

Significance
Inhibitors of microsomal prostaglandin E synthase-1 (mPGES-1) are being developed as analgesics. Although global depletion of mPGES-1 suggests they will be less likely to confer a cardiovascular hazard than NSAIDs selective for inhibition of COX-2, mPGES-1–derived PGE2 may have contrasting effects on discrete cellular components of the vasculature. mPGES-1 inhibition may result in substrate rediversion to other PG synthases, the products of which differ between cell types and exert contrasting biologies. Here, myeloid cell mPGES-1, reflective of the macrophage enzyme, promotes atherogenesis, fostering inflammation and oxidative stress. By contrast, mPGES-1 in endothelial and vascular smooth muscle cells has no discernable effect. Selective targeting of macrophage mPGES-1 may conserve cardiovascular benefit while avoiding adverse effects related to enzyme blockade in other tissues.
Keywords: atherosclerosis, prostanoid
Abstract
Microsomal prostaglandin E synthase-1 (mPGES-1) in myeloid and vascular cells differentially regulates the response to vascular injury, reflecting distinct effects of mPGES-1–derived PGE2 in these cell types on discrete cellular components of the vasculature. The cell selective roles of mPGES-1 in atherogenesis are unknown. Mice lacking mPGES-1 conditionally in myeloid cells (Mac-mPGES-1-KOs), vascular smooth muscle cells (VSMC-mPGES-1-KOs), or endothelial cells (EC-mPGES-1-KOs) were crossed into hyperlipidemic low-density lipoprotein receptor-deficient animals. En face aortic lesion analysis revealed markedly reduced atherogenesis in Mac-mPGES-1-KOs, which was concomitant with a reduction in oxidative stress, reflective of reduced macrophage infiltration, less lesional expression of inducible nitric oxide synthase (iNOS), and lower aortic expression of NADPH oxidases and proinflammatory cytokines. Reduced oxidative stress was reflected systemically by a decline in urinary 8,12-iso-iPF2α-VI. In contrast to exaggeration of the response to vascular injury, deletion of mPGES-1 in VSMCs, ECs, or both had no detectable phenotypic impact on atherogenesis. Macrophage foam cell formation and cholesterol efflux, together with plasma cholesterol and triglycerides, were unchanged as a function of genotype. In conclusion, myeloid cell mPGES-1 promotes atherogenesis in hyperlipidemic mice, coincident with iNOS-mediated oxidative stress. By contrast, mPGES-1 in vascular cells does not detectably influence atherogenesis in mice. This strengthens the therapeutic rationale for targeting macrophage mPGES-1 in inflammatory cardiovascular diseases.
Nonsteroidal anti-inflammatory drugs (NSAIDs) reduce pain and inflammation by suppressing the formation of proinflammatory prostaglandins (PGs), particularly prostaglandin E2 (PGE2) formed by cyclooxygenase-2 (COX-2) (1). However, the development of NSAIDs specific for inhibition of COX-2 revealed a cardiovascular hazard attributable to suppression of cardioprotective PGs, especially prostacyclin (PGI2) (2). This risk appears to extend to some of the older NSAIDs, like diclofenac, that also inhibit specifically COX-2 (3, 4). These developments prompted interest in microsomal PGE synthase (mPGES)-1 as a downstream alternative drug target to COX-2 (5): it is the dominant source among PGES enzymes in the biosynthesis of PGE2 (6). Unlike NSAIDs, inhibitors of mPGES-1 would spare PGI2 from suppression. Indeed, blockade or deletion of mPGES-1 results in accumulation of its PGH2 substrate, rendering it available for metabolism by other PG synthases, including PGI2 synthase (PGIS) (7).
Consistent with these observations, we have found that whereas deletion of COX-2 in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) renders mice susceptible to thrombosis and hypertension (2), deletion of mPGES-1 in vascular cells has no such effect (8). Indeed, global deficiency of mPGES-1 restrains atherogenesis (9), the proliferative response to vascular injury (10) and angiotensin-induced aortic aneurysm formation (11) in mice.
