

Significance
Understanding of the mechanisms of modulation of voltage-gated Na+ (NaV) channels by general anesthetic agents such as sevoflurane is critical to interpret their role in general anesthesia. By using the bacterial NaV channel analogue NaChBac plus a combination of computational and electrophysiological analyses, this work strongly suggests a multisite mechanism of sevoflurane action on NaV channels. Furthermore, computer simulations suggest specific putative anesthetic sites worthy of further investigation.
Keywords: anesthesia, MD simulations, anesthetics, membrane proteins
Abstract
Halogenated inhaled general anesthetic agents modulate voltage-gated ion channels, but the underlying molecular mechanisms are not understood. Many general anesthetic agents regulate voltage-gated Na+ (NaV) channels, including the commonly used drug sevoflurane. Here, we investigated the putative binding sites and molecular mechanisms of sevoflurane action on the bacterial NaV channel NaChBac by using a combination of molecular dynamics simulation, electrophysiology, and kinetic analysis. Structural modeling revealed multiple sevoflurane interaction sites possibly associated with NaChBac modulation. Electrophysiologically, sevoflurane favors activation and inactivation at low concentrations (0.2 mM), and additionally accelerates current decay at high concentrations (2 mM). Explaining these observations, kinetic modeling suggests concurrent destabilization of closed states and low-affinity open channel block. We propose that the multiple effects of sevoflurane on NaChBac result from simultaneous interactions at multiple sites with distinct affinities. This multiple-site, multiple-mode hypothesis offers a framework to study the structural basis of general anesthetic action.
General anesthetic agents have been in use for more than 160 y. However, we still understand relatively little about their mechanisms of action, which greatly limits our ability to design safer and more effective general anesthetic agents. Ion channels of the central nervous system are known to be key targets of general anesthetic agents, as their modulation can account for the endpoints and side effects of general anesthesia (1–4). Many families of ion channels are modulated by general anesthetic agents, including ligand-gated, voltage-gated, and nongated ion channels (2, 5–7). Mammalian voltage-gated Na+ (NaV) channels, which mediate the upstroke of the action potential, are regulated by numerous inhaled general anesthetic agents (8–14), which generally cause inhibition. Previous work showed that inhaled general anesthetic agents, including sevoflurane, isoflurane, desflurane, and halothane, mediate inhibition by increasing the rate of Na+ channel inactivation, hyperpolarizing steady-state inactivation, and slowing recovery from inactivation (11, 15–18). Inhibition of presynaptic NaV channels in the spinal cord is proposed to lead to inhibition of neurotransmitter release, facilitating immobilization—one of the endpoints of general anesthesia (14, 19, 20). Despite the importance of NaV channels as general anesthetic targets, little is known about interaction sites or the mechanisms of action.
What is known about anesthetic sites in NaV channels comes primarily from the local anesthetic field. Local anesthetic agent binding to NaV channels is well characterized. These amphiphilic drugs enter the channel pore from the intracellular side, causing open-channel block (21). Investigating molecular mechanisms of mammalian NaV channel modulation by general anesthetic agents has been complicated by the lack of high-resolution structures of these channels as a result of their large size and pseudotetrameric organization. However, the recent discovery of the smaller, tetrameric bacterial Na+ channel family has provided an invaluable tool to characterize the structural features of NaV channels and investigate their interactions with general anesthetic agents at the molecular level (22, 23). Several bacterial Na+ channels have been crystallized (24–27). These channels have a classical domain structure in which helices S1–S4 form the voltage sensor domain (VSD), S5 and S6 form the pore, and the S4–S5 linker connects the voltage sensor to the pore domain. One notable structural feature is the presence of “fenestrations” or hydrophobic tunnels through the pore domain (24).
Although crystal structures are not yet reported, the bacterial Na+ channel NaChBac has been extensively characterized by electrophysiology (22, 28–36). Additionally NaChBac exhibits conserved slow open channel block in response to local and general anesthetic agents (15, 37). These anesthetic agents reduce peak current and accelerate current decay, making it conceivable that local and general anesthetic agents could share a site of action in NaChBac. The local anesthetic binding site identified in the central cavity of the mammalian NaV1.2 channel, which mediates open channel block, is partially conserved in NaChBac (37, 38). A recent molecular dynamics (MD) modeling study found that isoflurane, which inhibits NaChBac (15), interacts with multiple regions of this channel, including the pore, the selectivity filter, and the S4–S5 linker/S6 interface (39). Although the importance of these interactions on the modulation of mammalian NaV channels remains to be determined, the available data indicate that NaChBac is currently one of the best starting points to investigate the mechanisms of action of sevoflurane.
