

Abstract
G-protein-coupled estrogen receptor-30 (GPR30), also known as G-protein estrogen receptor-1 (GPER1), is a putative extranuclear estrogen receptor whose precise functions in the brain are poorly understood. Studies using exogenous administration of the GPR30 agonist, G1 suggests that GPR30 may have a neuroprotective role in cerebral ischemia. However, the physiological role of GPR30 in mediating estrogen (E2)-induced neuroprotection in cerebral ischemia remains unclear. Also unclear is whether GPR30 has a role in mediating rapid signaling by E2 after cerebral ischemia, which is thought to underlie its neuroprotective actions. To address these deficits in our knowledge, the current study examined the effect of antisense oligonucleotide (AS) knockdown of GPR30 in the hippocampal CA1 region upon E2-BSA-induced neuroprotection and rapid kinase signaling in a rat model of global cerebral ischemia (GCI). Immunohistochemistry demonstrated that GPR30 is strongly expressed in the hippocampal CA1 region and dentate gyrus, with less expression in the CA3 region. E2-BSA exerted robust neuroprotection of hippocampal CA1 neurons against GCI, an effect abrogated by AS knockdown of GPR30. Missense control oligonucleotides had no effect upon E2-BSA-induced neuroprotection, indicating specificity of the effect. The GPR30 agonist, G1 also exerted significant neuroprotection against GCI. E2-BSA and G1 also rapidly enhanced activation of the prosurvival kinases, Akt and ERK, while decreasing proapototic JNK activation. Importantly, AS knockdown of GPR30 markedly attenuated these rapid kinase signaling effects of E2-BSA. As a whole, the studies provide evidence of an important role of GPR30 in mediating the rapid signaling and neuroprotective actions of E2 in the hippocampus.
Keywords: Estrogen, Estradiol, GPR30, GPER1, hippocampus, neuroprotection, extranuclear, ischemia
Introduction
17β-Estradiol (E2, estrogen) is a pleotropic hormone with multiple sites of action and effects throughout the body. In the brain, E2 has been implicated to regulate several key processes, including reproductive function, feeding and sexual behavior, cognition and memory, and to exert neurotrophic and neuroprotective functions [1–7]. Classically, E2 has been thought to exert its actions through a genomic mechanism involving binding to either estrogen receptor-α (ER-α) or estrogen receptor-β (ER-β) in the nucleus, with recruitment of cofactors and formation of a transcriptional regulatory complex that leads to regulation of the transcription of a variety of genes [1, 8].
In addition to a genomic mechanism of action, evidence has been mounting for a rapid signaling mechanism for E2 that is mediated by extranuclear estrogen receptors (ERs) [1, 8–11]. Along these lines, E2 has been shown to exert rapid (within minutes) activation of ion channel conductance and kinase signaling pathways in multiple tissues and cell types in the body [6, 12–19]. Furthermore, utilization of cell impermeable conjugates of E2, such as E2-BSA (bovine serum albumin) or E2 dendrimers (EDC), have been shown to rapidly activate kinase signaling pathways in vivo and in vitro, including pro-survival kinases such as extracellular regulated kinases-1,2 (ERK1,2) and Akt [20–24]. For instance, work by our laboratory showed that in vivo administration of E2-BSA or EDC into the lateral cerebroventricle rapidly enhances prosurvival ERK and Akt activation, while inhibiting activation of proapoptotic c-jun-n-terminal kinase (JNK). These rapid signaling effects of the E2 conjugates were correlated with robust neuroprotection in the rat hippocampal CA1 region against global cerebral ischemia (GCI) [20].
An important question is what is the identity of the extranuclear ER that mediates these rapid signaling and neuroprotective effects of E2 in the brain? While classical ERα and ERβ are primarily localized in the nucleus, a number of studies have shown that they can be trafficked and localized to the membrane in neurons [8, 11, 25]. Interestingly, both ERα and ERβ-specific agonists have been shown to exert neuroprotection against cerebral ischemia [26, 27]. In particular, ERα has been implicated to participate in mediating the neuroprotective effects of E2 as evidenced by ER knockout mouse studies [28–30], as well as by ERα antisense knockdown studies [31].
