

Significance
DNA methylation is essential for mammalian development. This modification is nearly completely erased and reestablished in early embryos, but specific classes of DNA elements escape such genome-wide changes via unknown mechanisms. In this study, we identified a likely factor responsible for lack of DNA methylation turnover on a large fraction of such sequences. By focusing on mouse embryonic stem cells depleted of de novo DNA methyltransferases, which exhibit widespread hypomethylation with the exception of particular loci, we show that regions retaining DNA methylation are associated with a specific chromatin state. In cells lacking the enzyme catalyzing this chromatin state, such regions begin to lose DNA methylation. Our results therefore advance the understanding of how DNA methylation turnover is regulated during development.
Keywords: epigenomics, histone modifications, repetitive elements, 5-hydroxymethylcytosine
Abstract
During mammalian development, DNA methylation patterns need to be reset in primordial germ cells (PGCs) and preimplantation embryos. However, many LTR retrotransposons and imprinted genes are impervious to such global epigenetic reprogramming via hitherto undefined mechanisms. Here, we report that a subset of such genomic regions are resistant to widespread erasure of DNA methylation in mouse embryonic stem cells (mESCs) lacking the de novo DNA methyltransferases (Dnmts) Dnmt3a and Dnmt3b. Intriguingly, these loci are enriched for H3K9me3 in mESCs, implicating this mark in DNA methylation homeostasis. Indeed, deletion of the H3K9 methyltransferase SET domain bifurcated 1 (Setdb1) results in reduced H3K9me3 and DNA methylation levels at specific loci, concomitant with increased 5-hydroxymethylation (5hmC) and ten-eleven translocation 1 binding. Taken together, these data reveal that Setdb1 promotes the persistence of DNA methylation in mESCs, likely reflecting one mechanism by which DNA methylation is maintained at LTR retrotransposons and imprinted genes during developmental stages when DNA methylation is reprogrammed.
Cytosine methylation (5mC) in the context of CpG dinucleotides (mCG) plays a significant role in transcriptional repression of genes and repetitive elements (1, 2). In mammals, this modification is established by the de novo DNA methyltransferases (Dnmts), Dnmt3a and Dnmt3b, and maintained through replication predominantly by Dnmt1 (3). During development, the genome undergoes two widespread waves of DNA methylation reprogramming. The first wave begins shortly after fertilization, where the parental genomes are broadly demethylated. The embryonic genome remains hypomethylated until approximately the time of implantation, when Dnmt3a/3b act in concert to reestablish mCG patterns (4). The second wave begins early in germ cell development, preceding and coincident with migration of primordial germ cells (PGCs) into the gonadal ridge (5). Following widespread demethylation, germ cells undergo de novo methylation at later embryonic or postnatal stages in male and female gametogenesis, respectively (6). Deletion of the Dnmts in mouse models results in embryonic lethality (1, 7), revealing that reestablishment of DNA methylation patterns is critical for postgastrulation development.
The recent discovery and characterization of the ten eleven translocation (Tet) family of dioxygenases has shed light on the molecular basis of DNA demethylation during mammalian development (8). Tet1, Tet2, and Tet3 catalyze the conversion of 5mC to 5-hydroxylmethyl-cytosine (5hmC) or further oxidation products, which act as intermediate steps in passive and/or active DNA demethylation pathways (9, 10). These proteins function in fertilized zygotes, PGCs, early embryos, and mouse embryonic stem cells (mESCs) to induce demethylation (11–15).
Although widespread hypomethylation is a hallmark of preimplantation blastocysts and postmigratory PGCs, several classes of sequences are apparently resistant to 5mC removal (5, 14, 15). A subset of endogenous retroviruses (ERVs), including intracisternal A-particle (IAP) elements in particular, retain relatively high levels of DNA methylation at both developmental stages (5, 16–18). In preimplantation embryos and mESCs, differentially methylated regions (DMRs) of imprinted loci, as well as a small number of nonimprinted genic promoters, also maintain higher levels of 5mC (16, 19). However, whether a common mechanism underlies the apparent resistance of such loci to demethylation remains to be determined.
