

Significance
The protective molecular chaperone protein heat shock protein 90 (HSP90) is required for the activity and stability of its client proteins, which play diverse roles in both normal cell homeostasis and diseases such as cancer. Pharmacologic inhibition of HSP90 leads to the deactivation and degradation of these clients. Here we report that depleting levels of the degradative enzyme Cullin-5 (CUL5) reduces both client breakdown and loss of signaling output normally observed upon HSP90 inhibitor treatment for five important oncogenic protein kinase clients in four different cancer types. Depleting CUL5 also reduces the sensitivity of a variety of human cancers to HSP90 inhibitors. Our results provide important translational as well as mechanistic insights into the molecular pharmacology of HSP90 inhibitors that are currently undergoing clinical trials.
Keywords: protein quality control, protein degradation, ERBB2, BRAF, oncoprotein
Abstract
The molecular chaperone heat shock protein 90 (HSP90) is required for the activity and stability of its client proteins. Pharmacologic inhibition of HSP90 leads to the ubiquitin-mediated degradation of clients, particularly activated or mutant oncogenic protein kinases. Client ubiquitination occurs via the action of one or more E3 ubiquitin ligases. We sought to identify the role of Cullin-RING family E3 ubiquitin ligases in the cellular response to HSP90 inhibition. Through a focused siRNA screen of 28 Cullin-RING ligase family members, we found that CUL5 and RBX2 were required for degradation of several HSP90 clients upon treatment of human cancer cells with the clinical HSP90 inhibitor 17-AAG. Surprisingly, silencing Cullin-5 (CUL5) also delayed the earlier loss of HSP90 client protein activity at the same time as delaying cochaperone dissociation from inhibited HSP90–client complexes. Expression of a dominant-negative CUL5 showed that NEDD8 conjugation of CUL5 is required for client degradation but not for loss of client activity or recruitment of clients and HSP90 to CUL5. Silencing CUL5 reduced cellular sensitivity to three distinct HSP90 inhibitors, across four cancer types driven by different protein kinases. Our results reveal the importance of CUL5 in multiple aspects of the cellular response to HSP90 inhibition.
Heat shock protein 90 (HSP90) is a molecular chaperone that facilitates the stabilization and activation of around 350 client proteins (see www.picard.ch/downloads/Hsp90interactors.pdf for an updated client protein list) (1). As well as being involved in a wide range of normal cellular processes, many HSP90 clients are oncogenic kinases that are hyperactivated by mutation or amplified/overexpressed in malignancies (2, 3). HSP90-mediated activation and stabilization of client proteins requires an ATP-driven chaperone cycle regulated by a number of cochaperones (4, 5). Pharmacologic inhibition of HSP90 disrupts this cycle and leads to the ubiquitin-mediated proteasomal degradation of client proteins (3, 6, 7).
The proposed model is that clients are ubiquitinated and thus targeted to the proteasome by the action of one or more E3 ubiquitin ligases (8). Some evidence suggests that the U box-containing ligase CHIP is involved in the degradation of certain HSP90 clients (9, 10). However, the stability of protein kinase clients such as ERBB2 is not increased in CHIP−/− cells treated with the first-in-class pharmacologic HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin [17-AAG, tanespimycin (11)] (10), suggesting that other E3 ubiquitin ligases are also involved.
One study showed that the Cullin-RING ligase Cullin-5 (CUL5) is recruited to HSP90-containing complexes and is involved in the ubiquitination and degradation of the client ERBB2 following HSP90 inhibition (12). Cullin-RING ligases function as modular, multisubunit complexes that consist of a Cullin scaffold, a RING-H2 finger protein, a substrate-recognition subunit, and, in most cases, an adaptor that links the Cullin to the substrate recognition subunit (13, 14). In the case of CUL5, functional complexes consist of RBX2, Elongin-B, Elongin-C, and a SOCS-containing substrate receptor.
Given the link between CUL5 and the HSP90 inhibitor-induced degradation of ERBB2 (12), we have investigated the role of Cullin-RING ligases with respect to HSP90’s protein kinase clients in human cancer cell lines. Our initial focused siRNA screen of 28 Cullin-RING ligase family members identified five genes, including CUL5, that were required for ERBB2 degradation following treatment with 17-AAG—which we use here as a representative HSP90 inhibitor and chemical tool to promote client protein turnover. We go on to show for the first time to our knowledge that RNAi silencing of CUL5 reduces the 17-AAG–induced degradation of four other structurally diverse protein kinase clients in addition to ERBB2. As well as reducing client ubiquitination, we were surprised to find that silencing CUL5 also delayed the much earlier loss of client activity upon HSP90 inhibition. This delayed loss of client activity correlated with the delay induced by CUL5 knockdown in cochaperone dissociation from inhibited HSP90–client protein complexes. A mutant CUL5 that cannot be neddylated impaired 17-AAG–induced client protein kinase degradation but did not affect loss of client activity or recruitment of clients and HSP90 to CUL5. Finally, silencing CUL5 decreased sensitivity to HSP90 inhibitors in cell lines encompassing four cancer types driven by five different oncogenic protein kinase clients of HSP90. We therefore conclude that CUL5 plays an important role in multiple aspects of the molecular and cellular response to HSP90 inhibition.
