg/mouse/day doses of apigenin in vehicle, respectively, for 8 weeks. The dose of 20 and 50μg/day apigenin used in our studies corresponds to consumption of approximately 50 and 120 mg/day of daily flavonoid consumption by an adult human, an intake that results in effective physiologically attainable serum concentrations of flavonoids in humans (31). Apigenin intake was started 2 weeks before cell inoculation and was continued for 8 weeks. Animals were monitored daily, and their body weights were recorded weekly throughout the studies. Once the tumors started growing, their sizes were measured twice weekly in two dimensions with calipers. At the termination of the experiment, tumors were excised and weighed to record wet tumor weight. A portion of the tumors from control and treated animals was used for preparation of tumor lysate used in further experiments.
Statistical analysis
Changes in tumor volume and body weight during the course of the experiments were visualized by scatter plot. Differences in tumor volume (mm3) and body weight at the termination of the experiment among various groups were examined using analysis of variance (ANOVA) followed by Tukey’s multiple comparison procedure. The statistical significance of differences between control and treatment group was determined by simple ANOVA followed by multiple comparison tests. All tests were two-tailed and P values less than 0.05 were considered to be statistically significant.
Results
Apigenin decreases HDAC activity in human prostate cancer cells
Dietary agents that inhibit HDAC enzyme activity and increase histone acetylation have promising chemopreventive potential (32, 33). We first sought to determine whether apigenin has the ability to act as an HDAC inhibitor in prostate cancer cells. The effects of apigenin on HDAC activity in prostate cancer cells were compared to those induced by a known HDAC inhibitor, TSA. Exposure of PC-3 cells to 20- and 80- ng/ml TSA resulted in 33% and 67% decrease in HDAC activity. Similar result although of lower magnitude was observed in 22Rv1 cells, in which TSA exposure caused 5% and 19% decrease in HDAC activity. Exposure of PC-3 cells to 20- and 40- μM apigenin resulted in 41% and 62% decrease in HDAC activity, whereas 8% and 22% reductions in HDAC activity were observed in 22Rv1 cells after apigenin treatment (Figure 1).
Figure 1.
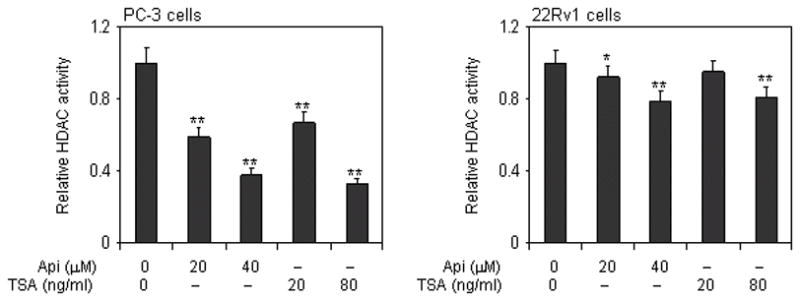
Effect of apigenin and HDAC inhibitor TSA on class I HDAC activity in human prostate cancer cells. Panel 1, Dose-dependent inhibition of HDAC activity in PC-3 cells exposed to 20 μM and 40 μM concentrations of apigenin and TSA (20 and 80 ng/ml) for 24 h Panel 2, Dose-dependent inhibition on HDAC activity in 22Rv1 cells after treatment with various concentrations of apigenin (20 μM and 40 μM) and TSA (20 and 80ng/ml) for 24 h. Data represents the mean ± SD of three different assays. **P < 0.001 versus control. The details are provided in ‘materials and methods’.
Apigenin causes cell cycle arrest and induces apoptosis in human prostate cancer cells
Inhibition of HDAC activity has been shown to cause cell cycle arrest and induction of apoptosis in cancer cells (34). We evaluated the effect of apigenin treatment on cell cycle arrest and apoptosis in prostate cancer cells. Exposure of PC-3 and 22Rv1 cells to apigenin caused a significant decrease in cell viability at these doses, an effect similar to that observed with cells exposed to TSA (data not shown). Analysis of the DNA content by flow cytometry showed that apigenin caused a marked decrease in the percentage of cells in S-phase of the cell cycle. Under the treatment conditions, apigenin treated cells were preferentially arrested in the G0-G1 phase, a finding similar to that observed after treatment of cells with HDAC inhibitor, TSA (35). Treatment of PC-3 cells with 20 μM and 40 μM apigenin resulted in 57.4% and 60.8% arrest of cells in G0-G1 phase, compared to 47.6% in untreated cells, whereas 61.3% and 62.1% G0-G1 phase arrest was observed in 22Rv1 cells compared to 54.7% in untreated control cells (Figure 2A). The percentage of cells in the G0-G1 phase arrest undergoing apoptosis was determined by measuring the annexin V-FITC staining. A significant increase of annexin V-FITC stained cells was observed after apigenin treatment in both cell lines. Treatment of PC-3 cells with 20 μM and 40 μM apigenin resulted in 5.2% and 10.1% annexin V-FITC positive cells (control, 0.4%) and 9.5% and 11.3% in 22Rv1 cells (control 1.2%), indicative of apoptosis (Figure 2B).