Recently, a novel G-protein-coupled estrogen receptor, GPR30 (also called GPER1 – G-protein estrogen receptor-1) was cloned in the human and implicated to participate in mediating estrogen signaling and actions in various tissues [32–36]. GPR30 has been shown to be localized at extranuclear sites, including the plasma membrane, endoplasmic reticulum, golgi, and dendritic spines in the brain [37–40]. The potential role of GPR30 in mediating rapid estrogen signaling and neuroprotection in the brain has only recently begun to be addressed. The majority of the studies to date have used a GPR30 agonist, G1, to explore the effect of exogenous activation of GPR30 in the brain. These studies have revealed that G1 can attenuate glutamate-induced cell death in cultured cortical and hippocampal neurons in vitro [33, 41] and attenuate focal and global cerebral ischemia-induced neuronal cell death in vivo [34, 41, 42]. While these studies clearly demonstrate that exogenous activation of GPR30 can exert neuroprotection, they do not prove a role for endogenous GPR30 in mediating E2-induced neuroprotection. Additional studies such as knockdown or knockout approaches are needed to more definitively confirm a role of GPR30 in mediating E2-induced rapid signaling and neuroprotection. To address this issue, the current study utilized an in vivo antisense oligodeoxynucleotide knockdown approach to attenuate GPR30 expression in the hippocampal CA1 region and determine the effect upon the ability of E2-BSA to induce rapid membrane-mediated kinase signaling and neuroprotection in an animal model of GCI. The results reveal an important role of GPR30 in mediating rapid kinase activation and neuroprotection by E2 in the hippocampal CA1 region.
Materials and Methods
Antibodies
GPR30 (SC-48525-R, rabbit), p-AKT1Ser473 (SC-7985-R, rabbit), ERK1/2 (SC-94-G, goat), p-ERK1/2Tyr204 (sc-7383, mouse), JNK (sc-572, rabbit), p-JNKThr183,Tyr185 (SC-6254, mouse) and β-actin (SC-5286, mouse) antibodies, and GPR30 blocking peptide (SC-48525 P) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, U.S.). NeuN (MAB377) antibody was from Merck Millipore (MA, U.S.). AKT (C67E7, rabbit) antibody was from Cell Signaling Technology. GPR30 agonist G1 was from Calbiochem/EMD Millipore (Darmstadt, Germany).
Animals
Adult female Sprague Dawley rats weighing 250–300g, aged 3 months, were used in this study. All procedures were approved by the local legislation for ethics of experiments on animals. All rats were allowed free access to food and water before the operation under optimal conditions (12 h light: 12 h darkness cycle, 22°C).
Global Cerebral Ischemia (GCI)
Female rats were bilaterally ovariectomized, and 1w later, GCI was induced by 4-vessel occlusion as described previously [31, 43]. Briefly, the rats were anesthetized with 10% chloral hydrate (350 mg/kg, i.p.), the vertebral arteries were electrocauterized and the common carotid arteries (CCA) were exposed. After 24 h, the rats were anesthetized using isoflurane anesthesia and the CCA were re-exposed and clipped by artery clips for 10 min followed by reperfusion. Rats that lost their righting reflex within 30 seconds and whose pupils were dilated and lost response to light during ischemia were selected for the experiments. Rectal temperature was maintained at 37±0.5°C using a thermal blanket during ischemia. Sham-operated animals underwent the same surgical procedures without occlusion of the CCA.
Drug Administration
To investigate the role of GPR30 in E2-BSA signaling and neuroprotection in the hippocampal CA1 region following GCI, 10 nmol of GPR30 anti-sense oligonucleotide (AS, synthesized by Integrated DNA Technologies, Inc.) or mismatch-sense oligonucleotide (MS) in 5μl TE buffer (10 mM Tris–HCl, 1 mM EDTA, PH 8.0) was unilaterally administrated by Intracerebroventricular (icv) injection of rats every 24h for 4 days. On the fourth day, 10 min GCI was performed 30 min after AS administration. The GPR30 agonist, G1 (50μg dissolved in 5μl DMSO) or E2-BSA (10 μM in 5μl 0.9% saline) were bilaterally administered by icv injection 60 min before induction of GCI. For icv injections, the rats were placed on ear bars of a stereotaxic instrument under isoflurane anesthesia. The needle of a Hamilton syringe was lowered into the right/left lateral ventricle using coordinates (from the Bregma: AP:−0.8, M/L:±1.5, D/V: :−3.5). A total of 5 μl were injected at a rate of 1 μl/min, and then the needle was left in place for 5 min.
Sample preparation
Tissue sample preparation was performed as described previously by our group [31, 43]. Briefly, the hippocampal CA1 region was microdissected from both sides of the hippocampal fissure and quickly frozen in liquid nitrogen. Tissues were homogenized in a 1:10 (w/v) ice-cold homogenization buffer for 10 min consisting of 50 mM MOPS (pH 7.4), 150 mM NaCl, 20 mM β-glycerophosphate, 3 mM DTT, 2 mM Na3VO4, 1 mM EGTA, 1mM EDTA, 1mM NaF, 1% Triton X-100, 1% NP-40 and inhibitors of proteases and enzymes (0.5 mM PMSF, 10mg/ml each of aprotinin, leupeptin, and pepstatin A). This was followed by centrifugation at 15,000g for 15 min. The supernatant was removed and stored at :−80°C until use. The protein concentrations were determined using a BCA protein assay kit with bovine serum albumin (BSA) as the standard.