Methylation of histone H3 at lysine K9 (H3K9) functions in concert with DNA methylation to maintain silencing of genes and repetitive elements in distantly related organisms. In mammals, the relationship between these pathways is less clear, as H3K9 specific histone methyltransferases (HMTases) apparently function in concert with or independent of DNA methylation, depending on the target sequence (20, 21). Indeed, all five bona fide H3K9 specific HMTases, Suv39h1, Suv39h2, G9a, GLP, and Setdb1, have been reported to affect DNA methylation at specific loci (20, 22–24). Unlike the other K9-specific HMTases, Setdb1 has been shown to repress many ERVs independent of DNA methylation (20, 24). Curiously however, simultaneous depletion of Dnmt1 and Setdb1 results in synergistic reactivation of a subset of ERVs, including IAP elements (20), indicating that both H3K9 and DNA methylation play a role in silencing of such parasitic elements.
Whereas the global level of DNA methylation is dramatically reduced in late passage Dnmt3a/3b double knockout (DKO) ESCs, analysis of specific genomic loci clearly reveals that a subset of regions, including IAP elements, retains relatively high levels of mCG in these cells (7, 23), reminiscent of the pattern observed in postmigratory PGCs and blastocysts. To ascertain the spectrum of genomic regions that remain methylated in the absence of de novo Dnmt activity, we conducted methylC-sequencing (methylC-seq) on Dnmt3a/3b DKO mESCs. Thousands of regions with higher than average mCG levels, which we term enriched residual methylation loci (ERML), were identified. We further show that persistence of mCG at a subset of these loci, including ERVs, germ-line genes, and a subgroup of imprinted DMRs, is dependent upon Setdb1. Finally, we provide evidence that Setdb1-mediated H3K9me3 promotes the stability of mCG at ERML at least in part by inhibiting Tet-dependent oxidation in these regions.
Results
Characterization of Enriched Residual Methylation Loci in Dnmt3a/3b DKO mESCs.
To comprehensively map the genomic regions that remain hypermethylated in the absence of the de novo Dnmts, we analyzed the genome-wide distribution of DNA methylation in late passage DKO (passage 33) and WT (passage 39) parental mESCs by methylC-seq. Consistent with previous studies, we found that with the exception of the majority of CpG islands, WT mESCs have high levels of mCG throughout the genome (average methylation level of ∼70%) (25). In contrast, mCG levels were dramatically reduced in Dnmt3a/3b DKO cells (average mCG level of 2.7%; Fig. S1 A and B). Non-CG methylation, an epigenetic mark primarily found in embryonic stem cells and neuronal cells, was reduced to background levels, indicating that Dnmt3a and/or Dnmt3b are responsible for deposition of these modifications, as previously reported (26). Although the vast majority of the Dnmt3a/3b DKO genome (>90%) showed a dramatic reduction in methylation, many regions retain relatively high levels of mCG. Using a hidden Markov model (HMM)-based algorithm developed for this study (details in SI Materials and Methods), we identified 6,115 such regions consisting of multiple contiguous bins with greater than 10% mCG, which is considerably higher than the genome average in DKO cells. The average mCG level of all these loci was 38%, with a median size of 5.4 kb. Examples of ERMLs are shown in Fig. 1A and Fig. S1B.
Fig. 1.
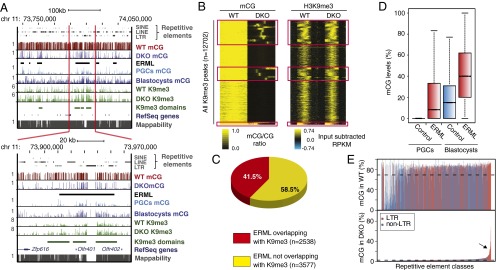
Genome-wide profile of mCG in wild-type and Dnmt3a,3b double knockout (DKO) mESCs. (A, Upper) University of California Santa Cruz (UCSC) genome browser screen capture showing the DNA methylation profiles for WT and DKO mESCs, mouse E13.5 male primordial germ cells (PGCs) (17), and blastocyst (16) across a region on chromosome 11. ERML are denoted with black bars. H3K9me3 ChIP-seq reads from WT and DKO are also included. H3K9me3-enriched domains in WT are called “ChromaBlocks.” Genes and repetitive elements as annotated by RefSeq and RepeatMasker, respectively, are shown. MethylC-seq reads from WT and DKO are mapped, with 100-bp binned data showing mCG ratios with bar heights between 0 and 1. H3K9me3 profiles are shown as reads per kilobase pair per million reads (RPKM). Mappability of reference genome from the ENCODE project for 50-bp segments is shown with the score between 0 and 1. (Lower) Screen capture of a narrowed segment within the same region as shown above. (B) Heat map generated by k-means clustering shows the H3K9me3 enrichment (Right) at all identified peaks and mCG (Left) in WT and DKO mESCs. Red boxes outline regions retaining mCG in the DKO cells, which we term ERML. These sites are enriched for strongest K9me3 peaks. (C) Pie chart summarizing the overlap between regions enriched for H3K9me3 in both WT and DKO mESCs and ERML. (D) Boxplot showing the difference in mCG levels at ERML and control shuffled regions in PGCs and blastocysts. (E) Barplot of mCG ratios of mappable LTR (red) and non-LTR (blue) repeats in WT (Upper) and DKO (Lower) mESCs. Arrow indicates families that retain higher levels of mCG.