Figure 2.

Effect of apigenin on cell cycle arrest and apoptosis in human prostate cancer cells. A, PC-3 and 22Rv1 cells treated with apigenin (20 μM and 40 μM) for 24h and distribution of cells in different stages of cell cycle was recorded and analyzed using FACS analysis. B, PC-3 and 22Rv1 cells were treated with apigenin (20 and 40μg/ml) for 24h and the number of cells undergoing apoptosis were determined using annexin-V staining. Data represents the mean ± SD of three different assays. *P <0.05, and **P <0.001 versus control. The details are provided in ‘materials and methods’.
Apigenin decreases HDAC expression in human prostate cancer cells
Next we determined whether inhibition of HDAC activity observed after treatment of prostate cancer cells with apigenin could be attributed to downregulation of HDAC1 and HDAC3 expression. We performed Western blotting on the lysates obtained from PC-3 and 22Rv1 cells with and without apigenin treatment. Cells treated with 20 ng/ml TSA were used as positive control. Exposure of cells to apigenin decreased the levels of HDAC1 and HDAC3 in a dose-dependent manner in both these cell lines. Treatment of PC-3 cells with 20- and 40- μM apigenin resulted in 28% and 46% reductions in HDAC1 protein expression, whereas 39% and 52% reductions in HDAC1 were noted in 22Rv1 cells. Similarly, 30% and 48% reductions in HDAC3 protein expression were observed in PC-3 cells, whereas 45% and 22% reductions in HDAC3 were observed in 22Rv1 cells after apigenin exposure (Figure 3A). Apigenin treatment also resulted in significant reductions in mRNA levels of HDAC1 and HDAC3 in PC-3 cells, in a dose-dependent manner. The magnitude of mRNA reductions in the levels of HDAC1 and HDAC3 was modest in 22Rv1 cells after apigenin treatment (Figure 3B).
Figure 3.

Effect of apigenin and HDAC inhibitor TSA on the expression of class I HDACs in human prostate cancer cells. A, Inhibition of HDAC1 and HDAC3 protein expression at 20 μM and 40 μM doses of apigenin and 20 ng/ml HDAC inhibitor TSA for 24 h Panel 1, PC-3 cells and Panel 2, 22Rv1 cells. B, Inhibition of mRNA levels of HDAC1 and HDAC3 at 20 μM and 40 μM doses of apigenin and 20 ng/ml HDAC inhibitor TSA for 24 h Panel 1, PC-3 cells and Panel 2, 22Rv1 cells. Data represents the mean ± SD of three different assays. *P <0.05, and **P <0.001 versus control. The details are provided in ‘materials and methods’.
Apigenin causes increase in histone acetylation in human prostate cancer cells
Next we determined whether decreased levels of HDAC after apigenin treatment resulted in increased acetylation of histone proteins in prostate cancer cells. Western blotting of acid extracted protein from PC-3 and 22Rv1 cells treated with 20 and 40μM apigenin exhibited significant increases in the acetylation of histone H3 and histone H4, compared to untreated controls. 7.4 and 8.2 fold increases were observed in acetylated histone H3, and 1.2 and 2.6 fold increases in acetylated histone H4 in PC-3 cells, after 20- and 40- μM apigenin treatments, respectively. In 22Rv1 cells, 8.5 and 9.6 fold increases in acetylated histone H3, and 1.5 and 1.6 fold increases in acetylated histone H4 were observed after 20 μM and 40 μM apigenin treatment, respectively. The increase in acetylation observed after apigenin exposure for both histone H3 and H4 was higher than the increase observed after treatment of cells with TSA (Figure 4A&B).
Figure 4.
