

Abstract
Double-stranded RNA-dependent protein kinase (PKR) regulates antiviral activity, immune responses, apoptosis and neurotoxicity. Gliatropic coronavirus infection induced PKR activation in infected as well uninfected cells within the central nervous system (CNS). However, PKR deficiency only modestly increased viral replication and did not affect IFN-α/β or IL-1β expression. Despite reduced Il-6, Ccl5, and Cxcl10 mRNA, protein levels remained unaltered. Furthermore, PKR deficiency selectively reduced IL-10 production in CD4, but not CD8 T cells, without affecting CNS pathology. The results demonstrate the ability of PKR to balance neuroinflammation by selectively modulating key cytokines and chemokines in CNS resident and CD4 T cells.
Keywords: Protein kinase RNA-dependent (PKR), Coronavirus, IL-10, CD4 T cells, Encephalomyelitis
Highlights
-
•
PKR is dispensable for antiviral activity against neurotropic coronavirus infection.
-
•
PKR regulates transcriptional and translational expression of CCL5 and CXCL10.
-
•
PKR promotes IL-10 production in virus specific CD4 T cells.
-
•
PKR mediated TIMP1 upregulation enhances CNS parenchymal entry of CD4 T cells.
1. Introduction
The serine and threonine protein kinase PKR was originally identified as an innate antiviral protein induced following type-1 IFN signaling (Meurs et al., 1990). Antiviral activity is mediated through phosphorylation of the eukaryotic translation initiation factor eIF2α, leading to inhibition of translation (Garcia et al., 2006, Sadler and Williams, 2007) and induction of apoptosis (Lee et al., 1997). Double stranded (ds) RNA, a common intermediate during viral replication, constitutes a primary signal inducing PKR dimerization and autophosphorylation, thereby converting latent PKR into the activated form (Taylor et al., 1996, Wu and Kaufman, 1997). However, PKR can also be activated by a variety of other stimuli, including stress, cytokines, RNA structures, bacterial infection, DNA damage, and release of damage associated molecular patterns (DAMPS) following cell injury and necrosis (Garcia et al., 2006, Sadler and Williams, 2007). Depending on the activating insult and cell type, activated PKR can also interact directly with inflammasome components to promote release of the cytokines IL-1β, IL-18 and high-mobility group box 1 (HMGB1) protein (Lu et al., 2012). Activated PKR also promotes induction of pro-inflammatory genes via NF-kB activation (Kumar et al., 1994), and can enhance IFN-α/β expression via both IRF3 activation (Zhang and Samuel, 2008) and maintaining IFN-α/β mRNA integrity (Schulz et al., 2010). In addition, PKR activation by poly I:C, LPS and various infections such as mycobacterium and Sendai virus induce the anti-inflammatory cytokine IL-10 in monocyte/macrophages (Cheung et al., 2005, Chakrabarti et al., 2008).
PKR also plays a role in regulating cellular immunity. For example, PKR reduces CD8 T cell proliferation in a contact hypersensitivity model (Kadereit et al., 2000) as well as during systemic lupus erythematosus (Grolleau et al., 2000). Furthermore, during lymphocytic choriomeningitis virus (LCMV) infection, PKR deficiency enhances expansion, but not effector function of virus specific CD8 T cells (Nakayama et al., 2010).
Finally, PKR is implicated in neuro-degenerative processes. It contributes to neurotoxicity by activating caspases 3 and 8 in beta amyloid treated cells as well as in a mouse model of Alzheimer's disease (Couturier et al., 2010). A detrimental role of PKR in neurotoxicity is supported by its implication as an early biomarker of neuronal cell death during Alzheimer's disease (Peel and Bredesen, 2003). The role of PKR in demyelinating disease remains unknown; however, PKR is activated in oligodendroglia, T cells and macrophages during experimental autoimmune encephalitis (EAE), a rodent model of multiple sclerosis (Chakrabarty et al., 2004).
Most studies analyzing PKR function during viral infections in vitro and in vivo have focused on its antiviral activity. How PKR regulates inflammation during infection, especially viral encephalitis is less well characterized. PKR plays a crucial role in antiviral defense following vesicular stomatitis virus (VSV) infection by directly diminishing virus replication in vitro and in vivo. Uncontrolled CNS VSV replication results in 100% mortality in PKR−/− mice (Stojdl et al., 2000). Increased peripheral virus replication was also associated with increased mortality of West Nile virus (WNV) infected dual RNaseL/PKR−/− compared to single RNaseL−/− mice. However, virus dissemination to the CNS was not enhanced (Samuel et al., 2006), suggesting PKR did not contribute to antiviral activity within the CNS. Poliovirus infected PKR−/− mice also showed no difference in CNS virus replication, but were protected from extensive spinal cord damage (Scheuner et al., 2003). While the effect of PKR on neurotropic coronaviruses has not been assessed, PKR modestly reduced replication of the hepato- and neuro-tropic coronavirus MHV-A59 in macrophage cultures (Zhao et al., 2012). These varying outcomes suggest that PKR activation and functions in vivo differ between infections and host cell types, thus regulating inflammatory responses and pathogenesis independent of antiviral activity.
Based on the limited knowledge of PKR mediated regulation of innate and adaptive responses during viral CNS infection, the present study sets out to characterize in vivo effects of PKR on immune modulation following infection with the sublethal, demyelinating gliatropic coronavirus JHMV. JHMV infection is initiated in the brain and spreads to the spinal cord, where it preferentially persists in oligodendrocytes (Fleming et al., 1986, Kapil et al., 2012). In wt mice infectious virus peaks between days 3–5 post infection (p.i.) and is reduced by T cells between days 7 and 14 p.i. to undetectable levels. Persistence is characterized by sustained viral RNA in the absence of infectious virus (Bergmann et al., 2006). Results herein show that Pkr mRNA is upregulated coincident with IFN-α/βfollowing JHMV infection and is sustained throughout T cell mediated viral control. Activated PKR was not only expressed in infected cells, but also in neighboring, uninfected cells. Nevertheless, the absence of PKR only modestly elevated virus early during infection, consistent with effective antiviral T cell control. Furthermore, PKR deficiency did not affect CNS IL-1β, CCL5 and CXCL10 expression, despite significantly reduced levels of the respective mRNAs. Moreover, these studies are the first to reveal a positive regulatory effect of PKR on TIMP-1, IL-21 and IL-10 expression, all prominently associated with CD4 T cells during JHMV encephalomyelitis. Overall the results highlight the potential for immune modulation by PKR in both CNS resident as well as infiltrating cells. Although these immune modulatory effects were insufficient to grossly influence JHMV pathogenesis, the results demonstrate PKR as a selective regulator of key cytokines and chemokines controlling neuroinflammation.
2. Materials and methods
2.1. Mice, viruses, titers and clinical disease
C57Bl/6 mice were purchased from the National Cancer Institute (Fredrick, MD). Homozygous PKR−/− mice on the C57Bl/6 background were previously described (Yang et al., 1995) and kindly provided by Dr. Ganes Sen (Cleveland Clinic, Cleveland, OH). All mice were housed under pathogen free conditions in accredited facility at the Cleveland Clinic Lerner Research Institute. All animal procedures were performed in compliance with protocols approved by the Cleveland Clinic Institutional Animal Care and Use Committee (PHS assurance number: A3047-01). Infections in vivo were carried out with the sublethal, gliatropic, monoclonal antibody (mAb) derived variant of JHMV, designated 2.2v-1 (Fleming et al., 1986). In vitro infections of bone marrow derived macrophages (BMDMs) were carried out with the MHV-A59 strain, kindly provided by Dr. Volker Thiel (Kantonal Hospital, St. Gallen, Switzerland). Mice at 6–7 weeks of age were infected intracranially in the left hemisphere with 1000 plaque forming units (PFU) of JHMV diluted in endotoxin-free Dulbecco's phosphate-buffered saline (PBS) in a final volume of 30 μl. Clinical disease severity was graded daily using the following scale: 0, healthy; 1, ruffled fur/hunched back; 2, inability to turn upright/partial hind limb paralysis; 3, complete hind limb paralysis; and 4, moribund or dead. Virus replication in cell free supernatants from the brain was determined by plaque assay on DBT astrocytoma cell monolayers as described. Briefly, individual brains or spinal cords were homogenized in 4 ml Dulbecco's PBS using chilled Tenbroeck glass homogenizers. Homogenates were centrifuged at 400 ×g for 7 min at 4 °C, and cell-free supernatants were stored at − 70 °C until use.
