

Significance
RNA viruses can contain a single (unsegmented) or multiple (segmented) genomic molecules. However, the evolutionary connection between these two fundamentally different forms of genome organization is unclear. We discovered a previously unidentified segmented RNA virus of ticks that, uniquely, contained two segments derived from an unsegmented flavivirus, as well as two highly divergent segments of unknown origin. In addition, copies of this virus were present in a nematode (dog roundworm). Collectively, these results demonstrate the remarkable diversity of viral genome structures as well as a previously unidentified evolutionary link between unsegmented and segmented viruses.
Keywords: evolution, segmentation
Abstract
Although segmented and unsegmented RNA viruses are commonplace, the evolutionary links between these two very different forms of genome organization are unclear. We report the discovery and characterization of a tick-borne virus—Jingmen tick virus (JMTV)—that reveals an unexpected connection between segmented and unsegmented RNA viruses. The JMTV genome comprises four segments, two of which are related to the nonstructural protein genes of the genus Flavivirus (family Flaviviridae), whereas the remaining segments are unique to this virus, have no known homologs, and contain a number of features indicative of structural protein genes. Remarkably, homology searching revealed that sequences related to JMTV were present in the cDNA library from Toxocara canis (dog roundworm; Nematoda), and that shared strong sequence and structural resemblances. Epidemiological studies showed that JMTV is distributed in tick populations across China, especially Rhipicephalus and Haemaphysalis spp., and experiences frequent host-switching and genomic reassortment. To our knowledge, JMTV is the first example of a segmented RNA virus with a genome derived in part from unsegmented viral ancestors.
Segmentation is a common form of genome organization in RNA viruses, although why it has evolved more than once and is maintained in such a diverse array of viruses, including those with both positive- and negative-sense genomes, are unclear (1). Segmented and unsegmented viruses usually belong to different viral families, such that the sequence divergence between them is often too great for any meaningful evolutionary inference. The only example of “recent” genome fragmentation in a single RNA molecule occurred in a laboratory strain of foot-and-mouth disease virus (2, 3), although that this occurred following extensive propagation in cell culture means that its relationship to segmentation in nature is uncertain. Hence, the evolutionary links between unsegmented and segmented viruses, as well as the mechanisms that underpin their genesis, are poorly understood.
The Flaviviridae are a family of unsegmented positive sense RNA viruses that infect vertebrate and invertebrate species, including the important human pathogens dengue virus, yellow fever virus, and hepatitis C virus. Despite the substantial sequence divergence between the Flavivirus, Pestivirus, and Hepacivirus genera that make up the Flaviviridae, they exhibit a similar genomic structure characterized by a single ORF with distinct clusters of structural and nonstructural genes. The ORF is translated into a single polyprotein, which is subsequently cleaved by cellular and viral proteases into structural and nonstructural proteins. Among the nonstructural protein products, NS3 and NS5 possess the enzymatic domains essential for RNA capping and genome replication (4), whereas the NS3 and NS2b proteins form a two-component serine protease involved in posttranslational cleavage of the viral polyprotein (5).
Herein we describe the discovery and characterization of an RNA virus with a four segment genome that encodes NS2b-NS3- and NS5-like proteins related to those found in the Flaviviridae, in turn revealing an unanticipated evolutionary link between the genomes of unsegmented and segmented RNA viruses.
Results
Discovery and Attempts to Isolate the Virus.
In 2010, a pan-Phlebovirus RT-PCR protocol was performed to determine the presence of Huaiyangshan virus in tick samples collected in China (6, 7). This screening identified flavivirus-like RNAs (segment 1 and segment 3) in a Rhipicephalus microplus ticks pool collected in the Jingmen region of Hubei province. Following the confirmation of its presence (as detailed later), we named this virus Jingmen tick virus (JMTV) after the location and the tick host in which it was initially found.
To access the presence of a virus, we infected mammalian and insect cell lines with the sample JMTV118 and (i) monitored the cell monolayer for a cytopathic effect, and (ii) attempted to detect viral genomic RNA by using RT-PCR with JMTV-specific primers. Following inoculation of the JMTV118 homogenate, neither C6/36 nor DH82 cells developed any characteristic cytopathic effect, although DH82 cells showed gradual detachment from the flask 7 d after inoculation. However, all four segments of the putative viral genome could be detected for the first two passages in both cell types (SI Appendix, Fig. S1), although the virus became untraceable after three passages. Collectively, these results provided evidence for viral replication in mosquito and mammalian cell lines. However, the ability of the virus to grow in both cell lines was limited. Consequently, in vitro purification and titration of the virus were unsuccessful. Cerebral injection into newborn mice produced results in accord with those of cell culture experiments: virus genomes were detected only during the first two passages.