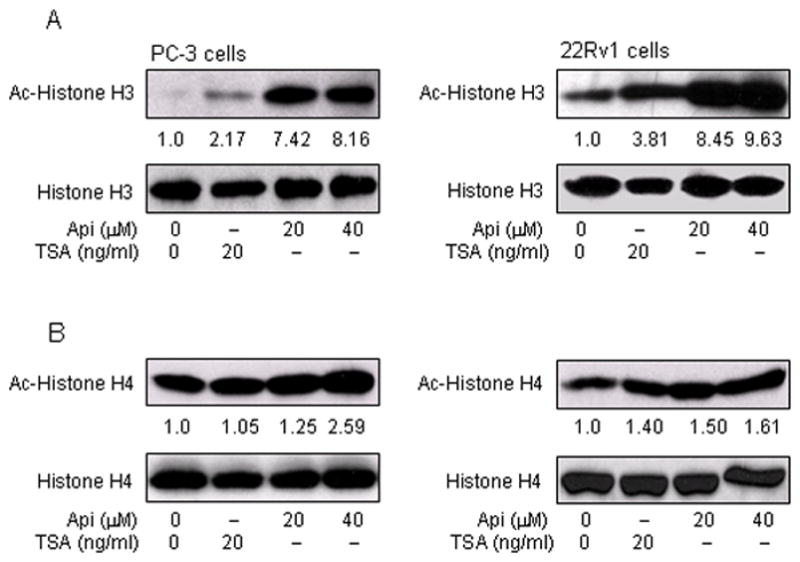
Effect of apigenin on histone H3 and H4 acetylation in human prostate cancer cells. A, Increase in acetylation of histone H3 Panel 1, PC-3 cells and Panel 2, 22Rv1 cells with 20 μM and 40μM doses of apigenin and 20 ng/ml HDAC inhibitor TSA exposed for 24 h. B, Increase in acetylation of histone H4 Panel 1, PC-3 cells and Panel 2, 22Rv1 cells with 20 μM and 40 μM doses of apigenin and 20 ng/ml HDAC inhibitor TSA after 24 h treatment. The details are provided in ‘materials and methods’.
Apigenin causes increase expression of p21/waf1 and bax in human prostate cancer cells
The cell cycle regulator protein p21/waf1and pro-apoptotic protein bax are well established targets of HDAC inhibitors (36). We sought to determine whether inhibition of HDAC by apigenin resulted in increased expression of these molecules. As shown in figure 5A, exposure of PC-3 and 22Rv1 cells to apigenin resulted in significant increases in p21/waf1 and bax protein expression in both cell lines. The level of p21/waf1 was increased by 1.8 and 2.0 fold in PC-3 cells and 3.6 and 3.4 fold in 22Rv1 cells after 20 μM and 40 μM doses of apigenin, respectively. A marked increase in bax protein expression of 3.9 and 11.2 fold in PC-3 cells and 1.7 and 1.8 fold was noted in 22Rv1 cells after 20 μM and 40 μM doses of apigenin treatment, respectively. The increase in p21/waf1 and bax correlated with simultaneous increase in p53 expression in 22Rv1 cells harboring functional p53 whereas no change was observed in PC-3 cells which lacks p53 expression. Furthermore, apigenin treatment resulted in significant increases in mRNA expression of p21/waf1 (2.8 and 5.0 fold in PC-3 cells, and 3.9 and 4.3 fold in 22Rv1 cells) and bax (1.2 and 1.5 fold in PC-3 cells, and 1.2 and 1.4 fold in 22Rv1 cells) in both cell lines in a dose-dependent manner. Similar results were noted after treatment of cells with HDAC inhibitor, TSA (Figure 5B).
Figure 5.

Effect of apigenin on the expression of p21/waf1 and bax in human prostate cancer cells. A, Increase in the protein expression of p21/waf1 and bax at 20 μM and 40 μM doses of apigenin and 20 ng/ml HDAC inhibitor TSA after 24 h treatment Panel 1, PC-3 cells and Panel 2, 22Rv1 cells. B, Increase in the mRNA levels of p21/waf1 and bax at 20 μM and 40 μM doses of apigenin and 20 ng/ml HDAC inhibitor TSA after 24 h exposure Panel 1, PC-3 cells and Panel 2, 22Rv1 cells C, Binding of acetylated H3 to the promoters of p21/waf1 in PC-3 cells. Chromatin immunoprecipitation (ChIP) assay was performed to determine the association of acetylated histone H3 with the promoters of p21/waf1. Apigenin treatment to PC-3 cells causes increased association of acetylated histone H3 to the promoters of p21/waf1. The details are provided in ‘materials and methods’.