Given our previous observations that specific genes and LTR retrotransposons are regulated by both H3K9me3 and DNA methylation, we analyzed the enrichment of H3K9me3 by ChIP-seq in WT and Dnmt3a/3b DKO cells. Consistent with our previous analysis of Dnmt TKO mESCs (20), we observed that H3K9me3 is generally preserved in DKO mESCs (Fig. 1B). Interestingly, many H3K9me3 peaks showed enrichment of mCG in the DKO cells, suggesting a possible relationship between ERML and this histone mark (Fig. 1B). Indeed, we found that a large proportion of ERML (41.5%, n = 2,538) overlap with H3K9me3-marked regions (Fig. 1 B and C). This is considerably higher than the overlap between H3K9me3 peaks and control shuffled regions (8.2%, n = 543) (Fig. S1C), generated by randomizing positions of ERML while maintaining the average region size. For subsequent analyses, we focused on the subset of ERML (n = 2451) found within the main clusters, generated by k-means clustering, which overlap with H3K9me3 (Fig.1B, red boxes).
Previous studies indicate that DNA methylation levels are substantially higher in mESCs than in blastocytes from which they are derived (16). Strikingly, we found that the ERML identified in DKO mESCs also show relatively higher levels of mCG in WT blastocysts (16) (Fig. 1D). Furthermore, we observed a similar retention of mCG in WT male embryonic day 13.5 (E13.5) PGCs at ERML compared with other genomic regions (17) (Fig. 1D). In fact, we observed a high co-occurrence of ERML and hypermethylated regions in both PGCs and blastocysts across large regions of the genome (Fig. 1A and Fig. S1D). Taken together, these data indicate that residual methylation in blastocysts, PGCs, and Dnmt3a/3b DKO mESCs may be regulated by a common H3K9me3-dependent pathway that promotes maintenance of DNA methylation and/or protects against DNA demethylation rather than promoting de novo DNA methylation per se.
To determine whether ERML are enriched for specific genomic features, we characterized these regions in greater detail. Interestingly, ERML are enriched for repetitive elements (Table S1). To address whether specific repeat subclasses are enriched, we calculated the average mCG levels of all mappable repeat classes in the Dnmt3a/3b DKO versus the WT parental line. Intriguingly, specific subsets of ERVs and their long terminal repeats (LTRs) retained significantly higher mCG levels than other repeat classes (Fig. 1E). We then compared the proportion of repeat subfamilies overlapping with ERML versus the percentage of base pairs of the same subfamilies overlapping with ERML (Fig. S1 E and F). This analysis allowed us to discriminate between specific repeat subfamilies showing substantial overlap with ERML and those found by chance to be near or minimally overlapping with ERML due to their high copy numbers in the genome. Repeat subfamilies showing the highest degree of overlap with ERML belong to class I or class II ERVs, including IAP, early mouse transposons (ETn), and Mus musculus ERV using tRNALys type 10 C (MMERVK10C) elements (Fig. S1 E and F and Table S1). These ERVs do not show significant overlap with shuffled negative control regions (Fig. S1G). We previously showed that MMERVK10C and ETn subfamilies are induced to a greater extent in Setdb1 KO cells than in Dnmt TKO mESCs, whereas IAP elements are only modestly induced in both KO lines (20, 24). Intriguingly however, simultaneous depletion of Setdb1 and Dnmt1 yielded synergistic activation of IAP ERVs (20), consistent with the hypothesis that Setdb1 and DNA methylation target the same ERV subfamilies.
Setdb1 Deletion Leads to Hypomethylation at a Subset of ERVs and ERML.