Results
A Focused siRNA Screen Identifies Five Cullin-RING Ligase Family Genes Involved in ERBB2 Degradation Upon HSP90 Inhibition.
To investigate the extent of Cullin-RING ligase involvement in client protein degradation, 28 representative members of the family were chosen as candidates to silence and test for their effect on ERBB2 depletion following HSP90 inhibition. Three different siRNA oligonucleotides were used individually for each gene to minimize false positives or false negatives due to off-target effects (see Table S1 for full list of siRNA sequences used). ERBB2 levels then were quantified by immunoassay (15).
Fig. 1 shows the effects on ERBB2 protein levels of treating HT29 human colon cancer cells for 48 h with each siRNA followed by 24 h exposure to 17-AAG or DMSO control (the mean signal per gene is shown in Fig. S1A). All three positive controls (siRNAs targeted to ERBB2 itself) caused over 90% decrease in ERBB2 compared with DMSO-treated All-Stars Negative Control (AS Neg) siRNA. Note that siRNAs targeting SKP2 and RAB40C were omitted from the analysis because these caused more than 50% cell death under mock-treated conditions (Fig. S1B). In the absence of 17-AAG, the average ERBB2 signal after silencing each of the 26 remaining genes was within 75% of the All-Stars Negative Control siRNA, indicating that these siRNAs do not significantly affect steady-state expression of ERBB2. Upon 17-AAG treatment, ERBB2 protein levels in 17-AAG– treated cells remained above 50% of the untreated negative control signal with five of the silenced genes, namely, CUL5, RBX1, SOCS5, CUL3, and RNF7 (hereafter referred to by its more common protein name RBX2), thus demonstrating stabilization. Note that RBX1 and RBX2 are physiological RING-finger protein binding partners of CUL3 and CUL5, respectively, whereas SOCS5 is a substrate-recognition module of CUL2/5-containing complexes (13, 14). When the 17-AAG–treated ERBB2 signal per silenced gene was expressed as a percentage of the DMSO-treated signal for the same gene, the stabilization observed was highly significant (P < 0.0001) for the same five genes compared with the AS Neg siRNA (Fig. S1C). Target knockdown for these five genes individually was confirmed by quantitative reverse transcriptase PCR of their respective mRNAs (Fig. S1D).
Fig. 1.
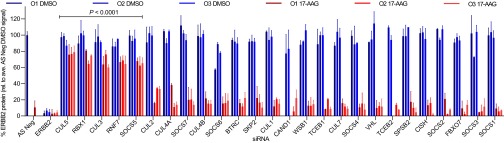
Focused siRNA screen to identify Cullin-RING ligase family members involved in 17-AAG–induced ERBB2 degradation. HT29 human colon cancer cells were transfected with one of three individual siRNAs targeted to a specific gene for 48 h and subsequently treated for 24 h with 5 × GI50 17-AAG (62.5 nM) or mock-treated with the equivalent volume of DMSO vehicle. Note that O1–O3 denote individual siRNA oligonucleotides, each with a unique sequence complementary to different parts of the target gene. ERBB2 protein levels were determined using the Total ERBB2 Whole Cell Lysate kit from Meso-Scale Diagnostics. After subtracting the mean background readout, the total ERBB2 signal for each siRNA was normalized by cell number and expressed as a percentage of the mean All-Stars Negative Control siRNA + DMSO signal. siRNAs that caused greater than 50% cytotoxicity under mock DMSO-treated conditions—individual oligonucleotides CAND1 O1 and TCEB2 O1 and all three SKP2 and RAB40C oligonucleotides—were excluded from the analysis. Bars represent mean signal of each individual siRNA for the same gene ± SD from two independent experiments. Following expression of data as a percentage of the normalized DMSO-treated signal for the same gene, those genes were identified for which statistically significant differences were observed compared with AS Neg siRNA by one-way ANOVA followed by Dunnett’s multiple comparisons test (see also Fig. S1C).
CUL5 and RBX2 Are Involved in the 17-AAG–Induced Degradation of Several Protein Kinase Clients of HSP90.
To determine if any of the identified genes were involved in the 17-AAG–induced depletion of protein kinase clients in addition to ERBB2, we analyzed the effects of silencing them on BRAFV600E, AKT, and CDK4 protein levels by Western blot in HT29 cells (Fig. 2A). Of these five proteins, silencing CUL3, RBX1, or SOCS5 did not affect depletion of any client tested other than ERBB2. In contrast, silencing RBX2 or CUL5 each reduced the degree of 17-AAG–induced depletion of BRAFV600E, AKT, and CDK4—all HSP90 clients belonging to different groups of the kinome, as defined by Manning et al. (16). These findings were found to be statistically significant by densitometry (Fig. 2B).