Apigenin causes increased binding of acetylated histone H3 with p21/waf1 promoter
Next we sought changes in the acetylation status of histone H3 associated with the promoter region of the p21/waf1 gene. Using anti-acetylated histone H3 antibody followed by PCR with the primers specific for p21/waf1 promoter, ChIP assay was performed. As shown in figure 5C, apigenin treatment resulted in an increase amount of acetylated histone H3 associated with the p21/waf1 promoter. These results correlated with the increase in p21/waf1 expression at the protein and message levels in PC-3 cells after apigenin treatment.
Apigenin intake inhibits tumor growth of PC-3 xenograft in athymic nude mice
Our previous studies have shown the effectiveness of apigenin in cell culture in causing cell cycle arrest and induction of apoptosis in human prostate cancer cells (27, 29). We extended these studies to determine whether these events occur in vivo using a xenograft model. We designed a protocol that simulates a chemoprevention regimen, wherein apigenin was provided at 20- and 50- μg/mouse/day through gavage, beginning 2 weeks prior to cell inoculation and continued for 8 weeks. In this experimental protocol, intake of apigenin inhibited the growth of tumor xenograft at both test doses. As shown in figure 6A&B, tumor volume was inhibited by 41.8% and 50.6% (P < 0.001 and 0.0001) and the wet weight of tumor was decreased by 38.5% and 74.6% (P < 0.001), respectively, at the termination of the experiment. There were no adverse effects of apigenin treatment on animal health, food intake or body weight (data not shown). The average daily intake of food did not differ between the control and treated groups. Moreover, body weight was not significantly different throughout the duration of the study, suggesting that apigenin was essentially non-toxic at the dietary concentrations used in the study.
Figure 6.
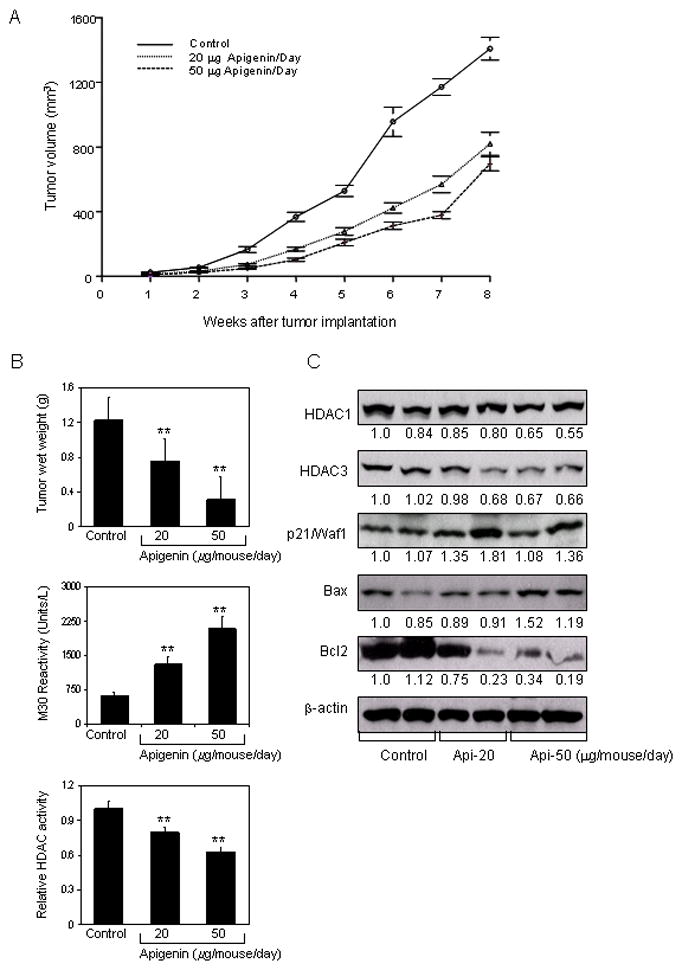
Effect of apigenin intake on PC-3 tumor growth in athymic nude mice and its correlation with downregulation of HDAC activity and expression. Approximately 1×106 cells were injected into both flanks of each mouse to initiate ectopic prostate tumor growth, and apigenin was provided to the animals 2 weeks before cell inoculation mimicking chemoprevention regimen. Mice were fed ad libitum with Teklad 8760 autoclaved high-protein diet. Apigenin was provided with 0.5% methyl cellulose and 0.025% Tween 20 as vehicle to these animals perorally on a daily basis. Group I, control, received 0.2 ml vehicle only, Group II received 20μg apigenin per mouse in 0.2 ml vehicle and Group III received 50μg apigenin per mouse in 0.2 ml vehicle daily for 8 weeks. Once the tumor xenografts started growing, their sizes were measured weekly in two dimensions throughout the study. A, Tumor volume (cubic millimeter) in control and treated groups B, Wet weight of tumors is represented as the mean of 6–8 tumors from each group, quantitative measurement of apoptosis as demonstrated by M30 reactivity, and relative HDAC activity in PC-3 tumors after apigenin intake at the indicated doses. Values are means ± SD, n = 6–8, repeated twice with similar results. **P < 0.001 versus control. C, Immunoblots for HDAC1, HDAC3, p21/waf1, bax and bcl2 in tumor lysates after apigenin intake at the indicated doses. The blots were stripped and reprobed with anti-β-actin antibody to ensure equal protein loading. The details are provided in ‘materials and methods’.