Despite this attractive cardiovascular profile, mPGES-1 is a complex drug target. The dominant prostanoid products of substrate rediversion differ among cell types. For example, whereas PGI2 might be augmented in vascular cells, the consequence of mPGES-1 blockade in other cells might be an increase in thromboxane (Tx)A2, a PG that promotes platelet activation, vasoconstriction, and atherogenesis (9). Even if an increase in PGI2 afforded a desirable cardiovascular profile, it might undermine the analgesic efficacy of mPGES-1 inhibitors. Although the impacts of global deletion of mPGES-1 and COX-2 in many mouse models of analgesia are indistinguishable (12, 13), in some, PGI2 rather than PGE2 predominates (14) and thus may be the dominant mediator in certain subtypes of human pain. Finally, the consequences of PGE2 suppression might differ between cell types. PGE2 activates four E prostanoid (EP) receptors with contrasting intracellular signaling and consequent biology (15, 16). Indeed, the contrasting effect of mPGES-1 deletion in myeloid vs. vascular cells on the proliferative response to vascular injury reflects the differential consequences of EP activation rather than substrate rediversion (8).
A potentially discriminating feature among inhibitors of COX-2 and mPGES-1 is their effect on atherosclerosis. Global postnatal deletion of COX-2 accelerates atherogenesis in hyperlipidemic mice (17), an observation that accords with a similar effect of deleting the PGI2 receptor (the IP) (18, 19) and with the delayed detection of a cardiovascular hazard in randomized trials of COX-2 inhibitors in patients initially selected for being at low cardiovascular risk (20). By contrast, global deletion of mPGES-1 restrains atherogenesis in mice; in this case suppression of PGE2 coincides with an increase in biosynthesis of PGI2 (9). Here, we wished to segregate the effects on atherosclerosis of mPGES-1 depletion in myeloid from vascular cells. Our results strengthen the rationale for targeting macrophage mPGES-1 in the treatment of inflammatory cardiovascular disease.
Results
mPGES-1 Deletion in Myeloid Cells Attenuates Atherogenesis.
Starting at 2 mo of age, mice of both genders were fed a high-fat diet (HFD) for 3 mo or 6 mo. Compared with either of the control groups, WT1 or WT2 (Materials and Methods), the atherosclerotic lesion burden was markedly reduced in Mac-mPGES-1-KOs in both male (Fig. 1A) and female mice (Fig. 1B). No difference was observed between WT1 and WT2; therefore for all of the following experiments, we used WT1 as controls, terming them Mac-mPGES-1-WT. Weight gain and plasma total cholesterol and triglyceride levels were not affected by myeloid mPGES-1 deletion in either sex at any time point (Fig. S1).
Fig. 1.

(A and B) Effects of myeloid cell mPGES-1 deletion on atherosclerotic lesion formation in male (A) and female (B) hyperlipidemic mice. (Upper) The extent of aortic atherosclerotic lesion formation, represented by the ratio of lesion area to total aortic area, was quantified by en face analysis of aortas from mice after 3 mo or 6 mo on a HFD. (Lower) Representative en face graphs are shown with each vertically matched to the corresponding dataset in the Upper panel (*P < 0.05, **P < 0.01, ***P < 0.001, n = 8–16).