EM of infected DH82 cells (approximately 5% in the inspected area) showed numerous virion-like particles in the cisternae of endoplasmic reticulum that were likely to be JMTV (Fig. 1A; as a comparison, DH82 cells without JMTV infections are also shown in SI Appendix, Fig. S2). Similar particles were observed from cell culture supernatant (Fig. 1B). These particles were spherical and enveloped and had clear protrusions. Despite their resemblance to a typical flavivirus virion, these particles were slightly larger: 70–80 nm in diameter compared with 45–60 nm noted in flaviviruses.
Fig. 1.

Transmission EM image of JMTV in (A) infected cells and (B) cell culture supernatant. The viral particles are indicated by the orange arrowheads.
Determination of the Complete Viral Genome Sequence.
Partial genome sequences resembling flavivirus NS3 and NS5 regions were initially recovered by using a “genome walking” approach. However, all attempts to bridge the two fragments failed. The high-throughput sequencing of the total RNA from tick homogenates extended the previous NS3- and NS5-like sequences to 2,744 bp and 3,017 bp, respectively, which were subsequently confirmed by Sanger sequencing. Surprisingly, the coding regions of both fragments were flanked by UTRs, which suggests that the NS3- and NS5-like proteins are encoded by separate ORFs and that JMTV genome is not translated as a single polyprotein. In addition, the 3′ UTRs included a poly(A) tail, which again distinguished JMTV from other flaviviruses (as detailed later).
To search for potential “structural” genes of JMTV, we undertook extensive BLAST (basic local alignment search tool) and HHblits (iterative homology detection by representing both query and database sequences by profile hidden Markov models) screening of the nonredundant (nr) protein sequences database. Strikingly, no flavivirus-like structural proteins were detected with the use of either algorithm. Next, we took advantage of the unusually high expression levels of JMTV NS3- and NS5-like contigs (comp197 and comp194, respectively; Table 1). From the assembled high-throughput sequencing contigs, we extracted those with expression levels similar to these two contigs. After excluding ribosomal RNA sequences, there were two target contigs (Table 1): one (comp202) showed homology to an unannotated gene from a cDNA library of Toxocara canis (dog roundworm; ANT3.1), whereas the other (comp196) had no BLAST or HHblits hits to the nr protein database. In addition, a nucleotide blast search against the host R. microplus (tick) genome (ADMZ02) yielded no hits for all four contigs, suggesting that these sequences were not transcribed from the host genome. To exclude other possible origins of these sequences, we performed regular and nested PCRs on the total DNA extracts by using primers designed to flank different regions of these contigs. Again, the results were negative, confirming the exclusively RNA form of the four segments.
Table 1.
Homology search results and expression levels of JMTV genome segments in the context of total RNA
| Contig | Top BLAST and HHblits hits | Length, nt* | No. of mapped reads | Expression level** | Total reads, % | Non-rRNA reads, % |
| comp194 | ANT5, NS5 (TABV) | 3,072 | 980,359 | 15,956.36 | 4.44 | 18.67 |
| comp196 | — | 2,785 | 961,719 | 17,266.05 | 4.36 | 18.32 |
| comp197 | ANT34, NS3 (TABV) | 2,793 | 767,020 | 13,731.11 | 3.48 | 14.61 |
| comp202 | ANT3.1 | 2,751 | 459,874 | 8,358.31 | 2.08 | 8.76 |
| Total | 11,401 | 3,168,972 | 51,696.34 | 14.37 | 60.36 |
Length (measured in nt) of genomic RNAs determined after cyclization RT-PCR, excluding poly(A) tail.
Expression level is calculated by using length of mapped reads/length of contig.
However, the most compelling evidence that these sequences belong to the same virus are the conserved nucleotide stretches near the 5′ and 3′ termini of all four segments: GCAAGUGCA and GGCAAGUGC, respectively (SI Appendix, Fig. S3). Indeed, alignment of the four segments revealed that the 5′ and 3′ UTRs regions were especially conserved. These results also strongly imply that the JMTV genome is segmented. As further evidence for segmentation, we were able to experimentally achieve successful cyclization of each segment, which extends segment length to both termini including the poly(A) tail. The total length of the JMTV RNA genome was determined to be 11,401 nt.
Proteomics of JMTV.
The segments of JMTV genome were numbered from 1 through 4 following the order of their high-throughput sequencing contig numbers. Among these, segments 1, 2, and 3 are monocistronic, whereas segment 4 is bicistronic (Fig. 2). Segment 1 encodes nonstructural protein 1 (NSP1), which was an NS5-like protein with the conserved motifs for methyltransferase and RNA-dependent RNA polymerase of a typical flavivirus (SI Appendix, Fig. S4). The JMTV NSP1 protein was slightly longer than the flavivirus NS5 in that it contained a predicted N-terminal transmembrane region or a signal peptide, which may be cleaved by serine protease or cellular signal peptidase (SI Appendix, Fig. S4). Segment 3 encodes the nonstructural protein that resembled the NS2b–NS3 complex of flaviviruses (i.e., NSP2). In the NS3 part of the protein, the flaviviral serine protease domain and the helicase domain are well conserved, whereas, in the NS2b part of the protein, the predicted transmembrane regions aligns with the flavivirus NS2b protein (SI Appendix, Fig. S5). Furthermore, a potential serine protease cleavage site between NS2b and NS3 is predicted in NSP2. Collectively, these results suggest the functional unit of NS2b–NS3 is conserved in JMTV NSP2.