Fig. 2.
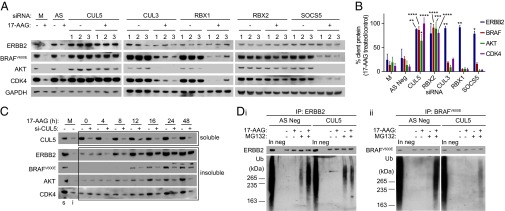
CUL5 and its functional partner RBX2 are required for the 17-AAG–induced ubiquitination and degradation of HSP90 protein kinase clients in HT29 human colon cancer cells. (A) Western blot analysis of protein levels in control mock lipid-transfected cells (M) or cells transfected with All-Stars Negative Control siRNA (AS) or one of three siRNAs (1–3) targeted to a specific gene for 48 h and subsequently treated for 24 h with 5 × GI50 17-AAG (62.5 nM) (+) or mock-treated with DMSO vehicle (−). GAPDH was used as a loading control. Bands from Western blots were quantified by densitometry (Figs. S6 and S7). (B) For each client protein, the mean GAPDH-normalized densitometry values from all three siRNAs targeted to each gene were calculated, and the 17-AAG–treated mean value was expressed as a percentage of the mock-treated mean value. Genes are shown for which statistically significant differences (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) were observed compared with AS Neg siRNA by one-way ANOVA followed by Dunnett’s multiple comparisons test. (C) HT29 colon cancer cells were transfected with pooled CUL5 siRNA oligonucleotides O2 and O4 (+) or All-Stars Negative Control siRNA (−) for 48 h and then treated with 5 × GI50 MG132 (200 nM) and 5 × GI50 17-AAG for between 0 and 48 h. Client protein levels present in the detergent-insoluble fraction (box labeled “insoluble”) were determined by Western blot. CUL5 levels in the detergent-soluble fraction (box labeled “soluble”) are shown to demonstrate protein knockdown. Samples representing the DMSO mock-treated (M) cells separated into detergent-soluble (s) and detergent-insoluble (i) fractions are also shown. (D) Immunoprecipitation of ERBB2 (D, i) and BRAFV600E (D, ii) in HT29 cells transfected with pooled CUL5 siRNA oligonucleotides or All-Stars Negative Control siRNA for 48 h and then treated for 24 h with 5 × GI50 17-AAG and/or MG132 (indicated by + or −). Whole-cell lysates were sonicated before immunoprecipitation. Total lysate protein levels are shown in Fig. S2C, iv. In, 5 µg input cell lysate; neg, IP negative control. See Fig. S6A for densitometry from three independent experiments.
Following blockade of protein synthesis using cyclohexamide, we found that silencing CUL5 delayed 17-AAG–induced turnover of the same four protein kinases (Fig. S2A), indicating that CUL5 is involved in their degradation at the protein level. To determine whether degradation occurred via the ubiquitin–proteasome system, we monitored HSP90 client accumulation and ubiquitination in the detergent-insoluble fraction of HT29 cells cotreated with 17-AAG and the proteasome inhibitor MG132. Silencing CUL5 delayed accumulation of the HSP90 clients ERBB2, BRAFV600E, AKT, and CDK4 by up to 32 h (Fig. 2C). The delay in ERBB2, AKT, and CDK4 accumulation was replicated in HCT116 human colon cancer cells (Fig. S2B). Note, however, that in contrast with the results for mutant BRAFV600E in HT29 cells, HSP90 inhibition did not induce accumulation of the wild-type BRAF (BRAFWT) in HCT116 cells. This was likely due to the low sensitivity of BRAFWT to HSP90 inhibitor-mediated depletion, as reported previously (17, 18).
Upon analysis of client protein immunoprecipitates from HT29 cells cotreated with 17-AAG and MG132, less ubiquitin was associated with ERBB2 and BRAFV600E in CUL5-silenced cells compared with AS Neg controls (Fig. 2D). Reduced ubiquitination was replicated with ERBB2 and AKT in HCT116 cells (Fig. S2C). Furthermore, CUL5-silenced cells displayed stronger and more diffuse cytoplasmic ERBB2 staining than AS Neg siRNA-treated cells upon HSP90 inhibition (Fig. S2D), indicative of impaired ERBB2 trafficking. Overall, our data indicate that CUL5 has a role in the ubiquitination and degradation of several protein kinase clients following HSP90 inhibition.
Silencing CUL5 Delays HSP90 Inhibitor-Induced Abrogation of Client Protein Signaling Output.
In addition to causing client degradation, HSP90 inhibitors also abrogate client protein signaling (11, 17, 19). Upon performing a 17-AAG exposure time course without CUL5 knockdown, we found that p-ERBB2 (Tyr1221/1222) and p-AKT (Ser479)—which represent active cellular pools of ERBB2 and AKT—as well as p-MEK1/2 (Ser217-221) and p-RB (Ser795)—which are direct downstream phosphorylated products of BRAF and CDK4, respectively—were all reduced within 4 h of 17-AAG treatment (Fig. 3A). Note that this was considerably earlier than total client protein levels were depleted (16–24 h).