Given that ERML are enriched for H3K9me3 and Setdb1-regulated ERV subfamilies (20, 24), we next addressed whether this HMTase is required to maintain mCG at ERML. As Setdb1-deficient mESCs are not viable, we used a previously described Setdb1 conditional knockout (CKO) cell line (24). Efficient depletion of Setdb1 was validated by Western blot (Fig. S2A). Analysis of the effects of Setdb1 deletion on H3K9me3 by ChIP-seq in predeletion (hereinafter referred to as WT) and postdeletion (Setdb1 KO) cells revealed a reduction at virtually all mappable H3K9me3 peaks (Fig. S2B) and especially at ERML marked with H3K9me3 (Fig. S2B), consistent with previously published data (20) (Fig. S2C). We then analyzed the methylomes of WT and Setdb1 KO cells by methylC-seq at day 6 postinduction with 4-hydroxytamoxifen (Fig. S3A). Global DNA methylation levels were not dramatically reduced upon Setdb1 KO, perhaps due to the limited number of cell divisions before harvest (24). Unfortunately, Setdb1 KO cells survive for only 7 d (24), precluding analysis at later time points. Nevertheless, we sought to identify specific regions where mCG is altered using Fisher’s exact test to calculate the significance of mCG differences across the genome between WT and Setdb1 KO (SI Materials and Methods). We found 2,395 mappable repetitive elements that showed significant hypomethylation. Strikingly, compared with all mappable repeats in the genome, LTR retrotransposons are significantly overrepresented among the hypomethylated elements identified in the Setdb1 KO cells (∼4.3-fold increase or 17% versus 73%) (Fig. 2A). Furthermore, compared with all mappable ERVs, class I and II ERVs in particular were overrepresented in Setdb1 KO cells, showing a ∼2.6-fold (7% versus 18%) and ∼2.5-fold increase (29% versus 72%), respectively (Fig. 2B). In contrast, class III ERVs, which are not marked by H3K9me3 (20), are underrepresented (64% versus 10%) (Fig. 2B). It is noteworthy that some regions are also hypermethylated in Setdb1 KO cells (Fig. S3 B–D). As the majority of these are non-LTR elements (Fig. S3C) or genes (Fig. S3D), which are not marked by K9me3 in WT mESCs (Fig. S3D) (27), we focused our subsequent analyses on the hypomethylated ERVs shown previously to be transcriptionally silenced by Setdb1, including IAP, ETn, RLTR4/murine leukemia virus (MLV), and MMERVK10C (20, 24) (Table S2). Bisulfite Sanger sequencing confirmed hypomethylation of MLV LTR regions upon Setdb1 deletion (Fig. S3E).
Fig. 2.

Subsets of endogenous retroviruses and ERML show significant mCG reduction in Setdb1 KO mESCs. (A) Pie charts demonstrating the distribution of repetitive element classes among all mappable repeats (n = 4,989,421) in the mouse mm9 reference genome [short interspersed nuclear elements (SINEs) = 30.5%, long interspersed nuclear elements (LINEs) = 19.4%, simple repeats = 21.3%, LTR = 17%, DNA transposons = 3.1%, and others = 8.4%] (Left) and significantly hypomethylated elements (n = 2,395) in Setdb1 KO mESCs (SINE = 14.7%, LINE = 8.4%, simple repeats = 1.6%, LTR = 73%, DNA transposons = 0.5%, and others = 1.7%) (Right). (B) Pie charts demonstrating the distribution of the three ERV classes among all mappable ERVs (n = 849,327) (Left) and significantly hypomethylated elements (n = 1,748) in Setdb1 KO mESCs. (C) Screen capture illustrating the overlap of ERML and an IAP-EY endogenous retroviral element. MethlyC-seq reads from WT (predeletion) and Setdb1 KO mESCs are mapped and divided into 100-bp bins, showing mCG ratios with bar heights between 0 and 1. Log P values (Fisher’s exact test) are shown for bins with significant difference in mCG ratio between WT and Setdb1 KO (P value cutoff of 0.001 or log = −3). RNA-seq (20) confirms substantial reactivation of this element in Setdb1 KO mESCs. Mappability or uniqueness of reference genome from the ENCODE project for 50-bp segments is shown with the score between 0 and 1.