Fig. 2.

Schematic genome organization and putative proteomic maps of JMTV, showing predicted ORFs (blue boxes), transmembrane regions (yellow boxes), signal peptides (red box), and N-linked glycosylation sites (upward-pointing pink triangles).
The group of nonflavivirus proteins in JMTV comprises viral protein 1 (VP1), VP2, and VP3. VP1 is encoded by segment 2 and contained three predicted transmembrane regions: one at the N part of the protein and two others located near the C terminus (Fig. 2 and SI Appendix, Fig. S6A). A potential serine protease site is located 24 aa residues downstream of the N-terminal transmembrane region (SI Appendix, Fig. S6A). In addition, VP1 carries two potential N-glycosylation sites at positions 171 and 225, respectively. VP2 and VP3 are encoded by overlapping ORFs of segment 4 (Fig. 2). VP2 is a small protein with one predicted signal peptide (SI Appendix, Fig. S6B), whereas VP3 is largely a membrane protein with 11 predicted transmembrane domains (SI Appendix, Fig. S6C). Neither of the two proteins encoded by segment 4 contains potential N-glycosylation sites.
Evolutionary History of JMTV.
Although the NSP1 and NSP2 proteins are related to the flaviviral NS3 and NS5 proteins, respectively, the top BLAST hits (i.e., highest e-values) for these proteins were not from the Flaviviridae. Instead, remarkably, BLASTx and BLASTp found matches in the transcripts of a larval cDNA library of T. canis (Table 1) (8, 9). This was confirmed in the sequence comparisons (SI Appendix, Table S1). Strikingly, and similar to JMTV, it was previously noted that the library of T. canis contained four “abundantly expressed” RNAs whose DNA form was not found. We can now show that these transcripts have genetic organizations that show a strong resemblance to the four segments of JMTV (8, 9). Specifically, of the four RNAs in T. canis, three are monocistronic and one was bicistronic with separate coding frames for two proteins (9). At the proteomic level, despite the lack of BLAST and HHblits hits for VP1 and VP3, each of the five JMTV proteins had a counterpart in T. canis with clear sequence similarity (SI Appendix, Table S1) and structural resemblance (SI Appendix, Figs. S4–S6). Indeed, independent predictions of the organization of JMTV and T. canis proteins demonstrated very similar distributions of signal peptide and transmembrane domains in the matching proteins. Therefore, the four RNAs identified from T. canis larva most likely belong to a virus related to JMTV, which we have tentatively named T. canis larva agent (TCLA).
Next, we explored the evolutionary relationships among the flaviviral nonstructural proteins and their homologs in JMTV and TCLA. In the NS3 and NS5 phylogenies, JMTV and TCLA formed a monophyletic group nested within the diversity of the Flaviviridae (Fig. 3), which suggests that these viral segments had a single origin from the (unsegmented) Flaviviridae. Specifically, JMTV and TCLA cluster together and show a closer relationship to members of the genus Flavivirus than to the Hepacivirus and Pestivirus genera (Fig. 3 and SI Appendix, Table S1). Furthermore, in the NS5 phylogeny, JMTV and TCLA constituted the most divergent members of genus Flavivirus. Although the NS3 phylogeny showed a slightly different branching order with respect to TABV, the basal position of the JMTV–TCLA cluster was unchanged.
Fig. 3.

ML phylogenetic trees of (A) NS3 and (B) NS5 proteins of JMTV and TCLA within the context of representative flaviviruses. The trees are midpoint-rooted, and all horizontal branches are drawn to a scale of amino acid substitutions per site. The branch and taxon labels of the JMTV–TCLA clade are shaded red. For clarity, bootstrap values are only shown for internal nodes.
Prevalence of JMTV in Nature.