Western Blot Analysis
Western blot analysis was performed as described previously [31, 43]. Briefly, 100 μg protein of each sample was heated at 100°C for 5 min with loading buffer containing 0.125 M Tris-HCL (PH 6.8), 20% glycerol, 4% SDS, 10% mercaptoethanol and 0.002% bromphenol blue, then separated by 10% SDS-PAGE. The proteins were transferred onto PVDF membranes (pore size, 0.25 μm). Blotting membranes were incubated with 3% BSA in TBST (10 mM Tris (PH 7.5), 150 mM NaCl, 0.05% Tween-20) and probed with corresponding primary antibodies at 4°C overnight. The membrane was conjugated with fluorescently-labeled corresponding secondary antibodies at room temperature for 1h and washed with washing buffer 3×10 min. Following Western blot analysis, bound proteins were visualized using the Odyssey Imaging System (LI-COR Bioscience, Lincoln, NB), and semi-quantitative analysis of the bands was performed with the Image J analysis software (Version 1.30v; NIH, USA). Phospho-protein signals were expressed as a ratio to the corresponding total protein, and the total proteins were expressed relative to β-actin in the same sample. Normalized means were then expressed relative to the ratio for sham-treated animals.
Quantification of Surviving Hippocampal CA1 Pyramidal Neurons
Seven days after GCI, anesthetized rats were perfused transcardially using 0.9% saline followed by ice cold 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB). Brains were removed, placed in the same formalin at 4°C overnight, fixed in 30% sucrose in PB and then the brains were cut longitudinally into 25μm sections with a cryostat. NeuN staining was performed as described previously by our lab [31]. Briefly, sections were washed with 0.1% PBS-Triton X-100 for 3×5 min and permeabilized with 0.4% Triton X-100 in PBS for 30 min. After incubation with blocking solutions containing 10% normal donkey serum for 1 h at room temperature in PBS containing 0.1% Triton X-100, sections were incubated in primary antibodies (mouse anti-NeuN, 1:1000) for 48 h at 4°C. Sections were washed for 3×10 min, followed by incubation with Alexa Fluor 488 donkey anti-mouse antibody (1:200) for 1 h at room temperature. Sections were then washed and mounted using water-based mounting medium. Images were captured on a Confocal Laser Scanning Microscope (Olympus FV1000). The number of NeuN-positive CA1 neurons per 1 mm length of the medial CA1 pyramidal cell layer was counted bilaterally in five sections per animal. Cell counts from the right and left hippocampus on each of the five sections were averaged to provide the mean value. A mean ± SD was calculated from the data in each group and statistical analysis performed as described below
Immunohistochemistry staining
Brain coronal sections (25μm) were washed in PBS for 3×10 min and permeabilized with 0.4% Triton X-100 in PBS for 30 min. To block endogenous peroxidase activity and reduce non-specific staining, the sections were incubated for 1 h in a solution containing 1% BSA, 2% normal horse serum and 0.1% H2O2 in 0.4% Triton X-100-PBS for 1 h at room temperature. The sections were then incubated in GPR30 primary antibody (1:50) for 48 h at 4°C. In order to address the immune-specificity of GPR30 antibody, sections from sham animals were incubated with competitive blocking peptide solution (1:1000) overnight and then with GPR30 primary antibody (1:50) for 48 h at 4°C. After being washed three times in PBS, the sections were incubated for 2 h in secondary antibody (1:200 dilution; Vector Laboratories, Burlingame, CA, USA) made up with 0.4% Triton X-100 in PBS. Sections were washed and incubated with avidin-conjugated horseradish peroxidase (diluted 1:200 in 0.3% Triton X-100) for 1 h at room temperature, followed by avidin–biotin-peroxidase (ABC, Vector Laboratories) at room temperature. The sections were examined by light microscopy.
Statistical Analysis
All data are expressed as mean±SD. Statistical analysis was performed using one-way analysis of variance (ANOVA) with SigmaStat 3.0 software (SPSS, Inc., Chicago, IL), followed by Student-Newman-Keuls post-hoc tests to determine group differences. A level of P < 0.05 was considered to be statistically significant.
Results
Localization of GPR30 in the Hippocampus
We first examined the localization of GPR30 in the ovariectomized adult female rat hippocampus using light microscopy. Immunohistochemistry revealed that GPR30 immunostaining was highest in the CA1 region and dentate gyrus, with less staining observed in the CA3 region (Figure 1). The specificity of the GPR30 antibody was determined by blocking with the commercially available GPR30 antigen peptide used to make the antibody. As shown in Figure 1, blocking with the GPR30 antigen peptide eliminated immunostaining in the hippocampal CA1 region.
