

Abstract
Oxidative damage from elevated production of reactive oxygen species (ROS) contributes to ischemia-reperfusion injury in myocardial infarction and stroke. The mechanism by which the increase in ROS occurs is not known, and it is unclear how this increase can be prevented. A wide variety of nitric oxide donors and S-nitrosating agents protect the ischemic myocardium from infarction, but the responsible mechanisms are unclear1–6. Here we used a mitochondria-selective S-nitrosating agent, MitoSNO, to determine how mitochondrial S-nitrosation at the reperfusion phase of myocardial infarction is cardioprotective in vivo in mice. We found that protection is due to the S-nitrosation of mitochondrial complex I, which is the entry point for electrons from NADH into the respiratory chain. Reversible S-nitrosation of complex I slows the reactivation of mitochondria during the crucial first minutes of the reperfusion of ischemic tissue, thereby decreasing ROS production, oxidative damage and tissue necrosis. Inhibition of complex I is afforded by the selective S-nitrosation of Cys39 on the ND3 subunit, which becomes susceptible to modification only after ischemia. Our results identify rapid complex I reactivation as a central pathological feature of ischemia-reperfusion injury and show that preventing this reactivation by modification of a cysteine switch is a robust cardioprotective mechanism and hence a rational therapeutic strategy.
Mitochondrial dysfunction is central to the cell death and tissue damage caused by ischemia-reperfusion injury during myocardial infarction and underlies this leading cause of mortality1–9. Nitric oxide and its metabolites are cardioprotective during ischemia-reperfusion1–6, and the S-nitrosation (or S-nitrosylation) of cysteines by nitric oxide metabolism correlates with damage prevention during ischemia-reperfusion injury10–13. However, the relative importance of mitochondrial and nonmitochondrial nitric oxide and S-nitrosation to cardioprotection and the mechanisms involved are unclear10–13. Here we sought to elucidate the cardioprotective mechanism of mitochondrial S-nitrosation in vivo. We used MitoSNO to examine mitochondrial S-nitrosation specifically, as it contains an S-nitrosating moiety attached to a lipophilic triphenylphosphonium cation that leads to its rapid and extensive uptake within mitochondria in vivo11.
As triphenylphosphonium cations drive accumulation within cardiac mitochondria within minutes of intravenous administration14, MitoSNO should rapidly S-nitrosate mitochondrial thiols in vivo (Supplementary Fig. 1). To determine the subcellular localization of S-nitrosation after in vivo MitoSNO administration during ischemiareperfusion injury, we used an established model of myocardial infarction4. We subjected mice to 30 min of left ventricular ischemia by occlusion of the left anterior descending coronary artery (LAD) followed by 5 min of reperfusion. We administered MitoSNO (100 ng per kg body weight) systemically by tail-vein injection just before reperfusion. This led to the selective S-nitrosation of mitochondrial proteins in the reperfused heart (Fig. 1a), as visualized by labeling with a red fluorescent probe15–17. In contrast, equal doses of the untargeted precursor S-nitrosating compound S-nitroso-N-acetylpenicillamine (SNAP) S-nitrosated only nonmitochondrial myocardial and plasma proteins (Fig. 1a).
Figure 1.

S-nitrosation of mitochondrial proteins is required for S-nitrosation–mediated protection from cardiac ischemia-reperfusion injury. (a) Protein S-nitrosation in mitochondrial and cytosolic fractions of the myocardium, as well as in blood plasma, of mice after tail-vein injection of MitoSNO or SNAP just before reperfusion. Top, S-nitrosated proteins, as detected by selective reduction of protein S-nitrosothiols in the presence of a red fluorescent maleimide. Bottom, protein loading of the scanned gels (top) assessed by Coomassie staining. SNO, S-nitrosation. (b) Top, quantification of myocardial infarct size after tail-vein injection of MitoSNO, SNAP or MitoNAP. Each open circle represents data from a single mouse, and filled circles represent the mean values of all mice for a particular condition. Bottom, representative images of cross-sections from mouse hearts treated as indicated above. Infarcted tissue is white, the rest of the area at risk is red, and nonrisk tissue is dark blue. n = 7 mice per group. (c) Mitochondrial hydrogen peroxide measured in vivo by the selective oxidation of the mitochondria-targeted mass spectrometric probe MitoB to its product MitoP during myocardial ischemia-reperfusion. MitoSNO, SNAP or MitoNAP was injected at reperfusion, and control hearts were collected from mice without intervention. n = 3–6 mice per group. Control, 60 min of normoxic perfusion; I 30, 30-min ischemia; R 30, 30-min reperfusion. (d,e) Assessment of protein oxidative damage (as assessed by protein carbonyl formation, d) and apoptosis (as assessed by caspase-3 and cleaved caspase-3 amounts, e) after myocardial ischemia-reperfusion (IR) with or without mitochondrial S-nitrosation by MitoSNO. The representative western blots in e show the amounts of caspase-3 and cleaved caspase-3, along with the amounts of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. n = 3 mice per group. (f) BN-PAGE analysis of heart mitochondria identifying S-nitrosated respiratory complexes in mice injected with MitoSNO during ischemia-reperfusion injury followed by S-nitrosothiol labeling. CI–CV indicate the locations of oxidative phosphorylation complexes I–V. (g) Complex I activity in vivo at baseline and after ischemia and reperfusion for 5 min (IR 5 min) or 30 min (IR 30 min) with or without MitoSNO injection. Following mitochondrial isolation from hearts, the S-nitrosothiol-selective reductant Cu/Asc, or the general thiol reductant DTT, were added where indicated in vitro to test for the reversibility of complex I inhibition. Complex I activity was normalized to citrate synthase activity. Cu/Asc, copper and ascorbate; DTT, dithiothreitol. n = 3 mice per group. *P < 0.05, **P < 0.01 determined by one-way analysis of variance (ANOVA). Data (c–e,g) are shown as the mean ± s.e.m. of at least three replicates.
The selective localization of MitoSNO-induced S-nitrosation enabled us to assess the importance of mitochondrial S-nitrosation specifically on myocardial ischemia-reperfusion injury in vivo. We subjected mice to 30 min of LAD ischemia followed by 120 min of reperfusion. MitoSNO administered at reperfusion significantly decreased infarct size (Fig. 1b and Supplementary Fig. 2). Neither SNAP nor the MitoSNO-targeting moiety (MitoNAP)11 had this effect (Fig. 1b). MitoSNO was protective only during reperfusion: injection 10 min after reperfusion was ineffective (Supplementary Fig. 3). MitoSNO has been reported to be protective in mice with cardiomyocyte deficiency of protein kinase G18, indicating that the cardio-protection conferred by MitoSNO is not due to nonmitochondrial pathways that operate through cyclic GMP–protein kinase G signaling3,6, and MitoSNO did not alter hemodynamics (Supplementary Fig. 4). These results are consistent with the notion that the cardioprotection is due to mitochondrial S-nitrosation.