Fig. 3.

CUL5 silencing delays inhibitor-induced loss of client activity. Western blot analysis of HT29 colon (A), BT474 breast (B), WM266.4 melanoma (C), and H1975 non-small cell lung (D) human cancer cells transfected with pooled CUL5 siRNAs O2 and O4 (+) or All-Stars Negative Control siRNA (−) for 48 h and then treated with 5 × GI50 17-AAG for between 0 and 24 h or mock-treated with DMSO for 24 h (M). The GI50 values for 96-h exposure were determined as 12.5 nM (HT29), 13.5 nM (BT474), 12.5nM (WM266.4), and 55 nM (H1975). GAPDH was used as a loading control. See Fig. S6B for densitometry from three independent experiments.
Although Cullin-RING ligases are proposed to be involved primarily with degradative pathways, other roles are emerging (20). Surprisingly, in addition to stabilizing client protein degradation (Fig. 2 and Fig. S2), we found that silencing CUL5 also delayed the loss of signaling output of these four client proteins by 6 h, i.e., from 2 h to 8 h following 17-AAG treatment (Fig. 3A).
Next, we performed the same experiment in three other cancer cell types driven by different hyperactivated HSP90 client proteins: ERBB2-positive breast cancer (BT474), BRAFV600E melanoma (WM266.4), and EGFRL858R/T790M non-small cell lung cancer (H1975) (Fig. 3 B–D). Again, loss of signaling output as measured by levels of p-ERBB2 (Tyr1221/1222), p-MEK1/2 (Ser217-221), and p-EGFR (Tyr1045), which we observed several hours earlier than the subsequent depletion of total ERBB2, BRAF, and EGFR levels, respectively, was delayed upon CUL5 silencing by 4–6 h. After 4 h 17-AAG treatment—at which time ERBB2 is dephosphorylated in AS Neg-treated cells but not in CUL5-silenced cells—we observed less ERBB2 internalization by immunofluorescence under the CUL5 knockdown condition (Fig. S2D). By contrast, in unsilenced cells ERBB2 localized in distinct cytoplasmic puncta at the same time point, consistent with previous studies in which this altered distribution was interpreted as an increase in receptor internalization and lysosomal processing following HSP90 inhibition (21, 22).
Taken together, our results indicate that CUL5 is involved not only in client protein degradation but also in the much earlier loss of client signaling output (and, in the case of ERBB2, receptor internalization and trafficking) following HSP90 inhibition for several oncoproteins in multiple cell lines from various cancer types with biological and clinical relevance to HSP90 inhibitors (3).
CUL5 Recruitment to HSP90-Protein Kinase Client Complexes Plays a Role in Dissociation of Cochaperones upon 17-AAG Treatment.
CUL5 was shown previously to coimmunoprecipitate with HSP90 and ERBB2 in HSP90 inhibitor-treated 293T human embryonic kidney cells (12). We found that CUL5 coimmunoprecipitated with ERBB2, BRAFV600E, AKT, and CDK4, as well as HSP90, in HT29 and HCT116 colon cancer cells following 17-AAG treatment (Fig. 4A and Fig. S3A). Strikingly, also shown is that the time points after HSP90 inhibition at which CUL5 was detected in the client protein immunoprecipitates correlated with the times when the representative HSP90 cochaperones CDC37 (23) and AHA1 (24) were lost from the immunoprecipitates (4 h onward), well before subsequent client degradation was observed. Although HSP90 did coimmunoprecipitate with CUL5, the HSP90 cochaperones CDC37 and AHA1 did not coimmunoprecipitate with CUL5 in either cell line, even after 17-AAG treatment (Fig. S3B).
Fig. 4.

CUL5 is recruited to HSP90-client complexes upon 17-AAG treatment in HT29 human colon cancer cells. (A) HT29 colon cancer cells were mock-treated with DMSO (M) or 5 × GI50 17-AAG (62.5 nM) and lysed between 0 and 24 h. ERBB2, BRAFV600E, AKT, or CDK4 immunoprecipitations were performed on these cell lysates (A, i–iv), and the resultant immunoblots were probed for the relevant proteins as indicated. Mock-treated cells were lysed after 24 h. GAPDH was used as a loading control. (B) ERBB2 (B, i) or BRAFV600E (B, ii) immunoprecipitation of HT29 colon cancer cells transfected with pooled CUL5 siRNAs O2 and O4 (+) or All-Stars Negative Control siRNA (−) for 48 h and then treated with 5 × GI50 17-AAG for between 0 and 16 h or mock-treated with DMSO for 16 h (M). In, 5 µg input cell lysate; neg, IP negative control. See Fig. S6C for densitometry from three independent experiments.