LysMCre mice express Cre recombinase in all myeloid cells, including macrophages, neutrophils, and some dendritic cells (21). Most recently, we reported that deficiency of mPGES-1 in macrophages results in PGE2 suppression, accompanied by an increased production of PGI2 and TxB2 (8). Here we examined the leukotriene (LT) profile. After lipopolysaccharide (LPS) exposure, a modest absolute increase in LTE4 was observed in the knockout cells. LTC4 was unaltered significantly by mPGES-1 deletion and LTB4 and LTD4 were undetectable (Fig. S2). A small capacity for PGE2 production in neutrophils was not detectably altered by enzyme deletion (Fig. S3A). Similarly, a suggestion of lower PGE2 in dendritic cells from knockouts (Fig. S3B) was tempered by the trivial amount formed. The absolute level (0.46 ± 0.08 ng⋅mL−1⋅1.5 × 106 cells−1) is ∼47-fold lower than in macrophages (14.61 ± 0.32 ng⋅mL−1⋅1 × 106 cells−1) (8). We failed to see any significant product rediversion to PGI2 or TxB2 in either neutrophils or dendritic cells and leukotriene formation (LTB4, -C4, -D4, and -E4) was not detected (Fig. S3 A and B).
Despite a reduction in macrophage PGE2 formation (8), systemic production of PGE2 and PGI2, reflected by their major urinary metabolites 11α-hydroxy-9,15-dioxo-2,3,4,5-tetranor-prostane-1,20-dioic acid (PGE-M) and 2,3-dinor 6-keto PGF1α (PGI-M), was not significantly affected by myeloid mPGES-1 deletion in hyperlipidemic mice (Fig. S4 A and B). Modest product rediversion to PGD2 (PGD-M: 11,15-dioxo-9-hydroxy-2,3,4,5-tetranor-prostan-1,20-dioic acid) and TxA2 (Tx-M: reflected by 2,3-dinor TxB2) was observed in 3-mo female and 6-mo male groups, respectively (Fig. S4 C and D). Notably, the female mice showed lower PGE-M and higher Tx-M excretion than males at both 3 mo and 6 mo (Fig. S4 A and D), consistent with our previous report (9). Lower PGD-M and PGI-M excretion was also observed in female mice after 6 mo HFD feeding (Fig. S4 B and C). The contribution of these differences to the sex-dependent rate of atherogenesis in hyperlipidemic mice is unknown.
mPGES-1 Deletion in Vascular Cells Does Not Influence Atherogenesis.
In contrast to the functional importance of mPGES-1 in myeloid cells, deletion of mPGES-1 in vascular cells (VSMCs, ECs, or both) had no detectable phenotypic impact on lesion burden in either sex (Fig. 2 A and B). Body weight, plasma lipid levels (Fig. S5), and the urinary production of prostanoid metabolites (Fig. S6) were all unchanged by vascular mPGES-1deletion.
Fig. 2.

(A and B) Effects of vascular cell mPGES-1 deletion on atherogenesis in male (A) and female (B) hyperlipidemic mice. The extent of aortic atherosclerotic lesion formation, represented by the ratio of lesion area to total aortic area, was quantified by en face analysis of aortas from mice with 3 mo HFD feeding (n = 9–15).
Morphology Consequent to Macrophage mPGES-1 Deletion in Low-Density Lipoprotein Receptor-Deficient Mice.
Genotypic distinctions in morphology are often less evident at more advanced stages of atherogenesis; thus we performed lesional analysis in the 3-mo cohort, using plaques of comparable size obtained at the aortic root. Staining with the pan-macrophage marker, CD11b, was not significantly altered by mPGES-1 deletion (Fig. 3 A, a and b, and B). By contrast, staining for inducible nitric oxide synthase (iNOS), a marker of classically activated type 1 macrophages (M1), was strikingly reduced in lesions from the Mac-mPGES-1-KO mice compared with WT controls (Fig. 3 A, c and d, and B). Robust iNOS staining was observed in almost all of the WT lesions, localized to areas rich in fibronectin staining (activated SMCs and fibrotic caps), and mixed regions contained fibronectin and macrophages (CD11b staining). Overall iNOS staining was found in the same types of lesions in the KO samples; however, the signal was much lower. No difference was observed between KOs and WTs in staining for the alternative activated type 2 macrophages (M2), as reflected by CD206 (Fig. 3 A, e and f, and B). VSMC content (contractile α-SMA or synthetic VCAM-1), fibronectin abundance, and collagen-rich areas were also comparable (Fig. S7). Despite the relevance of iNOS to blood pressure regulation in mice (22), systolic blood pressure was not changed in Mac-mPGES-1-KO mice, either at baseline or after 3 mo on a HFD (Fig. S8). The time to complete common carotid artery occlusion after photochemical injury was also unaltered by myeloid mPGES-1 deletion (Fig. S9). Plaque macrophage content through the whole aorta was significantly reduced in the KOs at both 3 mo and 6 mo on a HFD (Fig. 3C), as determined by fluorescence imaging of the distribution of 1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide (DiR-HDL) nanoparticles (23).