Here, we investigated NaChBac to gain structural insight into the mechanisms of inhaled anesthetic modulation of NaV channels. The focus of this work is sevoflurane because this anesthetic is commonly used in clinical settings and is a known inhibitor of several mammalian NaV channels (NaV 1.4, 1.7, and 1.8) (11, 13). A three-pronged approach incorporating MD simulation, whole-cell patch-clamp electrophysiology, and kinetic modeling suggests that sevoflurane acts on multiple sites to alter gating and permeation. Whereas the effect on gating results from modulating activation and inactivation gating at low concentrations (0.2 mM), the permeation effect is apparent at high concentrations (2 mM) and results from open channel block (2 mM). Although the net inhibitory effect of these multisite interactions is consistent with anesthetic-induced reduction of neuronal firing, general anesthesia does not simply result from a global reduction in firing. General anesthesia depends on complex mechanisms throughout the brain, which include increases and decreases in firing (3). Thus, precisely how Na+ channel activation by sevoflurane fits into the global effects of anesthesia remains to be seen. The present work helps elucidate the molecular mechanism of sevoflurane action on NaV channels.
Results and Discussion
MD Simulations.
We used the MD flooding method (40) to initially assess potential sevoflurane interactions on three structural states of NaChBac. The first one, the partially activated/closed state, resembles closely the X-ray structure of NaVAb (24) and is an intermediate of activation with the pore is closed (41). Hereafter, we will use a nomenclature whereby the state of the pore domain follows that of the VSD. We have also investigated the resting/closed and activated/open states, which are the two terminal states of the activation cycle (41). Worth noting is that the activated/open state undergoes pore closure during the flooding simulation as sevoflurane entry causes desolvation of the gate region. Overall, the structures were stable over the course of each simulation (Fig. S1). The initial model contained NaChBac, the membrane, a slab of water (25,310 molecules), and 100 sevoflurane molecules initially distributed randomly in the aqueous phase (the initial aqueous concentration is ∼200 mM). This concentration is much higher than clinically relevant concentrations, but is necessary to observe binding on the short time scales achievable in MD simulations. Sevoflurane then partitions almost completely into the lipid phase within the first 200 ns of simulation (Fig. S2), and the final aqueous concentration of sevoflurane is ∼2 mM. To analyze putative binding modes and prioritize regions of the channel exhibiting continuous sevoflurane occupancy, we applied a protocol for clustering described elsewhere (39). This analysis identified two major state-dependent binding sites present in the activated conformations (i.e., partially activated/closed and activated/open states): (i) a selectivity filter site and (ii) an S4–S5 linker site (Fig. 1). State-independent occupation of the central cavity and fenestrations was found in all conformations (Figs. 1 and 2 A–C). Additionally, the sevoflurane interaction pattern had two unique features in the closed state. First, an activation gate site not found in other conformations and numerous but diffuse sevoflurane interactions with the VSD (Fig. 2D). NaChBac’s central cavity was occupied by sevoflurane in all gating states, regardless of the state of the activation gate (Fig. 2 A–C). As previously found for isoflurane (39), sevoflurane enters the central cavity via the fenestrations. To enter, sevoflurane displaces the lipid tails that normally occupy the fenestrations, perhaps suggesting a basis for the well-known Meyer–Overton correlation. Despite being lipophilic, sevoflurane possesses a polar nature and can potentially interact electrostatically with its binding partners. In our simulations, sevoflurane is found often at contact distance from polar moieties.
Fig. 1.
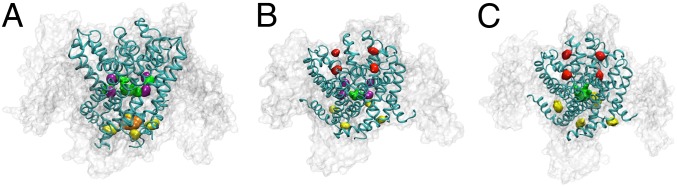
Putative sevoflurane binding sites in various states of NaChBac: (A) Resting/closed, (B) partially activated/closed and (C) activated/open. The three binding sites identified by clustering analysis are extracellular site (red), linker site (yellow), activation gate site (orange), and cavity/fenestration site (green/purple).
Fig. 2.

Zoom views of putative sevoflurane binding sites. (A–C) Shown are top views of sevoflurane binding in the cavity and fenestrations of the resting/closed, partially activated/closed, and activated/open states, respectively. (D and E) Top view (Left) and zoom view (Right) of the extracellular site in the partially activated/closed state (D) and the activated/open state (E). (F and G) Top view (Left) and zoom view (Right) of the linker site in the partially activated/closed state (F) and the activated/open state (G).