Based on these observations, we hypothesized that CUL5 recruitment to HSP90–protein kinase client complexes plays a role in the cochaperone dissociation that we observed upon HSP90 inhibition (Fig. 4A and Fig. S3A). Consistent with our hypothesis, we found that following 17-AAG treatment, CDC37 and AHA1 dissociated from ERBB2 and BRAFV600E considerably later in HT29 cells in which CUL5 had been silenced compared with AS Neg controls (Fig. 4B). CUL5 silencing also caused a similar, clear delay in cochaperone dissociation with the additional clients ERBB2 and AKT in HCT116 cells (Fig. S3C). The time points at which cochaperones did eventually dissociate (4–8 h) correlated precisely with the delayed times at which client activity was lost upon CUL5 silencing (Fig. 3).
Taken together, Figs. 3 and 4 show that recruitment of CUL5 to the HSP90–protein kinase complex can facilitate the dissociation of cochaperones from the complex and the accompanying loss of client signaling after HSP90 inhibition. However, the fact that silencing CUL5 delayed rather than blocked these events indicates that other factors are also likely involved with respect to these early effects of HSP90 inhibition.
CUL5-NEDD8 Conjugation Is Required for Client Protein Degradation but Not for Loss of Client Signaling Output.
Covalent attachment of the NEDD8 peptide to Cullins is required for the ubiquitination of their target substrates (25). To determine whether NEDD8-conjugated CUL5 was required for its effects on the HSP90 inhibition response, we silenced endogenous CUL5 in HT29 cells and transiently transfected them with either a positive control HA-tagged wild-type CUL5 construct (CUL5-WT), a validated HA-tagged CUL5 that cannot be neddylated (CUL5-∆NEDD8) (26), or an empty vector control (EV). The CUL5-∆NEDD8 construct we used has Lys to Ala mutations at the three amino acid residues where CUL5 neddylation could occur (724, 727, and 728) (27). No NEDD8-CUL5 band was detected in HT29 cells transfected with this construct together with a CUL5 siRNA targeted to the 3′ UTR of the CUL5 mRNA to silence specifically endogenous but not heterologous CUL5 expression (Fig. 5A). Supporting the hypothesis that CUL5 needs NEDD8 conjugation to mediate client depletion following HSP90 inhibition, client protein degradation was observed in CUL5-WT–transfected cells but not in those transfected with CUL5-∆NEDD8 following 17-AAG treatment. However, loss of client protein activity occurred similarly in cells transfected with either construct, as determined by phospho-protein levels 4 h after 17-AAG treatment to measure signaling output (Fig. 5B). Furthermore, immunoprecipitation of the CUL5 constructs via their HA tag showed that recruitment of HSP90 and protein kinases to CUL5-∆NEDD8 after 4 h 17-AAG treatment was not impaired compared with recruitment to CUL5-WT (Fig. 5C).
Fig. 5.

CUL5-NEDD8 conjugation is required for 17-AAG–induced client degradation but not for loss of client activity. HT29 human colon cancer cells were transfected with pcDNA5/FRT/TO plasmids containing HA-tagged CUL5 (CUL5-WT), an HA-tagged CUL5 triple mutant (S724A S727A S728A) (CUL5-∆NEDD8), or empty vector (EV) or mock-transfected with DMSO vehicle for 24 h and then transfected with CUL5 O1 siRNA (si-CUL5), All-Stars Negative Control siRNA (si-AS Neg), or DMSO vehicle for a further 48 h before compound treatment. Cells were then treated for 24 h (A) or 4 h (B) with 5 × GI50 17-AAG (62.5 nM) or DMSO vehicle, lysed, and analyzed for protein levels by Western blot. (C) HA-tag immunoprecipitation of HT29 colon cancer cells treated as in B. In, 5 µg input cell lysate; Neg, IP negative control. See Fig. S6D for densitometry from three independent experiments.
Overall, these results show that whereas NEDD8 conjugation is required for 17-AAG–induced client protein degradation mediated by CUL5, it is not necessary for the earlier formation of CUL5–HSP90–client protein complexes or loss of client activity.
Silencing CUL5 Reduces Cellular Sensitivity to HSP90 Inhibition.
Having demonstrated that silencing CUL5 affects the 17-AAG–induced loss of activity and stability of several HSP90 client proteins, we hypothesized that it might also reduce the cancer cell growth inhibitory effects of this pharmacologic perturbation. Silencing CUL5, or its functional binding partner RBX2, decreased cellular sensitivity to 17-AAG in HT29 and HCT116 cells (P < 0.01) (Fig. 6A and Fig. S4B). This significant decrease in sensitivity was reproduced with two additional, chemically distinct clinical HSP90 inhibitors, BIIB021 (28) and AUY922 (15, 29), in HT29 and HCT116 cells, respectively, indicating that the effect was likely general to HSP90 inhibitors. Silencing both CUL5 and RBX2 did not reduce 17-AAG sensitivity further compared with silencing either alone, consistent with their role as part of the same complex (13). In addition, reduced sensitivity to HSP90 inhibition was also observed upon silencing CUL5 in ERBB2-positive breast cancer, BRAFV600E melanoma, and EGFRL858R/T790M non-small cell lung cancer lines (Fig. S4 C–E).