We also observed that a subset of hypomethylated ERVs in Setdb1 KO mESCs overlapped with ERML defined in Dnmt3a/3b DKO cells (Fig. 2C). Transcriptional repression of ERV subfamilies, including ETn and IAP subfamily Ez (IAP-Ez) in particular, involves both DNA methylation and Setdb1-deposited H3K9me3 (20). At these elements, H3K9me3 may promote transcriptional silencing in part by facilitating DNA methylation. For example, methylation of specific CpGs within an IAP subfamily EY (IAP-EY) embedded within an ERML on chromosome 18 is clearly reduced in the Setdb1 KO, concomitant with a loss of H3K9me3 and transcriptional reactivation (Fig. 2C). Extending our analyses, we found a significantly higher overlap between H3K9me3-marked regions that exhibit hypomethylation in Setdb1 KO cells and ERML (as described in Fig. 1B, n = 2,451) than at shuffled control regions (n = 2,451) (Fig. S3F). Taken together, these data indicate that Setdb1 plays a role in maintaining mCG at specific sequences, including ERVs, marked with H3K9me3 in WT ESCs.
Setdb1 Antagonizes 5-Hydroxymethylcytosine-Mediated DNA Demethylation.
To determine whether Setdb1/H3K9me3 promotes the persistence of DNA methylation at least in part by inhibiting DNA demethylation, we studied the genome-wide distribution of 5hmC, an intermediate product of TET protein-dependent DNA demethylation pathways (9, 10). Using a method that selectively labels 5hmC for affinity capture, we produced genome-wide maps for 5hmC enrichment in WT and Setdb1 KO mESCs at day 6 postdeletion. Intriguingly, the mean 5hmC enrichment level was increased at ERML, showing a significant decrease in methylation in Setdb1 KO mESCs (n = 288) (Fig. 3A). Expanding our analyses to include all ERML that overlap with H3K9me3 (n = 2,451) in WT versus Setdb1 KO mESCs, we found a significantly greater number of loci showing a reduction of H3K9me3 upon Setdb1 deletion, coupled with an increase of 5hmC than at shuffled control regions (Fig. 3B and Fig. S4A). Furthermore, the genomic loci that showed the greatest increase of 5hmC (n = 169) were highly enriched for ERVs, including IAP and ETn elements (Fig. S4B). Analysis of the median levels of 5hmC enrichment revealed a clear increase at mappable ETn (n = 11,207) and IAP (n = 22,962) repeats (Fig. 3A). We validated the 5hmC increase observed in the ETn/Mus musculus type D retrovirus subfamily by hydroxymethylated DNA immunoprecipitation followed by quantitative PCR analyses (Fig. S4C). An example of a specific IAP-Ez element overlapping with an ERML and exhibiting a decrease of mCG concomitant with an increase of 5hmC upon Setdb1 deletion is shown in Fig. S5A. Importantly, no increase in 5hmC was observed at class III ERV loci (ERVL) elements, which are generally neither regulated by Setdb1 nor marked by H3K9me3 (20, 27). In fact, we detected a modest decrease in 5hmC enrichment at these ERVs (Fig. 3A).
Fig. 3.

ERML gain 5hmC in Setdb1 KO mESCs. (A) Boxplot showing an increase of 5hmC enrichment (RPKM) at ERML, which become hypomethylated upon Setdb1 depletion (n = 288). An increase is also observed at mappable ETn (n = 11,207) and IAP elements (n = 22,962). Mappable ERVL elements (n = 9,704), which are not generally marked by Setdb1-deposited K9me3, show a subtle decrease of 5hmC enrichment in Setdb1 KO mESCs. (B) Heat maps showing the difference of K9me3 and 5hmC enrichment (RPKM) between Setdb1 KO and WT mESCs at ERML marked by K9me3 (n = 2,451) (Left) and size-matched shuffled control regions (Right). (C) Boxplots illustrate the increase of Tet1 enrichment (RPKM) at ERML marked by K9me3 (n = 2,451) but not at control regions (n = 2,451), in Setdb1 KO mESCs. (*P < 0.05 and ***P < 0.001, respectively, as comparing WT versus Setdb1 KO with the Wilcoxon test.)
To determine whether Setdb1 protects against 5mC oxidation by antagonizing the binding of Tet1, we conducted ChIP-seq using a Tet1-specific ab. The enrichment patterns in WT mESCs were similar to previously published datasets (28) (Fig. S5B). Consistent with the increase in 5hmC levels at ERML, we detected a significant increase in Tet1 binding at these sites in Setdb1 KO mESCs (Fig. 3C and Fig. S5A). Taken together, these data reveal a previously unrecognized crosstalk between these epigenetic pathways, where Setdb1/H3K9me3 protects against Tet1/5hmC-associated DNA demethylation at many LTR elements.
Setdb1 Protects Against Demethylation of Germ-Line and Imprinted Genes.