A substantial amount of acute reperfusion damage is thought to be due to ROS production, but the source of this ROS is uncertain19. The difficulty of direct temporal and spatial determination of ROS production in vivo has made assessment of its role in ischemia-reperfusion injury challenging19. We recently developed MitoB, a mitochondria-targeted mass spectrometric hydrogen peroxide probe, which allows for quantification of mitochondrial hydrogen peroxide in vivo20. Using MitoB we found that the amount of mitochondrial hydrogen peroxide remained low during ischemia and increased only after reperfusion in vivo (Fig. 1c). MitoSNO administered at reperfusion blocked this rise in the amount of hydrogen peroxide, whereas administration of SNAP did not (Fig. 1c). The rise in the amount of mitochondrial hydrogen peroxide was associated with increased protein carbonylation (Fig. 1d), a marker of oxidative damage, as well as necrotic (Fig. 1b) and apoptotic (Fig. 1e) cell death, and these effects were all prevented by MitoSNO. Together these data indicate that cardioprotection by MitoSNO acts through mitochondrial S-nitrosation, which inhibits mitochondrial hydrogen peroxide production at reperfusion.
We next sought to determine the identity of the S-nitrosation target(s) mediating protection by MitoSNO. We first used the technique S-nitrosothiol difference in gel electrophoresis (SNO-DIGE), which utilizes fluorescent labeling of S-nitrosated proteins followed by two-dimensional electrophoresis12. We did not detect S-nitrosated mitochondrial proteins in MitoSNO-perfused hearts using this method (Supplementary Fig. 5), suggesting that the S-nitrosated mitochondrial proteins detected in vivo (Fig. 1a) were probably membrane-protein complexes, which resolve poorly by two-dimensional gel electrophoresis12. We confirmed this supposition by injecting MitoSNO into mice undergoing LAD ischemia-reperfusion, fluorescently labeling S-nitrosated proteins and separating mitochondrial membrane complexes by blue native PAGE (BN-PAGE). Using this method, we found that only respiratory complex I and the FoF1-ATP synthase were detectably S-nitrosated within mitochondria in vivo (Fig. 1f, Supplementary Fig. 6a and Supplementary Table 1). Although S-nitrosation did not affect FoF1-ATP synthase activity (Supplementary Fig. 6b), previous work has shown that complex I is inhibited by various nitric oxide donors in vitro and that this inhibition is associated with S-nitrosation2,10,11,21–24. However, to our knowledge, the existence and physiological importance of S-nitrosation of complex I in vivo has not previously been demonstrated.
To determine whether S-nitrosation of complex I by MitoSNO in vivo affects its activity, we subjected mice to 30 min of LAD occlusion followed by 5 min of reperfusion with or without MitoSNO treatment at reperfusion. Complex I activity was lower in heart mitochondria from MitoSNO-treated mice compared to untreated mice, and this decreased activity was normalized by selective S-nitrosothiol reduction using copper and ascorbate15–17 (Fig. 1g). In the MitoSNO-treated mice, complex I activity returned to its initial levels after 30 min, indicating that MitoSNO-mediated inhibition of complex I by S-nitrosation is reversible by endogenous mechanisms in vivo (Fig. 1g). In contrast, in the absence of MitoSNO treatment in vivo, complex I was not affected after 5 min of reperfusion, but its activity then decreased by ~50% during 30 min of reperfusion and could not be restored ex vivo by reductants, presumably because of irreversible oxidative damage (Fig. 1c,g). Notably, MitoSNO treatment did not inhibit complex I in the normoxic myocardium in vivo (Fig. 1g), indicating that the inhibitory S-nitrosation of complex I is dependent on an ischemic environment in the myocardium. Therefore, MitoSNO inhibits complex I by S-nitrosation only during ischemia.
As complex I was the only S-nitrosation target identified in our proteomic analysis whose activity was affected by this modification, we consider it to constitute a probable node for cardioprotection. We next sought to identify which of the 45 subunits comprising complex I (ref. 25) were modified by MitoSNO. We tagged S-nitrosated proteins in isolated bovine heart mitochondrial membranes with a red fluorescent dye (Supplementary Fig. 7a), isolated complex I by BN-PAGE and resolved the subunits by diagonal SDS-PAGE (Fig. 2a). MitoSNO reproducibly S-nitrosated three subunits: the 75 kDa, B8 and ND3 subunits (Fig. 2b,c, Supplementary Fig. 7b and Supplementary Table 2). To identify the S-nitrosated cysteines, we tagged unmodified thiols with N-ethylmaleimide (NEM) and S-nitrosated thiols with deuterated NEM (d5-NEM), generating a 5-Da shift detected by mass spectrometry (Fig. 2b,c). Using this method, we identified four cysteines that were S-nitrosated by MitoSNO: two on the 75 kDa subunit and one each on the ND3 and B8 subunits (Fig. 2c,d and Supplementary Table 3). Notably, one of these, Cys39 of the ND3 subunit, has been shown to become exposed under conditions of hypoxia or lack of NADH that result in low complex I activity due to the active-deactive transition26–28. As complex I was inhibited by S-nitrosation in vivo only after ischemia (Fig. 1g), we hypothesized that this cysteine may become susceptible to S-nitrosation only during ischemia when the lack of mitochondrial respiration leads to low complex I activity. We used d5-NEM labeling to quantify29 the status of the four S-nitrosated cysteines in mitochondrial membranes under conditions of low (−NADH) and high (+NADH) complex I activity (Fig. 2d). S-nitrosation of ND3 Cys39 was negligible under conditions of high activity but increased markedly under conditions of low activity (Fig. 2d). Conversely, S-nitrosation of the other three cysteines was unaffected by changes in complex I activity (Fig. 2d and Supplementary Figs. 8–10). Moreover, ND3 Cys39 was extensively S-nitrosated (>50%) by MitoSNO in isolated rat heart mitochondria during anoxia but was negligibly modified under normoxic conditions (Fig. 2e,f).
Figure 2.

Respiratory complex I ND3 Cys39 S-nitrosation is dependent on low complex I activity. (a) Resolution of the subunits of complex I by diagonal SDS-PAGE after treatment of bovine heart mitochondrial membranes with or without MitoSNO and processing to label S-nitrosothiols with a red fluorescent tag. Red spots, S-nitrosated subunits; yellow spots, subunits containing cysteine thiols that are occluded by other modifications. The red spot at ~37 kDa was not reproducible. Pr-SNO, protein S-nitrosothiol; Pr-SX, occluded cysteine thiol. (b) Scheme for the identification of peptides using mass spectrometry after ratiometric labeling of complex I cysteine residues with light NEM (d0-NEM (unmodified), blue) or heavy NEM (d5-NEM (S-nitrosated), red) followed by isolation of complex I by BN-PAGE electrophoresis, tryptic digestion and LC-MS. (c) Summary of complex I cysteines sensitive to S-nitrosation by MitoSNO. In the d0 column, `+' indicates that the NEM-modified cysteine residue could be detected by mass spectrometry. In the d5 column, `+' indicates that the cysteine was S-nitrosated by MitoSNO, and `−' indicates that it was not. Cys, cysteine. (d) The percentage S-nitrosation of sensitive complex I cysteines in mitochondrial membranes that underwent this modification after incubation with MitoSNO with or without NADH. n = 3 individual experiments on bovine heart mitochondrial membranes. (e) Representative mass spectroscopic traces of unmodified and S-nitrosated ND3 Cys39 peptides indicating their relative intensities from rat heart mitochondria incubated with or without O2 and MitoSNO. (f) Percentage of ND3 Cys39 in rat heart mitochondria that was S-nitrosated after incubation with or without O2 and MitoSNO. n = 3 individual experiments on rat heart mitochondria per group. *P < 0.05, **P < 0.01, ***P < 0.001 determined by one-way ANOVA. Data in d and f are shown as the mean ± s.e.m. of three replicates.