Fig. 6.

Silencing CUL5 reduces cellular sensitivity to HSP90 inhibition in HT29 human colon cancer cells. Growth inhibition of HT29 colon cancer cells upon treatment with HSP90 inhibitors 17-AAG (A) or BIIB021 (B) as determined by 96-h SRB assay. Cells were transfected 48 h before treatment. Conditions are no transfection (NT), DMSO transfection (Mock), 25 nM All-Stars Negative Control (Neg), pooled CUL5 siRNAs O2 and O4 (CUL5), pooled RBX2 siRNAs O1 and O9 (RBX2), and pooled CUL5 and RBX2 siRNAs at concentrations of 12.5 nM (DUAL 12.5) or 25 nM (DUAL 25) each. Dotted line represents 50% cell growth inhibition. Mean GI50 values from three independent experiments ± SEM and the corresponding Dunnett’s multiple comparisons test P values (following one-way ANOVA) are shown. *P < 0.01.
We conclude that the effects of silencing CUL5 on cochaperone dissociation, client activity loss, and client degradation result in an attenuation of the antiproliferative response caused by HSP90 inhibition, as shown with three different clinical HSP90 drugs, in several cancer cell lines of different histological origins and harboring diverse oncoprotein kinase drivers.
Discussion
The E3 ubiquitin ligase CUL5 was originally reported to be involved in the degradation of ERBB2 in human embryonic kidney 293T cells following HSP90 inhibition by geldanamycin (12). By silencing Cullin-RING ligase family members using RNAi, we investigated the extent to which CUL5 and other Cullin-RING family ligases are involved in the degradation of various HSP90 protein kinase clients. We show that in addition to ERBB2, CUL5 plays an important role in the 17-AAG–induced ubiquitination and degradation of the structurally and functionally diverse protein kinase clients BRAFV600E, AKT, and CDK4. Unexpectedly, we reveal that CUL5 is also involved in the dissociation of cochaperones from the inhibited HSP90–client protein complex that we observe and also in the loss of client signaling output that we demonstrate, both of which occur considerably earlier than subsequent client protein degradation. Immunofluorescence analysis also indicated that silencing CUL5 compromises 17-AAG–induced ERBB2 internalization and cytoplasmic trafficking. In addition, by expression of a validated CUL5 construct that cannot be neddylated, we show that NEDD8 conjugation of CUL5 is required for later degradation of client kinases but not for their early recruitment to CUL5 or loss of downstream signaling output following HSP90 inhibition. We demonstrate that silencing CUL5 expression reduces the sensitivity of four human cancer cell types, harboring diverse oncoprotein clients, to three structurally distinct HSP90 inhibitors.
Based on our data, we propose a possible model in which CUL5 recruitment to the inhibited HSP90–protein kinase complex is one of the factors involved in cochaperone dissociation and loss of client activity; CUL5 or some other associated factor could then potentially act as a scaffold for assembly of other components of the ubiquitin-proteasome system that ultimately results in client protein ubiquitination and degradation (Fig. S5). Some proteins involved in the degradation response may be specific to individual clients, as exemplified by our finding that CUL3, its functional binding partner RBX1, and the CUL2/5 substrate receptor SOCS5 affected the 17-AAG–induced depletion of ERBB2 but not the other protein kinases tested (Fig. 2A). This is in agreement with a recent quantitative analysis which identified 117 different E3 ubiquitin ligases that interacted to some extent with HSP90 in human 293T cells (1). We also found that silencing Elongin-B or Elongin-C did not affect 17-AAG–induced ERBB2 depletion, even though these proteins are classically required for formation of functional CUL5-containing E3 ubiquitin ligase complexes (13, 14). Our results are, however, consistent with previous findings that CUL5-mediated ERBB2 degradation is not impaired in cells with deficient Elongin-B/C activity (12). Further work is required to determine whether or not CUL5 forms typical Elongin-B/C–Cullin–SOCS-box protein complexes with respect to the degradative response following HSP90 inhibition. Precisely how CUL5 binding to HSP90 mediates early cochaperone loss and reduced client protein signaling and the mechanistic linkage between these events as well as to the subsequent client degradation also remain to be defined.
The chromosomal location of CUL5 is associated with loss of heterozygosity in breast cancer, suggesting that CUL5 may be a tumor suppressor (30, 31). Heterologous CUL5 expression has been reported to exert an antiproliferative effect (32, 33). Furthermore, a clinical study showed a significant decrease in CUL5 expression in breast tumors versus matched normal tissue (34). Based on these findings and on our demonstration that silencing CUL5 reduces cellular sensitivity to HSP90 inhibition (Fig. 6 and Fig. S4), it is possible that expression levels of this gene may affect tumor response to HSP90 inhibitors in patients. Loss of CUL5 is also a potential mechanism of acquired resistance. Given the clinical promise of HSP90 inhibitors (35), predictive biomarkers for patient response could prove invaluable.