We next investigated whether Setdb1/H3K9me3 plays a similar role in preserving DNA methylation at unique genic sequences. We analyzed the methylomes generated for WT and Setdb1 KO mESCs and found that 252 genes showed a significant loss of DNA methylation at their promoters. Gene ontology analysis revealed several classifications associated with meiosis (Fig. 4A). Interestingly, we previously showed that 30 germ-line–specific genes were transcriptionally repressed by both DNA methylation and Setdb1/H3K9me3 (20). Strikingly, there was a significant decrease in the median mCG level at these germ-line gene promoters in Setdb1 depleted cells, whereas no decrease in mCG levels was detected at the promoter regions of all Reference Sequence (RefSeq) annotated genes, which have a significantly lower median mCG methylation level in WT cells (Fig. 4B). This reduction in DNA methylation was clearly apparent at the promoter regions of the germ-line–specific genes Sycp3, Dazl, Mael, and Rhox13 (Fig. 4C and Fig. S6 A and B), and hypomethylation of the promoter region of several of these genes was validated by bisulfite Sanger sequencing and combined bisulfite restriction analysis (COBRA) (Figs. S6A, S7, and S8). Thus, Setdb1 is apparently also required for the persistence of DNA methylation at a subset of H3K9me3-marked germ-line–specific gene promoters.
Fig. 4.

Germ-line–specific and imprinted genes show reduction of mCG and/or increase in 5hmC. (A) Analysis of DNA hypomethylated regions in Setdb1 KO mESCs yielded three of seven Gene Ontology terms relating to meiosis. The values plotted are transformed P value [−log10(P value)] of each term. (B) Boxplot demonstrating a significant difference between the median mCG ratios at the promoters of 30 germline genes, which are up-regulated in both Setdb1 KO and Dnmt TKO mESCs, as previously shown (20) (Left) and all RefSeq genes (Right). (C) The mCG ratios (Left) of four germ-line–specific genes are significantly reduced upon Setdb1 deletion, concomitant with transcriptional up-regulation (Right). (D) Significant reduction of mCG was also observed at the differentially methylated regions of four of the five analyzed imprinted genes (Left). Four of the five genes also show dramatic increase in 5hmC (Right). (*P < 0.05 and ***P < 0.001, respectively, as comparing WT versus Setdb1 KO with the Wilcoxon test).
It is worth noting that although the loss of mCG is incomplete, it appears sufficient to perturb transcriptional repression. This observation is consistent with a recent study showing that, whereas the Dazl promoter is gradually demethylated during DNA methylation reprogramming in PGCs, the gene is induced at the initial onset of promoter hypomethylation (14). Surprisingly, no increase in 5hmC was detected at these loci at day 6 postdeletion. The kinetics of 5mC removal may vary between loci or between cells and these germ-line genes may be prone to relatively rapid DNA demethylation via hydroxylation, due to preloading of TET proteins. Indeed the Dazl promoter is highly enriched for Tet1 in WT mESCs (13). Further studies are needed to address this issue. Nevertheless, the promoters of these germ-line–specific genes clearly show a reduction in mCG and K9me3 upon Setdb1 depletion, coupled with increased expression (Fig. S6B).
A recent study revealed that H3K9me3 at differentially methylated regions (DMRs) of imprinted genes is dependent upon Kap1, which interacts directly with Setdb1 (29, 30). Consistent with this observation, we also found that H3K9me3 at a subset of DMRs is dependent upon Setdb1 (Fig. S6C). To address whether maintenance of DNA methylation at DMRs is also dependent on Setdb1 in mESCs, we focused on a subset of DMRs marked by H3K9me3, including those for both paternally and maternally imprinted genes. Consistent with previous reports, we found that the methylation level of DMRs in WT cells were not necessarily maintained at the expected 50%, possibly due to culture conditions of mESCs. Regardless, four of the five analyzed DMRs (Meg3, Nespas, Mest, and Peg3) showed a significant decrease of mCG levels, whereas H19 showed no significant change (Fig. 4D and Fig. S4C). Interestingly, four of the five DMRs (Meg3, Nespas, Mest, and H19) also showed a significant increase of 5hmC enrichment (Fig. 4D and Fig. S6C), indicating that Setdb1 may also play a role in protecting imprinted DMRs against 5hmC-dependent demethylation.