We next sought to determine the S-nitrosation profile of the sensitive complex I cysteines within the heart. Within the intact, ischemic heart perfused ex vivo in a Langendorff system, there was substantial (~40%) S-nitrosation of ND3 by MitoSNO that was not present in normoxic hearts; in contrast, the amount of S-nitrosation of the 75 kDa subunit cysteines was similar under normoxia and ischemia (Fig. 3a, Supplementary Table 4 and Supplementary Fig. 11). Intravenous injection of MitoSNO into mice also resulted in S-nitrosation of the ND3 cysteine in vivo during reperfusion of the ischemic heart, whereas MitoSNO injection did not have this effect in normoxic hearts (Fig. 3b). S-nitrosation of the ND3 subunit was lost over the subsequent 30 min both in vivo and in isolated mitochondria by a first-order process with a half-life of ~5 min (Fig. 3c). The kinetics of this loss are consistent with the time course of the restoration of complex I activity after MitoSNO treatment during reperfusion in vivo (Fig. 1g). S-nitrosation of the ND3 subunit in vitro was stable, suggesting that reduction in vivo occurs by thiol reductants of the mitochondrial glutathione and thioredoxin systems (Supplementary Fig. 12)30. Together these data suggest that ischemia exposes ND3 Cys39 for S-nitrosation and that this modification specifically mediates the reversible inhibition of complex I in vivo.
Figure 3.

ND3 Cys39 S-nitrosation mediates inhibition of complex I activity and ROS production. (a) The percentage S-nitrosation of sensitive complex I cysteines within the intact heart during normoxia and ischemia with or without MitoSNO treatment. n = 4–6 ex vivo hearts per group. (b) Quantification of the extent of ND3 S-nitrosation (top) and the ND3 subunit- complex I was isolated by BN-PAGE, and complex I subunits were resolved by SDS-PAGE. Data are shown as the means ± range. n = 2 hearts per group. (c) Left, time course showing the amount of S-nitrosated ND3 subunit after MitoSNO treatment in vivo after LAD ischemia and MitoSNO treatment of isolated mitochondria in vitro during anoxia. The inset shows the log10 of these data plotted against the time to calculate the half-life of ND3 S-nitrosation from the slope. Right, the ND3 subunit-containing region of representative fluorescent-scanned gels. The half-life of the ND3 S-nitrosothiol signal both in vivo and in vitro was determined by setting the fluorescence at 5 min to 100% and measuring the relative decrease in this signal at subsequent time points. The half-life values (in minutes) generated by this analysis for in vitro and in vivo conditions are shown next to the gels. – indicates non–fluorescently labeled sample. n = 3 experiments (in vitro), and n = 3 mice (in vivo). (d) The activity of complex I in bovine heart mitochondrial membranes with no modifications compared to the effects of S-nitrosating the 75 kDa and B8 subunits only (75 kDa/B8 SNO) by incubation with MitoSNO during high complex I activity and of S-nitrosating the ND3, 75 kDa and B8 subunits (ND3 SNO) by incubation with MitoSNO during high complex I activity generated by anoxia. n = 3–4 experiments per condition. (e) Rat heart mitochondria at baseline and after being subjected to anoxia and reoxygenation (AR) with and without MitoSNO and monitored for hydrogen peroxide production. n = 3 experiments. (f) A model of the core structure of mammalian complex I generated using orthologous bovine sequences modeled on the crystal structure of the entire complex I from Thermus thermophilus. Structural modeling places Cys39 near the matrix-facing ND3 loop region of mammalian complex I (blue, ND3; green, other modeled complex I subunits; gray, entire core complex I). FMN, flavin mononucleotide. (g) Expanded modeled structure from f indicating the functional components of complex I close to ND3 Cys39. ND3 Cys39 (yellow sphere) maps to the interface of the ubiquinone binding site (Q site, orange surface) and the mitochondrial matrix surface of the complex, as well as the half-channel element of one of the proposed34 proton translocation pathways within complex I (charged residues within the channel are shown in magenta, and an approximate proton translocation path34 is highlighted with a blue arrow). The N2 iron-sulfur cluster that transfers electrons to bound ubiquinone is also shown. *P < 0.05, **P < 0.01 determined by one-way ANOVA. Data (a–e) are shown as the mean ± s.e.m. of at least three replicates.
As the ND3 subunit is encoded by mitochondrial DNA, its cysteine cannot be replaced genetically, and there are no known natural mutations or polymorphisms in the gene encoding this subunit (MT-ND3)31. To test the functional consequences of S-nitrosation of ND3, we exploited the unique dependence of this S-nitrosation event on low complex I activity. Exposure of isolated bovine heart mitochondrial membranes to MitoSNO under conditions in which complex I is in a high activity state led to S-nitrosation of the 75 kDa and B8 subunit cysteines but not of the ND3 subunit, and this exposure did not affect complex I activity (Fig. 3d). In contrast, incubation with MitoSNO under conditions in which complex I is in a low activity state led to S-nitrosation of ND3 Cys39, as well as of the other three cysteines, and inhibited complex I activity (Fig. 3d). Copper and ascorbate treatment restored complex I activity, indicating that the inhibition of activity was due to S-nitrosation of ND3 Cys39 (Fig. 3d). The complex I–catalyzed reduction by NADH of the artificial electron acceptor hexaammineruthenium(III), which accepts electrons from the flavin mononucleotide, was unaffected by ND3 S-nitrosation (Supplementary Fig. 13). This result indicates that S-nitrosation of ND3 Cys39 does not disrupt complex I function by affecting the interaction of NADH with the flavin mononucleotide site and that the S-nitrosation of other residues in complex I does not affect its activity. Together these data suggest that the S-nitrosation of ND3 Cys39 affects complex I activity by disrupting the interaction of ubiquinone with complex I.
To assess directly whether ND3 S-nitrosation also inhibits mitochondrial ROS production by complex I, we mimicked ischemia-reperfusion injury in vitro by incubating mitochondria anaerobically and then rapidly reoxygenating them, a procedure that results in the generation of hydrogen peroxide. This ROS production was attenuated by MitoSNO after anaerobic incubation before reoxygenation (Fig. 3e). As the S-nitrosation of the ND3 subunit only occurs after anaerobic incubation, these findings suggest that MitoSNO decreases ROS production by S-nitrosating the ND3 subunit.
