

Abstract
Protein post‐translational modifications (PTMs) allow the cell to regulate protein activity and play a crucial role in the response to changes in external conditions or internal states. Advances in mass spectrometry now enable proteome wide characterization of PTMs and have revealed a broad functional role for a range of different types of modifications. Here we review advances in the study of the evolution and function of PTMs that were spurred by these technological improvements. We provide an overview of studies focusing on the origin and evolution of regulatory enzymes as well as the evolutionary dynamics of modification sites. Finally, we discuss different mechanisms of altering protein activity via post‐translational regulation and progress made in the large‐scale functional characterization of PTM function.
Keywords: acetylation, evolution, phosphorylation, post‐translational modifications, PTM cross‐talk
Introduction
Cells need to constantly sense changes in internal and external conditions, some of which have to be acted on very quickly. In such cases, conditional changes can be relayed from sensors to effectors via reversible post‐translational modifications (PTMs) of proteins. The abundance of PTMs is often regulated by enzymes that can add or remove the modifications (e.g. ‘writer’ and ‘eraser’ domains, respectively) and functional consequences can be mediated by domains that recognize and bind to the modifications (e.g. ‘reader’ domains; Seet et al, 2006). Not only in isolation but also in coordination, PTMs can influence numerous properties of proteins including enzymatic activity, protein interactions and subcellular location. Therefore, PTMs represent an important mechanism of regulation on protein function. Post‐translational regulatory networks require fine‐tuned crosstalk of individual players to coordinate various protein states in specific cellular conditions. While protein post‐translational modification has been studied for many years in the context of cellular signaling, very little is known about how these systems evolve when species occupy new niches. The knowledge on the respective evolutionary processes and adaptive constrains can, in turn, reveal what aspects are under selection and therefore more likely to be of high functional importance.
Recent developments in mass‐spectrometry (MS) methods now allow us to identify PTMs at an unprecedented scale (Choudhary & Mann, 2010). Thousands of PTM sites have been identified and novel enrichment strategies have uncovered the global cellular importance of several types of modifications (e.g. acetylation, ubiquitylation, O‐GlNac, N‐linked glycosylation). More than 200 different types of PTMs are currently known (Minguez et al, 2012), ranging from small chemical modifications (e.g. phosphorylation and acetylation) to the addition of complete proteins (e.g. ubiquitylation). Not all of these modifications have been extensively characterized. For example, 15 of these have at least 1,000 known modification sites, as annotated in Uniprot (Table 1), identified across a broad range of species (Fig 1). For a small subset of known PTM types, established enrichment protocols and mass‐spectrometry allow for the large‐scale identification of thousands of sites per study revealing important and broad functional roles (Choudhary & Mann, 2010). This increase in throughput is not only fueling a range of evolutionary studies but is also creating a bottleneck in the functional annotation and study of PTMs. We know the functional role of only a small fraction of all PTMs and functional studies cannot yet be carried out in high‐throughput. This bottleneck in the functional characterization of PTMs is reminiscent of earlier issues in the field of genomics when the dramatic increase in sequencing throughput created a need for the development of novel methods to study functional genomic elements (e.g. protein domains, transcriptional regulatory elements, transposable elements). Similarly, new methodologies are now needed to study the evolution and function of PTMs at a large‐scale and to decipher how single or combinatorial PTMs regulate cellular activities. Here we will review recent studies on these topics that were driven by this increased throughput in PTM identification.
Table 1. Top 15 most frequent PTMs annotated in UniProt.
| PTM id | Number of species | Total occurrences |
|---|---|---|
| Phosphoserine | 1701 | 71744 |
| Phosphothreonine | 505 | 14604 |
| Phosphotyrosine | 1023 | 8280 |
| N‐acetylalanine | 347 | 2971 |
| N‐acetylmethionine | 161 | 1711 |
| N‐acetylserine | 357 | 1767 |
| N6‐acetyllysine | 664 | 13570 |
| N‐palmitoyl cysteine | 577 | 2056 |
| S‐palmitoyl cysteine | 295 | 2628 |
| N6‐carboxylysine | 1178 | 1600 |
| 4‐carboxyglutamate | 97 | 1252 |
| N5‐methylglutamine | 776 | 1298 |
| 4‐hydroxyproline | 115 | 1796 |
| S‐diacylglycerol cysteine | 551 | 1940 |
| Sulfotyrosine | 188 | 1015 |
| Glycyl lysine isopeptide (with NEDD8) | 13 | 47 |
| Glycyl lysine isopeptide (with SUMO) | 106 | 1234 |
| Glycyl lysine isopeptide (with ubiquitin) | 252 | 2815 |
We downloaded UniProt on 14th of August 2013 which contained descriptions of 460 different post‐translational modifications, whereby modifications on different amino acids are counted individually (e.g. Phosphoserine and Phosphothreonine are two different PTMs). Of these, 218 of have been associated with at least one SwissProt entry. PTM types annotated in UniProt with at least 1,000 occurrences and with one annotated instance in at least 10 species (15 different PTM types) are listed here along with total counts for number of species and occurrences. Given their well‐studied functional importance, we also added information on Ubiquitin, SUMO and NEDD.
Figure 1. Phylogenetic distribution for the Top 15 most frequent PTMs annotated in UniProt.
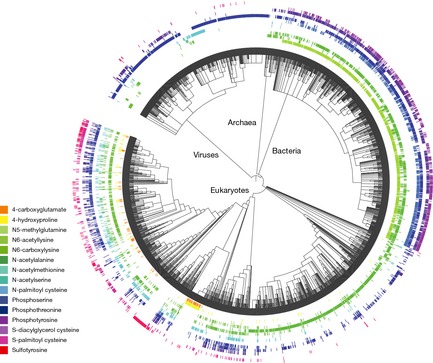
We selected the 15 PTM types annotated in UniProt with at least 1000 occurrences and with one annotated instance in at least 10 species. For these we obtained a list of all species with at least one occurrence. The species distribution of these 15 PTM types was mapped to a phylogenetic tree using the Interactive Tree of Life tool (http://itol.embl.de/). Presence or absence of at least one occurrence is displayed using a PTM color code next to each species leaf in the tree. Absence of a PTM occurrence in any given species has to be cautiously interpreted since this could be simply due to a lack of coverage.
Evolution of protein post‐translational regulation
The evolution of post‐translational regulation, and more specifically post‐translational modification, can be studied at multiple levels (Fig 2). On a very long timescale, PTMs can be traced to follow their origin and their diversification, starting from probably very few modification types in the last universal common ancestor (LUCA) to more than 200 known today. On an intermediate scale, the evolution of individual regulators can be dissected in the context of the enzymes that add and/or remove modifications from other proteins and the reader domains. On the shortest timescale, PTMs can be compared across species to study the conservation or divergence of individual regulatory interactions and modification sites.
Figure 2. Evolution of PTM regulation at different time scales.
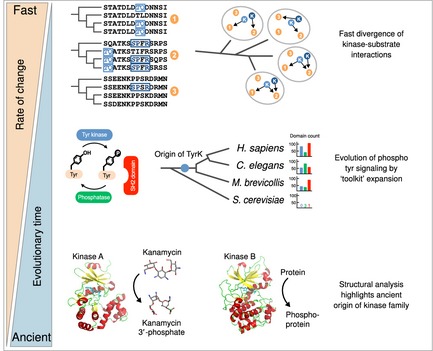
PTM evolution can be studied at different levels: from the origin of the PTMs and their enzymes (bottom) to the evolution of PTM enzymes and binding domains (middle) and their interactions (top). Many PTM types appear to be ancient and the origin of novel PTM type and their regulatory enzymes is likely to be a rare evolutionary event. The study of the ancient origin of protein phosphorylation has been aided by structural analysis where the conserved structural motifs of different phosphorylation enzymes can shed light on the origin of these enzymes. This is illustrated by the structural similarity between an aminoglycoside phosphotransferase (Kinase A, PDB: 1J7U) and a cAMP‐dependent protein kinase (Kinase B, PDB: 1CDK). Once a PTM type is established, the evolution of its regulators is likely to progress via gene‐duplication and divergence. Given the functional inter‐dependencies of the different effector domains (e.g. reader, writer and eraser) a high degree of co‐evolution is expected. This was observed for the phospho‐tyrosine effector domains (middle) where the three domain types typically display an all‐or‐none pattern of occurrence in the proteomes. The divergence of domain‐site interactions can occur at a faster rate than the divergence of effector domains since only a few mutations are required to create or destroy a PTM site. A hypothetical case illustrates how a few point mutations would be sufficient to significantly re‐wire a kinase‐substrate interaction network composed of three proteins and two kinases (top).