PCR-based surveillance studies revealed that JMTV is highly prevalent within ticks sampled from the Hubei province of China (Table 2). Of the eight tick species examined in this study, R. microplus (114 of 181), Haemaphysalis longicornis (50 of 91), and Haemaphysalis campanulata (18 of 24) had the highest positive rates, followed by Haemaphysalis flava (5 of 45). The remaining tick species—including Rhipicephalus sanguineus (1 of 3), Ixodide sinensis (1 of 1), and Ixodes granulatus (1 of 3)—all contained JMTV-positive samples. However, the sampling number was insufficient to infer viral prevalence in these species. In contrast to ticks, JMTV showed low prevalence in mosquitos (1 of 56). Remarkably, our immunofluorescence assay of cattle sera revealed reactivity in 54 of 145 samples from Hubei and Zhejiang, respectively, suggesting the presence of antibody against JMTV in the cattle blood (SI Appendix, Fig. S7). Additionally, the RT-PCR screening found JMTV RNAs in 13 seropositive samples, among which five contained the complete genome (i.e., all four segments). These sequences were closely related to the tick JMTVs sampled from the same geographic regions (SI Appendix, Fig. S8). Collectively, these data represent evidence for past and ongoing infections of JMTV in cattle.
Table 2.
Prevalence of JMTV in ticks, mosquitoes, and cattle
| Species | Location* | Mammalian host | No. of pools tested | No. of positive pools |
| R. microplus | 1–7 | Cattle | 181 | 114 |
| H. longicornis | 1 | Dog, goat, cattle | 91 | 50 |
| H. campanulata | 8 | Dog | 24 | 18 |
| H. flava | 7–9 | Hedgehog, badger | 45 | 5 |
| I. sinesis | 1 | Wild goat | 1 | 1 |
| I. granulatus | 10 | NA | 3 | 1 |
| R. sanguineus | 11 | NA | 3 | 1 |
| Haemaphysalis hystricis | 9 | Badger | 32 | 0 |
| Armigeres sp. (mosquito) | 1 | NK | 20 | 1 |
| Culex sp. (mosquito) | 1 | NK | 31 | 0 |
| Anopheles sp. (mosquito) | 1 | NK | 5 | 0 |
| Bos sp. (cattle) | 1, 6, 10 | NK | 145 | 5 |
| Total | — | — | 481 | 196 |
NA, not applicable; NK, not known.
Locations: 1, Wuhan; 2, Shiyan; 3, Shengnongjia; 4, Yichang; 5, Enshi; 6, Jingmen; 7, Xiangfan; 8, Xinzhou; 9, Huanggang; 10, Wenzhou; 11, Ningbo.
To explore the genetic diversity and evolution of JMTV in more detail, we performed a phylogenetic analysis of JMTV sampled from ticks in diverse geographical locations in China (SI Appendix, Table S2). Sequence comparisons based on NSP2 gene sequences revealed a remarkable diversity of JMTV circulating in ticks, with the most distant pair exhibiting only 92% nucleotide identity. R. microplus harbors the largest diversity for JMTV (SI Appendix, Fig. S9). Indeed, viruses sampled from other tick species and mosquitoes are nested within the diversity of R. microplus-associated JMTV, which suggests that R. microplus is a major reservoir host for JMTV. A more comprehensive analysis of host distribution on the JMTV phylogeny showed no significant clustering by host species [association index (AI) = 1.688, P = 0.79), indicating frequent transmission between different tick species. However, clustering is significant at the genus level (AI = 0.882, P = 0.003), such that the cross-species transmission of JMTV is not entirely unconstrained. Indeed, there are two clusters in the phylogeny that seemed to be mainly associated with Haemaphysalis but not Rhipicephalus genera ticks (SI Appendix, Fig. S9).
Geographically, JMTV has a wide distribution from central to eastern China. Because viruses were found wherever samples were taken, the true distribution is likely much wider. Geographic clustering was strongly supported in the phylogeny (AI = 3.179, P = 0.001). However, that the genetic similarity of these samples did not correlate with their geographic distance (Mantel test, r = 0.051, P = 0.191) implies that there frequent dispersals over long distances. Indeed, distance-independent dispersal can be directly observed in the phylogeny, in which sequences from Ningbo and Jingmen shared >99% sequence identity even though the two cities are hundreds of kilometers apart.
Reassortment in JMTV.
We examined whether reassortment had occurred in JMTV by sequencing all segments of depooled JMTV (SI Appendix, Table S3) and comparing their phylogenetic histories. All viral segments had comparable genetic diversity. Although they shared a similar backbone structure of three or four major lineages, several instances of reassortment were observed (Fig. 4). For example, in the segment 1 tree, samples 164, YS102-1, and X84-3 formed a sister clade to YJ3-3 and 85, whereas, in other segments, they are more closely related to samples 84, 10, and 204. Similarly, sample 85 also showed a variable grouping from SY84 (segment 3) to a cluster containing 164, YS102-1, X84-3, 84, 10, and 204 (segments 1, 2, and 4).
Fig. 4.