All four fenestrations are traversed/occupied by sevoflurane in each gating state. Over the equilibrated phase of trajectory for all gating states, at least two sevoflurane molecules were in the cavity; however, occasionally, three to four sevofluranes could be seen in this location (Fig. S3). Sevoflurane occupying the cavity displaces several water molecules, but not enough to prevent Na+ solvation in the cavity. Although sevoflurane is highly mobile in the central cavity, it forms stable interactions in all states. Namely, it interacts with conserved residues (T220 and F227) known to play a role in local anesthetic action on mammalian NaV channels (F1764 and Y1771 in NaV1.2) (37, 38). The observations of consistent cavity occupation by sevoflurane and interactions with residues known to be involved in open channel block by local anesthetic agents supports the hypothesis that sevoflurane may inhibit NaChBac function through an open channel block mechanism.
The sevoflurane extracellular site is located at the intersubunit interface between the P-loops (Fig. 2 D and E), similar to a previously identified isoflurane binding site on NaChBac (39). However, sevoflurane is found only in the extracellular site in the partially activated/closed and activated/open conformations. Nonpolar (Y172, L178, L181, Q185 M197, F201) and charged (E171, R198) residues participate in the interactions. Sevoflurane enters the four equivalent extracellular sites quickly during the anesthetic partitioning phase, and remains there for the duration of the simulations. The extracellular region of NaChBac could influence the conformation of the selectivity filter to regulate P/C type inactivation (28, 30). Therefore, sevoflurane interaction at the extracellular site could alter the stability of the filter region.
The linker site is located at the interface between the distal S6 helix and intersubunit “corner” formed by the N terminus of one linker and the C terminus of the linker from the adjacent subunit (Fig. 2 F and G). This linker site is found only in the partially activated/closed and activated/open states. In the partially activated/closed state, only one out of the four equivalent sites is continuously occupied by sevoflurane over the last half of the trajectory (Fig. 2F). The activated/open state linker site is continuously occupied in all four equivalent sites, sometimes by more than one sevoflurane (Fig. 2G). In the partially activated/closed state, the linker site primarily involves nonpolar interactions with sevoflurane, with residue contributions from the linker and S6 (L132, L142, I146, L225, I227, V234). Asparagines also participate (N145, N224), and could form polar interactions with sevoflurane. More residues were found interacting with sevoflurane in the activated/open state, and many sevoflurane molecules occupied each site. Although we do not know whether this high level of occupation is physiological, it is an indicator of a favorable interaction.
An additional site at the activation gate is also found in the resting state. Upon deactivation, this activation gate site emerges in the resting/closed state. This site involves residues of the S4–S5 linker, but sevoflurane is primarily situated at the intersubunit interface of all four S6 helices, below the hydrophobic seal of the activation gate (Fig. S4A). This activation gate site is also mainly nonpolar (L132, I230, V231, V234) and additionally contains an asparagine (N233).
Mutation of residues in the lower half of S6 can affect the rate of inactivation (29, 34), suggesting that this segment is implicated in inactivation. Furthermore, the S4–S5/S6 interface has been implicated as an inhibitory general anesthetic interaction region in K+ channels (41). Sevoflurane interactions in the S4–S5/S6 interface or directly below the activation gate could thus affect inactivation and voltage-dependent activation by altering the coupling between the VSD and the pore.
Electrophysiology.
In light of the MD simulation results suggesting multiple sevoflurane sites in NaChBac, we anticipated multiple distinguishable effects on the function of the channel. To test this hypothesis, we investigated the gating properties of NaChBac expressed in HEK-293 cells to establish a baseline (Fig. S5) and, subsequently, tested the effects of sevoflurane at various concentrations. Namely, we focused on the following properties: (i) conductance–voltage (Gp–V) relation, (ii) rate of macroscopic inactivation, (iii) prepulse inactivation, and (iv) recovery from inactivation.
Given a significant cell-to-cell variability in the gating properties of NaChBac (Fig. S6), all whole-cell current recordings were obtained in pairs from the same cell (before and after sevoflurane). A preliminary assessment of the effects of sevoflurane on NaChBac currents revealed apparent potentiation at low concentrations (<0.5 mM) and inhibition at high concentrations (>1 mM). To quantitatively characterize these effects, we investigated the channel’s gating properties at two sevoflurane concentrations differing by an order of magnitude, 0.2 and 2 mM. Whereas 0.2 mM sevoflurane is within the therapeutic concentration range, 2 mM is outside this range. We tested this concentration, however, to saturate putative relevant binding sites. The low concentration of sevoflurane produced the following effects on activation gating (Fig. 3, Fig. S6, and Table S1): (i) increased the peak current by ∼15% (at 0 mV) and (ii) shifted the Gp–V curve to the left (half-activation voltage ΔV1/2 ∼ −10 mV). Tail current kinetics were not significantly affected (Fig. S6 I and J). On inactivation gating, 0.2 mM sevoflurane also had the following effects (Fig. 3 and Table S1): (i) shifted the steady-state inactivation curve to the left (ΔV1/2 ∼ −14 mV), (ii) increased the rate constant of current decay by ∼20%, and (iii) increased the rate constant of recovery from inactivation by ∼36%.