The studies performed here indicate that CUL5 is a key component affecting multiple features of the complex molecular and cellular response to HSP90 inhibition with potential clinical relevance. Our findings warrant further investigation of CUL5 and other Cullin-RING family proteins, such as RBX2, with respect to this response in cancer cells.
Methods
Cell Culture and Compound Treatments.
All cell lines were obtained from ATCC (LGC Promochem). Cells were cultured in DMEM (Invitrogen) and supplemented with 10% vol/vol FCS (PAA Laboratories), 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 100 units of penicillin and streptomycin. Cells were maintained at 37 °C in a humidified incubator with 5% CO2 and subcultured at 70% confluency. Cells were confirmed as mycoplasma-free using the Venor Mycoplasma PCR Detection Kit (Minerva Biolaboratories). For analysis of cellular and molecular effects of compound treatments, appropriate concentrations of 17-AAG (Axxora), in-house synthesized AUY922 (29), BIIB021 (Selleck), MG132 (Millipore), MLN4924 (Active Biochem), cyclohexamide (Sigma-Aldrich), or DMSO vehicle (Sigma-Aldrich) were added to cells at 40% confluency or 48 h after siRNA transfection. For experiments where 17-AAG and MG132 or MLN4924 cotreatments were involved, MG132 or MLN4924 were added to cells 1 h before 17-AAG.
siRNA, Plasmids, and Transfection.
siRNA oligonucleotides were synthesized by Qiagen. CUL5 siRNA sequences were CAGCTGGTTATTGGAGTAAGA (O1), CTGGAGGACTTGATACCGGAA (O2), CAGGTTTGAATCAGTCACCTA (O3), and CCAGCTGATTCAGTTATTATA (O4). Target sequences for the siRNA screen are listed in Table S1. Oligonucleotides were transfected into HT29 cells using DharmaFECT-4 (Dharmacon), BT474 cells using DharmaFECT-2, or HCT116 and H1975 cells using HiPerFect (Qiagen) and 25 nM siRNA according to manufacturer’s instructions. pcDNA5/FRT/TO plasmids (Invitrogen) containing HA-EV, HA-CUL5-WT, and HA-CUL5-∆NEDD8 were provided by Arno Alpi (24) and transfected into cells using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions 24 h before siRNA transfection. For validation of gene silencing upon siRNA transfection, quantitative RT-PCR was performed. RNA was extracted and purified from silenced cells using the Cells-to-cDNA II Kit (Ambion) and synthesized into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA levels of the silenced genes were determined by performing the TAQman assay using predesigned gene-specific primers (Applied Biosystems).
Quantitation of ERBB2 Protein Levels.
The Total ERBB2 Whole Cell Lysate Kit (Meso-Scale Diagnostics) was used according to manufacturer’s instructions. Additional details are provided in SI Methods.
Western Blotting, Immunoprecipitation, and Immunofluorescence.
Western blotting, immunoprecipitation, and immunofluorescence are described in SI Methods. Antibodies used are listed in Table S2.
Cell Growth Inhibition Assay and Statistical Analysis.
Cellular sensitivity to compounds was measured by 96 h sulforhodamine B (SRB) assay (36). Statistical analyses were performed using Prism 6 (GraphPad Software).
Supplementary Material
Acknowledgments
We thank the P.W. laboratory members for helpful discussions and L. Howell for technical assistance with confocal microscopy. We also thank T. Kurz, A. Alpi, I. Kelsall, and J. Hastie at University of Dundee for help obtaining and using the CUL5 plasmids and CUL5 (S073D) antibody. P.W. acknowledges grant funding from Cancer Research UK (Program Grants C309/A8274 and C309/A11566) and is a Cancer Research UK Life Fellow. R.S.S. acknowledges a PhD studentship from Cancer Research UK (Grant C309/A8140).
Footnotes
Conflict of interest statement: All the authors are or were employees of The Institute of Cancer Research, which has a commercial interest in HSP90 inhibitors and operates a reward to inventors scheme. P.W. received funding from Vernalis for the discovery of HSP90 inhibitors, and intellectual property for this program was licenced to Vernalis Ltd. and Novartis. P.W. was previously involved in a research collaboration with AstraZeneca in the area of stress and chaperone pathways. P.W. has been a consultant to Novartis, is a founder of Chroma Therapeutics, and is scientific advisory board member of Astex Pharmaceuticals and Nextech Invest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322412111/-/DCSupplemental.