Figure 1. Localization of GPR30 protein in ovariectomized rat hippocampus.
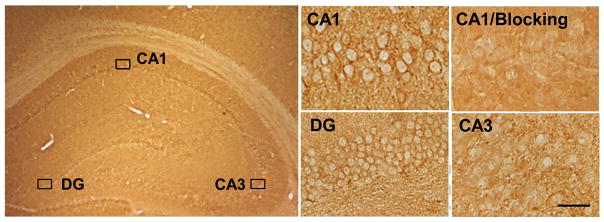
Immunohistochemistry staining shows GPR30 protein expression in hippocampal CA1, CA3 and DG regions. Blocking with the GPR30 antigen blocking peptide dramatically reduced immunostaining in the CA1 (the upper right section), indicating specificity of the primary antibody. Magnification, 40×; Scale bar, 25μm.
Antisense Knockdown of GPR30 in the Hippocampal CA1 Region
We next examined the role of GPR30 in mediating E2 rapid signaling and neuroprotection in the hippocampus following GCI. In these studies, we utilized an E2 conjugate, E2-BSA, which we have shown previously to exert rapid regulation of kinase activation and neuroprotection in the hippocampal CA1 region following GCI in ovariectomized adult rats [20]. To knockdown GPR30, antisense (AS) or missense (MS) oligodeoxynucleotides were administered by icv injection every 24 h for 4 days in animals. On the fourth day, 10 min GCI was performed 30 min after AS or MS administration. E2-BSA (10 μM in 5μl 0.9% saline) was bilaterally administered by icv injection 60 min before induction of GCI. Sham and Vehicle control groups were also included. As shown in Figure 2A, Western blot analysis revealed a major band at approximately 40–45 Kd, which is consistent with previous reports [37]. A lower molecular weight band was also observed. The identity of the lower band is unclear, but it most likely is a degradation product, as reported by others [44]. As shown in Figure 2A–B, Western blot analysis and immunohistochemistry revealed that GPR30 AS knockdown markedly attenuated GPR30 immunoprotein levels in the CA1 region in E2-BSA-treated rats (~60% decrease as compared to E2-BSA alone control group and MS control group). The specificity of the AS effect was indicated by the lack of effect of MS oligodeoxynucleotides on GPR30 immunoprotein levels in the CA1 region.
Figure 2. Effectiveness of GPR30-AS knockdown of GPR30 immunoprotein levels in the hippocampal CA1 region of E2-BSA treated rats.

(A) Ovariectomized rats were pretreated with E2-BSA, E2-BSA plus GPR30-AS, or E2-BSA plus GPR30-MS and subjected to ischemic reperfusion for 10 min following GCI. Western blot analyses of total GPR30 expression and β-Actin were performed with hippocampal CA1 protein in the indicated groups of ovariectomized rats. #P <0.05 versus GPR30-MS group or E2-BSA group. Data are mean ± SD from four different animals. (B) GPR30 immunoreactivity was detected by immunohistochemical staining in hippocampal CA1 region. Magnification, 40×; Scale bar, 25μm. n=4.
GPR30 Knockdown Attenuates Estrogen Neuroprotection Against GCI
We next examined the effect of GPR30 knockdown upon E2-BSA neuroprotective actions in the hippocampal CA1 region following GCI. Hippocampal sections were collected at 7d reperfusion after GCI and subjected to immunostaining with the neuronal marker, NeuN. Neuronal survival was quantified by counting the number of NeuN-positive cells per 1mm length in the medial CA1 area of all animals. Representative sections of NeuN staining results are presented in Figure 3A, while quantification of results from all animals is presented in Figure 3B. As shown in Figure 3A&B, GCI (Veh) resulted in a profound decrease in the number of surviving neurons in the CA1 region at 7d reperfusion, as compared to the Sham control group. E2-BSA treatment resulted in robust neuroprotection as indicated by a significant increase in surviving neurons in the CA1 region. GPR30 knockdown (AS) markedly attenuated the neuroprotective effect of E2-BSA in the CA1 region after GCI. In contrast, MS control oligodeoxynucleotides administration had no significant effect upon E2-BSA neuroprotection, indicating the specificity of the AS oligodeoxynucleotide effect. Finally, administration of the GPR30 agonist, G1, like E2-BSA, was found to exert a strong neuroprotective effect against GCI-induced neuronal cell loss in the CA1 region.
Figure 3. GPR30 knockdown attenuates E2-BSA neuroprotective effects in the hippocampal CA1 region following global cerebral ischemia.