Protein PTMs are of ancient origin
Some modifications, such as phosphorylation, acetylation and glycosylation, seem to be ubiquitous across all domains of life, suggesting that these represent ancient PTMs that were used already in the last universal common ancestor. Long believed to only play a role in eukaryotes, N‐glycosylation and O‐glycosylation have now also been demonstrated in bacteria (Abu‐Qarn et al, 2008). Intriguingly however, the variety of monosaccharides used for bacterial glycosylation is much wider than in eukaryotes and the evolutionary relatedness is not yet clear.
A well‐studied example of a small and ubiquitous modification is lysine acetylation. While first discovered in higher eukaryotes on histone tails, it is now becoming clear that both the species range and protein repertoire of K‐acetylation is broader (Choudhary et al, 2009; Zhang et al, 2009; Wang et al, 2010; Van Noort et al, 2012). The species for which K‐acetylation data are now available range from gram‐negative and gram‐positive bacteria to eukaryotic organisms and enzymes involved in central cellular metabolism are often targeted (Choudhary et al, 2009; Hayden et al, 2013; Kim et al, 2013; Lee et al, 2013; Wu et al, 2013; Zhang et al, 2013). The levels of acetyl‐coA can influence the levels of lysine‐acetylation in the cell as well as affect enzyme activity (Cai et al, 2011). Thus, the nutrient status of the cell reflected by acetyl‐coA levels may directly influence metabolic activities (Cai et al, 2011). Acetylation on lysines can be catalyzed by Lysine Acetyl Transferases (KATs) but can also occur non‐enzymatically (Weinert et al, 2013), which is one of the reasons to believe K‐acetylation is an ancient PTM. Similarly, protein phosphorylation can also occur non‐enzymatically by pyrophosphates (Saiardi et al, 2004).
The possibility of non‐enzymatic acetylation and phosphorylation suggests an evolutionary path, whereby, in an early evolutionary phase, the regulation of protein activity (e.g. metabolic enzymes) occurred in response to nutrient status and, in a later phase, modification enzymes evolved to actively change PTM status of target proteins. Examples of such modification enzymes are sirtuins, a family of proteins which deacetylate lysines. Interestingly, sirtuin targets include metabolic enzymes, such as, for instance, acetyl‐coA synthetase, which is activated by the deacetylation of lysine‐609 by cobB, a member of the class III type of sirtuins (Starai et al, 2002). Sirtuins are present in both prokaryotes and eukaryotes, supporting the notion that acetylation is an ancient PTM. In eukaryotes, sirtuins have radiated into different classes (Frye, 2000), some of which are specific to acetylated lysines but others to other acyl lysine modifications such as succinylation (Du et al, 2011) and myristoylation (Jiang et al, 2013). The nutrient status is not only reflected by acetyl‐coA but also by butyryl‐coA and propionyl‐coA. Propionyl‐coA is derived from fatty acid and amino acid catabolism, whereas butyryl‐coA is formed during beta‐oxidation of fatty acids. These coA moieties have also been discovered as modifications on histone proteins as propionyl lysine and butyryl lysine (Chen et al, 2007). It has therefore been hypothesized that lysine propionylation and butyrylation may also regulate cellular metabolic pathways in response to cellular physiological conditions and that these modifications might further be involved in cellular signaling pathways (Chen et al, 2007). For now, it is however unclear which enzymes regulate these PTMs, but it can be speculated that the activity spectrum of sirtuins might extend beyond acetylation, succinylation and myristoylation, to also include propionylation and butyrylation.
For PTMs that involve the addition of entire proteins, ubiquitylation represents the best‐studied example and it results in the addition of a large protein domain (ubiquitin) to protein lysine residues. Protein ubiquitylation is most commonly known as a PTM that targets proteins for degradation but it has been recently recognized that it also has a major regulatory role in protein homeostasis, DNA repair, signaling and protein trafficking (Mukhopadhyay & Riezman, 2007; Chen & Sun, 2009). This functional diversity has been further highlighted by the proteome‐wide characterization of human ubiquitinylation (Emanuele et al, 2011; Kim et al, 2011; Wagner et al, 2011). How the ubiquitylation system has evolved is not completely clear; it has been suggested that it is similar to the widespread bacterial sulfur activation and delivery system in enzymatic reaction mechanism (Hochstrasser, 2009). In particular, the activation of ubiquitin by E1 enzymes is analogous to the thiocarboxylation of C‐terminal glycines of the sulphur‐carrier proteins MoaD and ThiS (Hochstrasser, 2009). More recently, the search of ubiquitin and ubiquitin‐related domains has revealed simple versions of proteasome targeting systems in bacteria and archaea by protein conjugation of so‐called ‘prokaryotic ubiquitin‐like proteins’ (PUP) and ‘small archaeal modifier proteins’ (SAMP; Burroughs et al, 2012; Maupin‐Furlow, 2012). SAMPylation seems to be homologous to the ubiquitylation system with the conjugation of the ubiquitin‐like SAMP proteins performed by an E1‐like activating enzyme (Maupin‐Furlow, 2012). In contrast, PUPylation appears to be a convergent evolution for proteasomal targeting via protein conjugation that is performed via a mechanism that is not homologous to the ubiquitylation system (Festa et al, 2010; Poulsen et al, 2010; Maupin‐Furlow, 2012). Although domains that are analogous to E1 enzymes can be found in bacteria, evolutionary analysis suggests that a protein ubiquitylation system was only present in the last eukaryotic common ancestor (Koonin, 2010). Since the discovery of the ubiquitin conjugating system other evolutionary related enzyme cascades have been found to perform the conjugation of ubiquitin‐like proteins such as SUMO, NEDD8, ISG15, among others (reviewed in (Hochstrasser, 2009)).
Studying the evolutionary origin of PTM catalyzing enzymes (hereafter referred to as PTM enzymes) is particularly challenging as sequence similarity becomes less reliable with evolutionary distance. For ancient enzymes like protein kinases, structural studies can reveal conserved structural motifs and suggest an evolutionary trajectory (Fig 2, bottom; Scheeff & Bourne, 2005). Similar analyses have shed light on the evolution of different PTM enzymes including protein phosphatases (Moorhead et al, 2009), ubiquitin ligases (Burroughs et al, 2008, 2009; Grau‐Bové et al, 2013) and acetylases (Iyer et al, 2008). Such analyses have been reviewed elsewhere (Anantharaman et al, 2007) and provide a picture of the origin and early evolution of PTM signaling systems. Not all PTMs are likely to be as ancient in origin as the ones described above but most known PTMs have not been experimentally analyzed yet in a sufficiently systematic way across all domains of life to infer their evolutionary history. Improvements in enrichment strategies, MS sensitivity and phylogenetic coverage for different PTM types will be required to advance our understanding on the origin of novel PTMs.
Co‐evolution of a ‘toolkit’ of PTM writer, reader and eraser domains
The toolkit of writer, eraser and reader domains allow for specificity and spatial‐temporal control of target modification. The interplay between the three domains adds the possibility for regulated recruitment and complex non‐linear dynamics that might, for example, confer memory to signaling systems. Co‐evolution of these three domain types has been analyzed to shed more light onto their functional inter‐dependencies. In particular, the evolution of toolkit domains regulating tyrosine phosphorylation is one of the few well‐studied examples. In the context of phosphotyrosine signaling, tyrosine kinases and tyrosine phosphatases act as writers and erasers, respectively and proteins harboring Src Homology 2 (SH2) domains serve as readers that bind specifically to phosphotyrosine. Protein tyrosine kinases have extensive functions in cell‐to‐cell communication in metazoans and are thought to have been crucial for the development of multicellularity (Lim & Pawson, 2010). Two types of tyrosine kinases exist: receptor tyrosine kinases that bind ligands at extracellular domains and cytoplasmic tyrosine kinases that transmit the signals that they receive from the receptors. It has been suggested that the repertoire of metazoan cytoplasmic tyrosine kinases existed already before the split of filastereans, metazoans and choanoflagellates whereas the receptor tyrosine kinases have radiated independently in these lineages. This suggests that receptor tyrosine kinases in more primitive lineages were functioning as receivers for environmental changes and have been subsequently recruited for cell‐to‐cell communication (Suga et al, 2012). Tyrosine phosphatases and SH2 domains are thought to have been present in premetazoan lineages devoid of tyrosine kinases. In extant fungal model organisms, that lack dedicated tyrosine kinases, SH2 domains are not known to bind phosphotyrosine (Dengl et al, 2009) while the phosphatases are capable of erasing this modification (Zhan et al, 2000). This suggests that functional tyrosine phosphatases originated first while phosphotyrosine recognition by SH2 domains arose after the origin of tyrosine kinases (Lim & Pawson, 2010). In the absence of dedicated tyrosine kinases, it is likely that tyrosine residues can nevertheless be phosphorylated by promiscuous ser/thr kinases. Indeed, tyrosine phosphorylation in plants and fungi (Holt et al, 2009; Nakagami et al, 2010) have been shown to occur at a similar extent (~5%) as in human (Olsen et al, 2010; Rigbolt et al, 2011), even though these species do not have dedicated tyrosine kinases.