2.2. Cell isolation, flow cytometry and fluorescent activated cell sorting (FACS)
Cells for flow cytometric analysis were isolated from brains using chilled Tenbroeck glass homogenizers as described (Kapil et al., 2009). Following centrifugation of the cell suspension at 400 ×g for 7 min, cell pellets were resuspended at a final concentration of 30% Percoll (Pharmacia, Uppsala, Sweden), underlaid with 70% Percoll, and purified by centrifugation for 30 min at 800 ×g at 4 °C. Cells were collected from the 30%/70% Percoll interface, washed twice with RPMI-HEPES and viable cells counted based trypan blue exclusion. Cells were incubated with 10% mouse serum and anti-mouse CD16/32 (clone 2.4G2; BD Biosciences, San Diego, CA) for 15 min on ice to block non-specific binding prior to staining with fluorochrome conjugated mAbs specific for CD45 (30-F11), CD4 (RM4-5), CD8 (53-6.7) (all from BD Biosciences) and F4/80 (C1:A3-1; Serotec, Raleigh, NJ). Cells were analyzed on a flow cytometer (FACSCalibur, Becton Dickinson, Mountain View, CA) using FlowJo software (TreeStar, Inc., Ashland, OR). IFN-γ and IL-10 production by T cells was measured by intracellular flow cytometry following incubation of 5 × 105 CNS derived cells with 3 × 105 EL-4 or CHB3 feeder cells with or without virus specific peptide for 5 h as described. Briefly, CD8 and CD4 T cells were stimulated with 1 μM S510 or 10 μM M133 peptide, respectively, in a total volume of 200 μl of RPMI supplemented with 10% fetal calf serum (FCS) for 5 h at 37 °C with GolgiStop (BD Biosciences). Following surface staining for CD8, CD4 and CD45, cells were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's protocol. Intracellular cytokines were detected using FITC conjugated anti-IFN-γ mAb or APC conjugated anti-IL-10 mAb. Cells were analyzed by flow cytometry as described above.
For RNA analysis of distinct CNS derived cell populations, the brains were finely minced using razor blades and cells released by proteolysis in 0.25% trypsin for 30 min at 37 °C as described (Kapil et al., 2012). Live cells were separated from myelin debris by centrifugation on a 30/70% Percoll step gradient as described above. Cells were stained for expression of CD45, CD11b and O4 to separate infiltrating monocyte/macrophages (CD45hiCD11b+), microglia (CD45lo/intCD11b+) and oligodendrocytes (CD45−O4+). Representative flow plots depicting the sorting strategy are shown in Supplementary Fig. 1. Although CD45hiCD11b+ cells also comprise neutrophils, they constitute < 5% of CD45hi cells, while monocytes comprise up to 70% prior to T cell infiltration. FACS purified cell populations were recovered by centrifugation at 400 ×g for 7 min at 4 °C, resuspended in TRIzol (Invitrogen, Carlsbad, CA) and stored at − 80 °C for subsequent RNA extraction. Typical yields for 6–7 brains were 400,000–600,000 macrophages or microglia and 40,000–100,000 oligodendrocytes.
2.3. Bone marrow derived macrophages (BMDMs)
Mouse bone marrow was flushed from the femurs and tibiae with RPMI supplemented with 25 mM HEPES (pH 7.2). Following trituration cells were filtered using a 70 μm cell strainer, centrifuged at 400 ×g for 5 min, re-suspended in BMDM growth medium [Dulbecco's modified Eagles medium containing 10% FCS, 20% L929-conditioned medium as source of colony stimulating factor (CSF-1), 1% sodium pyruvate and 0.1% gentamicin], and adjusted to 2 × 106 cells/ml BMDM growth media. Cells were plated at 20 ml per 75 cm2 tissue culture flasks and differentiated in a humidified 37 °C, 5% CO2 incubator for 1 week. On alternate days flasks were washed with DMEM to remove non-adherent cells and fresh BMDM growth medium was added. Cells near confluency were removed by scraping and 2 × 106 cells plated into 60 mm × 15 mm petri dishes. After 3 days cells were washed and infected with MHV-A59 at a multiplicity of infection (MOI) of 1 for 1 h at 37 °C. After removing unattached virus, cells were further incubated in 3 ml DMEM and the medium was replaced at 4 h p.i. At indicated times p.i. supernatants were collected, cells lysed directly in 800 μl TRIzol (Invitrogen) and both stored at − 80 °C.
2.4. ELISA
IFN-γ in cell free CNS supernatants was measured by ELISA as described (Kapil et al., 2009). Briefly, 96-well plates were coated with rat anti-mouse IFN-γ mAb (R4-6A2; BD Biosciences) at 1 μg/ml overnight at 4 °C. Following blocking of nonspecific binding with PBS/10% FCS for 1 h, CNS supernatants and IFN-γ recombinant cytokine standard (BD Biosciences) were adsorbed at 4 °C overnight. Bound IFN-γ was detected following 1 h incubation at room temperature with biotinylated rat anti-mouse IFN-γ mAb (XMG1.2; BD Biosciences) and Avidin-Horseradish Peroxidase (Av-HRP) and subsequent development with 3,3′,5,5′ tetramethylbenzidine (TMB reagent set; BD Biosciences). Optical densities were read at 450 nm with a Bio-Rad model 680 microplate reader and analyzed using Microplate Manager 5.2 software (Bio-Rad Laboratories, Hercules, CA). IL-10 in brain supernatants was measured using a mouse IL-10 ELISA kit as per the manufacturer's instructions (eBioscience, catalog #88-7104-22). IL-1β, CCL5 and CXCL10 in the brain and BMDM supernatants were measured using mouse ELISA kits for IL-1β (R&D systems catalog # PMLB006), CCL5 (R&D systems catalog# MMR00), CXCL10 (R&D systems catalog# PMCX100) and TIMP1 (R&D systems catalog# MTM100) as per the manufacturer's instructions. Optical densities were read at 450 nm with a Bio-Rad model 680 microplate reader and analyzed using Microplate Manager 5.2 software (Bio-Rad Laboratories, Hercules, CA). Triplicate samples from individual mice (n = 6) and BMDM cultures from individual mice (n = 3), respectively, were averaged and standard error of the mean was calculated.
2.5. Histology and fluorescent staining
Spinal cords from PBS-perfused mice were fixed with 10% zinc formalin. The spinal cords were divided into six segments and embedded in paraffin for analysis of inflammation and pathology as described (Phares et al., 2012a). Cross sections from individual spinal cords were stained at each of 6 levels with hematoxylin and eosin (HE) and luxol fast blue (LFB) to assess inflammation and myelin loss, respectively. Axonal damage was determined by immunoperoxidase staining for phosphorylated and nonphosphorylated neurofilament using mAb SMI31 and SMI32 (SMI) (Covance, Princeton, NJ). High resolution images were obtained using a scanner (Aperio Scanscope, Vista, CA) with a 20 × objective and doubling lens. Sections were scored in a blinded fashion, and representative fields were identified based on average score of all sections in both experimental groups.
Brains from PBS-perfused mice were divided along the mid-sagittal plane and one half embedded in TissueTek O.C.T. compound, flash frozen in liquid nitrogen and stored at − 80 °C. Blocks were cut into 12 μm sections at 4 °C and stored at − 80 °C. Sections were thawed and fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized in 1% Triton-X in PBS for 30 min at room temperature and non-specific binding blocked using 1% bovine serum albumin and 10% normal goat serum. Phosphorylated PKR (PKR-p) was detected with rabbit anti-mouse PKR-p antibody (Cedarlane Laboratories, Ontario, Canada) and expression of viral nucleocapsid protein with anti-mouse J3.3 mAb (Phares et al., 2012a). The distribution of CD4 T cells in brain parenchyma and perivascular space was determined by staining brain sections with rabbit anti-mouse laminin Ab (Cedarlane Laboratories, Ontario, Canada) and rat anti-mouse CD4 mAb (BD Pharmingen). Sections were incubated with primary Ab overnight at 4 °C and subsequently incubated with AF488 conjugated anti rabbit or AF594 conjugated anti-mouse or anti-rat secondary Ab (Invitrogen) at room temperature for 1 h. Sections were mounted with ProLong Gold antifade mounting media containing 4′,6-diamidino-2-phenylindole (Invitrogen, Carlsbad, CA). Images were acquired on a SP-5 confocal microscope (Leica Microsystems, Wetzlar, Germany).