Discussion
Previous studies of Dnmt3a/3b DKO mESCs identified a few regions in the genome, including IAP elements and several imprinted DMRs, that retain near WT levels of DNA methylation (7, 23), presumably maintained by Dnmt1. Here, we conducted a genome-wide search for hypermethylated regions in these cells. Remarkably, many such ERML correspond to genomic regions that retain high mCG levels in PGCs and blastocysts. A significant fraction of ERML are marked by H3K9me3 in mESCs and are enriched for active class I and II ERVs, parasitic sequences with the potential to deregulate nearby genes and transpose to new genomic sites. Setdb1-deposited H3K9me3 is necessary for silencing of several such ERV subfamilies, including IAP, ETn, and MMERVK10C (20, 24). The overlap between ERML and H3K9me3-marked loci prompted us to investigate the relationship between these epigenetic pathways. Upon depletion of Setdb1 in mESCs, H3K9me3 and DNA methylation are concomitantly reduced at a subset of ERML, which simultaneously gain 5hmC and Tet1 binding. Given that oxidation of 5mC has been shown to act as an intermediate in DNA demethylation (9, 10), these results reveal an antagonistic relationship between Setdb1/K9me3 and 5mC removal. It is noteworthy that Setdb1 KO cells also show DNA hypermethylation at non-LTR retrotransposons and at a subset of single copy genes. However, as these sequences are not generally marked or transcriptionally regulated by H3K9me3, we focused our analyses on the sequences that lose DNA methylation upon Setdb1 depletion.
Reprogramming of DNA methylation patterns in PGCs was recently shown to be a gradual process involving sequential Tet-independent and Tet-dependent pathways. Whereas the reactivation of genes such as Dazl begins concomitantly with the onset of DNA demethylation, the mCG level decreases gradually over the next several days until the Dazl promoter is completely unmethylated (14). This temporal requirement for hydroxymethylation-dependent DNA methylation turnover likely reflects replication-coupled loss of mCG and may also apply in mESCs. The incomplete demethylation of ERML in Setdb1 null cells could result from a lack of sufficient cell divisions to detect replication-coupled loss of mCG. However, as Setdb1 null ESCs are not viable for more than 7 d postdeletion, it is not possible to determine whether DNA methylation is progressively lost in the absence of H3K9me3.
Interestingly, Uhrf1/Np95, which recruits Dnmt1 to replicating DNA and is essential for maintenance DNA methylation, was recently shown to bind cooperatively to hemimethylated CpG and H3K9me2/me3 (31, 32). Moreover, the interaction with methylated H3K9 promotes fidelitous maintenance DNA methylation (31). These observations indicate that in addition to antagonizing demethylation, Setdb1/K9me3 may also facilitate Dnmt1-dependent DNA methylation.
In the early zygote, H3K9me2 deposited by G9a, another H3K9 HMTase, on the maternal genome is recognized by PGC7, which in turn blocks Tet3 binding (33). The resulting conversion of 5mC to 5hmC exclusively in the paternal genome explains the asymmetric demethylation observed. We propose that in mESCs, Setdb1/H3K9me3 functions in a similar manner to protect against Tet-dependent DNA demethylation and perhaps to promote Dnmt1 activity at specific sites, such as LTR retrotransposons and imprinted DMRs, where loss of DNA methylation is detrimental. Whether G9a plays a similar role in mESCs has not been investigated in detail. Compared with Setdb1 depleted cells, G9a KO mESCs exhibit distinct phenotypes, in that dramatic loss of H3K9me2 and DNA methylation is observed at both genes and repetitive elements, with no detectable effect on H3K9me3 (23). As DNA hypomethylation appears to be independent of its catalytic activity (23), G9a may function in a manner distinct from Setdb1. It remains to be determined whether ERML not marked by H3K9me3 are protected through a G9a-mediated mechanism.
Analysis of published blastocyst and PGC methylC-seq data (16, 17) revealed that although many loci are hypomethylated compared with mESCs, ERML on average retain higher levels of mCG in these cells. This correlation indicates that H3K9me3 may also protect such regions against Tet-mediated oxidation of mCG and in turn DNA demethylation early in embryogenesis and in germ cell development, when global DNA methylation reprogramming occurs.
Our data reveal that H3K9me3 plays a role in preserving DNA methylation at many ERV subfamilies in mESCs and likely in preimplantation embryos. However, little proviral reactivation is observed in Dnmt TKO mESCs, which are devoid of 5mC (20, 24). This begs the question of why maintenance of this mark is necessary. Protection against DNA demethylation in preimplantation embryos may serve the purpose of preserving this epigenetic mark for subsequent developmental stages. Indeed, although IAP reactivation is negligible in Dnmt1 KO mESCs despite dramatic hypomethylation, these elements are up-regulated over 100-fold when differentiated in culture (34). Conversely, Setdb1 deletion does not lead to proviral derepression in mouse embryonic fibroblasts (24). Taken together, these observations indicate that in blastocysts and perhaps PGCs, preserving DNA methylation at specific genomic regions, including LTR retrotransposons, may be critical because this epigenetic mark assumes a primary role in repressing these parasitic sequences in differentiated tissues.