Apigenin intake causes apoptosis in PC-3 tumors through downregulation of HDAC expression
At the termination of the study, xenografts were examined for HDAC activity and the extent of tumor cell apoptosis in the xenografts was also evaluated. Compared to the untreated controls, HDAC activity was significantly inhibited in the xenograft mice fed with apigenin (P <0.001) at both treatment doses, and induction of apoptosis in tumor cells was also significantly increased in tumor xenografts in the same mice (P <0.001) (Figure 6B). We also measured the protein expression of HDAC1 and HDAC3 as an effect of apigenin feeding. As shown in figure 6C, oral intake of apigenin at doses of 20- and 50- μg/mouse/day resulted in marked reduction in the protein expression of HDAC1 and HDAC3 in PC-3 tumor xenografts. A dose-dependent decrease in HDAC1 and HDAC3 expression was observed along with increase in the levels of p21/waf1 and bax proteins. A decrease in the bcl2 protein was observed after apigenin intake and the shift in bax/bcl2 ratio was in favor of apoptosis in tumor xenograft cells.
Discussion
Considerable attention has been focused on the silencing and unsilencing of genes through changes in DNA methylation in cancer (37). However epigenetic modification in DNA is also influenced by events at the histone level. This has led to growing interest in the development of HDAC inhibitors capable of causing post-translational changes associated with histone modification, including deacetylation and acetylation of histone complex (38). In this study we demonstrate, for the first time, that apigenin downregulates class I HDACs and increases acetylation of histone H3 and H4, enhancing accessibility of the promoter region of the p21/waf1, resulting in increased transcription of this gene in prostate cancer cells.
The levels of HDAC activity within cells can be altered via direct inhibition of the HDAC enzyme and changes in HDAC protein levels. For example, crystallographic analysis of a complex formed between a homologue of mammalian HDAC and TSA or SAHA indicated that these inhibitors bind to the catalytic pocket of the enzyme (39). Some HDAC inhibitors such as valproic acid have been shown to mediate degradation of HDAC2 (40). In our studies, we demonstrate that apigenin has the ability to decrease HDAC enzyme activity and can cause downregulation of HDAC1 and HDAC3 at the protein and message levels. These properties may inhibit cancer cell growth in vitro and in vivo. However, more detailed studies are required to determine the precise mechanism of action and specificity of apigenin binding to class I HDACs.
HDAC inhibitors increase the levels of histone acetylation, which facilitates chromatin remodeling and recruitment of transcription factors to the promoters of target genes (41). Studies have shown that HDAC inhibitors butyrate and SAHA alter histone acetylation status and enhance Sp1/Sp3 binding to the promoter region of p21/waf1 (35, 42). Our studies demonstrate similar findings with increase in acetylation of histone H3 and H4 by apigenin. It is tempting to speculate that apigenin-induced p21/waf1 expression results in increased binding of Sp1/Sp3 to its promoter, and this hypothesis is worthy of future study. Simultaneously, deacetylation of histone results in increased acetylation of histone and non-histone proteins (6, 7). The p300/cyclic AMP response element binding protein CBP is associated with acetylation of the target genes with promoter activation upon treatment with HDAC inhibitors (38). The effects of apigenin on CBP/p300 and its associated factor PCAF with respect to their influences on intrinsic HAT activity are also worthy of future study.