Fig. 3.

Electrophysiological effects of sevoflurane on NaChBac. (A and C) Representative currents evoked from a step from −100 to 0 mV, showing the effect of 0.2 mM (A) and 2 mM (C) sevoflurane on NaChBac current amplitude and rate of inactivation. (B and D) Scatter plot comparing paired measurements of the time constants (τ) of current decay before and after application of 0.2 mM (B) and 2 mM (D) sevoflurane. (E and G) Representative peak Gp–V relationships from paired experiments before and after application of 0.2 mM (E) and 2 mM (G) sevoflurane. (F and H) Scatter plot from paired data showing the effect of 0.2 mM (F) and 2 mM (H) sevoflurane on the V1/2 of activation. (I and K) Representative steady-state inactivation vs. voltage relationships from paired experiments before and after application of 0.2 mM (I) and 2 mM (K) sevoflurane. (J and L) Scatter plot from paired data showing the effect of 0.2 mM (J) and 2 mM (L) sevoflurane on V1/2 of inactivation. (M and O) Representative recovery from inactivation curves from paired experiments before and after application of 0.2 mM (M) and 2 mM (O) sevoflurane. (N and P) Scatter plot from paired data showing the effect of 0.2 mM (N) and 2 mM (P) sevoflurane on the time constant (τ) of recovery from inactivation.
Generally, 2 mM sevoflurane produced qualitatively similar results (Fig. 3, Fig. S6, and Table S1): (i) shifted the Gp–V curve to the left (ΔV1/2 ∼ −6 mV) and slightly reduced the equivalent gating charge of activation, (ii) increased the rate constant of current decay by 70%, (iii) shifted the steady-state inactivation curve to the left (ΔV1/2 ∼ −19 mV), (iv) slightly reduced the equivalent gating charge of inactivation, and (v) increased the rate constant of recovery from inactivation by ∼50%. However, instead of increasing the peak current, as observed at low submillimolar concentrations, 2 mM sevoflurane slightly decreased peak current by ∼7%. Neither low nor high concentrations produced significant effects on the maximum value of the current, Gmax (Fig. S6 C and D).
The similarities in terms of the leftward shifts in the activation and inactivation curves and accelerated recovery from inactivation at low and high doses of sevoflurane suggest that the anesthetic might have modulated voltage-dependent activation and the stability of the inactivated state. Because these effects do not display substantial concentration dependence between 0.2 and 2 mM sevoflurane, gating modulation might be near saturation at submillimolar concentrations. Thus, it suggests relatively high-affinity interactions. Separately, however, accelerated current decay was dramatically enhanced at 2 mM sevoflurane. Such an effect suggests slow open-channel block involving a relatively low affinity interaction. This electrophysiological evidence for two distinct functional effects of sevoflurane is consistent with the simulation results suggesting multiple sevoflurane binding sites, which might separately account for effects on gating and permeation. A similar paradigm of distinct sites mediating activation and inhibition by anesthetic agents have been found in pentameric ligand-gated ion channels (42, 43).
These findings are also partly consistent with the effects of inhaled anesthetic agents on mammalian NaV channels (10, 11, 17, 18). An important difference, however, is in the recovery from inactivation. Whereas sevoflurane accelerates recovery from inactivation in NaChBac, inhaled anesthetic agents acting on other NaV channels typically slow recovery from inactivation (11, 15). The latter has been generally interpreted as resulting from anesthetic-induced stabilization of the channel’s inactivated state. Although leftward shifted prepulse inactivation curves alone would be consistent with this interpretation, accelerated recovery and other results from our study require an alternate explanation.
Kinetic Modeling.