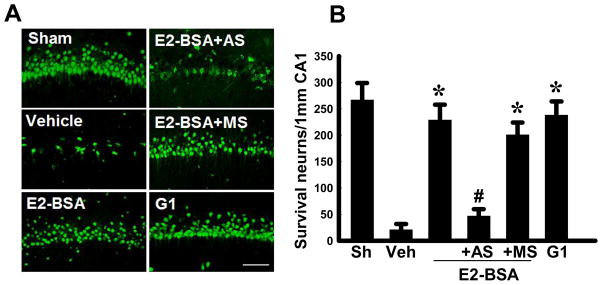
(A) Neuronal survival was quantitated using NeuN immunostaining of representative hippocampal CA1 sections from sham, vehicle, E2-BSA, E2-BSA plus GPR30-AS, E2-BSA plus GPR30-MS or G1-treated ovariectomized rats subjected to 10 min global cerebral ischemia followed by 7d reperfusion. Magnification, 40×; Scale bar, 100 μm. (B) Quantitative summary of data shows the number of surviving neurons per 1 mm length of medial CA1 sectors. Values are mean±SD of determinations from six individual rats per group (n=6). *P<0.05 versus vehicle group; #P<0.05 versus GPR30-MS group or E2-BSA group.
GPR30 Knockdown Attenuates Estrogen Rapid Signaling in the Hippocampus After GCI
In previous studies, we demonstrated that E2 can rapidly enhance activation of the prosurvival kinases, Akt and ERK in the hippocampal CA1 region after GCI, and can inhibit activation of JNK, which has been shown to facilitate apoptosis after cerebral ischemia [20]. Therefore, we next examined the effect of GPR30 knockdown on the ability of E2-BSA to enhance activation (phosphorylation) of Akt and ERK and inhibit JNK activation/phosphorylation after GCI. As shown in Figure 4A&B, E2-BSA significantly enhanced activation/phosphorylation of Akt and ERK in the CA1 region after GCI, while markedly inhibiting JNK activation/phosphorylation. GPR30 knockdown (AS) significantly attenuated these rapid kinase regulatory effects of E2-BSA on Akt, ERK and JNK activation in the CA1 region. MS oligonucleotides had no effect upon E2-BSA regulation of Akt, ERK or JNK activation in the CA1 region after CGI (data not shown). In addition, similar to the effects of E2-BSA, administration of the GPR30 agonist, G1 also robustly enhanced Akt and ERK activation in the CA1 region after GCI, while significantly inhibiting JNK activation.
Figure 4. GPR30 knockdown attenuates E2-BSA-induced activation of AKT and ERK, as well as inhibition of JNK activation in the hippocampal CA1 region after GCI.
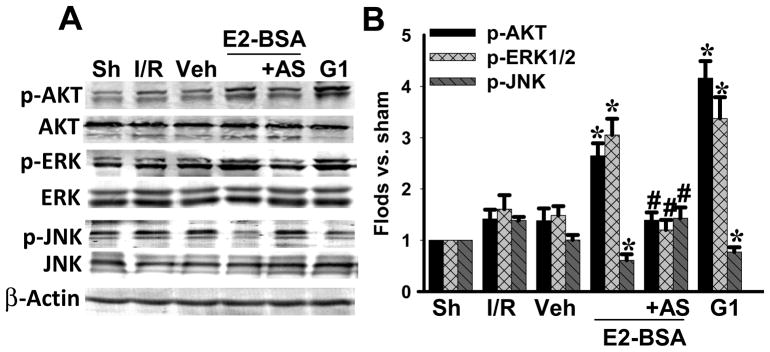
(A) Western blot analyses of the indicated proteins using hippocampal CA1 protein following 10 min reperfusion of ovariectomized rats. (B) Data are mean±SD of determinations from three to four individual rats. *P<0.05 versus vehicle group; #P<0.05 versus E2-BSA group.
Discussion
It is well known that GCI can result in significant neurological and cognitive deficits in humans. In particular, the hippocampal CA1 region in the brain has been shown to be highly vulnerable to damage by GCI [45, 46]. Clinically, GCI can be due to multiple causes, including cardiac arrest, asphyxiation, and hypotensive shock [45]. Of these, cardiac arrest is the most prevalent, and remains a leading cause of neurological damage, cognitive deficits and mortality. Previous studies have shown that a gender difference exists in both cardiac arrest survival and in GCI damage, with females being protected as compared to males [47–49]. Further work identified E2 as being strongly neuroprotective against GCI in females, an effect that is mediated, at least in part, by extranuclear ER [20, 29, 34, 50].
The current study enhances understanding in this important area by demonstrating that the newest member of the ER family, GPR30, participates in mediating the rapid signaling and neuroprotective effects of E2 in the hippocampus against GCI. Previous studies had used a GPR30 agonist (G1) to show that activation of GPR30 could be neuroprotective against GCI [34, 42]. While these studies provided important information that exogenous G1 activation of GPR30 is neuroprotective, the studies had a limitation in that they were not designed to address whether endogenous GPR30 actually mediates the neuroprotective effects of E2.