2.6. Real time PCR
RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions and cDNA subjected to real time PCR analysis as described (Kapil et al., 2009, Kapil et al., 2012, Butchi et al., 2014). Briefly, following RNA isolation, DNA contamination was removed with DNase I using a DNA Free™ kit (Ambion, Austin, TX) for 30 min at 37 °C, and cDNA synthesized using Murine Myeloid Leukemia virus (M-MLV) reverse transcriptase (Invitrogen) in buffer containing deoxynucleoside triphosphate mix, oligo(dT) (Promega, Madison, WI) and random hexamer primers (Invitrogen). Quantitative real-time PCR was performed using either SYBR green master mix (Applied Biosystems, Foster City, CA) or TaqMan technology. All PCR reactions were performed in duplicate in a 96-well plate in 10 μl volumes containing specific master mix, 1 mM of each primer, and 4 μl cDNA using the ABI 7500 fast PCR machine and 7500 software (Applied Biosystems). The primer sequences for SYBR green PCR analysis are as follows: Il-6, 5′-ACACATGTTCTCTGGGAAATCGT-3′ (sense) and 5′-AAGTGCATCATCGTTGTTCATACA-3′ (antisense); CCL2 5′-AGCAGGTGTCCCAAAGAA-3′ (sense) and 5′-TATGTCTGGACCCATTCCTT-3′ (antisense); Ccl5 5′-GCAAGTGCTCCAATCTTGCA-3′ (sense) and 5′-CTTCTCTGGGTTGGCACACA-3′ (antisense); CXCL10 5′-GACGGTCCGCTGCAACTG-3′ (sense) and 5′-GCTTCCCTATGGCCCTCATT-3′ (antisense); Il-10, 5′-TTTGAATTCCCTGGGTGAGAA-3′ (sense) and 5′-GCTCCACTGCCTTGCTCTTATT-3′ (antisense); Il-21 5′-GGACAGTATAGACGCTCACGAATG-3′ (sense) and 5′-CGTATCGTACTTCTCCACTTGCA-3′ (antisense); Timp1, 5′- CCAGAGCCGTCACTTTGCTT-3′ (sense) and 5′- AGGAAAAGTAGACAGTGTTCAGGCTT-3′ (antisense). Expression was compared relative to the endogenous control glyceraldehyde-3-phosphate dehydrogenase (Gapdh) as described (Kapil et al., 2012). SYBR green reaction conditions were as follows: 95 °C for 10 min, 40 cycles of denaturation at 94 °C for 10 s, annealing at 60 °C for 30 s, and elongation at 72 °C for 30 s. Dissociation curves were used to confirm amplification of a single product for each primer pair per sample. Expression levels of Ifn-β, Ifn-α4, Ifn-α5, Ifit1, Ifit2, Il-1β and Ifn-γ were determined using TaqMan primer-probe sets, and 2 × universal Taqman fast master mix (Applied Biosystems,). Reaction conditions for TaqMan were 95 °C for 20 s as holding temperature and then 40 cycles of denaturation at 95 °C for 3 s, annealing and extension at 60 °C for 30 s. Data were calculated relative to Gapdh using the formula 2[CT(GAPDH) − CT(Target Gene)] × 1000 where Ct is the threshold cycle.
2.7. Statistical analysis
Results are expressed as the mean ± SEM for each group of mice. In all cases, significance is considered at p ≤ 0.05 using Microsoft excel software.
3. Results
3.1. JHMV induces PKR activation in infected and uninfected cells
IFN-α/β dependent responses are vital to limit early JHMV spread throughout the CNS, stem neuronal infection, and prevent mortality (Ireland et al., 2008). Furthermore, the kinetics, diversity and magnitude of gene expression of IFN-α/β and IFN-α/β stimulated genes (ISGs) are regulated in a cell type specific manner during JHMV infection in vivo (Kapil et al., 2012). However, the antiviral and regulatory roles of ISGs within the CNS remain poorly characterized (Ireland et al., 2008, Ireland et al., 2009, Rose et al., 2010). Due to the diverse PKR functions in regulating antiviral activity and inflammation, we initially assessed the expression kinetics of Pkr relative to Ifn-α/β mRNA in the brains of wt mice following JHMV infection (Fig. 1 ). Ifn-β and Ifn-α4 mRNA, barely detectable in naïve mice, were modestly upregulated by days 3 and 5 p.i. and already reduced by day 7 p.i. Pkr mRNA, abundant in naïve brains, was further increased by ~ 3 and ~ 10 fold at days 3 and 5 p.i., respectively, consistent with early Ifn-α/β expression However, unlike the decline of Ifn-α/β mRNAs after day 5 p.i., Pkr mRNA only decreased after day 7 p.i. While microglia/macrophages are the early targets of infection, virus replicates prominently in oligodendrocytes by day 7 p.i. However, Ifn-α/β mRNA is only induced in microglia/macrophages and not in oligodendrocytes (Kapil et al., 2012). Transcripts were thus also monitored in CNS derived cell populations preferentially infected by JHMV to assess cell type specific Pkr mRNA regulation in vivo (Fig. 1). Basal Pkr mRNA was higher in microglia compared to that in oligodendrocytes, consistent with various other ISGs (Kapil et al., 2012). In infected mice Pkr mRNA levels rose by 3-fold in microglia at day 3 p.i. but declined thereafter. Similarly, Pkr mRNA was high in CNS infiltrating monocytes, but subsided after day 5 p.i. By contrast, infection induced a ~ 11–18 fold increase in Pkr mRNA in oligodendrocytes by days 3 and 5 p.i., which further rose to 30-fold above basal values by day 7 p.i., at which time T cell effector function and IFN-γ production peaks (Bergmann et al., 2006). These data revealed that microglia/macrophages as well as oligodendrocytes contribute to overall elevated Pkr mRNA throughout acute infection, but that maximal expression in oligodendrocytes coincides with peak adaptive rather than innate immune responses.
Fig. 1.

Kinetics of PKR mRNA expression following JHMV infection. Ifn-α/β mRNA expression and Pkr mRNA expression in brains of wt infected mice were determined relative to Gapdh by real time PCR. Upper panels show the average ± standard error mean (SEM) mRNA levels from individual brains with n = 3–6 mice/time-point. Lower panels show Pkr mRNA levels in CNS derived microglia, macrophages and oligodendrocytes purified by FACS. Arrows indicate virus infected cells. Data for cell populations are average values ± SEM derived from 2 to 3 separate experiments, each using pooled brain samples from n = 5–6 per timepoint.
PKR exerts activity following autophosphorylation (Taylor et al., 1996), with dsRNA as a primary, but not exclusive activation signal during viral infection (Garcia et al., 2006). To determine if PKR activation is restricted to infected cells, expression of phosphorylated PKR (PKR-p) and viral antigen were assessed by immune fluorescence (Fig. 2 ). PKR-p was abundant in infected brains at both days 5 and 7 p.i. (Fig. 2), but undetectable in naïve brains (data not shown). Quantification revealed that > 50% of cells expressed PKR-p, while it remained undetectable in infected PKR−/− mice at matched time points (data not shown). Furthermore, the vast majority of virus infected cells exhibited PKR activation at day 5 p.i., but not at day 7 p.i. (Fig. 2), when T cells start controlling virus replication (Bergmann et al., 2006). PKR-p was also detected in a large population of uninfected cells localized not only proximal, but also distal to foci of virus infection at both 5 and 7 days p.i. (Fig. 2); however we cannot exclude the possibility of proximal infected cells below detection or outside the focus field. Overall, these results suggest that non-virus associated signals, such as stress, pro-inflammatory factors, or death associated molecular stimuli contribute to PKR activation during acute viral encephalitis.
Fig. 2.

PKR activation is not restricted to infected cells. Expression of activated PKR was determined in the brain at days 5 and 7 p.i. relative to total and virus infected cells. Brain sections were stained with antibody specific for phosphorylated PKR (PKR-p; green) in combination with either DAPI (blue) or mAb J3.3 (red) specific for viral nucleocapsid protein and analyzed by confocal microscopy. Images are representative of 6–9 fields/brain with n = 6 mice/group/time-point. Graphs show numbers of PKR-p+ within total DAPI+ cells/field and numbers of PKR-p+ cells within viral antigen+ cells.
3.2. Reduced virus control early in PKR−/− mice is not due to defects in IFN-α/β signals
A crucial function of PKR-p is the reduction of viral replication by blocking translation (Garcia et al., 2006). Antiviral activity of PKR was thus assessed by comparing CNS viral replication in PKR−/− and wt mice. PKR−/− mice harbored ~ 5 and 10-fold elevated virus in the brains throughout days 3 and 5 p.i., respectively (Fig. 3 ). Virus load was still 10-fold higher in the absence of PKR at day 7 p.i., but was reduced to similar levels as in wt mice by day 10 p.i. These data indicated an early protective effect of PKR, which nevertheless appeared redundant for ultimate viral control by adaptive immune effector function (Bergmann et al., 2006).