The evolution of functional reader/writer/eraser PTM toolkits provided new mechanisms to encode information and to build new signaling systems orthogonal to the other cellular regulatory systems. Thus, after the emergence of tyrosine kinases, the reader/writer/eraser system was complete and the three domains begun to expand significantly. This expansion was apparently very rapid since extant species either have no tyrosine kinases and few readers/erasers or have an expanded set of the three domains (Fig 2, middle; Pincus et al, 2008). This expanded toolkit might have facilitated the development of multicellularity (Pincus et al, 2008). The addition of a new modification type to an organism's repertoire may not only provide benefits to this species but is also associated with a cost, since it is likely to add new constraints on proteome evolution. For example, the origin of tyrosine phosphorylation and the expansion of tyrosine kinases are correlated with a decrease in the tyrosine content within proteins (Tan et al, 2009b). This result suggests that the amount and location of modifiable amino‐acids within the proteome might be constrained by the set of PTM enzymes used by a species.
The tyrosine phosphorylation system represents one example for which evolutionary studies could trace the origin of a novel PTM type and follow the coordinated evolution of its cognate PTM toolkit domains and of the modifiable residues in target proteins. Future work on other PTM types will be required to verify the generality of these findings.
Evolution of PTM toolkit proteins by duplication and divergence
Once a PTM writer/eraser/reader toolkit exists, its expansion within the proteome is expected to correlate with the evolution of substrate recognition. When a duplicate pair of these domains are retained in the genome for a large amount of time, it indicates that their function is unlikely to be redundant. Although there are many studies of specificity for writer/eraser/reader domains (Miller et al, 2008; Persaud et al, 2009; Mok et al, 2010), these have yet to be done within an evolutionary context. By analogy with linear‐motif interactions and TF‐promoter interactions, one might speculate that substrate recognition will follow similar evolutionary trends in the case of PTM domains. From previous studies on SH3, PDZ and TFs specificity and evolution (Tonikian et al, 2008; Tuch et al, 2008; Xin et al, 2013), we expect that orthologous PTM domains will often have conserved substrate recognition. Since these domains usually have many interaction partners, a change in specificity would have very drastic effects by simultaneous disrupting tens to hundreds of interactions. Therefore, specificity changes are expected to be largely driven by divergence after duplication, as it has been observed for SH3 and PDZ domains (Tonikian et al, 2008, 2009; Xin et al, 2013). It is important to note that recognition might be also determined independently from the substrate recognition site by additional factors such as localization, expression or interaction with adaptor proteins. Therefore, divergence after duplication could also be achieved by a change in these other factors. The main cell cycle kinases are an interesting example of how substrate recognition and localization work together to ensure specificity (Alexander et al, 2011). While the Aurora kinases and Plk1 share sub‐cellular localizations during the cell cycle, they have mutually exclusive substrate recognition. Conversely, Nek2 and Plk1 or Nek2 and the Aurora kinases have partially overlapping substrate recognition motifs but would not target the same proteins due to non‐overlapping localizations (Alexander et al, 2011).
Although we expect that orthologous PTM enzymes will have mostly conserved substrate recognition, exceptions have been studied in transcription‐factor recognition (Baker et al, 2011) and are thus possible also for PTM enzymes or binding domains.
Fast divergence of enzyme‐protein interactions and PTM site position
Large‐scale identification of PTMs across multiple species has opened the door to comparative studies regarding the evolutionary conservation of the modification sites, interactions and function. These studies have primarily focused on protein phosphorylation given its broad functional role and well established detection methods. Most of the comparative analyses performed to date have studied the conservation of the modified residues in alignments of orthologous proteins (Holt et al, 2009; Landry et al, 2009; Nguyen Ba & Moses, 2010; Gray & Kumar, 2011). There is some debate regarding the extent of conservation: some studies report little to no conservation of the modified residues when compared to unmodified amino‐acids (Holt et al, 2009; Landry et al, 2009; Nguyen Ba & Moses, 2010), whereas others observe a significant constraint due to the phosphorylation (Gray & Kumar, 2011). Overall, a reconciled view on these finding is that phosphorylated residues show a significant but small increase in conservation when compared to the similar unmodified residues within the same proteins when taking into account ordered and disordered regions separately. Similar conclusions were derived from the conservation of the phosphorylation state by comparing different phosphoproteomes (Beltrao et al, 2009, 2012; Tan et al, 2009a).
Two caveats regarding PTM site conservation are worth noting: mimetic amino‐acids and PTM abundance. Some amino‐acids can sometimes mimic PTMs but few studies have taken this into account in comparative analysis. For example, the negatively charged Asp/Glu can often mimic phospho‐Ser/Thr and analysis of sequence alignments of highly conserved proteins suggest that on the order of 5% of phosphosites occur in positions that likely were Asp/Glu in the ancestral state (Pearlman et al, 2011). These positions may highlight phosphosites that positively regulate proteins by conditionally restoring the negative charges present in the ancestral states (Pearlman et al, 2011). Only a fraction of the total pool of a given protein is likely to be modified at any given time but only a few studies have been able to measure PTM stoichiometry at a large scale (Wu et al, 2011). Using these measurements it has been shown that phosphosites that are more abundant are more likely to be conserved (Levy et al, 2012; Tan & Bader, 2012). It is therefore possible that a fraction of sites are experimentally correct but are in‐vivo ‘off‐targets’ of a PTM catalyzing enzyme.
The recent availability of data for other types of modifications has allowed for comparative studies to be extended to different PTMs beyond phosphorylation (Hagai et al, 2012; Zielinska et al, 2012). The conservation of the modification state of phosphorylation, lysine acetylation and ubiquitylation has been recently compared (Beltrao et al, 2012). Although lysine residues are overall more conserved than the common phospho‐acceptor residues, the constraints imposed by these two lysine modifications appear also to be significant but weak when compared to the unmodified lysines. The conservation of amino‐acid residues for 13 different PTMs has revealed some variation in the levels of constraint for different PTMs with higher conservation of carboxylation followed by N‐glycosylation and C‐glycosylation and lowest conservation of sumoylation (Minguez et al, 2012). Overall, many types of PTMs analyzed to date show weak evolutionary constraints in that the modified residues are only somewhat more conserved than equivalent unmodified ones.
Two justifications have been proposed for the observed low constraints due to PTMs. One possibility would be that a significant fraction of the sites are not functional and have no impact on fitness when mutated. In support of this, phosphosites with a known kinase regulator or characterized functional role show a significantly higher conservation when compared with sites from high‐throughput studies (Landry et al, 2009; Nguyen Ba & Moses, 2010; Beltrao et al, 2012). A second hypothesis is that phosphorylation sites could diverge without an impact on function through redundant intermediates. Consistent with this theory, it has been shown that the average number of phosphorylation sites per protein within functional modules (i.e. pathways or complexes) is similar across species, although different residues are modified in different species (Beltrao et al, 2009). Within a single protein, clusters of kinase target sites can act as a functional unit whereby the position of each site within the cluster might not be strongly constrained. This could be the case if the role of the modifications were to regulate the bulk electrostatics of a protein region (Strickfaden et al, 2007) or to achieve a non‐linear regulatory outcome (Kõivomägi et al, 2011). Phosphosites are often found in clusters within proteins (Schweiger & Linial, 2010; Christian et al, 2012) and the clustering of sites matching a kinase motif can serve as a predictor for kinase‐target interactions further highlighting the functional importance of these clusters (Moses et al, 2007a). In addition, there are well‐characterized examples where even the regulation of a crucial process like DNA re‐replication is conserved but implemented via different regulatory phosphorylation sites (Moses et al, 2007b; Drury & Diffley, 2009). Either of the two hypotheses described above would predict that the kinase‐protein interactions should be more conserved than the individual sites. Although this prediction has not been experimentally tested, analysis of predicted kinase networks supports this hypothesis (Tan et al, 2009a).