To explain the results more quantitatively and gain insights into the biophysical basis of the interactions of sevoflurane with multiple sites, we used kinetic modeling of NaChBac gating in the absence and presence of the anesthetic. We assumed a sequential five-state gating scheme based on the study by Kuzmenkin et al. (28). This model consists of four closed (C) states, one open (O) state, and one inactivated (I) state (Fig. 4A). In this scheme, C<>C and C<>O transitions are governed by rate constants that strongly depend on membrane potential and the O<>I transition, which is weakly voltage-dependent (Table S1). This relatively simple scheme was able to reproduce normal NaChBac gating and recapitulate the effects of sevoflurane on NaChBac by making the following minimal assumptions (Table S1): (i) accelerated forward rate constant of voltage-dependent activation, (ii) slightly slowed forward rate constant of inactivation and accelerated backward rate constant of inactivation, and (iii) the presence of low-affinity open channel block. Parameter optimization yielded changes that semiquantitatively accounted for leftward shifted Gp–V and prepulse inactivation curves, accelerated recovery from inactivation, and accelerated current decay at submillimolar and millimolar concentrations of sevoflurane (Table S2 and Fig. S7).
Fig. 4.

Kinetic modeling of NaChBac modulation by sevoflurane. (A and B) Kinetic schemes in the absence (A) and presence (B) of sevoflurane. Parameter values are summarized in Table S1. Note that the rate constants α1 and β1 are strongly voltage-dependent, whereas the rate constants α2 and β2 are only weakly voltage-dependent. Only the binding rate constant kon depends on the concentration of sevoflurane. (C) Simulated NaChBac currents in the absence (black) and presence of 0.2 mM (blue) and 2 mM (red). The depolarizing step is from −100 to 0 mV. (D) Comparison of observed vs. simulated time constants of current decay. Observed data are shown with hatch marks: control (gray), 0.2 mM (blue), and 2 mM (red). (E) Simulated Gp–V relations before (black) and after application of 0.2 mM (blue) and 2 mM (red) sevoflurane. (F) Comparison of observed vs. simulated V1/2 shifts estimated from the corresponding Gp–V relations. Color scheme is as in C. (G) Simulated steady-state inactivation vs. voltage relations before (black) and after application of 0.2 mM (blue) and 2 mM (red) sevoflurane. (H) Comparison of observed vs. simulated V1/2 shifts estimated from the corresponding steady-state inactivation curves. Color scheme is as in B. (I) Simulated trajectories of recovery from inactivation before (black) and after application of 0.2 mM (blue) and 2 mM (red) sevoflurane. (J) Comparison of observed vs. simulated time constants of recovery from inactivation estimated from the corresponding trajectories of recovery.
The combination of MD simulations, electrophysiological investigation, and kinetic modeling strongly support a multisite mechanism of sevoflurane action. Sevoflurane may bind with high affinity to activation gating sites (e.g., sites involving S4–S5 linker sites and the activation gate) and inactivation gate sites (e.g., extracellular sites that influence the selectivity filter). Consequently, NaChBac shifts to a gating mode exhibiting more favorable voltage-dependent activation and inactivation, and a modestly destabilized inactivated state. Sevoflurane may also bind with low affinity to pore sites along the central cavity. This binding is essentially responsible for the acceleration of current decay reflecting slow open channel block. Consistent with anesthetic action responsible for reduced neuronal firing, the combined net impact of these changes might be inhibitory because sevoflurane shifts the prepulse inactivation curve to the left (decreasing channel availability at more negative membrane potentials) and induces pore block. Further analysis by site-directed mutagenesis would help determine the precise contributions of each putative sevoflurane binding site to the effects of sevoflurane on NaChBac function. Also, the eventual direct structural analysis of mammalian NaV channels would be necessary to help validate the significance of these binding sites in general anesthesia.
Conclusion
Understanding of the molecular mechanisms of general anesthetic modulation of NaV channels is critical to interpret their role in general anesthesia. In rat neurohypophysial nerve terminal preparations, isoflurane inhibits Na+ currents and dampens action potentials (44). This inhibition reduces neurotransmitter release (45–48), which may play a critical role in mediating key physiological features of general anesthesia in vivo. In addition, NaV channel modulation may explain anesthetic agent-induced immobilization (19, 20). This study paves the way to map relevant general anesthetic binding regions in NaV channels and helps understand how their modulation by sevoflurane might influence physiological processes implicated in general anesthesia. Apparent activation at low concentrations of sevoflurane is particularly intriguing in light of the observation that patients display excitation during the induction phase, before experiencing the endpoints of anesthesia (49). Perhaps this excitatory phase is mediated in part by activation of NaV channels. Our results also raise the fascinating possibility of a general paradigm whereby modulation of ion channels by small molecules relies on a complex interplay of several modes of action involving distinct binding sites. This speculative generalization is in part corroborated by the fact that an analogous behavior is observed in Cys-loop receptors (50).
Methods
MD Simulations.