The current study overcame this limitation by utilizing a GPR30 AS knockdown approach to study the role of GPR30 in E2-BSA rapid signaling and neuroprotection. GPR30 knockdown in the hippocampal CA1 region essentially abolished the rapid kinase activation and neuroprotective ability of E2-BSA following CGI. These findings suggest a critical role for GPR30 in the hippocampus to mediate the beneficial rapid signaling and neuroprotective effects of E2. It is important to note that the GPR30 AS knockdown effect observed in our study was specific, as control missense (MS) oligodeoxynucleotides had no significant effect upon E2-BSA rapid signaling or neuroprotection after GCI. Furthermore, exogenous activation of GPR30 by the agonist, G1 in our studies enhanced activation of the prosurvival factors, Akt and ERK, inhibited proapototic JNK activation, and exerted profound neuroprotection against GCI.
The neuroprotective effect of G1 agonist in our study agrees well with the results of previous studies by other investigators [34, 42]. However, our study extends these observations by demonstrating the rapid signaling effects of G1 on activation of the prosurvival kinases, Akt and ERK, and upon inhibition of the proapototic signaling kinase, JNK in the hippocampal CA1 region after GCI. Importantly, our study also provides evidence that GPR30 mediates the rapid signaling effects of E2-BSA upon Akt, ERK and JNK activation in the hippocampus after GCI. Along these lines, GPR30 AS knockdown abolished the rapid kinase signaling effects of E2-BSA on Akt, ERK and JNK activation in the CA1 region after GCI. The loss of the rapid signaling ability of E2-BSA in GPR30 AS knockdown animals correlated with a loss of neuroprotection by E2-BSA against GCI, suggesting that these two effects may be linked. In support of this possibility, previous work by our lab has shown that blocking activation of Akt or ERK via use of specific PI3K or MEK inhibitors results in significant attenuation of E2-BSA neuroprotection against GCI [20]. Furthermore, in vitro studies in developing hippocampal neurons similarly found that administration of a PI3K inhibitor blocks the neuritogenic effect of G1 and E2, and that GPR30 silencing by GPR30 siRNA also blocked the neuritogenic effect of G1 and E2 [51].
While our study provides important new evidence supporting GPR30 in mediation of the neuroprotective effects of E2 in the hippocampus, the effect of E2 is clearly complex and likely involves multiple ERs. In support of this contention, previous work by our laboratory demonstrated that antisense knockdown of ERα, but not ERβ, can also attenuate E2 rapid signaling and neuroprotection against GCI [31]. As a whole, these results suggest that both ERα and GPR30 participate in mediation of E2-induced rapid kinase signaling and neuroprotection in the hippocampus following GCI. A similar dual role for ERα and GPR30 has been implicated in mediation of E2-induced islet survival in the pancreas [52]. E2 activation of multiple ERs may thus be an important mechanism for enhancing cell survival in a variety of tissues in the body.
Finally, in addition to GCI, activation of GPR30 has also been shown to be neuroprotective in models of Parkinson’s disease [53] and multiple sclerosis [54]. Thus, targeting GPR30 may represent a new therapeutic modality for multiple neurological disorders. Since GPR30 is distinct from the other ERs, targeting GPR30 for therapeutic intervention has the potential advantage that it may avoid negative side effects currently associated with estrogen therapy. Further research is needed to address this important possibility.
In conclusion, the current study provides important new evidence that GPR30 participates in mediating E2 rapid signaling and neuroprotection in the hippocampus following GCI. As such, the study adds to a growing literature suggesting that GPR30 may be an important mediator of extranuclear signaling by E2 in various tissues and processes throughout the body.
Highlights.
GPR30 is highly expressed in hippocampal CA1 region and dentate gyrus.
Activation of GPR30 induces rapid kinase signaling and neuroprotection.
GPR30 is critical for mediating estrogen rapid kinase signaling and neuroprotection in hippocampus.