Underlying either of the two arguments proposed above is a capacity to generate novel PTM sites quickly during evolution. While novel protein‐protein interactions among protein complex subunits tend to evolve via gene‐duplication and divergence (van Dam & Snel, 2008; Pereira‐Leal et al, 2007), new PTM sites can be created by a few point mutations (Fig 2, top). This is possible because PTM enzyme recognition of the target modification site is often mediated by a few amino‐acids within a linear peptide stretch (i.e. linear‐motifs; Diella et al, 2008). In fact, it might be expected that low binding specificity (high promiscuity) should in general correlate with high divergence rates for physical interactions (Neduva & Russell, 2005; Beltrao & Serrano, 2007; Shou et al, 2011; Sun et al, 2012). Many of the evolutionary properties of post‐translational modification sites and their enzyme interactions are reminiscent of those observed for transcription factor (TF) binding sites and interactions between TFs and promoters/enhancers. TFs have also interactions that are mediated by a small number of determinant sites, which diverge quickly. Thus, similar arguments about the functional importance and co‐evolution of transcriptional interactions have been made (Moses & Landry, 2010). This analogy has been well described and allow us to generalize these evolutionary properties (e.g. divergence rates, degree of functional constraint, existence of non‐functional interactions) and their functional consequences to other interaction types and layers of regulation that are determined by degenerated sequence motifs such as splicing, miRNA‐binding sites, protein localization signals or protease cleavage sites.
Functional importance of protein post‐translational modifications
As we described above, a significant fraction of PTM sites within proteins appear to be under low evolutionary constraints and many are not conserved across species. In order to understand if these modifications are in fact not functional or if they are diverging but keeping their function through redundant evolutionary intermediates, we need to determine their functional role (Fig 3). It is possible to use the conservation of PTM sites (Budovskaya et al, 2005; Lam et al, 2010) or the conservation of predicted enzyme‐PTM interactions (Tan et al, 2009a) to highlight sites that are more likely functionally important. However, identifying important PTMs through conservation does not predict the function of the modifications and cannot identify species‐specific functionally important sites. In order to address this and to cope with the large amounts of PTM data being generated by mass‐spectrometry, several groups have recently tried to develop computational and experimental methods that can rank PTMs according to functional importance and assign a putative functional role.
Figure 3. Functional role of post‐translational modifications.
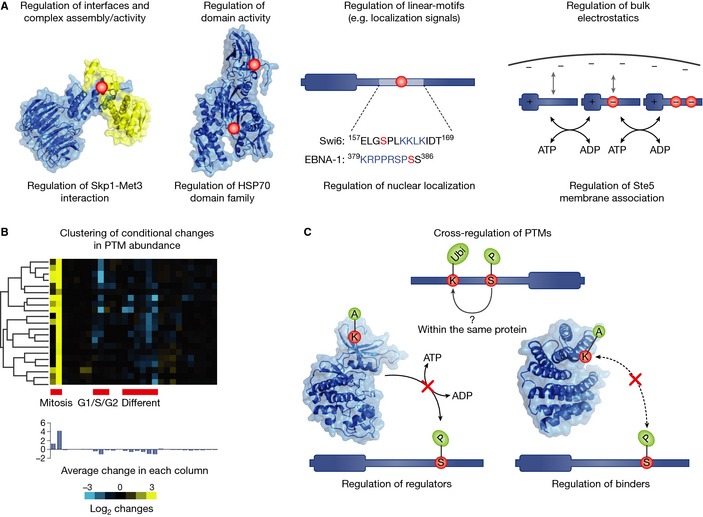
PTMs act to change the activity of proteins through different mechanism and in response to different conditions. (A) Different mechanism used by PTMs to regulate protein activity. (B) Example of conditional regulation of phosphorylation sites. A hypothetical dataset of conditional regulation of phosphosites under different conditions was subjected to hierarchical clustering. The cluster shows a set of co‐regulated sites that is up‐regulated during mitosis and down‐regulated during stem‐cell differentiation and G1/S/G2. This cluster illustrates how patterns of co‐regulation provide additional functional annotation to PTMs. (C) Mechanism of cross‐regulation between different PTM types. Two different PTM types have been observed to cross‐regulate each other in the same protein where for example a phosphosite may recruit an E3‐ligase promoting protein ubiquitylation. The regulatory enzyme of one type can be regulated by modification of another type such as the regulation of protein kinases via acetylation (regulation of regulators). Additionally, a binder for one PTM type may be regulated by a modification site of a different type. This has been observed for 14‐3‐3 domains which can bind phosphosites and have been shown to be regulated by acetylation (regulation of regulators).
Conditional regulation of PTMs
One approach to identify functionally relevant sites is to study their dynamical regulation using quantitative mass‐spectrometry approaches that can measure changes in PTM abundance under different conditions (Choudhary & Mann, 2010; Fig 3B). Sites that are regulated under specific conditions are more likely to have a functional role when compared to unregulated sites. In line with this, regulated phosphosites show an above average conservation across species (Beltrao et al, 2012). These quantitative experiments also place the role of the modification in the context of a specific condition, allowing for more focused experimental follow up characterization. There are several recent studies that have used quantitative approaches to find PTMs involved in different biological processes such as cell cycle (Olsen et al, 2010), DNA damage (Matsuoka et al, 2007; Bennetzen et al, 2010; Bensimon et al, 2010; Beli et al, 2012) and stem‐cell differentiation (Rigbolt et al, 2011). This concept was recently taken in an exciting research direction by integrating quantitative phosphosite information with changes in metabolic fluxes obtained under the same conditions. The correlation between these two different types of dynamical information allowed for the identification of phosphosites that regulate metabolic enzyme activity (Oliveira et al, 2012). Correlation of changes in PTM abundances with quantitative changes in other meaningful read‐outs (e.g. metabolic fluxes, gene expression, protein localization, genetic interactions) under different experimental conditions (Ideker & Krogan, 2012) should allow for large‐scale identification of functionally relevant modifications.
Regulation of activity and interactions
PTMs are known to regulate protein activity through different mechanisms, including regulating protein‐protein interactions, protein localization, degradation, cleavage or allosterically regulating enzyme activity (Fig 3A). In addition to studying dynamical regulation, several groups have developed methods that predict the mechanistic role of PTMs. For example, it was shown that the conservation of PTMs across members of a domain family could be used to predict functionally important regulatory regions (i.e. PTM regulatory hot‐spots; Beltrao et al, 2012). Some of these predicted regions were previously known to allosterically regulate domain activity such as the activation loop or glycine rich region of protein kinases and specific segments of the heat‐shock protein 90. PTMs found within these putative regulatory regions are therefore more likely to regulate protein activity. PTMs that might regulate inter‐domain contacts or protein‐protein interfaces have been predicted based on structural information (Nishi et al, 2011; Zanzoni et al, 2011; Beltrao et al, 2012). It has been shown that phosphorylation sites tend to be enriched at interface regions and that in‐silico mutations predict a significant impact on binding stability for about one‐third of the complexes analyzed in this way (Nishi et al, 2011). It was also observed that phosphosites and lysine acetylation (but not ubiquitylation) sites at interface regions show an above average conservation (Beltrao et al, 2012). These results suggest that PTMs are a common way by which the cell tunes the binding affinity of protein interactions and suggests approaches to identify these regulatory PTMs at a large scale. Another analysis has focused on the interplay between phosphorylation and protease cleavage (Dix et al, 2012). A novel MS approach was developed that could find protein cleavage events that exposed phosphosites, many of which could either promote or inhibit protease cleavage. Many other molecular mechanisms exist to regulate protein function via modifications. For example, phosphorylation is often used to regulate protein localization (Nardozzi et al, 2010) or protein degradation (see below). Several examples of PTMs regulating protein activity by the regulation of linear‐motifs, such as localization and cleavage peptide signals, has been recently cataloged and analyzed in the switches.ELM resource (Van Roey et al, 2013).