Simulations were initialized by using theoretical models of NaChBac in the resting/closed, activated/open, and partially activated/closed conformations obtained previously (51). These homology models were built on the basis of the X-ray crystal structure of NaVAb (Protein Data Bank ID code 3RVY) (26). Each model, embedded in a fully hydrated bilayer of 1-palmytoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine, was equilibrated by brief MD simulation runs. Sevoflurane molecules were initially placed in the aqueous phase with random positions and orientation. MD trajectories were collected for 0.4–0.5 µs using the CHARMM27 force field. Further details are given in SI Methods.
Electrophysiology.
Current recordings were obtained from calcium phosphate transfected HEK-293 cells in the whole-cell patch-clamp configuration. The NaChBac-GFP bicistronic plasmid was a gift from D. Ren (University of Pennsylvania, Philadelphia, PA). All experiments were performed at room temperature (22–25 °C). After fire polishing, patch pipette resistance in bath solution was 1.5–2 MΩ. Passive leak current and transients were subtracted online by using a P/4 procedure. Bath solution contained (in mM): 140 NaCl, 4 KCl, 1.5 CaCl2, 1.5 MgCl2, 10 Hepes, and 5 d-glucose, pH 7.3 adjusted with NaOH. Pipette solution contained (in mM): 15 NaCl, 80 CsF, 40 CsCl, 10 EGTA, and 10 Hepes, pH 7.3 adjusted with CsOH. Sevoflurane containing external solution was prepared by sonication and delivered by a gastight perfusion system as previously described (52). To eliminate inaccuracies caused by membrane lipid retention of anesthetic molecules, each cell was exposed to only one dose of anesthetic agent and washout data were not used. Additional details are provided in SI Methods.
Kinetic Modeling.
Kinetic modeling was performed in IonChannelLab (53) based on the NaChBac kinetic model described previously by Kuzmenkin et al. (28). The goal of kinetic modeling was to approximate the following observations: leftward shifts in the V1/2 values of activation and inactivation, increased rate of recovery from inactivation, and increased rate of inactivation at high anesthetic agent concentrations. Individual rate constants were adjusted manually, and all observed gating properties were evaluated. This process was repeated until best fits to all observations were achieved. Initially, the model parameters were adjusted to approximate the control data in the absence of sevoflurane, and subsequently refined to create a model of sevoflurane modulation at 0.2 mM and 2 mM sevoflurane.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grant National Institute of General Medical Sciences (NIGMS) P01 55876 (to R.G.E., M.C., and M.L.K.), National Institute of Neurological Disorders and Stroke (NINDS) Grant F31 077689 (to A.F.B.), and the Commonwealth of Pennsylvania. The computations were performed using resources from Extreme Science and Engineering Discovery Environment (XSEDE, www.xsede.org/high-performance-computing) Grant MCA93S020 (to M.L.K.) and the Temple University High-Performance Computing System purchased in part with NSF Grant MRI-R2 0958854 (to M.L.K.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405768111/-/DCSupplemental.
References
- 1.Hemmings HC, Jr, et al. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26(10):503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Franks NP. Molecular targets underlying general anaesthesia. Br J Pharmacol. 2006;147(Suppl 1):S72–S81. doi: 10.1038/sj.bjp.0706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9(5):370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- 4.Urban BW. The site of anesthetic action. Handbook Exp Pharmacol. 2008;183(182):3–29. doi: 10.1007/978-3-540-74806-9_1. [DOI] [PubMed] [Google Scholar]
- 5.Krasowski MD, Harrison NL. General anaesthetic actions on ligand-gated ion channels. Cell Mol Life Sci. 1999;55(10):1278–1303. doi: 10.1007/s000180050371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel AJ, et al. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci. 1999;2(5):422–426. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- 7.Cacheaux LP, et al. Impairment of hyperpolarization-activated, cyclic nucleotide-gated channel function by the intravenous general anesthetic propofol. J Pharmacol Exp Ther. 2005;315(2):517–525. doi: 10.1124/jpet.105.091801. [DOI] [PubMed] [Google Scholar]
- 8.Ratnakumari L, Hemmings HC., Jr Inhibition of presynaptic sodium channels by halothane. Anesthesiology. 1998;88(4):1043–1054. doi: 10.1097/00000542-199804000-00025. [DOI] [PubMed] [Google Scholar]
- 9.Ouyang W, Wang G, Hemmings HC., Jr Isoflurane and propofol inhibit voltage-gated sodium channels in isolated rat neurohypophysial nerve terminals. Mol Pharmacol. 2003;64(2):373–381. doi: 10.1124/mol.64.2.373. [DOI] [PubMed] [Google Scholar]