Fig. 3.
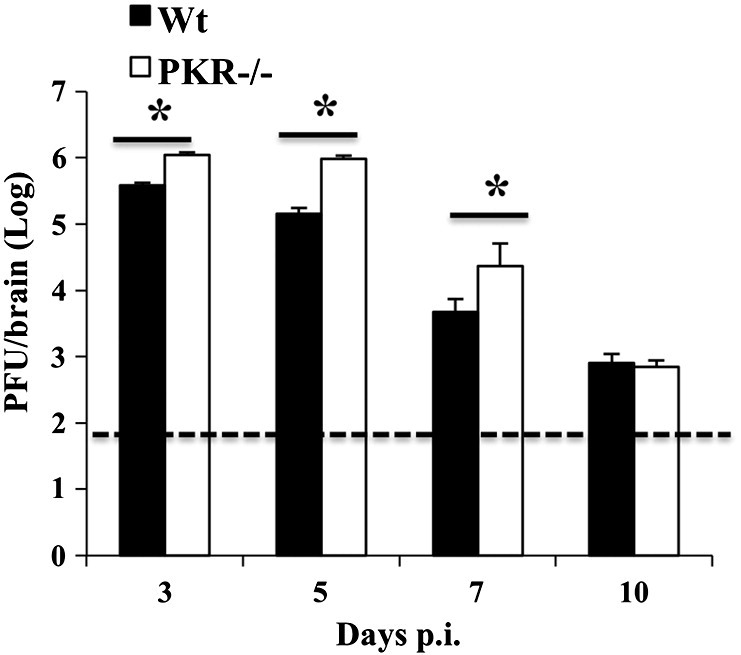
Virus replication is initially enhanced in PKR−/− mice but eventually controlled. Virus replication was determined by plaque assay of brain supernatants at the indicated time points p.i. Data represent the average of three separate experiments ± SEM (n = 9 per group and timepoint). *p ≥ .05. Dotted line represents detection limit.
As activated PKR can directly promote IFN-α/β expression in response to select stimuli (Der and Lau, 1995, Mcallister et al., 2012), we evaluated whether reduced viral control early was indirectly due to reduced IFN-α/β induction. PKR deficiency did not alter the kinetics or expression of Ifn-β, Ifn-α4 and Ifn-α5 mRNA in the brains of infected mice (Fig. 4 ). Ifn-β mRNA was induced to similar levels at days 3 and 5 p.i. and did not decline until day 7 p.i., while Ifn-α4 and Ifn-α5 mRNAs peaked at day 3 p.i. and declined by day 5 p.i. in both groups. Induction of the ISGs Ifit1 and Ifit2, which are highly sensitive to IFN-α/β during JHMV infection (Ireland et al., 2008), was used as a readout for IFN-α/β signaling, as coronaviruses only induce barely detectable levels of IFN-α/β within the CNS (Butchi et al., 2014). No differences in ifit1 or ifit2 mRNA levels supported similar IFN-α/β activity in both groups (Fig. 4). These data suggest that PKR contributes modestly to innate antiviral immunity by directly interfering with virus replication.
Fig. 4.

PKR deficiency does not alter expression of IFN-α/β or responsive genes. Expression of Ifn-α/β, Ifit1 and Ifit2 in brains of naïve and JHMV infected wt and PKR−/− mice was determined using real time PCR. Data represented the average ± SEM of n = 6 mice/group/time-point and are pooled from two separate experiments. *p ≥ .05. Naïve values are shown for wt mice.
3.3. PKR deficiency reduces mRNA but not protein expression of select pro-inflammatory factors
PKR-p regulates NF-kB activity by degrading the NF-kB inhibitor Ik-B, thereby promoting expression of pro-inflammatory genes (Kumar et al., 1994). However, to our knowledge this has not been investigated extensively in vivo. Several proinflammatory factors were therefore monitored at the transcriptional level to reveal potential impairment in the absence of PKR. IL-1β and IL-6, commonly upregulated following neuroinflammation, both promote disruption of the blood brain barrier (BBB) and recruitment of lymphocytes (Hopkins and Rothwell, 1995, Erta et al., 2012). CCL2 is essential for monocyte recruitment (Savarin et al., 2010), while CXCL10 and CCL5 attract T cells to the CNS (Glass et al., 2004, Liu et al., 2001a). Il-1β, Il-6, Ccl2, and Ccl5 mRNAs were all induced to comparable levels in the brains of infected PKR−/− relative to wt mice at day 3 p.i. (Fig. 5A). After day 3 p.i. Ccl2, Il-6, and Il-1β mRNA declined progressively, with Il-6 mRNA dropping to 50% of wt levels in PKR−/− brains at days 5 and 7 p.i. (Fig. 5A). Ccl5 mRNA in PKR−/− brains was sustained throughout days 3–10 p.i., contrasting the significant increase in wt mice at days 7 and 10 p.i. (Fig. 5A). The selective increase in Ccl5 mRNA in the CNS of infected wt mice at day 7 p.i. suggested expression by a newly infiltrating population, possibly T cells (Glass and Lane, 2003), which may be dysregulated in the absence of PKR. Cxcl10 mRNA was not induced until day 5 p.i. in either group, but was significantly reduced in the absence of PKR, reaching only ~ 30% of the levels in wt mice at both days 5 and 7 p.i. (Fig. 5A). These data support PKR-p as a positive regulator of only a subset of genes regulated by NF-kB.
Fig. 5.

PKR deficiency impairs mRNA but not protein expression of pro-inflammatory genes in the CNS. (A) Expression of NF-kB induced pro-inflammatory genes Il-1β, Il-6, Ccl2, Ccl5, and Cxcl10 relative to Gapdh in brains of infected PKR−/− and wt mice. Data represent the average of n = 6 mice/group/time-point from two separate experiments ± SEM. Data from day 3 p.i. represents n = 3. (B) IL-1β, CCL5 and CXCL10 proteins in cell free supernatants from brains from PKR−/− and wt mice were measured by ELISA. Data are the average of n = 6 mice/group/time-point ± SEM. *p ≥ .05.
The opposing functions of PKR in enhancing cytokine and chemokine expression at the transcriptional level, yet also interfering with host cell translation makes it difficult to predict the net outcome on cytokine/chemokine release. We therefore measured IL-1β, CCL5 and CXCL10 protein in the CNS at various times p.i. (Fig. 5B). IL-1β levels were similar at day 3 p.i. in both groups and correlated with the mRNA levels. However, despite the decline in Il-1β mRNA, protein levels remained high and even increased by 2-fold in the absence of PKR relative to wt levels at day 5 p.i. (Fig. 5B). IL-1β remained similarly high in both groups at day 7 p.i. and only declined at day 10 p.i. Similar CCL5 levels in both groups also reflected equal mRNA levels at day 5 p.i. By day 7 p.i. the ~ 3-fold increase in CCL5 in wt mice also coincided with elevated mRNA. Unexpectedly however, CCL5 in PKR−/− mice reached similar levels observed in wt mice at days 7 and 10 p.i., despite the absence of enhanced transcriptional activation after day 5 p.i. (Fig. 5B). Similarly, irrespective of significantly reduced mRNA levels in the absence of PKR (Fig. 5A), CXCL10 protein was comparable to wt mice at days 5 and 7 p.i. These data are consistent with the notion that reduced transcriptional activation of these cytokines in vivo in the absence of PKR is compensated by enhanced translation.
Total CNS expression analysis does not address whether reduced transcription of pro-inflammatory genes, yet no defects in protein production, is attributed to the same cell type in vivo. Pro-inflammatory factors are induced in multiple cell types during JHMV infection, including astrocytes and microglia/macrophages early and T cells later during infection (Glass et al., 2002, Bergmann et al., 2006). We therefore assessed whether PKR deficiency results in similarly dysregulated chemokine expression in vitro. The MHV-A59 strain, which infects primary cells more efficiently (> 70%) than the JHMV variant, was used to infect BMDM as a defined myeloid cell population known to induce both CCL5 and CXCL10. While infected wt BMDM strongly upregulated both Ccl5 and Cxcl10 mRNA between 12 and 18 h p.i., induction of both mRNAs was significantly reduced in PKR−/− BMDM (Suppl. Fig. 2). By contrast, Ccl2 mRNA levels were not affected by PKR deficiency (Suppl. Fig. 2). Furthermore, infected PKR−/− BMDM secreted increased CCL5 and similar CXCL10 compared to wt BMDM (Suppl. Fig. 2). Expression patterns of mRNAs and proteins thus mirrored the in vivo findings.