We next sought to determine whether the local environment of the ND3 cysteine might explain the dependence of its S-nitrosation on complex I activity and the subsequent effects on ROS production. We modeled the location of ND3 Cys39 using structures of prokaryotic complex I (refs. 32–34). This modeling led to an orthologous mammalian core structure containing ND3 Cys39 (Fig. 3f,g and Supplementary Figs. 14 and 15). Our model shows that Cys39 is located at the hinge region of complex I facing the mitochondrial matrix. Furthermore, Cys39 is adjacent to the half-channel element of one of the four proposed proton translocation pathways of complex I and is close to the ubiquinone binding pocket, where ubiquinone receives electrons from the N2 iron-sulfur center (Fig. 3g)34. The location of ND3 at a crucial region in complex I and the position of Cys39 in the ND3 loop region (Fig. 3g) are consistent with the observed activity-dependent exposure and reactivity of this cysteine.
We next assessed the importance of the S-nitrosation of ND3 Cys39 for the protective action of MitoSNO in cardiomyocytes exposed to ischemia-reperfusion injury in vitro, an assay that can be used to reliably assess cardioprotective agents35. We selectively knocked down expression of the complex I assembly factor NDUFAF1 (ref. 36) by RNAi, which decreased protein amounts by 80% (Supplementary Fig. 16a–c) and lowered complex I activity (Supplementary Fig. 16d) while leaving the amount of complex II unaffected (Supplementary Fig. 16a,e). MitoSNO protected control cardiomyocytes treated with a scrambled RNAi construct from ischemia-reperfusion injury, but decreasing complex I activity by NDUFAF1-targeted RNAi completely abolished MitoSNO protection (Fig. 4a). These data suggest that complex I ND3 Cys39 is the crucial functional S-nitrosation target and that other interactions of MitoSNO with mitochondria do not contribute to protection.
Figure 4.
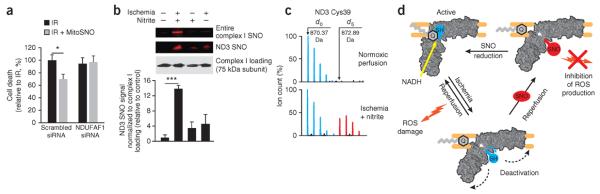
S-nitrosation of ND3 Cys39 underlies protection from ischemia-reperfusion injury by mitochondrial S-nitrosation in vivo and represents a general mechanism for cardioprotection. (a) Cell death of rat heart–derived H9C2 cells subjected to ischemia-reperfusion injury with or without MitoSNO or NDUFAF1-targeted RNAi. n = 8 experiments per group. (b) S-nitrosation of complex I ND3 Cys39 in the intact heart followed by exposure to nitrite during ischemia as determined by fluorescent labeling of the entire complex after its resolution by BN-PAGE (top gel). In a separate experiment, after the isolation of complex I by BN-PAGE, the constituent subunits of complex I were resolved, allowing for direct assessment of the S-nitrosation state of ND3 (middle gel). Loading of complex I was assessed by western blotting of the 75 kDa subunit (bottom gel). Bottom, quantification of ND3 Cys39 S-nitrosation signals relative to those of complex I loading as determined by western blotting. n = 4 hearts per group. (c) A representative mass spectrum of the Cys39-containing tryptic peptide of ND3 indicating S-nitrosation of ND3 Cys39 after exposure of the heart to nitrite during ischemia by labeling of the ND3 S-nitrosothiol with d5-NEM. (d) A model showing how S-nitrosation of ND3 Cys39 protects from ischemia-reperfusion injury. During normal complex I activity, ND3 Cys39 is occluded. The low activity of complex I after ischemia causes this cysteine to become exposed. Reperfusion of the ischemic tissue rapidly reactivates complex I and generates superoxide and hydrogen peroxide, causing oxidative damage and cell death. When MitoSNO, or other S-nitrosating species, are present during reperfusion, the exposed ND3 cysteine becomes S-nitrosated, temporarily holding complex I in a low activity state and decreasing ROS production. The S-nitrosothiol is subsequently reduced by glutathione and thioredoxin, gradually reactivating complex I. SH, unmodified ND3 Cys39; Q, ubiquinone. *P < 0.05, ***P < 0.001 determined by one-way ANOVA. Data (a,b) are shown as the mean ± s.e.m.
We next determined whether ND3 Cys39 S-nitrosation is a general mechanism of cardioprotection. The nitrite (NO2−) anion protects from ischemia-reperfusion injury in vivo1–3,37 and is thought to act as an S-nitrosating agent in the ischemic myocardium2. We infused mouse hearts ex vivo in a Langendorff perfusion apparatus with a dose of nitrite that is known to protect from reperfusion damage38 and measured S-nitrosation of complex I ND3 Cys39. Nitrite infusion during prolonged ischemia resulted in substantial S-nitrosation of this cysteine, which was negligibly modified by nitrite under normoxic conditions (Fig. 4b,c and Supplementary Table 5). Protection against ischemia-reperfusion injury afforded by ischemic preconditioning (IPC) is another cardioprotective strategy in which nitric oxide has been recently shown to have an essential role10,39. Subjecting hearts to an IPC protocol that protects against subsequent ischemia-reperfusion injury40 also led to S-nitrosation of ND3 Cys39 (Supplementary Table 6). Therefore, the reversible modification of ND3 Cys39 by a number of endogenous and exogenous processes is probably a general mechanism to limit ischemia-reperfusion damage. These findings suggest that the mechanism of cardioprotection we discovered using MitoSNO also applies to a range of established methods of protection from ischemia-reperfusion injury (Supplementary Fig. 17).
We propose the following scenario to explain the mechanism of protection by S-nitrosation of complex I ND3 Cys39 in myocardial ischemia-reperfusion injury (Fig. 4d). During normal complex I activity, the crucial cysteine is occluded, but during ischemia, the low activity of complex I causes this cysteine to become exposed. Reperfusion of the ischemic tissue rapidly reactivates complex I and generates hydrogen peroxide, which causes oxidative damage that leads to the cell death underlying ischemia-reperfusion injury. When S-nitrosating species are present during reperfusion, the exposed ND3 cysteine becomes S-nitrosated, locking complex I in a low activity state and decreasing ROS production during the reperfusion phase. The S-nitrosothiol is subsequently reduced by glutathione and thioredoxin, gradually reactivating complex I.
Cys39 in ND3 is conserved throughout metazoans, consistent with a physiological role for this residue. Possibilities for this role include protecting tissues that regularly become ischemic in vivo from ischemia-reperfusion damage, such as the muscle or kidney, or lowering ROS production when complex I is being assembled, repaired or degraded.