- 10.OuYang W, Hemmings HC., Jr Isoform-selective effects of isoflurane on voltage-gated Na+ channels. Anesthesiology. 2007;107(1):91–98. doi: 10.1097/01.anes.0000268390.28362.4a. [DOI] [PubMed] [Google Scholar]
- 11.Ouyang W, Herold KF, Hemmings HC., Jr Comparative effects of halogenated inhaled anesthetics on voltage-gated Na+ channel function. Anesthesiology. 2009;110(3):582–590. doi: 10.1097/ALN.0b013e318197941e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herold KF, Nau C, Ouyang W, Hemmings HC., Jr Isoflurane inhibits the tetrodotoxin-resistant voltage-gated sodium channel Nav1.8. Anesthesiology. 2009;111(3):591–599. doi: 10.1097/ALN.0b013e3181af64d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yokoyama T, et al. Effects of sevoflurane on voltage-gated sodium channel Na(v)1.8, Na(v)1.7, and Na(v)1.4 expressed in Xenopus oocytes. J Anesth. 2011;25(4):609–613. doi: 10.1007/s00540-011-1167-7. [DOI] [PubMed] [Google Scholar]
- 14.Herold KF, Hemmings HC., Jr Sodium channels as targets for volatile anesthetics. Front Pharmacol. 2012;3:50. doi: 10.3389/fphar.2012.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouyang W, Jih T-Y, Zhang T-T, Correa AM, Hemmings HC., Jr Isoflurane inhibits NaChBac, a prokaryotic voltage-gated sodium channel. J Pharmacol Exp Ther. 2007;322(3):1076–1083. doi: 10.1124/jpet.107.122929. [DOI] [PubMed] [Google Scholar]
- 16.Horishita T, Eger EI, 2nd, Harris RA. The effects of volatile aromatic anesthetics on voltage-gated Na+ channels expressed in Xenopus oocytes. Anesth Analg. 2008;107(5):1579–1586. doi: 10.1213/ane.0b013e318184b966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bean BP, Shrager P, Goldstein DA. Modification of sodium and potassium channel gating kinetics by ether and halothane. J Gen Physiol. 1981;77(3):233–253. doi: 10.1085/jgp.77.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rehberg B, Xiao YH, Duch DS. Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anesthetics. Anesthesiology. 1996;84(5):1223–1233. doi: 10.1097/00000542-199605000-00025. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, et al. Intrathecal veratridine administration increases minimum alveolar concentration in rats. Anesth Analg. 2008;107(3):875–878. doi: 10.1213/ane.0b013e3181815fbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, et al. Bidirectional modulation of isoflurane potency by intrathecal tetrodotoxin and veratridine in rats. Br J Pharmacol. 2010;159(4):872–878. doi: 10.1111/j.1476-5381.2009.00583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hille B. Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer; 2001. [Google Scholar]
- 22.Ren D, et al. A prokaryotic voltage-gated sodium channel. Science. 2001;294(5550):2372–2375. doi: 10.1126/science.1065635. [DOI] [PubMed] [Google Scholar]
- 23.Koishi R, et al. A superfamily of voltage-gated sodium channels in bacteria. J Biol Chem. 2004;279(10):9532–9538. doi: 10.1074/jbc.M313100200. [DOI] [PubMed] [Google Scholar]
- 24.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature. 2012;486(7401):130–134. doi: 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486(7401):135–139. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCusker EC, et al. Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat Commun. 2012;3:1102. doi: 10.1038/ncomms2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzmenkin A, Bezanilla F, Correa AM. Gating of the bacterial sodium channel, NaChBac: Voltage-dependent charge movement and gating currents. J Gen Physiol. 2004;124(4):349–356. doi: 10.1085/jgp.200409139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y, Scheuer T, Catterall WA. Reversed voltage-dependent gating of a bacterial sodium channel with proline substitutions in the S6 transmembrane segment. Proc Natl Acad Sci USA. 2004;101(51):17873–17878. doi: 10.1073/pnas.0408270101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavlov E, et al. The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys J. 2005;89(1):232–242. doi: 10.1529/biophysj.104.056994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanchet J, Pilote S, Chahine M. Acidic residues on the voltage-sensor domain determine the activation of the NaChBac sodium channel. Biophys J. 2007;92(10):3513–3523. doi: 10.1529/biophysj.106.090464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, Catterall WA. Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci USA. 2008;105(39):15142–15147. doi: 10.1073/pnas.0806486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paldi T, Gurevitz M. Coupling between residues on S4 and S1 defines the voltage-sensor resting conformation in NaChBac. Biophys J. 2010;99(2):456–463. doi: 10.1016/j.bpj.2010.