3.4. PKR regulates CNS CD4 T cell function and perivascular accumulation
The absence of PKR correlated with increased CD8 T cell proliferation in other models (Kadereit et al., 2000, Nakayama et al., 2010). Effective virus control, despite elevated replication during the early phase of JHMV infection, supported the concept that PKR deficiency may enhance T cell recruitment or antiviral function. The magnitude and composition of CNS infiltrating cells were thus compared between infected PKR−/− and wt mice by flow cytometry (Suppl. Fig. 3). Total CD45hi infiltrating cells and the magnitude of CD45hi F4/80+ infiltrating monocytes were similar in both groups (Suppl. Fig. 3). CD4 and CD8 T cells both accumulated in the CNS to maximal levels between days 7–10 p.i. and accounted for ~ 60% of total CD45hi infiltrates in both groups. Furthermore, CD8 T cells were consistently higher at day 7 p.i. and CD4 T cells at both days 7 and 10 p.i. within the brains of PKR−/− relative to wt mice (Suppl. Fig. 3). No differences or less than 2-fold higher numbers of monocytes and T cells, respectively, supported the absence of defects in chemokine mediated leukocyte recruitment in the absence of PKR.
PKR has been implicated in regulating CD8 T cell antiviral function and IL-10 production in monocytes (Cheung et al., 2005, Nakayama et al., 2010). During acute JHMV encephalomyelitis, perforin mediated cytolysis and IFN-γ produced by T cells are essential antiviral mediators (Bergmann et al., 2006). PKR deficiency enhanced IFN-γ mRNA and protein early, but did not affect IFN-γ during peak T cell accumulation in the CNS (Fig. 6 ). To rule out a reduction in other T cell functions in the absence of PKR, we further assessed expression of IL-10. During JHMV infection IL-10 in the CNS is predominantly produced by CD4 T cells (Trandem et al., 2011) and is critical to minimize demyelination. IL-10 is also produced by a small fraction of CD8 T cells with high cytolytic capacity, yet it inhibits clearance of infectious virus (Trandem et al., 2011). Il-10 mRNA was prominently increased at day 7 in wt mice, but was reduced by 6–7 fold in PKR−/− mice (Fig. 6). Moreover, IL-10 protein levels were also significantly impaired (Fig. 6). The kinetics and breadth of IL-10 reduction, despite unimpaired T cell infiltration suggest a CD4 T cell specific functional defect. CNS CD4 T cells are also the main producers of IL-21 and TIMP1, but do not express IL-17 during JHMV infection (Zhou et al., 2005, Kapil et al., 2009, Phares et al., 2012b). While IL-21 enhances humoral responses during JHMV infection (Phares et al., 2013a), TIMP1 deficiency impairs migration of CD4 T cells from the perivascular space to the brain parenchyma (Savarin et al., 2013). Reduced Il-21 and Timp1 transcripts in the CNS of PKR−/− mice supported selectively impaired CD4 T cell function (Fig. 6). Furthermore, reduced IL-21 protein expression was supported by reduced levels of IgG mRNA at day 10 p.i. (Fig. 6), when IL-21 dependent antibody secreting cells start accumulating in the CNS (Phares et al., 2013a). Moreover, TIMP1 levels were also significantly reduced (Fig. 6), suggesting that impaired IL-10, IL-21 and TIMP1 transcription was not overcome by enhanced translation.
Fig. 6.

PKR deficiency impairs IL-10, Il-21 and TIMP1 expression. Cytokines prominently produced by T cells during JHMV infection were monitored by analysis of brain derived mRNA and protein in cell free supernatants. Upper panel: Expression of Ifn-γ, Il-10, Timp1 and Il-21 mRNA by real time PCR and lower panel: IFN-γ, IL-10 and TIMP1 in cell free supernatants was determined by ELISA and IgG mRNA by real time PCR. All PCR data are calculated relative to Gapdh and represent the average ± SEM of n = 6/time point/group. ELISA data are the average ± SEM of n = 6/time point/group with values from naïve mice subtracted.
A role of PKR in specifically modulating CD4 T cell function was confirmed by stimulating brain derived T cells with immunodominant viral peptides, followed by analysis of IFN-γ and IL-10 production. The ability of virus specific CD4 T cells to produce IL-10 was indeed reduced by ~ 50% in the absence of PKR (Fig. 7 ). By contrast, CD8 T cells demonstrated no affects of PKR deficiency on IL-10 production (Fig. 7). Consistent with similar IFN-γ protein in CNS supernatants, IFN-γ production was not impaired in either CD4 or CD8 T cells (Fig. 7). These results demonstrate that PKR specifically regulates IL-10, but not IFN-γ, in virus specific CD4 T cells during JHMV induced encephalomyelitis. Furthermore, unlike CXCL10 and CCL5, IL-10 appeared prominently regulated at the transcriptional level.
Fig. 7.

PKR deficiency impairs IL-10 expression by virus specific CD4, but not CD8 T cells. Brain infiltrating CD4 and CD8 T cells were tested for production of IL-10 and IFN-γ after stimulation with class II restricted M133 or class I restricted S510 peptide by intracellular staining. Representative density plots show unstimulated (−) and M133 stimulated (+) CD4 T cells at day 10 p.i. Numbers above boxed cells are percentages of IL-10 secreting cells. Bar graphs show average frequencies ± SEM of IL-10 and IFN-γproducing CD4 or CD8 T cells from 3 individual mice; *p ≥ .05.
The relevance of reduced TIMP1 expression in PKR deficient mice was tested by comparing CD4 T cell distribution in the perivascular space and parenchyma (Fig. 8A). Numbers of CD4 T cells were increased at days 7 and 10 p.i. in PKR−/− compared to wt mice (data not shown), consistent with flow cytometric analysis (Suppl. Fig. 3). Although there were no differences in parenchymal CD4 T cell numbers between the groups, PKR−/− mice harbored higher numbers of perivascular CD4 T cells (Fig. 8). In wt mice overall distribution of total CD4 T cells dropped from ~ 40% at day 7 to ~ 15% at day 10 p.i. in perivascular cuffs (Fig. 8A). By contrast, ~ 30–40% of total CD4 T cells remained perivascular in PKR−/− mice at day 10 p.i. (Fig. 8A). These results support a role for TIMP1 in promoting CD4 T cell access to the parenchyma and demonstrate PKR enhances TIMP1 expression following JHMV induced encephalitis. To confirm PKR effects on CD4 T cells are CD4 T cell intrinsic, PKR activation in CNS localized wt CD4 T cells was assessed using immunohistochemistry. PKR-p expression was detected in CD4 T cells in the brain at days 7 p.i. (Fig. 8B), supporting a direct role of activated PKR-p in CD4 T cell function following viral CNS infection.
Fig. 8.

PKR deficiency results in increased perivascular accumulation of CD4 T cells. (A) Localization of CD4 T cells was assessed at days 7 and 10 p.i. by dual staining of brain sections for laminin (green) and CD4 (red) and analyzed by confocal microscopy. Bar graphs show average of CD4 T cells in perivascular space and in brain parenchyma of 6–9 fields/brain with n = 6 mice/group/time-point ± SEM. (B) Staining of brain sections at day 7 p.i. for PKR-p (green), CD4 (red) and nuclei (DAPI, blue) was analyzed by florescent microscopy. Images for both panels are representative of 6–9 fields/brain with n = 6 mice/group/time-point. *p ≥ .05.
3.5. PKR deficiency does not alter demyelination, axonal damage, or clinical disease
Based on the protective role of IL-10 in limiting demyelination following JHMV infection (Trandem et al., 2011), we assessed whether reduced IL-10 expression correlated with enhanced demyelination. Although Il-10 transcripts were also reduced by 70–80% in spinal cords of PKR−/− mice at days 7 and 10 p.i. (data not shown), the extent of inflammation, demyelination and axonal damage was similar in wt and PKR−/− mice at day 14 p.i. (Fig. 9A). Moreover, demyelination was also similar at day 21 pi. reaching 38 ± 6% in wt and 44 ± 6% in PKR deficient spinal cords. Although increased perivascular cuffs were still noted at day 14 p.i., differences had resolved by day 21 p.i. and the distribution and number of inflammatory cells were indistinguishable in the two groups (data not shown). The absence of overt pathological differences was consistent with the observation that PKR deficiency did not affect severity or progression of clinical disease (Fig. 9B).
Fig. 9.

PKR deficiency does not alter inflammation, demyelination, axonal damage or disease severity. (A) Cross sections of spinal cords from infected wt and PKR−/− mice at day 14 p.i. stained with hematoxylin and eosin (HE) for inflammation, luxol fast blue (LFB) for demyelination, or SMI31/32 (SMI) for axonal loss (bar = 500 μm; inset = 100 μm). (B) Clinical disease of infected wt and PKR−/− mice. Data are representative of three separate experiments (n ≥ 9-15).