The methods developed here for the spatial, temporal and quantitative assessment of S-nitrosation in vivo allowed us to identify the ND3 Cys39 of complex I as a key node in protection against acute reperfusion injury. Together these findings provide a unifying mechanism for many of the cardioprotective effects of mitochondrial S-nitrosation in vivo. This mechanism probably contributes to cardioprotection by untargeted nitric oxide donors and alterations to nitric oxide metabolism in general, although nonmitochondrial pathways may also contribute. The capacity of MitoSNO to protect against ischemia-reperfusion injury when administered systemically at low doses before reperfusion and the lack of secondary hemodynamic effects make it a potential therapeutic candidate for myocardial infarction, stroke, elective surgery and organ transplantation. More generally, the development of methods for assessing the modification of ND3 Cys39 in vivo will help determine the mechanisms of action of other cardioprotective interventions.
ONLINE METHODS
In vivo treatments
We used an open-chest, in situ mouse (C57BL/6) heart model as recently described41. Briefly, male mice (8–10 weeks of age; Charles River Laboratories, UK) were anesthetized with sodium pentobarbital (70 mg per kg body weight intraperitoneally (i.p.)), intubated endotracheally and ventilated with 3 cm H2O positive-end expiratory pressure. We monitored the adequacy of the anesthesia using corneal and withdrawal reflexes, and additional anesthesia was administered as needed throughout the experiment. We kept the ventilation frequency at 240 breaths per minute with a tidal volume between 125 μl and 150 μl. We performed a small thoracotomy, and the heart was exposed by stripping of the pericardium. All hearts underwent 30 min of regional ischemia by ligation of a main branch of the left coronary artery. We gave MitoSNO, MitoNAP or SNAP (100 ng per kg body weight each) 5 min before reperfusion as a slow bolus intravenously into a tail vein.
We assessed infarct size after 120 min of reperfusion using triphenyltetrazolium chloride staining and expressed it as a percentage of the risk zone as described recently40. For various experiments on treated tissues, we removed the left ventricle at various time points after reperfusion, as indicated in the corresponding figure legends. If isolated mitochondria were needed, hearts were excised, and the left ventricle was isolated and homogenized before mitochondrial isolation as described below. All animal experiments described here were carried out in accordance with the UK Home Office Guide on the Operation of Animal (Scientific Procedures) Act of 1986 and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication 85–23, revised 1996). All procedures were approved under the Project License number PPL 80/2393.
Mitochondrial preparations and incubations
We prepared rat and mouse heart mitochondria (RHM and MHM, respectively) and bovine heart mitochondrial membranes (BHM) as described previously42 by homogenization of tissue in STE (250 mM sucrose, 5 mM Tris-HCl and 1 mM ethylene glycol tetraacetic acid (EGTA), pH 7.4), followed by differential centrifugation. For the rat and mouse heart mitochondria preparations, STE was supplemented with 0.1% fatty acid–free BSA. Protein concentration was measured with the biuret assay using BSA as a standard. When protein S-nitrosothiols were to be assessed in mitochondria isolated from hearts that had been subjected to in vivo treatments, 100 mM NEM was included in the isolation buffers.
We incubated RHM and BHM in a 120 mM KCl solution containing 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1 mM EDTA, 1 mM diethylene triamine pentaacetic acid (DTPA) and 10 μM neocuproine, pH 7.4, at 37 °C with or without the following respiratory substrates: for RHM, 5 mM glutamate and 5 mM malate or 10 mM succinate; for BHM, 10 mM NADH. When using BHM, normoxic high complex I activity conditions were generated by preincubating with NADH for 20 min at 4 °C, and for low complex I activity conditions NADH was omitted. We generated anoxia for both BHM and RHM by incubating with the indicated substrate along with 1 mM ADP at 37 °C in the stirred, sealed 3-ml chamber of a Clark-type O2 electrode (Rank Brothers, UK). When the chamber was sealed, mitochondrial respiration was used to bring the solution to anoxia, which was assessed by continuous monitoring of the O2 concentration, and conditions were maintained as anoxic for 30 min. Normoxic incubations were initially carried out in the O2 electrode chamber by stirring when it was open to the atmosphere without the lid, but as these incubations gave the same results as those in 3 ml of medium in a 15-ml Falcon tube with continuous rotation at 37 °C, the latter procedure was used for all data reported here. To distinguish the functional consequences of normoxic compared to anoxic S-nitrosation of complex I, we incubated mitochondrial membranes under either normoxic or anoxic conditions for 25 min, then added MitoSNO or MitoNAP (50 μM of each) followed by a further 5-min incubation under the same conditions. This was followed by pelleting and washing of the mitochondria or membranes before analysis. All samples were shielded from light throughout the experiment to preserve S-nitrosothiols.
Differential labeling of respiratory complex I S-nitrosothiols
We incubated mitochondria or mitochondrial membranes from both the in vitro and in vivo treatment groups in 100 mM NEM to quench unreacted thiols. Mitochondria from mice treated in vivo with an S-nitrosating agent were isolated as described above in buffer supplemented with 100 mM NEM before differential labeling. Both in vivo– and in vitro–treated samples were transferred to opaque micro-fuge tubes, and the mitochondria or membranes were pelleted by centrifugation at 16,000g for 2 min. We resuspended pellets in 100 mM NEM in water and briefly homogenized them on ice using a blender to rupture intact mitochondria before pelleting them once more. Pelleted samples were washed three times in Chelex 100–treated PBS before resuspension in 100 μl of the same buffer containing 1 mM ascorbate and 10 μM CuSO4 that was further supplemented with 100 mM d5-NEM (for mass spectrometry labeling; Cambridge Isotope Laboratories, Inc, USA) or 40 μM of CyDye DIGE Fluor Cy3 saturation dye (Cy3, GE Healthcare) or CyDye DIGE Fluor Cy5 saturation dye (Cy5, GE Healthcare) for fluorescent labeling. We incubated resuspended samples for 30 min at 37 °C, mixed them with the appropriate loading buffer for BN-PAGE or SDS-PAGE and further analyzed them by electrophoresis.
Band excision and Orbitrap mass spectrometry
We separated complex I subunits by gel electrophoresis, stained the gel with Coomassie blue, excised subunits from the gels and digested the proteins by `in gel' cleavage with either trypsin (Roche, UK) or chymotrypsin (Roche, UK). We analyzed the peptides by mass spectrometry using an LTQ Orbitrap XL mass spectrometer (Thermo) after chromatography on a nanoscale reverse-phase column (75 μm inner diameter × 100 mm; Nanoseparations, Nieukoop, Netherlands) using a gradient of 0–35% buffer B over 40 min at 300 nl min−1 (starting buffer A, 5% acetonitrile and 0.1% formic acid; buffer B, 95% acetonitrile and 0.1% formic acid). Proteins were identified by comparison of peptide mass and, if possible, peptide fragmentation data with the UniProt sequence database using the MASCOT algorithm. All peptide assignments from the resulting mass spectrometry data were made using the bioinformatics search engine Mascot version 2.2.04 (Matrix Sciences, London, UK)43. We used the following search parameters: enzyme specificity was defined as trypsin or chymotrypsin, and d0-NEM and d5-NEM fixed modifications were chosen. The variable modifications chosen were: oxidation (Met). The peptide ion (precursor) and the fragment ion mass tolerance were set at 5 p.p.m. and 0.5 Da, respectively. The maximum number of missed cleavages was set at 1. Ions scores were defined as (−10 × log(p)), where p is the probability that the observed match is a random event. Amounts of cysteine S-nitrosation were calculated as the percentage of the precursor ion peak areas of the S-nitrosated peptide labeled with d5-NEM for each condition over the sum of the reduced peptide labeled with d0-NEM plus S-nitrosated d5-NEM-labeled peptide: (S-nitrosated peptide d5-NEM)/(reduced peptide d0-NEM + S-nitrosated peptide d5-NEM) × 100.