In summary, we show that in mESCs, Setdb1-deposited H3K9me3 functions to block the removal of 5mC at loci where preservation of DNA methylation may be essential, at least in part by antagonizing hydroxylation-dependent demethylation. These findings reveal a previously unrecognized dependence of DNA methylation homeostasis on H3K9 trimethylation and provide insight into the molecular mechanism by which specific genomic regions escape DNA methylation turnover during global reprogramming in the mouse.
Materials and Methods
ChIP-seq, RNA-seq, and methylC-seq were conducted as previously described. Detailed experimental protocols can be found in SI Materials and Methods. Sequences and applications of all primers used in this study are provided in Table S3. Methods applied to analysis of methylC-seq, ChIP-seq, and RNA-seq data are described in detail in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. Yoichi Shinkai for sharing the Setdb1 CKO cell line, Dr. Guoping Fan for sharing the Dnmt3a/3b DKO cells, and members of the B.R. and M.C.L. laboratories for helpful comments during preparation of the manuscript. This work was supported by the National Institutes of Health Epigenome Roadmap Project (U01 ES017166), California Institute for Regenerative Medicine RN2-00905-1 (to B.R.), and the Canadian Institutes of Health Research Grant 77805 (to M.C.L.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE47894).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322273111/-/DCSupplemental.
References
- 1.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20(2):116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 3.Lei H, et al. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122(10):3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 4.Santos F, Dean W. Epigenetic reprogramming during early development in mammals. Reproduction. 2004;127(6):643–651. doi: 10.1530/rep.1.00221. [DOI] [PubMed] [Google Scholar]
- 5.Hajkova P, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117(1-2):15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki H, Matsui Y. Epigenetic events in mammalian germ-cell development: Reprogramming and beyond. Nat Rev Genet. 2008;9(2):129–140. doi: 10.1038/nrg2295. [DOI] [PubMed] [Google Scholar]
- 7.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 8.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333(6047):1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334(6053):194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iqbal K, Jin SG, Pfeifer GP, Szabó PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci USA. 2011;108(9):3642–3647. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koh KP, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8(2):200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams K, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347):343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hackett JA, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339(6118):448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vincent JJ, et al. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12(4):470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi H, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8(1):e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi H, et al. High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res. 2013;23(4):616–627. doi: 10.1101/gr.148023.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guibert S, Forné T, Weber M. Global profiling of DNA methylation erasure in mouse primordial germ cells. Genome Res. 2012;22(4):633–641. doi: 10.1101/gr.130997.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borgel J, et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet. 2010;42(12):1093–1100. doi: 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 20.Karimi MM, et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8(6):676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, et al. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281(28):19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- 22.Lehnertz B, et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol. 2003;13(14):1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 23.Dong KB, et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 2008;27(20):2691–2701. doi: 10.1038/emboj.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsui T, et al. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464(7290):927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- 25.Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 26.Arand J, et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012;8(6):e1002750. doi: 10.1371/journal.pgen.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu H, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473(7347):389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delaval K, et al. Differential histone modifications mark mouse imprinting control regions during spermatogenesis. EMBO J. 2007;26(3):720–729. doi: 10.1038/sj.emboj.7601513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quenneville S, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44(3):361–372. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun. 2013;4:1563. doi: 10.1038/ncomms2562. [DOI] [PubMed] [Google Scholar]
- 32.Cheng J, et al. Structural insight into coordinated recognition of trimethylated histone H3 lysine 9 (H3K9me3) by the plant homeodomain (PHD) and tandem tudor domain (TTD) of UHRF1 (ubiquitin-like, containing PHD and RING finger domains, 1) protein. J Biol Chem. 2013;288(2):1329–1339. doi: 10.1074/jbc.M112.415398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura T, et al. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486(7403):415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 34.Hutnick LK, Huang X, Loo TC, Ma Z, Fan G. Repression of retrotransposal elements in mouse embryonic stem cells is primarily mediated by a DNA methylation-independent mechanism. J Biol Chem. 2010;285(27):21082–21091. doi: 10.1074/jbc.M110.125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.