4. Discussion
The anti-viral mediators of IFN-α/β mediated protection during coronavirus encephalomyelitis are largely unknown. The 2′-5′-oligoadenylate synthetase (OAS)/RNaseL pathway plays only a minor direct antiviral role in the CNS (Ireland et al., 2009, Zhao et al., 2013), but protects from lethal pathology during JHMV infection (Ireland et al., 2009). Ifit2 mediates innate antiviral activity against MHV-A59 via promoting IFN-α/β (Butchi et al., 2014). The present study investigated PKR as a candidate contributing to protection based on its ability to not only block translation, but also promote IFN-α/β production and pro-inflammatory mediators (Garcia et al., 2006). Following JHMV infection PKR was activated in the majority of CNS infected cells at 5 day p.i., supporting virus mediated activation. However, PKR-p detection in neighboring uninfected cells suggested that stimuli other than viral RNA also activate PKR. Higher CNS virus replication early during JHMV infection in PKR−/− relative to wt mice, in the absence of overt defects in Ifn-α/β mRNA or ISGs induction, supported PKR mediated antiviral activity in vivo. However, the effects were modest and increased replication was ultimately controlled by adaptive immunity.
PKR can influence inflammatory responses by promoting NF-kB transcriptional activation in a variety of settings in vitro, including poly I:C activation of mouse embryonic fibroblasts (Garcia et al., 2006). However, poly I:C-induced NF-kB activation in mouse peritoneal macrophages does not require PKR (Maggi et al., 2000), suggesting that the function of PKR in promoting NF-kB transcriptional activation depends on the activating stimulus and cell type. MHV induces IFN-α/β via the intracellular recognition receptor MDA5 in macrophages (Roth-Cross et al., 2008) and both MDA5 and RIGI in an oligodendrocyte cell line (Li et al., 2010). Although the pathway underlying induction of NF-kB dependent chemokines and cytokines has not been studied extensively, abrogation of RIGI partially blocked NF-kB activity following MHV-A59 infection (Li et al., 2010). This finding supported a potential link between viral mediated MDA5 activation and NF-kB transcriptional activation. Reduced expression of Il-6, Ccl5 and Cxcl10 but not Il-1β, and Ccl2 mRNAs in vivo in the absence of PKR during JHMV infection is thus consistent with PKR mediated promotion of NF-kB activation. Although the cell types involved remain to be identified, MHV-A59 infected BMDM exhibited similar selective defects in proinflammatory gene induction. TMEV infected PKR−/− astrocytes also showed limited activation of NF-kB dependent transcription, but also impaired IFN-α/β expression (Palma et al., 2003, Carpentier et al., 2007). By contrast, no differences were evident in Ifn-α/β, Ifit1 or Ifit2 mRNA following MHV infection of PKR−/− BMDM (data not shown). IL-6, CXCL10 and CCL5 are all implicated in promoting leukocyte infiltration into the CNS parenchyma. Specifically CXCL10 and CCL5 facilitate CD4 T cell recruitment to the CNS during JHMV infection (Liu et al., 2001, Glass and Lane, 2003). Reduced mRNA levels of these proinflammatory factors were thus inconsistent with the increase in CNS infiltrating T cells in PKR−/− mice. This discrepancy was resolved by the demonstration of similar or even increased CCL5, CXCL10 and IL-1β protein in the brains. The notion that impaired transcription of select genes was balanced by increased translation was supported by MHV infected BMDM from wt and PKR−/− mice. Moreover, astrocytes, which are not or rarely infected by JHMV (Ireland et al., 2009), not microglia/macrophages, are the predominant source of CXCL10 following MHV infection (Lane et al., 1998, Phares et al., 2013b). These results indicate that PKR dependent regulation of CXCL10 expression is not dependent on infection and not necessarily cell type specific. Although CD4 T cells are implicated as a dominant source of CCL5 based on reduced CCL5 mRNA levels in CD4−/− mice (Lane et al., 2000), CD4 T cells may also indirectly affect CCL5 production by other cellular sources (Lane et al., 2000).
The most surprising effect of PKR deficiency following JHMV infection was the impaired CNS expression of selective CD4 T cell dependent functions, especially the anti-inflammatory cytokine IL-10. Consistent with CD4 T cells as the main producers of IL-10 in our model (Puntambekar et al., 2011), the frequency of CNS derived virus specific PKR−/− CD4 T cells capable of IL-10 production was reduced by ~ 50% compared to that of wt CD4 T cells. By contrast, PKR−/− CD8 T cells showed no evidence for reduced IL-10 production, and IFN-γ production was not impaired in either T cell subset, consistent with effective viral control. Decreased Il-21 mRNA and TIMP1, also mainly produced by CD4 T cells (Zhou et al., 2005, Phares et al., 2012b), support the concept that PKR primarily affects CD4 T cell function. In this context it is interesting to note that Il-10 expression is regulated by various transcription factors, including NF-kB, Jnk, cMaf, AP1, and IRF4, while Il-21 transcription in CD4 T cells is linked to cMaf activation (Hiramatsu et al., 2010), suggesting a possible in vivo link between PKR and cMaf activation. While our results specifically demonstrate an effect on virus specific CD4 T cells, it is not clear if PKR also affects IL-10 production by other CD4 regulatory T cell subsets. Irrespectively, detection of activated PKR-p in CD4 T cells, consistent with PKR activation in CNS infiltrating CD3 T cells during EAE (Chakrabarty et al., 2004), supports a direct effect.
PKR upregulates IL-10 in an NF-kB dependent manner in response to mycobacterium, bacterial LPS, dsRNA and Sendai virus (Chakrabarti et al., 2008). While microglia/macrophages only sparsely produce IL-10 in vivo during JHMV infection, analysis of MHV-A59 infected PKR−/− BMDM confirmed a 70–80% decrease in Il-10 mRNA, coincident with a 44% reduction in IL-10 secretion (data not shown). As CD4 T cells are not infected, the mechanisms inducing IL-10 in macrophages and CD4 T cells are clearly distinct, but suggest that activated PKR-p acts on a common intermediate signaling factor, such as NF-kB. The upstream signals activating PKR remain to be identified, but may involve a variety of factors, including stress inducing molecules (Sadler and Williams, 2007). Similarly, the promotion of CXCL10 mRNA expression in astrocytes, independent of direct infection, suggests that intrinsic cellular not virus mediated signals regulates PKR activation. While no evidence for dysregulation of CD8 T cells contrasted the apparent PKR effects on CD8 T cell expansion following LCMV infection (Nakayama et al., 2010), the effects of PKR deficiency in the latter studies appeared to be indirectly mediated by innate defects in antigen presenting cells rather than intrinsic CD8 T cell effects (Nakayama et al., 2010).
PKR is also associated with exacerbated pathology in various neurological diseases. PKR activation in hippocampal neurons is associated with neuronal loss and during Alzheimer's, Huntington, and Parkinson's diseases (Bando et al., 2005). Similarly, the absence of PKR provides protection from extensive spinal cord damage during poliomyelitis (Scheuner et al., 2003). In contrast to these reports, pathological changes in JHMV infected PKR−/− and wt mice was similar with respect to inflammation, demyelination and axonal damage. Thus, while impaired IL-10 expression has been associated with enhanced demyelination during JHMV encephalomyelitis (Trandem et al., 2011), the partial loss of IL-10 in the absence of PKR appeared insufficient to exacerbate pathology.
In summary, our results demonstrate that PKR has a modest direct antiviral role in the CNS during the innate phase of JHMV infection, but does not affect adaptive immune control. Paradoxically, impaired mRNA, but not protein expression of CXCL10 and CCL5 in the absence of PKR confirmed that it selectively regulates pro-inflammatory cytokines at both the transcriptional and translational levels in vivo and in vitro. Importantly these results are the first to demonstrate that PKR promotes IL-10, IL-21 and TIMP1 production in virus specific CD4 T cells. While the imbalance between pro- and anti-inflammatory responses in the CNS did not affect severity or progression of clinical disease or tissue damage, the data highlight the potential of PKR to modulate numerous pro- as well as anti-inflammatory molecules in both infected and non-infected cells, including T cells.
The following are the supplementary data related to this article.