Acknowledgments
This research was supported by Research Grants (NS050730) to DWB from the National Institutes of Neurological Disorders and Stroke, National Institutes of Health, and from the Natural Science Foundation of China (30970664, 31171354) to RW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids. 2007;72:381–405. doi: 10.1016/j.steroids.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahesh VB, Brann DW. Neuroendocrine mechanisms underlying the control of gonadotropin secretion by steroids. Steroids. 1998;63:252–6. doi: 10.1016/s0039-128x(98)00031-2. [DOI] [PubMed] [Google Scholar]
- 3.McEwen BS. Invited review: Estrogens effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol. 2001;91:2785–801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- 4.Spencer JL, Waters EM, Romeo RD, Wood GE, Milner TA, McEwen BS. Uncovering the mechanisms of estrogen effects on hippocampal function. Front Neuroendocrinol. 2008;29:219–37. doi: 10.1016/j.yfrne.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, et al. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–30. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- 6.Simpkins JW, Singh M, Brock C, Etgen AM. Neuroprotection and estrogen receptors. Neuroendocrinology. 2012;96:119–30. doi: 10.1159/000338409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang QG, Han D, Wang RM, Dong Y, Yang F, Vadlamudi RK, et al. C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E617–24. doi: 10.1073/pnas.1104391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levin ER. Integration of the Extranuclear and Nuclear Actions of Estrogen. Mol Endocrinol. 2005;19:1951–9. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–53. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- 10.Milner TA, McEwen BS, Hayashi S, Li CJ, Reagan LP, Alves SE. Ultrastructural evidence that hippocampal alpha estrogen receptors are located at extranuclear sites. J Comp Neurol. 2001;429:355–71. [PubMed] [Google Scholar]
- 11.Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocr Rev. 2007;28:726–41. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- 12.Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology. 1997;138:4030–3. doi: 10.1210/endo.138.9.5489. [DOI] [PubMed] [Google Scholar]
- 13.Mermelstein PG, Becker JB, Surmeier DJ. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci. 1996;16:595–604. doi: 10.1523/JNEUROSCI.16-02-00595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y, Huang YL, Zhang S, Zhu YC, Yao T. Estradiol acutely attenuates glutamate-induced calcium overload in primarily cultured rat hippocampal neurons through a membrane receptor-dependent mechanism. Brain Res. 2004;1026:254–60. doi: 10.1016/j.brainres.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 15.Beyer C, Raab H. Nongenomic effects of oestrogen: embryonic mouse midbrain neurones respond with a rapid release of calcium from intracellular stores. European J Neurosci. 1998;10:255–62. doi: 10.1046/j.1460-9568.1998.00045.x. [DOI] [PubMed] [Google Scholar]
- 16.Singh M, Setalo G, Jr, Guan X, Warren M, Toran-Allerand CD. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci. 1999;19:1179–88. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mannella P, Brinton RD. Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J Neurosci. 2006;26:9439–47. doi: 10.1523/JNEUROSCI.1443-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu TW, Wang JM, Chen S, Brinton RD. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 19.Jover-Mengual T, Zukin RS, Etgen AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology. 2007;148:1131–43. doi: 10.1210/en.2006-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang LC, Zhang QG, Zhou CF, Yang F, Zhang YD, Wang RM, et al. Extranuclear estrogen receptors mediate the neuroprotective effects of estrogen in the rat hippocampus. PLoS One. 2010;5:e9851. doi: 10.1371/journal.pone.0009851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, et al. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol. 2006;20:491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- 22.Kousteni S, Almeida M, Han L, Bellido T, Jilka RL, Manolagas SC. Induction of osteoblast differentiation by selective activation of kinase-mediated actions of the estrogen receptor. Mol Cell Biol. 2007;27:1516–30. doi: 10.1128/MCB.01550-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madak-Erdogan Z, Kieser KJ, Kim SH, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22:2116–27. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286:14737–43. doi: 10.1074/jbc.R110.191791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evinger AJ, 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70:361–3. doi: 10.1016/j.steroids.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Carswell HV, Macrae IM, Gallagher L, Harrop E, Horsburgh KJ. Neuroprotection by a selective estrogen receptor beta agonist in a mouse model of global ischemia. Am J Physiol Heart Circ Physiol. 2004;287:H1501–4. doi: 10.1152/ajpheart.00227.2004. [DOI] [PubMed] [Google Scholar]
- 27.Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology. 2005;146:3070–9. doi: 10.1210/en.2004-1515. [DOI] [PubMed] [Google Scholar]