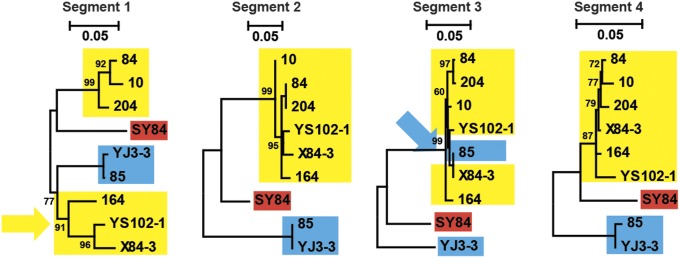
ML phylogenetic trees of the four genome segments obtained from nine depooled JMTV samples. The trees are midpoint-rooted, and all horizontal branches are drawn to a scale of nucleotide substitutions per site. Major clade/lineages are shaded with blue, yellow, and red, respectively. Phylogenetic incongruence, suggestive of reassortment, is indicated with colored arrowheads.
Discussion
We have described a segmented RNA virus that contains two genomic segments derived from an unsegmented flavivirus. The genome sequences of a distant relative of JMTV—i.e., TCLA—were first reported by Tetteh et al. (8) from T. canis and later extended by Callister et al. (9), although they were not regarded as of viral origin. Although those authors could not find evidence for the cellular origin of these RNAs, their data provided compelling evidence of a typical virus genome, including bicistronic RNAs and frame-shifting. Notably, TCLA represents, to our knowledge, the first evidence of a flavivirus-line organism in a member of the Nematoda. Recently, NS3 and NS5-like sequences were also identified in a salivary gland cDNA library of R. microplus (10). Short sequences available in GenBank show 97% and 100% amino acid similarity with JMTV NSP1 and NSP2, respectively, suggesting they belong to the same virus species. Although Maruyama et al. (10) collected evidence of virus replication in cell culture, their attempts to recover a flavivirus-like full genome were unsuccessful.
In addition to these partial proofs of the presence of a virus, we were able to view the JMTV particle by using EM and characterized all four of its genome segments. In the nonstructural proteins, the flavivirus-like NS2b–NS3 and NS5 domains are sufficient for the enzymatic activities required for RNA capping and genome replication. In the presumed structural proteins of JMTV, VP3 contains multiple transmembrane regions and is most likely the membrane protein, VP1 has the C-terminal transmembrane element and two glycosylation sites such that it resembles an envelope protein, and the short VP2 protein contains a signal peptide and hence is a good candidate for a capsid protein, although these will need to be verified with more data. In addition, JMTV has characteristics of a typical viral pattern of transmission: there is substantial viral genetic diversity within a given species of tick, an evolutionary history of frequent cross-species transmission events, and reassortment among the four genome segments. Furthermore, the lack of DNA form of the sequences suggests they are not part of a bacterial genome.
Based on the phylogenetic analyses, it is evident the JMTV-like viruses possess two segments that originated in the genus Flavivirus, although their genomes have been radically reorganized from the flavivirus prototype and, uniquely, have experienced fragmentation (as detailed later). One theory for the evolutionary advantage of segmentation is that it allows better gene regulation than unsegmented viruses (1), such that segments can be viewed as independent transcriptional units. Interestingly, in both JMTV and TCLA, the four genome segments are abundantly expressed, and it is possible that genome segmentation helps these viruses to efficiently produce high copy numbers. This hypothesis is supported by the observation that there are conserved runs of nucleotides in the UTRs of the four segments, which are likely gene regulatory components acquired after the genome segmentation, although this clearly requires further study. Alternatively, it has been proposed that smaller genomic molecules, as represented by segments, increase the stability of viral particles (3, 11). Although we were unable to test this theory on our data, it opens an important line of future research.
Crucially, not all JMTV proteins are flavivirus-like. Indeed, the putative structural proteins of JMTV and TCLA share no apparent sequence or structural homology with the envelope, membrane, or capsid proteins of flavivirus, nor to any other virus or cellular sequence. This could be a result of rapid evolution of these structural proteins, or because they are “orphan” proteins derived from an as yet uncharacterized virus. In light of these observations and the genome structure of JMTV-like viruses, we suggest that the stages in its genesis comprised coinfection of a flavivirus and a currently uncharacterized virus, followed by reassortment/repackaging to establish the genome structure described here. Interestingly, viral genesis involving the shuffling of nonstructural and structural components has previously been documented between an RNA and a (single-strand) DNA virus (12), indicating that large-scale genetic exchanges can occur between unrelated viruses. Also of importance is that, at some point, an ancestral flavivirus genome must have “fragmented” to produce the individual NS3- and NS5-like “segments” present in JMTV. This is obviously a highly unusual process, as flaviviruses are transcribed as a single mRNA and subgenomic RNAs are only generated from noncoding regions (13), although functional and defective capsid proteins of dengue virus have been observed to cocirculate in nature (14, 15). Unfortunately, determining exactly how the flavivirus genome formed the individual segments subsequently observed in JMTV is beyond the scope of this study.