PTM cross‐regulation is pervasive
Different PTMs do not only function by themselves but can coordinately determine the activity and/or function of proteins in multiple ways (Fig 3C). An example of this is the interplay between phosphorylation and acetylation in the regulation of metabolic flux. As described above, phosphorylation changes in glycolytic enzymes are correlated to metabolic flux changes in yeast. However, glycolytic enzymes are acetylated in yeast as well (Henriksen et al, 2012) and in bacteria, changes in lysine‐acetylation patterns have been shown to redirect metabolic fluxes (Wang et al, 2010). The question arises whether and how these two types of modifications are coordinated. It is yet unclear if they exert changes on the same enzyme molecules or if each have their specific target proteins within the protein pool of a given enzyme. It is also not known if the phosphorylation changes cause acetylation changes or vice versa. In histone tails this kind of coordination between PTMs has been studied in‐depth to a point where a histone PTM code has been proposed (Jenuwein & Allis, 2001). The examples of PTM co‐regulation on histone tails (Latham & Dent, 2007) and in p53 (Brooks & Gu, 2003) have been used to suggest that most eukaryotic proteins are under the control of cooperative PTMs to determine their molecular functions and a general PTM code has been postulated (Benayoun & Veitia, 2009). The integration of multiple PTMs in such central proteins as glycolytic enzymes also suggest that co‐regulation between PTMs is very common.
Cross‐regulation of PTM regulators
PTMs can activate or deactivate other regulators thereby changing the PTM status of downstream proteins. Cyclin‐dependent kinases have lysines within the first 50 amino acids that are important for the coordination of adenosine 5′‐triphosphate (ATP; Choudhary et al, 2009). These lysines are critical for the proper functioning of the Cdk proteins, are conserved and have also been shown to be acetylated. In fact, the increase of acetylation has been shown in Cdk9 to reduce the kinase activity (Sabò et al, 2008). This shows that lysine acetylation has a direct role in regulating phosphorylation levels through inhibition of the kinase. Another example of regulation via acetylation involves Polypeptide GalNAc transferases (ppGalNAc‐Ts), a family of enzymes that catalyze initiation of mucin‐type O‐glycosylation. Specific glycosyltransferase activity of ppGalNAc‐T2 was reduced 95% by acetylation of five lysines in the catalytic core. Acetylation of lysines in the lectin domain results in alteration of the carbohydrate‐binding ability of ppGalNAc‐T2 (Zlocowski et al, 2011).
PTM cross‐talk within and across proteins
Multiple PTMs on the same protein molecule can together determine a functional outcome. For example, combinations of phophorylation, acetylation, methylation and ubiquitination determine the regulatory activity of histones. One such case involves the regulation of chromatin dynamics via cross‐talk between phosphorylation and acetylation on histone H3, carried out by the Snf1 kinase and the HAT Gcn5, respectively (Lo et al, 2001). More recently, evidence of cooperativity has been collected for other proteins. Phosphorylation of the transcription factor MEF2D on S444 is required for subsequent SUMOylation of K439 (Grégoire et al, 2006). Hydroxylation and O‐linked glycosylation seem to be cooperative PTMs as well. For instance, in O2‐dependent signaling in Dictyostelium, hydroxylation of Skp1 enhances its subsequent O‐linked glycosylations (Wang et al, 2011). In collagen proteins, there is an interplay between glycosylated residues and nearby hydroxylated prolines to determine collagen conformation and stability. (Bann & Bächinger, 2000). Coordination between different PTMs can also occur across different proteins. For example, acetylation of 14‐3‐3 disrupts binding of phosphopeptides of RAF1 and KIF1c (Choudhary et al, 2009). Furthermore, this cross‐talk can extend to different proteins within the nucleosome, as it has been shown that ubiquitination on histone H2B by Rad6/Bre1 is a prerequisite for methylation of histone H3 K4 and K79 by the histone methyltransferases, COMPASS and Dot1, respectively (Shukla et al, 2009).
Large‐scale determination of co‐regulation between PTMs remains a hard challenge. Computational analysis of different PTM types within human proteins has shown that these often co‐occur within proteins and form clusters of putative signal integration (or PTMi spots; Woodsmith et al, 2013). Another step in this direction was developed using the concept of correlated evolution to discover novel types of PTM co‐regulation within proteins that are not necessarily close in sequence space (Minguez et al, 2012). The nature of those co‐regulations will still have to be determined but it has so far indicated a vast amount of co‐regulation in proteins that has been understudied (Minguez et al, 2012). Large‐scale simultaneous quantification of PTM changes for two or more PTM types after perturbation or during the course of a time dependent process may be a useful approach to detect PTM cross‐talk within or across proteins. Recent studies have measured changes in phosphorylation and acetylation after DNA damage (Beli et al, 2012) or phosphorylation and ubiquitylation after proteasome inhibition (Swaney et al, 2013). In the latter case, pairs of phosphorylation and ubiquitylation sites found to increase in abundance after proteasome inhibition were indicative of potential phospho‐degron function (Swaney et al, 2013). Together, these studies suggest that there is an extensive and understudied degree of cross‐talk between different PTMs and substantial effort will be required to map‐out and carefully dissect these interactions.
Summary and future challenges
Over the past 15 years, remarkable progress has been made in understanding the functional elements within genomes and their evolution. This progress has largely been made possible due to advances in sequencing technologies coupled with comparative studies. Although studies on protein function and evolution have historically preceded genome studies, large‐scale methods to study protein abundance, interactions and modifications have lagged behind genome sequencing and gene‐expression measurements. Only recently have these proteomic measurements started to reach a scale that approaches a genome‐wide level, raising interesting challenges and providing exciting research opportunities. We are now able to measure PTMs in large‐scale and these measurements brought a renewed interest in the study of PTM evolution. Although many questions remain unanswered, a broad picture is starting to emerge. The few well characterized PTMs to date appear to be, in general, very ancient and studying their origin is therefore challenging. Metabolic proteins are very commonly modified by small modifications like lysine acetylation and phosphorylation and these PTMs can be linked to the energy level of the cell. These observations suggest that acetylation and phosphorylation might have an origin that relates to energy sensing and have since then been co‐opted to other functions. Most PTM types have yet to be studied with large‐scale proteomics approaches due to the lack of enrichment strategies. Even for the types that can be studied, there is a clear need to increase the phylogenetic coverage of the species studied. As these data become available, we will have a much clearer view regarding the origin and evolution of PTM types.
The extended use of a PTM is linked to the origin of a dedicated set of writer/eraser/reader domains. The expansion of these domains with diversification of their substrate recognition are then likely driven and at the same time limited by the need to maintain specific recognition. Uri Alon and others have likened regulatory interactions to information channels that are bounded in information capacity by their binding specificity (Itzkovitz et al, 2006). A novel regulatory interaction, in the form of a new PTM or new TF family, would be analogous to a new information channel that the cell can use, free from interference with other channels. For example, in this context, different protein kinases are most useful if they can be sub‐divided into sub‐families with non‐overlapping substrate specificity. The extent that this subdivision is possible is limited by the kinase fold itself and perhaps to a lesser extent by the trade‐off between substrate specificity and evolvability. Even if a PTM enzyme fold was capable of recognizing substrates with a much higher specificity, improving communication fidelity, the interactions and function of this enzyme would be also less evolvable and therefore perhaps less likely to be highly duplicated during evolution. High‐throughput methods to study PTMs and the specificity of PTM enzymes will allow for these questions to be studied in detail. Specificity of interactions is determined in‐vivo by many factors besides domain binding specificity. For this reason, the diversification of PTM toolkit domains by duplication can be achieved also by divergence of others factors like localization or time/condition dependent expression. A great example of this type of divergence is seen for cell‐cycle kinases (Alexander et al, 2011).