Measurement of mitochondrial hydrogen peroxide flux in vivo
We measured mitochondrial hydrogen peroxide in vivo using the MitoB mass spectro-metric probe as described previously20. Briefly, 75 nmol MitoB (synthesized as described in ref. 20) (~3 μmol per kg body weight for 25- to 30-g mice) in 50 μl saline was administered by tail-vein injection to the mouse 1 h before initiating ischemia-reperfusion. At the end of the procedure, the ischemic heart tissue was identified visually, dissected out, frozen on liquid nitrogen and then stored at −80 °C. For analysis, we spiked the tissues with deuterated internal standards, extracted the MitoB and its product MitoP from the reaction with hydrogen peroxide and determined the amounts of MitoB and MitoP present in the tissue by liquid chromatography and tandem mass spectrometry as described previously20.
Chemical syntheses
We synthesized MitoSNO and MitoNAP as described previously11. SNAP was from Sigma. Solutions were stored in absolute ethanol at −80 °C. MitoSNO stability was assessed before use from its absorbance at 340 nm.
Sample preparation for SNO-DIGE
After incubating hearts ex vivo with or without MitoSNO, we isolated mitochondria as described above, resuspended them in an assay medium containing 10 mM HEPES, 1 mM EGTA, 100 μM neocuproine, pH 7.4, and 50 mM NEM, and incubated them for 5 min at 37 °C. We then added 1% SDS and incubated them for a further 5 min. NEM was removed from the samples using three Micro Bio-Spin 6 chromatography columns (Bio-Rad), and a 1% SDS concentration was maintained throughout the assay.
SNO-DIGE experimental design
To assess differences in protein fluorescence using two-dimensional DIGE, we used an experimental design that included biological variance analysis (BVA)12. In each DIGE experiment, we used four biological replicates, where a biological replicate is an independently prepared heart tissue subjected to the CyDye sample preparations described above. As is the custom with BVA experiments, one fluorescent component of each gel (in this case, Cy3) comprised a pooled standard of equal amounts of protein from each biological replicate, and the Cy5 component of each gel was an individual treatment (either untreated or treated with MitoSNO). Each DIGE analysis therefore consisted of eight gels from four biological replicates: four gels compared the control condition to the pooled standard and four gels compared the MitoSNO-treated condition to the standard.
Two-dimensional electrophoresis
We resolved mitochondrial proteins as described previously12. Briefly, equal volumes of 2× sample buffer (7 M urea, 2 M thiourea, 2% amidosulfobetaine-14, 20 mg ml−1 DTT and 2% Pharmalytes 3–10 NL) (GE Healthcare) were added to the fluorescently labeled samples (25 μg total protein per gel) and incubated for 15 min at room temperature. We then made up the sample volume to 250 μl using rehydration buffer (7 M urea, 2 M thiourea, 2% amidosulfobetaine-14, 2 mg ml−1 DTT and 1% Pharmalytes 3–10 NL) before isoelectric focusing using 13-cm immobilized pH gradient (IPG) strips (pH 3–10 NL, GE Healthcare). We rehydrated strips with the samples for 10 h at 20 °C and applied 20 V using the IPGPhor II apparatus following the manufacturer's instructions (GE Healthcare). Isoelectric focusing was performed for a total of 41,700 V-h at 20 °C and 50 μA. Before SDS-PAGE, we washed the strips in two equilibrating buffers: (i) 100 mM Tris-HCl, pH 6.8, 30% glycerol, 8 M urea, 1% SDS and 5 mg ml−1 DTT for 15 min and (ii) 100 mM Tris-HCl, pH 6.8, 30% glycerol, 8 M urea, 1% SDS and 20 mg ml−1 iodoacetamide for 15 min. We then loaded the strips onto a 12%, 1-mm acrylamide gel and overlaid the gel with 1% agarose in SDS running buffer containing bromophenol blue. The gels were run at 20 °C with 20 mV applied for 15 min, and then 40 mV was applied until the bromophenol blue dye front had run off the bottom of the gels.
Gel imaging and analysis
We transferred gels containing CyDye-labeled proteins to a Typhoon 9410 imager (GE Healthcare). Cy3 fluorescent images were obtained using a 532-nm laser and a 580-nm emission filter (band pass of 30 nm). Cy5 fluorescent images were obtained using a 633-nm laser and a 670-nm emission filter (band pass of 30 nm). We scanned all gels at 100- to 200-μm pixel size, and the photomultiplier tube was set to ensure a maximum pixel intensity within the linear acquisition range for the region of the gel being analyzed (most commonly the sub–20 kDa region of gels containing the complex I ND3 subunit) and thereby ensuring that fluorescent signal intensities were in the linear range.
We performed analysis of the two-dimensional fluorescent images using DeCyder BVA version 6.5 (GE Healthcare), a two-dimensional gel analysis software package designed specifically for DIGE. The software was used following the manufacturer's recommendations. Briefly, we set the estimated number of spots to 2,500, and each protein spot across eight gel images was matched automatically and then checked and matched manually. Successfully matched spots were compared across groups according to standardized abundances, where a sample spot volume (Cy5) is divided by the standard spot volume (Cy3), normalized with ratiometric normalization and transformed with a log10 function. Comparing the standardized abundances, differences across the two groups were considered significant at >2, P < 0.05.
Resolution of complex I subunits by gel electrophoresis
We first separated respiratory complexes from mitochondrial samples by BN-PAGE, as described previously44, using a 5–12% acrylamide gel. After electrophoresis, we excised complex I bands and incubated them in SDS-PAGE loading buffer supplemented with 100 mM NEM for 30 min at 37 °C. Samples resolved by diagonal SDS-PAGE were prepared as described previously45 using a 10% acrylamide and 6 M urea gel for the first dimension, after which the gel lane was excised, annealed with a 16% acrylamide gel and run in the second dimension. We separated samples for quantification of the ratio of d0-NEM to d5-NEM by mass spectrometry analysis through a 12% or 12–22% gradient SDS-PAGE gel after complex I BN-PAGE isolation. Fluorescence analysis of ND3 S-nitrosation from in vivo samples was performed by isolation of complex I by BN-PAGE as described above followed by resolution of complex I subunits by SDS-PAGE. The ND3 subunit resolved to ~15 kDa, and the SNO fluorescence in this region was confirmed to be due specifically to ND3 using diagonal SDS-PAGE experiments (Fig. 2a). We corroborated this further by mass spectrometry identification43 of ND3 from gel slices containing the ND3 fluorescent signal. When complex I loading in the SDS-PAGE phase was assessed by immunoblotting, an antibody to the 75 kDa subunit was used (Proteintech Europe, 12444-1-AP, 1:2,000).