It is also noteworthy that JMTV has a wide range of hosts, and that viral transmission among ticks appears to be efficient. Tick transmission most likely occurs through “cofeeding” as reported in tick-borne flaviviruses (16), and occurs when noninfected ticks and infected ticks feed at the same time on the same mammal. As different species of ticks tend to feed in clumps on the same cattle, viruses can also be transmitted from one species to another. Another important feature of JMTV epidemiology is the presence of long distance dispersals. As it is unlikely for ticks themselves to move over long distance, the most plausible route is human-mediated transportation of infected cattle and sheep, or the migration of wild animals (17, 18).
Finally, although JMTV is in part related to the flaviviruses, it possesses a very different genome organization. Future research on the biological properties of this virus is required for an in-depth understanding of its host interactions, as well as the precise evolutionary pathway by which two flavivirus proteins came to be resident within a highly divergent segmented virus.
Materials and Methods
Sample Preparation.
Between 2010 and 2011, 2,136 ticks were collected from various sites in Hubei and Zhejiang provinces in China (SI Appendix, Fig. S10). Most ticks were directly picked from infested wild and domestic animals, although a few were captured by using a tick drag-flag method. In some sampling locations, 672 mosquitoes were also trapped by using baited light traps. In addition, 145 cattle blood samples were obtained from local cattle farms or slaughterhouses before butchery. All samples were directly stored in liquid nitrogen upon capture and were later transferred to −80 °C for storage. Before RNA extraction, all samples were identified by trained field biologists and further confirmed by sequencing the mitochondrial cytochrome oxidase subunit I gene (19). They were placed in pools of 5–20 by species, sex, host, location, and stage of development. All pools were then homogenized with a mortar and pestle in 0.5 mL of PBS solution for RNA extraction and virus isolation.
Virus Discovery and Early Sanger Sequencing.
A pan-phlebovirus RT-PCR protocol was initially designed to examine the presence of Huaiyangshan virus in the tick samples (6, 7). The primer sets which were used to recover the initial fragments of NS3- and NS5-like genes are available upon request. The extension of the NS5 and NS3 genomic regions was performed by using an anchored PCR method, which included one specific primer designed from known fragments and a combination of nonspecific oligonucleotides. The resulting PCR products were cloned and sequenced with ABI-PRISM Dye Termination Sequencing kit on ABI 3730 genetic analyzer (Applied Biosystems).
Virus Isolation and EM.
An insect cell line C6/36 derived from Aedes albopictus and the canine macrophage cell line DH82 were used for virus isolation. Cells were cultured in Eagle minimum essential medium (MEM; HyClone) with 5% FBS (HyClone), 100 U/mL penicillin, 100 µg/mL streptomycin, and 2 mM l-glutamine (HyClone), and maintained at 28 °C and 5% CO2. A JMTV-positive tick homogenate (JMTV118) was filtered through a syringe-driven 0.22-μm filter and then diluted 10-fold before inoculation onto cell monolayer for 1 h. After inoculation, the cell monolayer was washed twice with 5 mL PBS solution and maintained in MEM supplemented with 2% FCS, 2 mM l-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin. Following the wash, cell cultures were incubated at the same conditions until 50% of cells were observed detached from the flask. For each passage, the cells and cell culture supernatant were harvested for RNA extraction, and the presence of JMTV in the cell culture was examined by using specific primers (available upon request) targeting at a conserved domain within each ORF of JMTV. The JMTV-positive pools were also subjected to intracerebral infection of suckling mice.
For EM, the JMTV-positive DH82 cells and supernatant were harvested 14 d after inoculation. The cells were washed three times with 1× PBS solution, trypsinized, and subjected to centrifuge at 1,000 × g for 5 min. The cell pellets were fixed with 2.5% (wt/vol) glutaraldehyde overnight at 4 °C and postfixed with 1% OsO4 in cacodylate buffer for 1 h at room temperature. Following dehydration with ethanol, the pellets were rinsed with propylene oxide for 30 min at room temperature and then embedded in resin for sectioning. Images of thin sections were observed under a transmission electron microscope (JEM 1230; JEOL). The supernatant was also harvested and subject to negative staining EM as described previously (20).
Immunofluorescence Assay.
An immunofluorescence assay was performed by using JMTV-infected DH82 cells. Cells were fixed with acetone for 30 min before being permeabilized with 0.1% Triton X-100 in PBS solution for 5 min. After rinsing with PBS solution, they were blocked with 5% horse serum in PBS solution for 30 min, and then washed with PBS solution for another 3 min. The washing steps were repeated three times. Subsequently, bovine sera (diluted 1:20 in PBS solution) were incubated with cells for 30 min and washed as described earlier. Reactions were detected with fluorescein isothiocyanate conjugated goat anti-bovine IgG (1:100; Boaosen).
Genome Sequencing.