Strategy for FACS purification of oligodendrocytes, microglia and myeloid cells from the CNS of infected mice. Brain cells from JHMV infected wt mice at 5 days p.i. were stained with anti CD45, CD11b and O4 mAb. Representative plots depict gating strategy: All events were gated to exclude debris (P1); doublets were excluded based on FSC-W (P2; FSC-Area/FSC-Height). Oligodendrocytes (P3) were separated from CD45lo microglia (P5), CD45hi infiltrating leukocytes (P6) and other CNS resident cells (P4; CD45−O4−) by O4 staining (CD45−O4+). Microglia (P5) were separated from infiltrating and resident cells by their CD45lo CD11b+ phenotype. Infiltrating myeloid cells (P7) were separated from other CD45hi CNS infiltrating cells by their CD45hiCD11b+ phenotype.
PKR deficiency impairs mRNA but not protein expression of pro-inflammatory chemokines in virus infected BMDM. BMDM from wt and PKR−/− mice were infected with MHV-A59 at a MOI of 1. Transcript expression of Ccl2, Ccl5 and Cxcl10 and secreted protein levels of CCL5 and CXCL10 were determined at 12 and 18 h p.i. Data are the average of 3 separate experiments ± SEM. *p ≥ .05.
PKR deficiency enhances T cell inflammation. Numbers of total brain infiltrating leukocytes (CD45hi), macrophages (CD45hi F480+), CD8 and CD4 T cells derived from infected wt and PKR−/− mice were determined by flow cytometry. Data represent the average of three separate experiments ± SEM with n = 3/group per timepoint and experiment. *p ≥ .05.
Acknowledgment
This work was supported by the National Institutes of Health Grant P01 NS064932. We sincerely thank Dr. Robert H. Silverman for helpful comments on this manuscript. We also thank Wenqiang Wei, Eric Barron, Mi Widness and Dr. Shweta Puntambekar for exceptional technical assistance, Jennifer Powers for FACS purification, and Dr. Niranjan Butchi for help with BMDM experiments.
References
- Bando Y., Onuki R., Katayama T., Manabe T., Kudo T., Taira K., Tohyama M. Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson's disease and Huntington's disease. Neurochem. Int. 2005;46:11–18. doi: 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Bergmann C.C., Lane T.E., Stohlman S.A. Coronavirus infection of the central nervous system: host-virus stand-off. Nat. Rev. Microbiol. 2006;4:121–132. doi: 10.1038/nrmicro1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchi N.B., Hinton D.R., Stohlman S.A., Kapil P., Fensterl V., Sen G.C., Bergmann C.C. Ifit2 deficiency results in uncontrolled neurotropic coronavirus replication and enhanced encephalitis via impaired alpha/beta interferon induction in macrophages. J. Virol. 2014;88:1051–1064. doi: 10.1128/JVI.02272-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier P.A., Williams B.R., Miller S.D. Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia. 2007;55:239–252. doi: 10.1002/glia.20450. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A., Sadler A.J., Kar N., Young H.A., Silverman R.H., Williams B.R. Protein kinase R-dependent regulation of interleukin-10 in response to double-stranded RNA. J. Biol. Chem. 2008;283:25132–25139. doi: 10.1074/jbc.M804770200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty A., Danley M.M., Levine S.M. Immunohistochemical localization of phosphorylated protein kinase R and phosphorylated eukaryotic initiation factor-2 alpha in the central nervous system of SJL mice with experimental allergic encephalomyelitis. J. Neurosci. Res. 2004;76:822–833. doi: 10.1002/jnr.20125. [DOI] [PubMed] [Google Scholar]
- Cheung B.K., Lee D.C., Li J.C., Lau Y.L., Lau A.S. A role for double-stranded RNA-activated protein kinase PKR in mycobacterium-induced cytokine expression. J. Immunol. 2005;175:7218–7225. doi: 10.4049/jimmunol.175.11.7218. [DOI] [PubMed] [Google Scholar]
- Couturier J., Morel M., Pontcharraud R., Gontier V., Fauconneau B., Paccalin M., Page G. Interaction of double-stranded RNA-dependent protein kinase (PKR) with the death receptor signaling pathway in amyloid beta (Abeta)-treated cells and in APPSLPS1 knock-in mice. J. Biol. Chem. 2010;285:1272–1282. doi: 10.1074/jbc.M109.041954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der S.D., Lau A.S. Involvement of the double-stranded-RNA-dependent kinase PKR in interferon expression and interferon-mediated antiviral activity. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8841–8845. doi: 10.1073/pnas.92.19.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erta M., Quintana A., Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012;8:1254–1266. doi: 10.7150/ijbs.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming J.O., Trousdale M.D., El-Zaatari F.A., Stohlman S.A., Weiner L.P. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J. Virol. 1986;58:869–875. doi: 10.1128/jvi.58.3.869-875.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M.A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev.: MMBR. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass W.G., Chen B.P., Liu M.T., Lane T.E. Mouse hepatitis virus infection of the central nervous system: chemokine-mediated regulation of host defense and disease. Viral Immunol. 2002;15:261–272. doi: 10.1089/08828240260066215. [DOI] [PubMed] [Google Scholar]
- Glass W.G., Hickey M.J., Hardison J.L., Liu M.T., Manning J.E., Lane T.E. Antibody targeting of the CC chemokine ligand 5 results in diminished leukocyte infiltration into the central nervous system and reduced neurologic disease in a viral model of multiple sclerosis. J. Immunol. 2004;172:4018–4025. doi: 10.4049/jimmunol.172.7.4018. [DOI] [PubMed] [Google Scholar]
- Glass W.G., Lane T.E. Functional expression of chemokine receptor CCR5 on CD4+ T cells during virus-induced central nervous system disease. J. Virol. 2003;77:191–198. doi: 10.1128/JVI.77.1.191-198.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grolleau A., Kaplan M.J., Hanash S.M., Beretta L., Richardson B. Impaired translational response and increased protein kinase PKR expression in T cells from lupus patients. J. Clin. Invest. 2000;106:1561–1568. doi: 10.1172/JCI9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu Y., Suto A., Kashiwakuma D., Kanari H., Kagami S., Ikeda K., Hirose K., Watanabe N., Grusby M.J., Iwamoto I., Nakajima H. c-Maf activates the promoter and enhancer of the IL-21 gene, and TGF-beta inhibits c-Maf-induced IL-21 production in CD4+ T cells. J. Leukoc. Biol. 2010;87:703–712. doi: 10.1189/jlb.0909639. [DOI] [PubMed] [Google Scholar]
- Hopkins S.J., Rothwell N.J. Cytokines and the nervous system. I: expression and recognition. Trends Neurosci. 1995;18:83–88. [PubMed] [Google Scholar]
- Ireland D.D., Stohlman S.A., Hinton D.R., Atkinson R., Bergmann C.C. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 2008;82:300–310. doi: 10.1128/JVI.01794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland D.D., Stohlman S.A., Hinton D.R., Kapil P., Silverman R.H., Atkinson R.A., Bergmann C.C. RNase L mediated protection from virus induced demyelination. PloS Pathog. 2009;5:e1000602. doi: 10.1371/journal.ppat.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadereit S., Xu H., Engeman T.M., Yang Y.L., Fairchild R.L., Williams B.R. Negative regulation of CD8+ T cell function by the IFN-induced and double-stranded RNA-activated kinase PKR. J. Immunol. 2000;165:6896–6901. doi: 10.4049/jimmunol.165.12.6896. [DOI] [PubMed] [Google Scholar]
- Kapil P., Atkinson R., Ramakrishna C., Cua D.J., Bergmann C.C., Stohlman S.A. Interleukin-12 (IL-12), but not IL-23, deficiency ameliorates viral encephalitis without affecting viral control. J. Virol. 2009;83:5978–5986. doi: 10.1128/JVI.00315-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapil P., Butchi N.B., Stohlman S.A., Bergmann C.C. Oligodendroglia are limited in type I interferon induction and responsiveness in vivo. Glia. 2012;60:1555–1566. doi: 10.1002/glia.22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Haque J., Lacoste J., Hiscott J., Williams B.R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. U. S. A. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane T.E., Asensio V.C., Yu N., Paoletti A.D., Campbell I.L., Buchmeier M.J. Dynamic regulation of alpha- and beta-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J. Immunol. 1998;160:970–978. [PubMed] [Google Scholar]
- Lane T.E., Liu M.T., Chen B.P., Asensio V.C., Samawi R.M., Paoletti A.D., Campbell I.L., Kunkel S.L., Fox H.S., Buchmeier M.J. A central role for CD4+ T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 2000;74:1415–1424. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.B., Rodriguez D., Rodriguez J.R., Esteban M. The apoptosis pathway triggered by the interferon-induced protein kinase PKR requires the third basic domain, initiates upstream of Bcl-2, and involves ICE-like proteases. Virology. 1997;231:81–88. doi: 10.1006/viro.1997.8494. [DOI] [PubMed] [Google Scholar]