While new PTM types arise only rarely and PTM domain sub‐families and specificity diverge by duplication and divergence, new PTM sites and interactions have much faster evolutionary dynamics. Given the promiscuous nature of PTM toolkit domains, novel binding sites can be created in existing proteins by a few point mutations. Many PTM sites of broadly studied PTM types (phosphorylation, acetylation and ubiquitylation) identified to date are weakly constrained and are often not conserved. Additional studies will be required to increase the coverage of known PTM sites for other species and for other PTM types, as well as determining their conditional regulation and abundance. Evolutionary studies have suggested that a significant fraction of PTM sites are unlikely to have a biological role and some might change position while retaining function via redundant intermediates. These hypotheses are difficult to test experimentally and much more effort needs to be directed to the experimental study of specific signaling systems in different species and/or individuals of the same species. This view of high evolutionary plasticity of enzyme‐PTM interactions with a significant fraction of non‐functional PTMs is in stark contrast with the neatly organized signaling cascades often found in textbooks (Fig 4, electronic circuit). Signalling interactions are highly cooperative and dynamic and very often are spatially organized (Gibson, 2009). A paradigm of highly logic circuits of information cascades has initially been useful to conceptualize major signaling pathways but might also hinder our progress in a more unbiased study of signalling networks (Gibson, 2009). Large‐scale studies of cellular interactions have provided us with a different paradigm for reasoning about cell‐decision making, in which signaling components operate as part of a dense network of molecular interactions (Fig 4, hairball). This ‘nodes and edges’ network view of cell biology provides a good representation of the high degree of cooperativity between cellular components. However, this network paradigm does not convey the logic and design principles so often observed in cell biology. We suggest that an appropriate idealization of a cell must reside at the convergence of these two paradigms and will certainly be informed by evolutionary studies. Given that post‐translational and transcriptional interactions can rapidly explore novel functional space and that natural selection constrains only the emerging function and not the implementations, we expect that the same signaling function will be achieved by different species in different ways. Examples of this include the conserved timing of cell‐cycle regulation of protein complexes (Jensen et al, 2006), the regulation of mating (Tsong et al, 2006), regulation of DNA re‐replication (Kearsey & Cotterill, 2003; Moses et al, 2007b; Drury & Diffley, 2009) and SH3 domain function (Xin et al, 2013) despite changes in the underlying interactions. Comparing different implementations of crucial functions across species should highlight the important design principles underlying the function under study.
Figure 4. A depiction of cell‐decision making at the convergence of different approaches to cell biology.
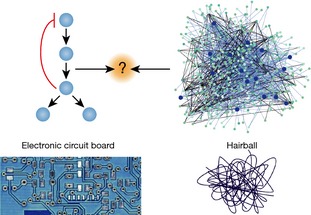
Cell signaling systems are often depicted as highly logical and engineered circuits in a representation that tries to capture the main design principles of a cellular function. However, this representation may hinder our progress in studying systems that are not engineered but are highly cooperative and evolved. A second paradigm has recently been put forward, based on large scale studies of cellular networks. A view where each component is a node and each association between components is an edge. This simple network view has been useful in describing the high degree of complexity and cooperativity inside the cell but is not informative of the logic observed in these systems. We suggest that a useful and realistic description of cell biology must be informed by these two view‐points.
The novel high‐throughput methods to identify PTMs are creating challenges and opportunities in the functional characterization of these modifications. Novel computational and experimental methods are required to facilitate future functional studies. Some studies have attempted to achieve this by highlighting conserved PTM‐domain interactions or attempting to identify sites that have regulatory potential. Still, a clear need for additional methodologies is expected to drive research in this area. In analogy to gene‐expression studies, PTM abundance can also be measured in different conditions. The correlation of changes of PTM abundance in different conditions with measurements of other functional outputs (e.g. metabolic fluxes, protein localization, gene‐expression, protein abundance, abundance of other PTM types, etc.) under the same conditions is expected to be a powerful and large‐scale approach to identify functional PTMs. It is also becoming increasingly apparent that different PTMs tend to act in coordinated fashion. Several small‐scale studies have provided us with examples of this interplay between different PTMs and some large‐scale approaches have recently attempted to identify these functional connections on a more global scale. It is likely that many more types and instances of these cross‐regulatory interactions remain to be identified.
Taken together, this is a challenging and exciting time and analogous to the very early days of genome sequencing where tools and concepts had to build around the incoming data to transform it into knowledge. With the generation of PTM‐relevant data at large‐scale and a corresponding global analysis, we expect that many of the most exciting discoveries in the study of PTM regulation still remain ahead of us. We believe that a better understanding of the evolution and function of PTMs will result in a much more reliable picture of how these signaling proteins integrate and relay information inside the cell.
Acknowledgements
N.J.K. is supported by the US National Institutes of Health (P50GM082250, R01GM084448, P01AI090935, P50GM081879, R01GM098101, R01GM084279 and P01AI091575) and the Defense Advanced Research Projects Agency (DARPA‐10‐93‐Prophecy‐PA‐008). P.B. is supported by the Human Frontier Science Program (CDA00069/2013‐C). We thank Michael Shales for graphics work.
Mol Syst Biol. 9: 714
References
- Abu‐Qarn M, Eichler J, Sharon N (2008) Not just for Eukarya anymore: protein glycosylation in Bacteria and Archaea. Curr Opin Struct Biol 18: 544–550 [DOI] [PubMed] [Google Scholar]
- Alexander J, Lim D, Joughin BA, Hegemann B, Hutchins JRA, Ehrenberger T, Ivins F, Sessa F, Hudecz O, Nigg EA, Fry AM, Musacchio A, Stukenberg PT, Mechtler K, Peters J‐M, Smerdon SJ, Yaffe MB (2011) Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context‐dependent mitotic signaling. Sci Signal 4: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, Iyer LM, Aravind L (2007) Comparative genomics of protists: new insights into the evolution of eukaryotic signal transduction and gene regulation. Annu Rev Microbiol 61: 453–475 [DOI] [PubMed] [Google Scholar]
- Baker CR, Tuch BB, Johnson AD (2011) Extensive DNA‐binding specificity divergence of a conserved transcription regulator. Proc Natl Acad Sci USA 108: 7493–7498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bann JG, Bächinger HP (2000) Glycosylation/Hydroxylation‐induced stabilization of the collagen triple helix. 4‐trans‐hydroxyproline in the Xaa position can stabilize the triple helix. J Biol Chem 275: 24466–24469 [DOI] [PubMed] [Google Scholar]
- Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C (2012) Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol Cell 46: 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, Lim WA, Fraser JS, Frydman J, Krogan NJ (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150: 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Serrano L (2007) Specificity and evolvability in eukaryotic protein interaction networks. PLoS Comput Biol 3: e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Trinidad JC, Fiedler D, Roguev A, Lim WA, Shokat KM, Burlingame AL, Krogan NJ (2009) Evolution of phosphoregulation: comparison of phosphorylation patterns across yeast species. PLoS Biol 7: e1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Veitia RA (2009) A post‐translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol 19: 189–197 [DOI] [PubMed] [Google Scholar]
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS (2010) Site‐specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics 9: 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang S‐Y, Chen DJ, Aebersold R, Shiloh Y (2010) ATM‐dependent and ‐independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal 3: rs3. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W (2003) Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 15: 164–171 [DOI] [PubMed] [Google Scholar]
- Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK (2005) An evolutionary proteomics approach identifies substrates of the cAMP‐dependent protein kinase. Proc Natl Acad Sci USA 102: 13933–13938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Iyer LM, Aravind L (2009) Natural history of the E1‐like superfamily: implication for adenylation, sulfur transfer, and ubiquitin conjugation. Proteins 75: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Iyer LM, Aravind L (2012) The natural history of ubiquitin and ubiquitin‐related domains. Front Biosci 17: 1433–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Jaffee M, Iyer LM, Aravind L (2008) Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub‐like protein conjugation. J Struct Biol 162: 205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, Tu BP (2011) Acetyl‐CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 42: 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y (2007) Lysine propionylation and butyrylation are novel post‐translational modifications in histones. Mol Cell Proteomics 6: 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33: 275–286 [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science 325: 834–840 [DOI] [PubMed] [Google Scholar]
- Choudhary C, Mann M (2010) Decoding signalling networks by mass spectrometry‐based proteomics. Nat Rev Mol Cell Biol 11: 427–439 [DOI] [PubMed] [Google Scholar]
- Christian J‐O, Braginets R, Schulze WX, Walther D (2012) Characterization and prediction of protein phosphorylation hotspots in Arabidopsis thaliana. Front Plant Sci 3: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam TJ, Snel B (2008) Protein complex evolution does not involve extensive network rewiring. PLoS Comput Biol 4: e1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengl S, Mayer A, Sun M, Cramer P (2009) Structure and in vivo requirement of the yeast Spt6 SH2 domain. J Mol Biol 389: 211–225 [DOI] [PubMed] [Google Scholar]
- Diella F, Haslam N, Chica C, Budd A, Michael S, Brown NP, Trave G, Gibson TJ (2009) Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci 13: 6580–6603 [DOI] [PubMed] [Google Scholar]
- Dix MM, Simon GM, Wang C, Okerberg E, Patricelli MP, Cravatt BF (2012) Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell 150: 426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury LS, Diffley JFX (2009) Factors affecting the diversity of DNA replication licensing control in eukaryotes. Curr Biol 19: 530–535 [DOI] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H (2011) Sirt5 is a NAD‐dependent protein lysine demalonylase and desuccinylase. Science 334: 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AEH, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen Y‐N, Rush J, Hsu PW‐C, Yen H‐CS, Elledge SJ (2011) Global identification of modular cullin‐RING ligase substrates. Cell 147: 459–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa RA, McAllister F, Pearce MJ, Mintseris J, Burns KE, Gygi SP, Darwin KH (2010) Prokaryotic ubiquitin‐like protein (Pup) proteome of Mycobacterium tuberculosis. PLoS ONE 5: e8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2‐like proteins. Biochem Biophys Res Commun 273: 793–798 [DOI] [PubMed] [Google Scholar]
- Gibson TJ (2009) Cell regulation: determined to signal discrete cooperation. Trends Biochem Sci 34: 471–482 [DOI] [PubMed] [Google Scholar]
- Grau‐Bové X, Sebé‐Pedrós A, Ruiz‐Trillo I (2013) A genomic survey of HECT ubiquitin ligases in eukaryotes reveals independent expansions of the HECT system in several lineages. Genome Biol Evol 5: 833–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray VE, Kumar S (2011) Rampant purifying selection conserves positions with posttranslational modifications in human proteins. Mol Biol Evol 28: 1565–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire S, Tremblay AM, Xiao L, Yang Q, Ma K, Nie J, Mao Z, Wu Z, Giguère V, Yang X‐J (2006) Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J Biol Chem 281: 4423–4433 [DOI] [PubMed] [Google Scholar]
- Hagai T, Tóth‐Petróczy A, Azia A, Levy Y (2012) The origins and evolution of ubiquitination sites. Mol BioSyst 8: 1865–1877 [DOI] [PubMed] [Google Scholar]
- Hayden JD, Brown LR, Gunawardena HP, Perkowski EF, Chen X, Braunstein M (2013) Reversible acetylation regulates acetate and propionate metabolism in Mycobacterium smegmatis. Microbiology 159: 1986–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen P, Wagner SA, Weinert BT, Sharma S, Bacinskaja G, Rehman M, Juffer AH, Walther TC, Lisby M, Choudhary C (2012) Proteome‐wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae. Mol Cell Proteomics 11: 1510–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M (2009) Origin and function of ubiquitin‐like proteins. Nature 458: 422–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Krogan NJ (2012) Differential network biology. Mol Syst Biol 8: 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzkovitz S, Tlusty T, Alon U (2006) Coding limits on the number of transcription factors. BMC Genomics 7: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Anantharaman V, Wolf MY, Aravind L (2008) Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int J Parasitol 38: 1–31 [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Jensen TS, de Lichtenberg U, Brunak S, Bork P (2006) Co‐evolution of transcriptional and post‐translational cell‐cycle regulation. Nature 443: 594–597 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H (2013) SIRT6 regulates TNF‐α secretion through hydrolysis of long‐chain fatty acyl lysine. Nature 496: 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey SE, Cotterill S (2003) Enigmatic variations: divergent modes of regulating eukaryotic DNA replication. Mol Cell 12: 1067–1075 [DOI] [PubMed] [Google Scholar]
- Kim D, Yu BJ, Kim JA, Lee Y‐J, Choi S‐G, Kang S, Pan J‐G (2013) The acetylproteome of gram‐positive model bacterium Bacillus subtilis. Proteomics 13: 1726–1736 [DOI] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP (2011) Systematic and quantitative assessment of the ubiquitin‐modified proteome. Mol Cell 44: 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kõivomägi M, Valk E, Venta R, Iofik A, Lepiku M, Balog ERM, Rubin SM, Morgan DO, Loog M (2011) Cascades of multisite phosphorylation control Sic1 destruction at the onset of S phase. Nature 480: 128–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV (2010) The origin and early evolution of eukaryotes in the light of phylogenomics. Genome Biol 11: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HYK, Kim PM, Mok J, Tonikian R, Sidhu SS, Turk BE, Snyder M, Gerstein MB (2010) MOTIPS: automated motif analysis for predicting targets of modular protein domains. BMC Bioinformatics 11: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Levy ED, Michnick SW (2009) Weak functional constraints on phosphoproteomes. Trends Genet 25: 193–197 [DOI] [PubMed] [Google Scholar]
- Latham JA, Dent SYR (2007) Cross‐regulation of histone modifications. Nat Struct Mol Biol 14: 1017–1024 [DOI] [PubMed] [Google Scholar]
- Lee D‐W, Kim D, Lee Y‐J, Kim J‐A, Choi JY, Kang S, Pan J‐G (2013) Proteomic analysis of acetylation in thermophilic Geobacillus kaustophilus. Proteomics 13: 2278–2282 [DOI] [PubMed] [Google Scholar]
- Levy ED, Michnick SW, Landry CR (2012) Protein abundance is key to distinguish promiscuous from functional phosphorylation based on evolutionary information. Philos Trans R Soc Lond B Biol Sci 367: 2594–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, Pawson T (2010) Phosphotyrosine signaling: evolving a new cellular communication system. Cell 142: 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Duggan L, Emre NC, Belotserkovskya R, Lane WS, Shiekhattar R, Berger SL (2001) Snf1–a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293: 1142–1146 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166 [DOI] [PubMed] [Google Scholar]
- Maupin‐Furlow J (2012) Proteasomes and protein conjugation across domains of life. Nat Rev Microbiol 10: 100–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ML, Jensen LJ, Diella F, Jørgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz‐Ponten T, Olhovsky M, Pasculescu A, Alexander J, Knapp S, Blom N, Bork P, Li S, Cesareni G, Pawson T, Turk BEet al (2008) Linear motif atlas for phosphorylation‐dependent signaling. Sci Signal 1: ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguez P, Parca L, Diella F, Mende DR, Kumar R, Helmer‐Citterich M, Gavin A‐C, van Noort V, Bork P (2012) Deciphering a global network of functionally associated post‐translational modifications. Mol Syst Biol 8: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J, Kim PM, Lam HYK, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma J‐LN, Sheu Y‐J, Sassi HE, Sopko R, Chan CSM, De Virgilio Cet al (2010) Deciphering protein kinase specificity through large‐scale analysis of yeast phosphorylation site motifs. Sci Signal 3: ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead GBG, De Wever V, Templeton G, Kerk D (2009) Evolution of protein phosphatases in plants and animals. Biochem J 417: 401–409 [DOI] [PubMed] [Google Scholar]
- Moses AM, Hériché J‐K, Durbin R (2007a) Clustering of phosphorylation site recognition motifs can be exploited to predict the targets of cyclin‐dependent kinase. Genome Biol 8: R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses AM, Landry CR (2010) Moving from transcriptional to phospho‐evolution: generalizing regulatory evolution? Trends Genet 26: 462–467 [DOI] [PubMed] [Google Scholar]
- Moses AM, Liku ME, Li JJ, Durbin R (2007b) Regulatory evolution in proteins by turnover and lineage‐specific changes of cyclin‐dependent kinase consensus sites. Proc Natl Acad Sci USA 104: 17713–17718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D, Riezman H (2007) Proteasome‐independent functions of ubiquitin in endocytosis and signaling. Science 315: 201–205 [DOI] [PubMed] [Google Scholar]