Measurement of complex I activity
After mitochondrial treatment in vitro, we resuspended mitochondria or mitochondrial membranes in a 10 mM HEPES and 120 mM KCl assay buffer containing 100 μM NADH, 10 μM decylubiquinone, 300 nM antimycin and 2 mM potassium cyanide. We incubated the samples for 5 min at 32 °C while monitoring absorbance at 340 nm (A340) and 380 nm (A380) in a 96-well plate using a SpectraMax (Molecular Devices) plate reader. We determined the maximum linear rate as (A340 −A380) s−1, subtracted the background rate after adding 4 μg ml−1 rotenone from this value and expressed the rates as a percentage of those in the control incubations. We incubated mitochondria isolated from in vivo treatments under the same conditions as described above in a 1-ml cuvette, and 30 μg ml−1 alamethicin was added to permeabilize the mitochodrial inner membrane. The rotenone-sensitive rate of absorbance change was then determined at 340 nm. In parallel, we determined the citrate synthase activity of each sample46. We then normalized the complex I rate to the citrate synthase rate and expressed it as a percentage of the appropriate control. To assess the effect of reversing S-nitrosation, we treated mitochondria with 10 μM CuSO4 and 1 mM ascorbate for 5 min. We determined complex I activity using hexaammine-ruthenium (HAR) as an electron receptor as described previously using 3.5 mM HAR47.
Ex vivo heart treatment with MitoSNO and IPC
Mice were anesthetized with avertin (0.5 mg per kg body weight i.p.), the chest was opened and the aorta was cannulated in situ with a 22.5G needle filled with 37 °C Krebs-Henseleit buffer comprising 118 mM NaCl, 4.7 mM KCl, 25 mM NaHCO3, 10 mM glucose, 1.2 mM MgSO4, 1.2 mM KH2PO4 and 2.5 mM CaCl2. We rapidly transferred (<10 s) the heart to a perfusion apparatus with 37 °C Krebs-Henseleit buffer and initiated perfusion with gassed 37 °C Krebs-Henseleit buffer (95% O2, 5% CO2) in constant flow mode (4 ml min−1). We measured coronary pressure at the cannula and measured hemodynamics using a transducer-linked water-filled balloon in the left ventricle. Initial diastolic pressure was set to 5–10 mm Hg by inflating the balloon. Transducers were linked through an A/D converter (Dataq) to a computer, and data were collected at 1 kHz. We separated hearts into five groups: (i) control perfusion, (ii) MitoSNO infusion (100 nM) during normoxic perfusion, (iii) ischemia (25 min), (iv) ischemia plus MitoSNO and (v) IPC comprising 5-min ischemia followed immediately by reperfusion for 5 min, and this cycle was repeated two more times.
Ex vivo heart treatment with nitrite
We treated mouse hearts with nitrite using previously established protocols known to elicit cardioprotection38. We heparinized mice (200 U i.p.) and anesthetized them with sodium pentobarbital (175 mg per kg body weight i.p.), then opened the chest, excised the heart and arrested it in cold Krebs-Henseleit buffer comprising 0.5 mM EDTA, 118 mM NaCl, 4.7 mM KCl, 25 mM NaHCO3, 11 mM glucose, 1.2 mM MgSO4, 1.2 mM KH2PO4 and 2 mM CaCl2. We then cannulated the aorta with a 22G blunt needle, transferred the heart to a perfusion apparatus and perfused it with 37 °C Krebs-Henseleit buffer (95% O2 and 5% CO2) in a constant flow mode of 2 ml min−1. Drugs were added by infusion pump through a side port directly above the aortic cannula. After 15 min of equilibration, hearts were separated into four groups: (i) control normoxic perfusion, (ii) NO2− infusion (100 μM) during normoxic perfusion, (iii) control global ischemia (30 min) and (iv) NO2− infusion (100 μM) plus global normothermic ischemia. NO2− and control saline infusions began 15 min before ischemia and continued throughout the ischemic phase so that a constant final concentration of NO2− was maintained in the heart. When administering NO2− or saline solutions to ischemic hearts, we deoxygenated the solutions during preparation by bubbling with 95% N2 and 5% CO2.
In vitro ischemia reperfusion injury to cells
In vitro ischemia-reperfusion was performed as described previously35. Briefly, rat heart–derived H9C2 cells (American Type Culture collection, Manassas, VA, USA) were grown in Complete Growth Media (CGM) comprising DMEM, 4 mM glutamine, 10% FBS and 1% penicillin and streptomycin. Seventy-two hours after transfection, we removed the CGM and replaced it with normoxic buffer (DMEM powder (Gibco), 10 mM glucose and 10 mM HEPES, pH 7.4, at 37 °C). After 30 min, we transferred the plate into a hypoxic chamber (Coy Inc., Grass Lake, MI); the medium was then replaced with hypoxic buffer (DMEM powder and 10 mM HEPES, pH 6.5, at 37 °C), and MitoSNO (500 nM) was added to half of the control and siRNA wells. After 2 h, we removed the plate from the hypoxic chamber and replaced the medium with normoxic buffer. We protected cells from light during hypoxia (2 h) and reperfusion (1.5 h) and measured cell death using a lactate dehydrogenase (LDH) release assay (Cytotoxicity Detection, Roche Applied Science) according to the manufacturer's instructions. We present the data as LDH release during ischemia and reperfusion as a percentage of total LDH.
Statistical analyses
Error bars represent the s.e.m. from at least three replicates unless otherwise stated. We quantified P values using one-way ANOVA. P < 0.05 was considered statistically significant.
Additional methods. Detailed methodology is described in the Supplementary Methods.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the UK Medical Research Council and grants from the UK Biotechnology and Biological Sciences Research Council (BB/I012923 to M.P.M. and R.C.H.), the Gates Cambridge Trust and the Canadian Institutes of Health Research (doctoral scholarship and postdoctoral fellowship to E.T.C.), the British Heart Foundation (PG/12/42/29655 to T.K.), the US National Institutes of Health (R01-HL071158 to P.S.B.) and the International Society for Heart Research (ISHR-ES/SERVIER research fellowship to C.M.). We thank L. Sazanov and J. Hirst for helpful discussions.
Footnotes
AUTHOR CONTRIBUTIONS E.T.C. designed research, carried out mass spectrometry, fluorescence and biochemical experiments, analyzed data from in vivo experiments and cowrote the paper. C.M., S.M.N. and V.R.P. carried out and analyzed data from in vivo experiments. J.R. assisted with DIGE experiments. A.L. and S.D. carried out mass spectrometry experiments. A.M.J. and H.M.C. assisted with research design and data interpretation. A.J.R. designed and performed bioinformatic experiments. L.P., I.M.F., R.C.H., K.S.L., R.A.J.S., T.K. and P.S.B. assisted with research design. M.P.M. designed and directed the project and cowrote the paper.