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irie K, et al. Comparative study of the gating motif and C-type inactivation in prokaryotic voltage-gated sodium channels. J Biol Chem. 2010;285(6):3685–3694. doi: 10.1074/jbc.M109.057455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarov-Yarovoy V, et al. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA. 2012;109(2):E93–E102. doi: 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeCaen PG, Yarov-Yarovoy V, Scheuer T, Catterall WA. Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci USA. 2011;108(46):18825–18830. doi: 10.1073/pnas.1116449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee S, Goodchild SJ, Ahern CA. Local anesthetic inhibition of a bacterial sodium channel. J Gen Physiol. 2012;139(6):507–516. doi: 10.1085/jgp.201210779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265(5179):1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 39.Raju SG, Barber AF, LeBard DN, Klein ML, Carnevale V. Exploring volatile general anesthetic binding to a closed membrane-bound bacterial voltage-gated sodium channel via computation. PLOS Comput Biol. 2013;9(6):e1003090. doi: 10.1371/journal.pcbi.1003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vemparala S, Domene C, Klein ML. Interaction of anesthetics with open and closed conformations of a potassium channel studied via molecular dynamics and normal mode analysis. Biophys J. 2008;94(11):4260–4269. doi: 10.1529/biophysj.107.119958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barber AF, Liang Q, Amaral C, Treptow W, Covarrubias M. Molecular mapping of general anesthetic sites in a voltage-gated ion channel. Biophys J. 2011;101(7):1613–1622. doi: 10.1016/j.bpj.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauguet L, et al. Structural basis for potentiation by alcohols and anaesthetics in a ligand-gated ion channel. Nat Commun. 2013;4:1697. doi: 10.1038/ncomms2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brömstrup T, Howard RJ, Trudell JR, Harris RA, Lindahl E. Inhibition versus potentiation of ligand-gated ion channels can be altered by a single mutation that moves ligands between intra- and intersubunit sites. Structure. 2013;21(8):1307–1316. doi: 10.1016/j.str.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ouyang W, Hemmings HC., Jr Depression by isoflurane of the action potential and underlying voltage-gated ion currents in isolated rat neurohypophysial nerve terminals. J Pharmacol Exp Ther. 2005;312(2):801–808. doi: 10.1124/jpet.104.074609. [DOI] [PubMed] [Google Scholar]
- 45.Westphalen RI, Hemmings HC., Jr Volatile anesthetic effects on glutamate versus GABA release from isolated rat cortical nerve terminals: 4-aminopyridine-evoked release. J Pharmacol Exp Ther. 2006;316(1):216–223. doi: 10.1124/jpet.105.090662. [DOI] [PubMed] [Google Scholar]
- 46.Westphalen RI, Yu J, Krivitski M, Jih T-Y, Hemmings HC., Jr Regional differences in nerve terminal Na+ channel subtype expression and Na+ channel-dependent glutamate and GABA release in rat CNS. J Neurochem. 2010;113(6):1611–1620. doi: 10.1111/j.1471-4159.2010.06722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westphalen RI, Kwak N-B, Daniels K, Hemmings HC., Jr Regional differences in the effects of isoflurane on neurotransmitter release. Neuropharmacology. 2011;61(4):699–706. doi: 10.1016/j.neuropharm.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westphalen RI, Desai KM, Hemmings HC., Jr Presynaptic inhibition of the release of multiple major central nervous system neurotransmitter types by the inhaled anaesthetic isoflurane. Br J Anaesth. 2013;110(4):592–599. doi: 10.1093/bja/aes448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vlajkovic GP, Sindjelic RP. Emergence delirium in children: Many questions, few answers. Anesth Analg. 2007;104(1):84–91. doi: 10.1213/01.ane.0000250914.91881.a8. [DOI] [PubMed] [Google Scholar]
- 50.Brannigan G, LeBard DN, Hénin J, Eckenhoff RG, Klein ML. Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc Natl Acad Sci USA. 2010;107(32):14122–14127. doi: 10.1073/pnas.1008534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barber AF, et al. Hinge-bending motions in the pore domain of a bacterial voltage-gated sodium channel. Biochim Biophys Acta. 2012;1818(9):2120–2125. doi: 10.1016/j.bbamem.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barber AF, Liang Q, Covarrubias M. Novel activation of voltage-gated K(+) channels by sevoflurane. J Biol Chem. 2012;287(48):40425–40432. doi: 10.1074/jbc.M112.405787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santiago-Castillo JAD, Covarrubias M, Sánchez-Rodríguez JE, Perez-Cornejo P, Arreola J. Simulating complex ion channel kinetics with IonChannelLab. Channels (Austin) 2010;4(5):422–428. doi: 10.4161/chan.4.5.13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.