References
- 1.Taipale M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150(5):987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: The story unfolds. Expert Opin Biol Ther. 2002;2(1):3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 3.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Panaretou B, et al. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998;17(16):4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 6.Schulte TW, An WG, Neckers LM. Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem Biophys Res Commun. 1997;239(3):655–659. doi: 10.1006/bbrc.1997.7527. [DOI] [PubMed] [Google Scholar]
- 7.Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol. 1996;10(6):705–712. doi: 10.1210/mend.10.6.8776730. [DOI] [PubMed] [Google Scholar]
- 8.Sepp-Lorenzino L, Ma Z, Lebwohl DE, Vinitsky A, Rosen N. Herbimycin A induces the 20 S proteasome- and ubiquitin-dependent degradation of receptor tyrosine kinases. J Biol Chem. 1995;270(28):16580–16587. doi: 10.1074/jbc.270.28.16580. [DOI] [PubMed] [Google Scholar]
- 9.Connell P, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3(1):93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 10.Xu W, et al. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci USA. 2002;99(20):12847–12852. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61(10):4003–4009. [PubMed] [Google Scholar]
- 12.Ehrlich ES, et al. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc Natl Acad Sci USA. 2009;106(48):20330–20335. doi: 10.1073/pnas.0810571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 14.Zimmerman ES, Schulman BA, Zheng N. Structural assembly of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol. 2010;20(6):714–721. doi: 10.1016/j.sbi.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eccles SA, et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68(8):2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 16.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 17.da Rocha Dias S, et al. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65(23):10686–10691. doi: 10.1158/0008-5472.CAN-05-2632. [DOI] [PubMed] [Google Scholar]
- 18.Grbovic OM, et al. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc Natl Acad Sci USA. 2006;103(1):57–62. doi: 10.1073/pnas.0609973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basso AD, Solit DB, Munster PN, Rosen N. Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene. 2002;21(8):1159–1166. doi: 10.1038/sj.onc.1205184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dikic I, Robertson M. Ubiquitin ligases and beyond. BMC Biol. 2012;10:22. doi: 10.1186/1741-7007-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tikhomirov O, Carpenter G. Geldanamycin induces ErbB-2 degradation by proteolytic fragmentation. J Biol Chem. 2000;275(34):26625–26631. doi: 10.1074/jbc.M003114200. [DOI] [PubMed] [Google Scholar]
- 22.Lerdrup M, Hommelgaard AM, Grandal M, van Deurs B. Geldanamycin stimulates internalization of ErbB2 in a proteasome-dependent way. J Cell Sci. 2006;119(Pt 1):85–95. doi: 10.1242/jcs.02707. [DOI] [PubMed] [Google Scholar]
- 23.Smith JR, Clarke PA, de Billy E, Workman P. Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene. 2009;28(2):157–169. doi: 10.1038/onc.2008.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2008;68(4):1188–1197. doi: 10.1158/0008-5472.CAN-07-3268. [DOI] [PubMed] [Google Scholar]
- 25.Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: Building an expressway to protein destruction. Oncogene. 2004;23(11):1985–1997. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- 26.Kelsall IR, et al. TRIAD1 and HHARI bind to and are activated by distinct neddylated Cullin-RING ligase complexes. EMBO J. 2013;32(21):2848–2860. doi: 10.1038/emboj.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 28.Lundgren K, et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol Cancer Ther. 2009;8(4):921–929. doi: 10.1158/1535-7163.MCT-08-0758. [DOI] [PubMed] [Google Scholar]
- 29.Brough PA, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51(2):196–218. doi: 10.1021/jm701018h. [DOI] [PubMed] [Google Scholar]
- 30.Byrd PJ, et al. Identification and analysis of expression of human VACM-1, a cullin gene family member located on chromosome 11q22-23. Genome Res. 1997;7(1):71–75. doi: 10.1101/gr.7.1.71. [DOI] [PubMed] [Google Scholar]
- 31.Carter SL, et al. Loss of heterozygosity at 11q22-q23 in breast cancer. Cancer Res. 1994;54(23):6270–6274. [PubMed] [Google Scholar]
- 32.Van Dort C, et al. VACM-1, a cul-5 gene, inhibits cellular growth by a mechanism that involves MAPK and p53 signaling pathways. Am J Physiol Cell Physiol. 2003;285(6):C1386–C1396. doi: 10.1152/ajpcell.00338.2002. [DOI] [PubMed] [Google Scholar]
- 33.Burnatowska-Hledin MA, et al. T47D breast cancer cell growth is inhibited by expression of VACM-1, a cul-5 gene. Biochem Biophys Res Commun. 2004;319(3):817–825. doi: 10.1016/j.bbrc.2004.05.057. [DOI] [PubMed] [Google Scholar]
- 34.Fay MJ, et al. Analysis of CUL-5 expression in breast epithelial cells, breast cancer cell lines, normal tissues and tumor tissues. Mol Cancer. 2003;2:40. doi: 10.1186/1476-4598-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holford J, Sharp SY, Murrer BA, Abrams M, Kelland LR. In vitro circumvention of cisplatin resistance by the novel sterically hindered platinum complex AMD473. Br J Cancer. 1998;77(3):366–373. doi: 10.1038/bjc.1998.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.