Fig. 3.
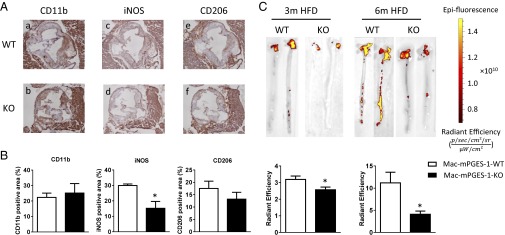
Effects of myeloid cell mPGES-1 deletion on macrophage abundance. (A) Lesion morphology analysis in aortic roots from mice on a HFD for 3 mo. Shown are representative images for CD11b (a), iNOS (c), and CD206 (e) staining in Mac-mPGES-1-WT aortic root sections and CD11b (b), iNOS (d), and CD206 (f) staining in Mac-mPGES-1-KO sections. (B) Quantification of immunohistochemistry staining of CD11b, iNOS, and CD206 (*P < 0.05, n = 4–5). (C, Upper) Representative pictures of the Spectrum fluorescence imaging of aortas from mice injected with DiR-HDL nanoparticles. (Lower) Quantification data (*P < 0.05, n = 2–5).
Macrophage mPGES-1 Deletion Coincides with Reduced Oxidative Stress During Atherogenesis.
Coincident with reduced iNOS immunoreactivity and impaired macrophage accumulation in Mac-mPGES-1-KO mice, freshly isolated thoracic aortas from the KOs revealed decreased expression of iNOS mRNA and the macrophage marker CD68 compared with the controls (Fig. 4A). Expression of TNFα and monocyte chemoattractant protein 1 (MCP-1) was also significantly reduced in the KOs (Fig. 4A). Myeloid cell depletion of mPGES-1 also depressed the oxidant stress attendant to atherogenesis. Thus, RT-PCR analysis revealed decreased expression in the KOs of the gp91phox (NOX-2), p47phox, and p22phox subunits of NADPH oxidase (Fig. 4B), whereas expression of NOX-1 and NOX-4 was unchanged. The antioxidant effect of myeloid mPGES-1 deletion was also reflected systemically by a reduction in urinary 8,12-iso-iPF2α-VI (Fig. 4C), a sensitive index of lipid peroxidation in vivo (24).
Fig. 4.

Myeloid cell mPGES-1 deletion coincides with a reduced proinflammatory response and reduced oxidative stress during atherogenesis. (A) Real-time RT-PCR analysis of CD68, iNOS, TNFα, and MCP-1 expression in thoracic aortas from mice treated with HFD for 3 mo (n = 4, *P < 0.05). (B) Real-time RT-PCR analysis of NADPH oxidase subunits expression in thoracic aortas from mice treated with HFD for 3 mo (n = 4, *P < 0.05, **P < 0.01, ***P < 0.01). (C) Urinary 8,12-iso-iPF2α-VI in mice treated with a HFD for 3 mo (n = 12–13, *P < 0.05).
mPGES-1–Deficient Macrophages Showed Reduced Potential for Migration and Unaltered Lipid Handling.