A common target for HDAC inhibitors is p21/waf1, which controls transition through the cell cycle via the inhibition of cyclin-dependent kinases (15, 16). In the present study, induction of p21/waf1 by apigenin was associated with increase in p53 expression and arrest in G1 phase of the cell cycle in 22Rv1 cells. Apigenin-mediated p21/waf1 activation was also observed in PC-3 cells, which lack p53, demonstrating that apigenin can cause induction of p21/waf1 which is independent of p53 regulation. Since apigenin has been shown to affect other cell cycle regulators, such as Akt, checkpoint kinase1, c-Jun NH2-terminal kinase, and MAPK signaling axis, it seems reasonable to hypothesize that it may regulate p21/waf1 expression through other mechanisms in p53 null cells.
HDAC inhibitors such as TSA and SAHA at nanomolar concentration inhibit HDAC activity by selectively targeting cancer cells, causing an increase in histone acetylation and re-expression of target genes such as p21/waf1 and bax, triggering cell cycle arrest and apoptosis in cancer cells (34–36). Similar findings have been reported with some dietary polyphenols viz. sulforaphane, phenethyl isothiocyanate, 3, 3′-diindolylmethane, organosulfur compounds derived from garlic and green tea polyphenols which act as weak HDAC inhibitors (43–47). In our studies, dose-dependent reductions in HDAC activity and expression along with increase in the expression of p21/waf1 and bax were observed after apigenin treatment of human prostate cancer cells at micromolar concentrations. These data provide support for the use of apigenin as a dietary HDAC inhibitor and chemopreventive agent which selectively targets cancer cells without affecting normal cells. This selectivity opens new avenues for targeting cancer cells at multiple steps in the carcinogenesis pathway and supports the notion that apigenin may prove to be both an effective chemopreventive as well as a chemotherapeutic agent.
In our cell culture studies, we observed induction of apoptosis, as well as impairment in cell survival, by exposure of prostate cancer cells to apigenin at 20 μM and 40 μM concentrations. Although these findings provide mechanistic insights, demonstration that these effects are also operative in vivo is required to establish a potential for clinical development. Our in vivo studies using 20- and 50- μg/day apigenin administration to mice with prostate cancer xenografts confirmed that apigenin intake significantly inhibited tumor growth, without any apparent signs of toxicity. Consistent with the findings in cell culture, apigenin intake resulted in downregulation of HDAC activity and expression, inactivation of bcl2 and, increase in the levels of p21/waf1 and bax favoring apoptosis, compared with vehicle-treated animals. The doses of apigenin which is effective in vivo is much less that the concentrations used in cell culture studies. Previous studies from our group has shown that oral intake of apigenin at similar doses circulate ~1% of apigenin in the mouse serum, which is consistent with other reported studies documenting ~1.2% of radioactivity after single dose administration of [3H] apigenin to Wistar rats (48). Several studies suggest that flavonoids have limited bioavailability and actively metabolized upon ingestion (49 and references therein). However, regular consumption of flavonoids leads to build up of effective tissue concentration which results in tumor growth inhibition. Because doses that induce antitumor effects in vitro and in vivo often differ, additional studies are needed to determine the specific plasma and intratumoral levels in order to directly correlate reduction in tumor with in vivo apigenin concentration.
In summary we provide the first evidence that apigenin inhibits HDAC1 and HDAC3 in vitro and in vivo with increase in global histone acetylation, as well as localized hyperacetylation of histone H3 on the p21/waf1 promoter. The demonstration that apigenin induces downregulation of constitutive class I HDAC activity and expression provides support for further investigation of its role as a chemopreventive agent, and perhaps as a chemotherapeutic agent, particularly in prostate cancers that manifest HDAC-mediated therapeutic resistance.
Acknowledgments
This work was supported by grants from United States Public Health Services RO1 CA108512 and RO1 AT002709 to SG. We are thankful to Pamela Steele at the Athymic Animal Core Facility for technical assistance and support from the Case Comprehensive Cancer Center grant (P30 CA43703).