Total DNA and RNA of the pool SY84 was extracted with AllPrep DNA/RNA mini kit (Qiagen) according to the manufacturer’s instructions. Total RNA was subjected to library preparation following a standard protocol provided by Illumina. Briefly, after an rRNA removal step, the rest of the RNA was fragmented, reverse-transcribed, adaptored, purified, and examined by the Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System. Single-end sequencing (50 bp) of the library was then performed on the HiSEq 2000 platform (Illumina). All library preparation and sequencing steps were handled by BGI Tech (Shenzhen, China). The resulting sequencing reads were cleaned and assembled de novo by the Trinity program (21) into 1,259 contigs (>200 bp). The assembled contigs were subsequently translated and aligned to the nr database (October 2013) by BLASTx. In addition, an HHblits search (22) against a clustered version of the nonredundant database from NCBI (nr20) was performed to identify more distant homologs of these proteins (23). For the BLASTx and HHblits searches, the e-value cutoff was set to 1.
The sequences of JMTV genomes were further confirmed by Sanger sequencing with primers designed based on deep-sequencing results. The 5′ and 3′ end of each JMTV genome fragment was determined by RNA circularization, followed by RT-PCR amplification across the ligated termini and sequencing of the cloned PCR products. All sequences generated here have been submitted to GenBank and assigned accession numbers KJ001579–KJ001582.
Sequence Analyses.
For each of the predicted JMTV protein sequences, we used the SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui/sosui_submit.html) and TMHMM (version 2.0; www.cbs.dtu.dk/services/TMHMM/) programs to predict the transmembrane helical segments, SignalP (version 4.0; www.cbs.dtu.dk/services/SignalP/) to determine signal sequences, and NetNGlyc (version 1.0; www.cbs.dtu.dk/services/NetNGlyc/) to identify N-linked glycosylation sites. These proteins were then aligned with their corresponding homologs by using MAFFT version 7 (24). Phylogenetic trees were inferred by using the maximum likelihood method (ML) implemented in PhyML version 3.0 (25), with a Subtree Pruning and Regrafting tree searching algorithm, a WAG substitution model with discrete gamma distributed rate variation among sites (Γ), and 1,000 bootstrap replicates. Phylogenies were also inferred by using the Bayesian method implemented in MrBayes version 3.2 (26), with the same amino acid substitution model as used in ML tree inference.
Prevalence of JMTV in Ticks, Mosquitoes, and Cattle.
RNA extracted from 193 pools of ticks, 56 pools of mosquitoes, and 145 cattle blood samples were screened for the presence of JMTV by using nested virus-specific primers. Gel electrophoresis was performed on second-round PCR products, and the bands with target size (360 bp) were then purified and sequenced with the PCR primers on an ABI 3730 genetic analyzer (Applied Biosystems). The sample was denoted as JMTV-positive if the sequencing results showed homology to our reference segment 1 sequence obtained from isolated virus. To avoid cross-contamination, all JMTV-positive RNA samples were screened for the presence of rest of the segments.
Because the initial short amplicons did not resolve the phylogenetic relationships, we obtained longer amplicons of segment 3 for RNA samples that are representative of host and geographic location. The subsequent phylogenetic trees were inferred using ML and Bayesian methods by using the time-reversible (GTR) model with discrete gamma distributed rate variation among sites (Γ) and a proportion of invariable sites (I). To determine the extent to which the viral population was structured by host species and by geography (i.e., place of sampling), we computed, for each trait, values for the AI statistic (27) by using Bayesian tip-significance testing version 1.0 (28). To account for phylogenetic uncertainty, the AI statistics were summarized over the posterior distribution of trees obtained by MrBayes. To characterize quantitative associations between virus genetic and geographical distance, we performed a Mantel test (29) by using the ecodist package implemented in R version 3.0.1 (30).
Reassortment Analyses of JMTV.
To compare the evolutionary histories of the four segments of JMTV, we obtained additional samples for RNA extraction. All samples were depooled (i.e., use single tick for RNA extraction) to avoid the case in which multiple JMTV strains are present within the same RNA pool. We subsequently screened for the presence of four segments by using primer sets flanking 1,266-bp, 1,120-bp, 1,107-bp, and 885-bp regions of segments 1–4, respectively. The resulting PCR products were then purified and sequenced. Finally, ML phylogenies were inferred (as described earlier) and compared among segments, with phylogenetic incongruence providing evidence for potential genomic reassortment events.
Supplementary Material
Acknowledgments
This study was supported by National Natural Science Foundation of China Grants 81290343 and 81273014 and by State Key Laboratory for Infectious Disease Prevention and Control Grants 2011SKLID101, 2011SKLID304, and 2012SKLID309. E.C.H. is supported by an Australian National Health and Medical Research Council Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. KJ001547–KJ001634).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1324194111/-/DCSupplemental.