- 28.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, et al. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–7. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- 30.Elzer JG, Muhammad S, Wintermantel TM, Regnier-Vigouroux A, Ludwig J, Schutz G, et al. Neuronal estrogen receptor-alpha mediates neuroprotection by 17beta-estradiol. J Cereb Blood Flow Metab. 2009 doi: 10.1038/jcbfm.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, et al. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci. 2009;29:13823–36. doi: 10.1523/JNEUROSCI.3574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346:904–10. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 33.Gingerich S, Kim GL, Chalmers JA, Koletar MM, Wang X, Wang Y, et al. Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17beta-estradiol in novel murine hippocampal cell models. Neuroscience. 2010;170:54–66. doi: 10.1016/j.neuroscience.2010.06.076. [DOI] [PubMed] [Google Scholar]
- 34.Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM. Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids. 2009;74:555–61. doi: 10.1016/j.steroids.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. 2013;154:4136–45. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenhart PM, Broselid S, Barrick CJ, Leeb-Lundberg LM, Caron KM. G-protein-coupled receptor 30 interacts with receptor activity-modifying protein 3 and confers sex-dependent cardioprotection. J Mol Endo. 2013;51:191–202. doi: 10.1530/JME-13-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akama KT, Thompson LI, Milner TA, McEwen BS. Post-synaptic density-95 (PSD-95) binding capacity of G-protein-coupled receptor 30 (GPR30), an estrogen receptor that can be identified in hippocampal dendritic spines. J Biol Chem. 2013;288:6438–50. doi: 10.1074/jbc.M112.412478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem Biophys Res Commun. 2006;346:904–10. doi: 10.1016/j.bbrc.2006.05.191. [DOI] [PubMed] [Google Scholar]
- 39.Hammond R, Nelson D, Gibbs RB. GPR30 co-localizes with cholinergic neurons in the basal forebrain and enhances potassium-stimulated acetylcholine release in the hippocampus. Psychoneuroendocrinology. 2011;36:182–92. doi: 10.1016/j.psyneuen.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, et al. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–21. doi: 10.1677/JOE-07-0017. [DOI] [PubMed] [Google Scholar]
- 41.Liu SB, Zhang N, Guo YY, Zhao R, Shi TY, Feng SF, et al. G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors. J Neurosci. 2012;32:4887–900. doi: 10.1523/JNEUROSCI.5828-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kosaka Y, Quillinan N, Bond C, Traystman R, Hurn P, Herson P. GPER1/GPR30 activation improves neuronal survival following global cerebral ischemia induced by cardiac arrest in mice. Transl Stroke Res. 2012;3:500–7. doi: 10.1007/s12975-012-0211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang R, Tu J, Zhang Q, Zhang X, Zhu Y, Ma W, et al. Genistein attenuates ischemic oxidative damage and behavioral deficits via eNOS/Nrf2/HO-1 signaling. Hippocampus. 2013;23:634–47. doi: 10.1002/hipo.22126. [DOI] [PubMed] [Google Scholar]
- 44.Sanden C, Broselid S, Cornmark L, Andersson K, Daszkiewicz-Nilsson J, Martensson UE, et al. G protein-coupled estrogen receptor 1/G protein-coupled receptor 30 localizes in the plasma membrane and traffics intracellularly on cytokeratin intermediate filaments. Mol Pharmacol. 2011;79:400–10. doi: 10.1124/mol.110.069500. [DOI] [PubMed] [Google Scholar]
- 45.Neumann JT, Cohan CH, Dave KR, Wright CB, Perez-Pinzon MA. Global cerebral ischemia: synaptic and cognitive dysfunction. Curr Drug Targets. 2013;14:20–35. doi: 10.2174/138945013804806514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. 2006;24:1–21. doi: 10.1016/j.ncl.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 47.Hall ED, Pazara KE, Linseman KL. Sex differences in postischemic neuronal necrosis in gerbils. J Cerebral Blood Flow Metab. 1991;11:292–8. doi: 10.1038/jcbfm.1991.61. [DOI] [PubMed] [Google Scholar]
- 48.Kofler J, Hurn PD, Traystman RJ. SOD1 overexpression and female sex exhibit region-specific neuroprotection after global cerebral ischemia due to cardiac arrest. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2005;25:1130–7. doi: 10.1038/sj.jcbfm.9600119. [DOI] [PubMed] [Google Scholar]
- 49.Kitamura T, Iwami T, Nichol G, Nishiuchi T, Hayashi Y, Nishiyama C, et al. Reduction in incidence and fatality of out-of-hospital cardiac arrest in females of the reproductive age. Eur Heart J. 2010;31:1365–72. doi: 10.1093/eurheartj/ehq059. [DOI] [PubMed] [Google Scholar]
- 50.Shughrue PJ, Merchenthaler I. Estrogen prevents the loss of CA1 hippocampal neurons in gerbils after ischemic injury. Neuroscience. 2003;116:851–61. doi: 10.1016/s0306-4522(02)00790-x. [DOI] [PubMed] [Google Scholar]
- 51.Ruiz-Palmero I, Hernando M, Garcia-Segura LM, Arevalo MA. G protein-coupled estrogen receptor is required for the neuritogenic mechanism of 17beta-estradiol in developing hippocampal neurons. Mol Cell Endocrinol. 2013;372:105–15. doi: 10.1016/j.mce.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 52.Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, et al. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes. 2009;58:2292–302. doi: 10.2337/db09-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bourque M, Morissette M, Cote M, Soulet D, Di Paolo T. Implication of GPER1 in neuroprotection in a mouse model of Parkinson’s disease. Neurobiol Aging. 2013;34:887–901. doi: 10.1016/j.neurobiolaging.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 54.Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, et al. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol. 2009;214:67–77. doi: 10.1016/j.jneuroim.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]