- Nakagami H, Sugiyama N, Mochida K, Daudi A, Yoshida Y, Toyoda T, Tomita M, Ishihama Y, Shirasu K (2010) Large‐scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants. Plant Physiol 153: 1161–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardozzi JD, Lott K, Cingolani G (2010) Phosphorylation meets nuclear import: a review. Cell Commun Signal 8: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neduva V, Russell RB (2005) Linear motifs: evolutionary interaction switches. FEBS Lett 579: 3342–3345 [DOI] [PubMed] [Google Scholar]
- Nguyen Ba AN, Moses AM (2010) Evolution of characterized phosphorylation sites in budding yeast. Mol Biol Evol 27: 2027–2037 [DOI] [PubMed] [Google Scholar]
- Nishi H, Hashimoto K, Panchenko AR (2011) Phosphorylation in protein‐protein binding: effect on stability and function. Structure 19: 1807–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AP, Ludwig C, Picotti P, Kogadeeva M, Aebersold R, Sauer U (2012) Regulation of yeast central metabolism by enzyme phosphorylation. Mol Syst Biol 8: 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3: ra3. [DOI] [PubMed] [Google Scholar]
- Pearlman SM, Serber Z, Ferrell JE (2011) A mechanism for the evolution of phosphorylation sites. Cell 147: 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira‐Leal JB, Levy ED, Kamp C, Teichmann SA (2007) Evolution of protein complexes by duplication of homomeric interactions. Genome Biol 8: R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud A, Alberts P, Amsen EM, Xiong X, Wasmuth J, Saadon Z, Fladd C, Parkinson J, Rotin D (2009) Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4‐2 using proteome arrays. Mol Syst Biol 5: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus D, Letunic I, Bork P, Lim WA (2008) Evolution of the phospho‐tyrosine signaling machinery in premetazoan lineages. Proc Natl Acad Sci USA 105: 9680–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen C, Akhter Y, Jeon AH, Schmitt‐Ulms G, Meyer HE, Stefanski A, Stühler K, Wilmanns M, Song YH (2010) Proteome‐wide identification of mycobacterial pupylation targets. Mol Syst Biol 13: 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigbolt KTG, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, Kassem M, Mann M, Olsen JV, Blagoev B (2011) System‐wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal 4: rs3. [DOI] [PubMed] [Google Scholar]
- Sabò A, Lusic M, Cereseto A, Giacca M (2008) Acetylation of conserved lysines in the catalytic core of cyclin‐dependent kinase 9 inhibits kinase activity and regulates transcription. Mol Cell Biol 28: 2201–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH (2004) Phosphorylation of proteins by inositol pyrophosphates. Science 306: 2101–2105 [DOI] [PubMed] [Google Scholar]
- Scheeff ED, Bourne PE (2005) Structural evolution of the protein kinase‐like superfamily. PLoS Comput Biol 1: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger R, Linial M (2010) Cooperativity within proximal phosphorylation sites is revealed from large‐scale proteomics data. Biol Direct 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seet BT, Dikic I, Zhou M‐M, Pawson T (2006) Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol 7: 473–483 [DOI] [PubMed] [Google Scholar]
- Shou C, Bhardwaj N, Lam HYK, Yan K‐K, Kim PM, Snyder M, Gerstein MB (2011) Measuring the evolutionary rewiring of biological networks. PLoS Comput Biol 7: e1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Chaurasia P, Bhaumik SR (2009) Histone methylation and ubiquitination with their cross‐talk and roles in gene expression and stability. Cell Mol life Sci 66: 1419–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Celic I, Cole RN, Boeke JD, Escalante‐Semerena JC (2002) Sir2‐dependent activation of acetyl‐CoA synthetase by deacetylation of active lysine. Science 298: 2390–2392 [DOI] [PubMed] [Google Scholar]
- Strickfaden SC, Winters MJ, Ben‐Ari G, Lamson RE, Tyers M, Pryciak PM (2007) A mechanism for cell‐cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell 128: 519–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga H, Dacre M, de Mendoza A, Shalchian‐Tabrizi K, Manning G, Ruiz‐Trillo I (2012) Genomic survey of premetazoans shows deep conservation of cytoplasmic tyrosine kinases and multiple radiations of receptor tyrosine kinases. Sci Signal 5: ra35. [DOI] [PubMed] [Google Scholar]
- Sun MGF, Sikora M, Costanzo M, Boone C, Kim PM (2012) Network evolution: rewiring and signatures of conservation in signaling. PLoS Comput Biol 8: e1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villén J (2013) Global analysis of phosphorylation and ubiquitylation cross‐talk in protein degradation. Nat Methods 10: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CSH, Bader GD (2012) Phosphorylation sites of higher stoichiometry are more conserved. Nat Methods 9: 317; author reply 318 [DOI] [PubMed] [Google Scholar]
- Tan CSH, Bodenmiller B, Pasculescu A, Jovanovic M, Hengartner MO, Jørgensen C, Bader GD, Aebersold R, Pawson T, Linding R, Soon C, Tan H (2009a) Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci Signal 2: ra39. [DOI] [PubMed] [Google Scholar]
- Tan CSH, Pasculescu A, Lim WA, Pawson T, Bader GD, Linding R (2009b) Positive selection of tyrosine loss in metazoan evolution. Science 325: 1686–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonikian R, Xin X, Toret CP, Gfeller D, Landgraf C, Panni S, Paoluzi S, Castagnoli L, Currell B, Seshagiri S, Yu H, Winsor B, Vidal M, Gerstein MB, Bader GD, Volkmer R, Cesareni G, Drubin DG, Kim PM, Sidhu SSet al (2009) Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol 7: e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh J‐H, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagiri S, Lasky LA, Sander C, Boone C, Bader GD, Sidhu SS (2008) A specificity map for the PDZ domain family. PLoS Biol 6: e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsong AE, Tuch BB, Li H, Johnson AD (2006) Evolution of alternative transcriptional circuits with identical logic. Nature 443: 415–420 [DOI] [PubMed] [Google Scholar]
- Tuch BB, Li H, Johnson AD (2008) Evolution of eukaryotic transcription circuits. Science 319: 1797–1799 [DOI] [PubMed] [Google Scholar]
- Van Noort V, Seebacher J, Bader S, Mohammed S, Vonkova I, Betts MJ, Kühner S, Kumar R, Maier T, O'Flaherty M, Rybin V, Schmeisky A, Yus E, Stülke J, Serrano L, Russell RB, Heck AJR, Bork P, Gavin A‐C (2012) Cross‐talk between phosphorylation and lysine acetylation in a genome‐reduced bacterium. Mol Syst Biol 8: 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Roey K, Dinkel H, Weatheritt RJ, Gibson TJ, Davey NE (2013) The switches.ELM resource: a compendium of conditional regulatory interaction interfaces. Sci Signal 6: rs7. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C (2011) A proteome‐wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 10: M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning Z‐B, Zeng R, Xiong Y, Guan K‐L, Zhao S, Zhao G‐P (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327: 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZA, Singh D, van der Wel H, West CM (2011) Prolyl hydroxylation‐ and glycosylation‐dependent functions of Skp1 in O2‐regulated development of Dictyostelium. Dev Biol 349: 283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Iesmantavicius V, Wagner SA, Schölz C, Gummesson B, Beli P, Nyström T, Choudhary C (2013) Acetyl‐phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell 51: 265–272 [DOI] [PubMed] [Google Scholar]
- Woodsmith J, Kamburov A, Stelzl U (2013) Dual coordination of post translational modifications in human protein networks. PLoS Comput Biol 9: e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Haas W, Dephoure N, Huttlin EL, Zhai B, Sowa ME, Gygi SP (2011) A large‐scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods 8: 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Vellaichamy A, Wang D, Zamdborg L, Kelleher NL, Huber SC, Zhao Y (2013) Differential lysine acetylation profiles of Erwinia amylovora strains revealed by proteomics. J Proteomics 79: 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin X, Gfeller D, Cheng J, Tonikian R, Sun L, Guo A, Lopez L, Pavlenco A, Akintobi A, Zhang Y, Rual J‐F, Currell B, Seshagiri S, Hao T, Yang X, Shen YA, Salehi‐Ashtiani K, Li J, Cheng AT, Bouamalay Det al (2013) SH3 interactome conserves general function over specific form. Mol Syst Biol 9: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanzoni A, Carbajo D, Diella F, Gherardini PF, Tramontano A, Helmer‐Citterich M, Via A (2011) Phospho3D 2.0: an enhanced database of three‐dimensional structures of phosphorylation sites. Nucleic Acids Res 39: D268–D271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan XL, Hong Y, Zhu T, Mitchell AP, Deschenes RJ, Guan KL (2000) Essential functions of protein tyrosine phosphatases PTP2 and PTP3 and RIM11 tyrosine phosphorylation in Saccharomyces cerevisiae meiosis and sporulation. Mol Biol Cell 11: 663–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu C‐F, Grishin NV, Zhao Y (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics 8: 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zheng S, Yang JS, Chen Y, Cheng Z (2013) Comprehensive profiling of protein lysine acetylation in Escherichia coli. J Proteome Res 12: 844–851 [DOI] [PubMed] [Google Scholar]
- Zielinska DF, Gnad F, Schropp K, Wiśniewski JR, Mann M (2012) Mapping N‐glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol Cell 46: 542–548 [DOI] [PubMed] [Google Scholar]
- Zlocowski N, Sendra VG, Lorenz V, Villarreal MA, Jorge A, Núñez Y, Bennett EP, Clausen H, Nores GA, Irazoqui FJ (2011) Catalytic and glycan‐binding abilities of ppGalNAc‐T2 are regulated by acetylation. Biochem Biophys Res Commun 410: 140–145 [DOI] [PubMed] [Google Scholar]