Abbreviations
- CBP
CREB-binding protein
- ChIP
chromatin immunoprecipitation
- DNA
deoxyribonucleic acid
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- HGPIN
high-grade prostatic intraepithelial neoplasia
- HIF
hypoxia inducible factor
- IGF
insulin-like growth factor
- MAPKs
Mitogen-activated protein kinases
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PI3K
Phosphatidylinositol 3-kinase
- PSA
prostate-specific antigen
- RIPA
radio-immunoprecipitation assay
- SAHA
suberoylanilide hydroxamic acid
- Sir2
silent mating type information regulation 2
- Sirt
sirtuin
- TSA
trichostatin A
References
- 1.Kanwal R, Gupta S. Epigenetics and cancer. J Appl Physiol. 2010;109:598–605. doi: 10.1152/japplphysiol.00066.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21:502–17. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim MK, Shin JM, Eun HC, Chung JH. The role of p300 histone acetyltransferase in UV-induced histone modifications and MMP-1 gene transcription. PLoS One. 2009;4:e4864. doi: 10.1371/journal.pone.0004864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–95. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abbas A, Gupta S. The role of histone deacetylases in prostate cancer. Epigenetics. 2008;3:300–9. doi: 10.4161/epi.3.6.7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1:19–25. doi: 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–74. [PubMed] [Google Scholar]
- 10.Song Y, Shiota M, Tamiya S, Kuroiwa K, Naito S, Tsuneyoshi M. The significance of strong histone deacetylase 1 expression in the progression of prostate cancer. Histopathology. 2011;58:773–80. doi: 10.1111/j.1365-2559.2011.03797.x. [DOI] [PubMed] [Google Scholar]
- 11.Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M, Kristiansen G. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98:604–10. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waltregny D, North B, Van Mellaert F, de Leval J, Verdin E, Castronovo V. Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. Eur J Histochem. 2004;48:273–90. [PubMed] [Google Scholar]
- 13.Patra SK, Patra A, Dahiya R. Histone deacetylase and DNA methyltransferase in human prostate cancer. Biochem Biophys Res Commun. 2001;287:705–13. doi: 10.1006/bbrc.2001.5639. [DOI] [PubMed] [Google Scholar]
- 14.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–89. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 15.Zupkovitz G, Grausenburger R, Brunmeir R, Senese S, Tischler J, Jurkin J, Rembold M, Meunier D, Egger G, Lagger S, Chiocca S, Propst F, Weitzer G, Seiser C. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010;30:1171–81. doi: 10.1128/MCB.01500-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neuhouser ML. Dietary flavonoids and cancer risk: evidence from human population studies. Nutr Cancer. 2004;50:1–7. doi: 10.1207/s15327914nc5001_1. [DOI] [PubMed] [Google Scholar]
- 18.Lewis JE, Soler-Vilá H, Clark PE, Kresty LA, Allen GO, Hu JJ. Intake of plant foods and associated nutrients in prostate cancer risk. Nutr Cancer. 2009;61:216–24. doi: 10.1080/01635580802419756. [DOI] [PubMed] [Google Scholar]
- 19.Birt DF, Pelling JC, Nair S, Lepley D. Diet intervention for modifying cancer risk. Prog Clin Biol Res. 1996;395:223–34. [PubMed] [Google Scholar]
- 20.Shukla S, Gupta S. Apigenin: a promising molecule for cancer prevention. Pharm Res. 2010;27:962–78. doi: 10.1007/s11095-010-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu L, Zhang L, Bertucci AM, Pope RM, Datta SK. Apigenin, a dietary flavonoid, sensitizes human T cells for activation-induced cell death by inhibiting PKB/Akt and NF-kappaB activation pathway. Immunol Lett. 2008;121:74–83. doi: 10.1016/j.imlet.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shukla S, Gupta S. Apigenin suppresses insulin-like growth factor I receptor signaling in human prostate cancer: an in vitro and in vivo study. Mol Carcinog. 2009;48:243–52. doi: 10.1002/mc.20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang BH. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005;19:342–53. doi: 10.1096/fj.04-2175com. [DOI] [PubMed] [Google Scholar]
- 24.Mirzoeva S, Kim ND, Chiu K, Franzen CA, Bergan RC, Pelling JC. Inhibition of HIF-1 alpha and VEGF expression by the chemopreventive bioflavonoid apigenin is accompanied by Akt inhibition in human prostate carcinoma PC3-M cells. Mol Carcinog. 2008;47:686–700. doi: 10.1002/mc.20421. [DOI] [PubMed] [Google Scholar]
- 25.Shukla S, MacLennan GT, Flask CA, Fu P, Mishra A, Resnick MI, Gupta S. Blockade of beta-catenin signaling by plant flavonoid apigenin suppresses prostate carcinogenesis in TRAMP mice. Cancer Res. 2007;67:6925–35. doi: 10.1158/0008-5472.CAN-07-0717. [DOI] [PubMed] [Google Scholar]