References
- 1.Holmes EC. The Evolution and Emergence of RNA Viruses. New York: Oxford Univ Press; 2009. pp. 111–113. [Google Scholar]
- 2.García-Arriaza J, Manrubia SC, Toja M, Domingo E, Escarmís C. Evolutionary transition toward defective RNAs that are infectious by complementation. J Virol. 2004;78(21):11678–11685. doi: 10.1128/JVI.78.21.11678-11685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ojosnegros S, et al. Viral genome segmentation can result from a trade-off between genetic content and particle stability. PLoS Genet. 2011;7(3):e1001344. doi: 10.1371/journal.pgen.1001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson AD. Chapter 2. New insights into flavivirus nonstructural protein 5. Adv Virus Res. 2009;74:41–101. doi: 10.1016/S0065-3527(09)74002-3. [DOI] [PubMed] [Google Scholar]
- 5.Bollati M, et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antiviral Res. 2010;87(2):125–148. doi: 10.1016/j.antiviral.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang YZ, et al. The ecology, genetic diversity, and phylogeny of Huaiyangshan virus in China. J Virol. 2012;86(5):2864–2868. doi: 10.1128/JVI.06192-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang YZ, et al. Hemorrhagic fever caused by a novel tick-borne Bunyavirus in Huaiyangshan, China. Zhonghua Liu Xing Bing Xue Za Zhi. 2011;32(3):209–220. [PubMed] [Google Scholar]
- 8.Tetteh KK, Loukas A, Tripp C, Maizels RM. Identification of abundantly expressed novel and conserved genes from the infective larval stage of Toxocara canis by an expressed sequence tag strategy. Infect Immun. 1999;67(9):4771–4779. doi: 10.1128/iai.67.9.4771-4779.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Callister DM, Winter AD, Page AP, Maizels RM. Four abundant novel transcript genes from Toxocara canis with unrelated coding sequences share untranslated region tracts implicated in the control of gene expression. Mol Biochem Parasitol. 2008;162(1):60–70. doi: 10.1016/j.molbiopara.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Maruyama S, et al. Characterisation of divergent flavivirus NS3 and NS5 protein sequences detected in Rhipicephalus microplus ticks from Brazil. Mem Inst Oswaldo Cruz. 2013;108:1–13. doi: 10.1590/0074-0276130166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iranzo J, Manrubia SC. Evolutionary dynamics of genome segmentation in multipartite viruses. Proc Biol Sci. 2012;279(1743):3812–3819. doi: 10.1098/rspb.2012.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diemer GS, Stedman KM. A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol Direct. 2012;7(13):13. doi: 10.1186/1745-6150-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pijlman GP, et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe. 2008;4(6):579–591. doi: 10.1016/j.chom.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science. 2006;311(5758):236–238. doi: 10.1126/science.1115030. [DOI] [PubMed] [Google Scholar]
- 15.Ke R, Aaskov J, Holmes EC, Lloyd-Smith JO. Phylodynamic analysis of the emergence and epidemiological impact of transmissible defective dengue viruses. PLoS Pathog. 2013;9(2):e1003193. doi: 10.1371/journal.ppat.1003193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labuda M, et al. Non-viraemic transmission of tick-borne encephalitis virus: a mechanism for arbovirus survival in nature. Experientia. 1993;49(9):802–805. doi: 10.1007/BF01923553. [DOI] [PubMed] [Google Scholar]
- 17.Lin XD, et al. Migration of Norway rats resulted in the worldwide distribution of Seoul hantavirus today. J Virol. 2012;86(2):972–981. doi: 10.1128/JVI.00725-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weidmann M, et al. Molecular phylogeography of tick-borne encephalitis virus in central Europe. J Gen Virol. 2013;94(Pt 9):2129–2139. doi: 10.1099/vir.0.054478-0. [DOI] [PubMed] [Google Scholar]
- 19.Lu X, et al. Molecular survey of hard ticks in endemic areas of tick-borne diseases in China. Ticks Tick Borne Dis. 2013;4(4):288–296. doi: 10.1016/j.ttbdis.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Cook S, et al. Isolation of a novel species of flavivirus and a new strain of Culex flavivirus (Flaviviridae) from a natural mosquito population in Uganda. J Gen Virol. 2009;90(Pt 11):2669–2678. doi: 10.1099/vir.0.014183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remmert M, Biegert A, Hauser A, Söding J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9(2):173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 23.Kuchibhatla DB, et al. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J Virol. 2014;88(1):10–20. doi: 10.1128/JVI.02595-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 26.Ronquist F, et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang TH, Donaldson YK, Brettle RP, Bell JE, Simmonds P. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J Virol. 2001;75(23):11686–11699. doi: 10.1128/JVI.75.23.11686-11699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker J, Rambaut A, Pybus OG. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect Genet Evol. 2008;8(3):239–246. doi: 10.1016/j.meegid.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27(2):209–220. [PubMed] [Google Scholar]
- 30.Goslee SC, Urban DL. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw. 2007;22(7):1–19. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.