- Li J., Liu Y., Zhang X. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J. Virol. 2010;84:6472–6482. doi: 10.1128/JVI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.T., Keirstead H.S., Lane T.E. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J. Immunol. 2001;167:4091–4097. doi: 10.4049/jimmunol.167.7.4091. [DOI] [PubMed] [Google Scholar]
- Lu B., Nakamura T., Inouye K., Li J., Tang Y., Lundback P., Valdes-Ferrer S.I., Olofsson P.S., Kalb T., Roth J., Zou Y., Erlandsson-Harris H., Yang H., Ting J.P., Wang H., Andersson U., Antoine D.J., Chavan S.S., Hotamisligil G.S., Tracey K.J. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L.B., Jr., Heitmeier M.R., Scheuner D., Kaufman R.J., Buller R.M., Corbett J.A. Potential role of PKR in double-stranded RNA-induced macrophage activation. EMBO J. 2000;19:3630–3638. doi: 10.1093/emboj/19.14.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcallister C.S., Taghavi N., Samuel C.E. Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF-2alpha-mediated translational control. J. Biol. Chem. 2012;287:36384–36392. doi: 10.1074/jbc.M112.390039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs E., Chong K., Galabru J., Thomas N.S., Kerr I.M., Williams B.R., Hovanessian A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- Nakayama Y., Plisch E.H., Sullivan J., Thomas C., Czuprynski C.J., Williams B.R., Suresh M. Role of PKR and Type I IFNs in viral control during primary and secondary infection. PloS Pathog. 2010;6:e1000966. doi: 10.1371/journal.ppat.1000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma J.P., Kwon D., Clipstone N.A., Kim B.S. Infection with Theiler's murine encephalomyelitis virus directly induces proinflammatory cytokines in primary astrocytes via NF-kappaB activation: potential role for the initiation of demyelinating disease. J. Virol. 2003;77:6322–6331. doi: 10.1128/JVI.77.11.6322-6331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel A.L., Bredesen D.E. Activation of the cell stress kinase PKR in Alzheimer's disease and human amyloid precursor protein transgenic mice. Neurobiol. Dis. 2003;14:52–62. doi: 10.1016/s0969-9961(03)00086-x. [DOI] [PubMed] [Google Scholar]
- Phares T.W., Disano K.D., Hinton D.R., Hwang M., Zajac A.J., Stohlman S.A., Bergmann C.C. IL-21 optimizes T cell and humoral responses in the central nervous system during viral encephalitis. J. Neuroimmunol. 2013;263:43–54. doi: 10.1016/j.jneuroim.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares T.W., Stohlman S.A., Hinton D.R., Bergmann C.C. Enhanced CD8 T-cell anti-viral function and clinical disease in B7-H1-deficient mice requires CD4 T cells during encephalomyelitis. J. Neuroinflammation. 2012;9:269. doi: 10.1186/1742-2094-9-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares T.W., Stohlman S.A., Hinton D.R., Bergmann C.C. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J. Virol. 2013;87:3382–3392. doi: 10.1128/JVI.03307-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares T.W., Stohlman S.A., Hwang M., Min B., Hinton D.R., Bergmann C.C. CD4 T cells promote CD8 T cell immunity at the priming and effector site during viral encephalitis. J. Virol. 2012;86:2416–2427. doi: 10.1128/JVI.06797-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puntambekar S.S., Bergmann C.C., Savarin C., Karp C.L., Phares T.W., Parra G.I., Hinton D.R., Stohlman S.A. Shifting hierarchies of interleukin-10-producing T cell populations in the central nervous system during acute and persistent viral encephalomyelitis. J. Virol. 2011;85:6702–6713. doi: 10.1128/JVI.00200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K.M., Elliott R., Martinez-Sobrido L., Garcia-Sastre A., Weiss S.R. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J. Virol. 2010;84:5656–5669. doi: 10.1128/JVI.00211-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth-Cross J.K., Bender S.J., Weiss S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 2008;82:9829–9838. doi: 10.1128/JVI.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler A.J., Williams B.R. Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 2007;316:253–292. doi: 10.1007/978-3-540-71329-6_13. [DOI] [PubMed] [Google Scholar]
- Samuel M.A., Whitby K., Keller B.C., Marri A., Barchet W., Williams B.R., Silverman R.H., Gale M., Jr., Diamond M.S. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarin C., Bergmann C.C., Hinton D.R., Stohlman S.A. MMP-independent role of TIMP-1 at the blood brain barrier during viral encephalomyelitis. ASN Neuro. 2013;5(5):321–331. doi: 10.1042/AN20130033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarin C., Stohlman S.A., Atkinson R., Ransohoff R.M., Bergmann C.C. Monocytes regulate T cell migration through the glia limitans during acute viral encephalitis. J. Virol. 2010;84:4878–4888. doi: 10.1128/JVI.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D., Gromeier M., Davies M.V., Dorner A.J., Song B., Patel R.V., Wimmer E.J., Mclendon R.E., Kaufman R.J. The double-stranded RNA-activated protein kinase mediates viral-induced encephalitis. Virology. 2003;317:263–274. doi: 10.1016/j.virol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Schulz O., Pichlmair A., Rehwinkel J., Rogers N.C., Scheuner D., Kato H., Takeuchi O., Akira S., Kaufman R.J., Reis E Sousa C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. 2010;7:354–361. doi: 10.1016/j.chom.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl D.F., Abraham N., Knowles S., Marius R., Brasey A., Lichty B.D., Brown E.G., Sonenberg N., Bell J.C. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 2000;74:9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D.R., Lee S.B., Romano P.R., Marshak D.R., Hinnebusch A.G., Esteban M., Mathews M.B. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol. Cell. Biol. 1996;16:6295–6302. doi: 10.1128/mcb.16.11.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trandem K., Zhao J., Fleming E., Perlman S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J. Immunol. 2011;186:3642–3652. doi: 10.4049/jimmunol.1003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Kaufman R.J. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem. 1997;272:1291–1296. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- Yang Y.L., Reis L.F., Pavlovic J., Aguzzi A., Schafer R., Kumar A., Williams B.R., Aguet M., Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Samuel C.E. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 2008;283:34580–34587. doi: 10.1074/jbc.M807029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Birdwell L.D., Wu A., Elliott R., Rose K.M., Phillips J.M., Li Y., Grinspan J., Silverman R.H., Weiss S.R. Cell-type-specific activation of the oligoadenylate synthetase-RNase L pathway by a murine coronavirus. J. Virol. 2013;87:8408–8418. doi: 10.1128/JVI.00769-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Jha B.K., Wu A., Elliott R., Ziebuhr J., Gorbalenya A.E., Silverman R.H., Weiss S.R. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe. 2012;11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Marten N.W., Bergmann C.C., Macklin W.B., Hinton D.R., Stohlman S.A. Expression of matrix metalloproteinases and their tissue inhibitor during viral encephalitis. J. Virol. 2005;79:4764–4773. doi: 10.1128/JVI.79.8.4764-4773.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strategy for FACS purification of oligodendrocytes, microglia and myeloid cells from the CNS of infected mice. Brain cells from JHMV infected wt mice at 5 days p.i. were stained with anti CD45, CD11b and O4 mAb. Representative plots depict gating strategy: All events were gated to exclude debris (P1); doublets were excluded based on FSC-W (P2; FSC-Area/FSC-Height). Oligodendrocytes (P3) were separated from CD45lo microglia (P5), CD45hi infiltrating leukocytes (P6) and other CNS resident cells (P4; CD45−O4−) by O4 staining (CD45−O4+). Microglia (P5) were separated from infiltrating and resident cells by their CD45lo CD11b+ phenotype. Infiltrating myeloid cells (P7) were separated from other CD45hi CNS infiltrating cells by their CD45hiCD11b+ phenotype.
PKR deficiency impairs mRNA but not protein expression of pro-inflammatory chemokines in virus infected BMDM. BMDM from wt and PKR−/− mice were infected with MHV-A59 at a MOI of 1. Transcript expression of Ccl2, Ccl5 and Cxcl10 and secreted protein levels of CCL5 and CXCL10 were determined at 12 and 18 h p.i. Data are the average of 3 separate experiments ± SEM. *p ≥ .05.
PKR deficiency enhances T cell inflammation. Numbers of total brain infiltrating leukocytes (CD45hi), macrophages (CD45hi F480+), CD8 and CD4 T cells derived from infected wt and PKR−/− mice were determined by flow cytometry. Data represent the average of three separate experiments ± SEM with n = 3/group per timepoint and experiment. *p ≥ .05.