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details are available in the online version of the paper.
Note: Supplementary information is available in the online version of the paper.
References
- 1.Duranski MR, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J. Clin. Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shiva S, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J. Exp. Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite–nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 4.Methner C, Schmidt K, Cohen MV, Downey JM, Krieg T. Both A2a and A2b adenosine receptors at reperfusion are necessary to reduce infarct size in mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H1262–H1264. doi: 10.1152/ajpheart.00181.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen MV, Yang XM, Liu YP, Solenkova NV, Downey JM. Cardioprotective PKG-independent NO signaling at reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H2028–H2036. doi: 10.1152/ajpheart.00527.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa AD, et al. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ. Res. 2005;97:329–336. doi: 10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- 7.Bolli R, et al. Myocardial protection at a crossroads. Circ. Res. 2004;95:125–134. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 8.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N. Engl. J. Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 9.Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am. J. Cardiol. 2010;106:360–368. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ. Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 11.Prime TA, et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA. 2009;106:10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chouchani ET, et al. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J. 2010;430:49–59. doi: 10.1042/BJ20100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray CI, Kane LA, Uhrigshardt H, Wang SB, Van Eyk JE. Site-mapping of in vitro S-nitrosation in cardiac mitochondria: implications for cardioprotection. Mol. Cell Prot. 2011;10 doi: 10.1074/mcp.M110.004721. M110.004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porteous CM, et al. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: implications for mitochondria-specific therapies and probes. Biochim. Biophys. Acta. 2010;1800:1009–1017. doi: 10.1016/j.bbagen.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Kettenhofen NJ, Shiva S, Hogg N, Gladwin MT. Copper dependence of the biotin switch assay: modified assay for measuring cellular and blood nitrosated proteins. Free Radic. Biol. Med. 2008;44:1362–1372. doi: 10.1016/j.freeradbiomed.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kettenhofen NJ, Wang X, Gladwin MT, Hogg N. In-gel detection of S-nitrosated proteins using fluorescence methods. Methods Enzymol. 2008;441:53–71. doi: 10.1016/S0076-6879(08)01204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chouchani ET, James AM, Fearnley IM, Lilley KS, Murphy MP. Proteomic approaches to the characterization of protein thiol modification. Curr. Opin. Chem. Biol. 2011;15:120–128. doi: 10.1016/j.cbpa.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Methner C, Lukowski R, Hofman F, Murphy MP, Krieg T. Protection through postconditioning or a mitochondria-targeted S-nitrosothiol is unaffected by cardiomyocyte-selective ablation of protein kinase G. Basic Res. Cardiol. 2013;108:337. doi: 10.1007/s00395-013-0337-1. [DOI] [PubMed] [Google Scholar]
- 19.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cochemé HM, et al. Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab. 2011;13:340–350. doi: 10.1016/j.cmet.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite. J. Biol. Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- 24.Galkin A, Moncada S. S-nitrosation of mitochondrial complex I depends on its structural conformation. J. Biol. Chem. 2007;282:37448–37453. doi: 10.1074/jbc.M707543200. [DOI] [PubMed] [Google Scholar]
- 25.Carroll J, et al. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 26.Galkin A, et al. Identification of the mitochondrial ND3 subunit as a structural component involved in the active/deactive enzyme transition of respiratory complex I. J. Biol. Chem. 2008;283:20907–20913. doi: 10.1074/jbc.M803190200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciano M, Fuszard M, Heide H, Botting CH, Galkin A. Conformation-specific crosslinking of mitochondrial complex I. FEBS Lett. 2013;587:867–872. doi: 10.1016/j.febslet.2013.02.039. [DOI] [PubMed] [Google Scholar]
- 28.Gorenkova N, Robinson E, Grieve DJ, Galkin A. Conformational change of mitochondrial complex I increases ROS sensitivity during ischemia. Antioxid. Redox Signal. 2013 Mar 29; doi: 10.1089/ars.2012.4698. published online, http://dx.doi.org/10.1089/ARS.2012.4698. [DOI] [PMC free article] [PubMed]
- 29.Held JM, et al. Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol. Cell. Proteomics. 2010;9:1400–1410. doi: 10.1074/mcp.M900643-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Requejo R, et al. Measuring mitochondrial protein thiol redox state. Methods Enzymol. 2010;474:123–147. doi: 10.1016/S0076-6879(10)74008-8. [DOI] [PubMed] [Google Scholar]
- 31.Bridges HR, Birrell JA, Hirst J. The mitochondrial-encoded subunits of respiratory complex I (NADH:ubiquinone oxidoreductase): identifying residues important in mechanism and disease. Biochem. Soc. Trans. 2011;39:799–806. doi: 10.1042/BST0390799. [DOI] [PubMed] [Google Scholar]
- 32.Sazanov LA, Hinchcliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 33.Efremov RG, Sazanov LA. Structure of the membrane domain of respiratory complex I. Nature. 2011;476:414–420. doi: 10.1038/nature10330. [DOI] [PubMed] [Google Scholar]
- 34.Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo S, et al. A cell-based phenotypic assay to identify cardioprotective agents. Circ. Res. 2012;110:948–957. doi: 10.1161/CIRCRESAHA.111.263715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mimaki M, et al. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta. 2012;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Shiva S, Gladwin MT. Nitrite mediates cytoprotection after ischemia/reperfusion by modulating mitochondrial function. Basic Res. Cardiol. 2009;104:113–119. doi: 10.1007/s00395-009-0009-3. [DOI] [PubMed] [Google Scholar]
- 38.Webb A, et al. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc. Natl. Acad. Sci. USA. 2004;101:13683–13688. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun J, et al. Essential role of nitric oxide in acute ischemic preconditioning: S-nitros(yl)ation versus sGC/cGMP/PKG signaling? Free Radic. Biol. Med. 2013;54:105–112. doi: 10.1016/j.freeradbiomed.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc. Res. 2011;89:643–649. doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt K, et al. Cardioprotective effects of mineralocorticoid receptor antagonists at reperfusion. Eur. Heart J. 2010;31:1655–1662. doi: 10.1093/eurheartj/ehp555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chappell JB, Hansford RG. In: Subcellular Components: Preparation and Fractionation. Birnie GD, editor. Butterworths; London: 1972. p. 77. [Google Scholar]
- 43.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 44.Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 45.Rais I, Karas M, Schagger H. Two-dimensional electrophoresis for the isolation of integral membrane proteins and mass spectrometric identification. Proteomics. 2004;4:2567–2571. doi: 10.1002/pmic.200400829. [DOI] [PubMed] [Google Scholar]
- 46.Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–10. [Google Scholar]
- 47.Birrell JA, Yakovlev G, Hirst J. Reactions of the flavin mononucleotide in complex I: a combined mechanism describes NADH oxidation coupled to the reduction of APAD+, ferricyanide, or molecular oxygen. Biochemistry. 2009;48:12005–12013. doi: 10.1021/bi901706w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.