- 26.Shukla S, Gupta S. Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radic Biol Med. 2008;44:1833–45. doi: 10.1016/j.freeradbiomed.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shukla S, Gupta S. Apigenin-induced cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and loss of cyclin D1 associated retinoblastoma dephosphorylation in human prostate cancer cells. Cell Cycle. 2007;6:1102–14. doi: 10.4161/cc.6.9.4146. [DOI] [PubMed] [Google Scholar]
- 28.Franzen CA, Amargo E, Todorovi V, Desai BV, Huda S, Mirzoeva S, Chiu K, Grzybowski BA, Chew TL, Green KJ, Pelling JC. The chemopreventive bioflavonoid apigenin inhibits prostate cancer cell motility through the focal adhesion kinase/Src signaling mechanism. Cancer Prev Res. 2009;2:830–41. doi: 10.1158/1940-6207.CAPR-09-0066. [DOI] [PubMed] [Google Scholar]
- 29.Gupta S, Afaq F, Mukhtar H. Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene. 2002;21:3727–38. doi: 10.1038/sj.onc.1205474. [DOI] [PubMed] [Google Scholar]
- 30.Lu HF, Chie YJ, Yang MS, Lee CS, Fu JJ, Yang JS, Tan TW, Wu SH, Ma YS, Ip SW, Chung JG. Apigenin induces caspase-dependent apoptosis in human lung cancer A549 cells through Bax- and Bcl-2-triggered mitochondrial pathway. Int J Oncol. 2010;36:1477–84. doi: 10.3892/ijo_00000634. [DOI] [PubMed] [Google Scholar]
- 31.Hollman PC, Katan MB. Dietary flavonoids: intake, health effects and bioavailability. Food Chem Toxicol. 1999;37:937–42. doi: 10.1016/s0278-6915(99)00079-4. [DOI] [PubMed] [Google Scholar]
- 32.Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344–9. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meeran SM, Ahmed A, Tollefsbol TO. Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin Epigenetics. 2010;1:101–116. doi: 10.1007/s13148-010-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang MR, Kang JS, Han SB, Kim JH, Kim DM, Lee K, Lee CW, Lee KH, Lee CH, Han G, Kang JS, Kim HM, Park SK. A novel delta-lactam-based histone deacetylase inhibitor, KBH-A42, induces cell cycle arrest and apoptosis in colon cancer cells. Biochem Pharmacol. 2009;78:486–94. doi: 10.1016/j.bcp.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 35.Li GC, Zhang X, Pan TJ, Chen Z, Ye ZQ. Histone deacetylase inhibitor trichostatin A inhibits the growth of bladder cancer cells through induction of p21WAF1 and G1 cell cycle arrest. Int J Urol. 2006;13:581–6. doi: 10.1111/j.1442-2042.2006.01344.x. [DOI] [PubMed] [Google Scholar]
- 36.Lindemann RK, Gabrielli B, Johnstone RW. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle. 2004;3:779–88. [PubMed] [Google Scholar]
- 37.Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22:4632–42. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- 38.Villar-Garea A, Esteller M. Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int J Cancer. 2004;112:171–8. doi: 10.1002/ijc.20372. [DOI] [PubMed] [Google Scholar]
- 39.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–93. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 40.Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Göttlicher M. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003;22:3411–20. doi: 10.1093/emboj/cdg315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steele N, Finn P, Brown R, Plumb JA. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br J Cancer. 2009;100:758–63. doi: 10.1038/sj.bjc.6604932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ammanamanchi S, Brattain MG. Restoration of transforming growth factor-beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J Biol Chem. 2004;279:32620–5. doi: 10.1074/jbc.M402691200. [DOI] [PubMed] [Google Scholar]
- 43.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–74. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 44.Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, Feng J, Liu D, Chiao JW. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol. 2008;33:375–80. [PubMed] [Google Scholar]
- 45.Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70:646–54. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pandey M, Shukla S, Gupta S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int J Cancer. 2010;126:2520–33. doi: 10.1002/ijc.24988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duée PH, Martel P, Chaumontet C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004:251227–36. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 48.Gradolatto A, Basly JP, Berges R, Teyssier C, Chagnon MC, Siess MH, Canivenc-Lavier MC. Pharmacokinetics and metabolism of apigenin in female and male rats after a single oral administration. Drug Metab Dispos. 2005;33:49–54. doi: 10.1124/dmd.104.000893. [DOI] [PubMed] [Google Scholar]
- 49.Ross JA, Kasum CM. Dietary flavonoids: bioavailability, metabolic effects, and safety. Annu Rev Nutr. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]