Although random macrophage migration in a Boyden chamber assay was unaltered by mPGES-1 depletion, migration induced by MCP-1 was markedly reduced (Fig. 5A). By contrast, uptake of acetylated LDL was unaltered (Fig. 5B), consistent with unaltered expression of several genes relevant to foam cell formation and cholesterol efflux, such as CD36, SR-A, ABCA1, and ABCG1 (Fig. 5C).
Fig. 5.

Mac-mPGES-1-KO macrophages showed reduced migration potential and unaltered lipid handling in vitro. (A) Cells were placed in modified Boyden chambers and chemotaxis was assessed with or without exposure to 10 ng/mL MCP-1; the KO cells showed significantly reduced migration to MCP-1 (n = 6, *P < 0.05). (B) Cells were treated with 10 µg/mL DiI-Ac-LDL for 4 h and uptake was assessed. Representative fluorescence images showed no difference between WT and KO cells. DAPI staining was used as a control (n = 4). (C) Cells were treated with or without 50 µg/mL ox-LDL for 24 h; real-time RT-PCR showed no difference between WT and KO cells for CD36, SR-A, ABCA1, and ABCG1 expression (n = 5–10).
Discussion
NSAIDs are among the most commonly consumed drugs worldwide, affording relief from pain and inflammation by suppressing COX-dependent PG formation, particularly PGE2. However, placebo-controlled trials reveal that NSAIDs specifically developed for inhibition of COX-2 confer a cardiovascular hazard, consisting of thrombosis, heart failure, hypertension, and sudden cardiac death (25). Indirect lines of evidence suggest that this risk extends to some of the older NSAIDs, such as diclofenac (3, 4), and is attributable to suppression of COX-2–derived PGI2 (2, 18, 26).
These observations increased interest in mPGES-1 as an alternative drug target. In principle, targeting this enzyme, downstream of the COXs and often coregulated by inflammatory cytokines with COX-2 (27, 28), has two principal advantages. First, it is the dominant source of PGE2 formation in vivo and, unlike NSAIDs, inhibition of mPGES-1 would leave PGI2 formation unscathed. Indeed, PGH2 substrate rediversion might enhance formation of this cardioprotective compound and neutralize the cardiovascular risk observed with NSAIDs. Results obtained in mice globally deficient in mPGES-1 have been encouraging. Analgesic effects have often been indistinguishable from those of NSAIDs or those consequent to COX-2 inhibition (12, 13). The predisposition to thrombosis and hypertension observed in mice treated with COX-2 inhibitors (29, 30) or lacking the IP (31) is absent in mPGES-1 KO mice (32) and a similar contrast is evident when deletion of COX-2 vs. mPGES-1 is confined to ECs or VSMCs (2, 8). It is possible that the distinction with respect to hypertension is relative rather than absolute as there have been some reports of evoked hypertension consequent to mPGES-1 deletion (33–36). Also, inhibition of mPGES-1 may be undesirable in the setting of acute myocardial infarction, when inflammation appears to contribute to the healing process (37). Finally, deletion of mPGES-1 restrains atherogenesis, the response to vascular injury, and angiotensin-induced aneurysm formation (9–11), whereas atherogenesis is accelerated but aneurysm formation and vascular injury responses are attenuated by disruption of COX-2 (17, 38, 39).
Despite these observations, mPGES-1 is a complicated drug target. Due to the possibilities of substrate rediversion to PGs with contrasting biological effects and to varied EP expression in different tissues, global deletion of mPGES-1 or systemic drug administration may conceal quite distinct tissue-dependent consequences of mPGES-1 blockade. For example, recently we found that whereas myeloid cell mPGES-1 promoted the tissue response to vascular injury, this was attenuated by EC and VSMC mPGES-1 (8). In this case, the results were attributable to differential consequences of EP activation in the two cell types, rather than contrasting products of PGH2 diversion.
Here we show that myeloid cell mPGES-1 promotes atherogenesis, fostering M1 macrophage lesional infiltration, oxidant stress, and attendant proinflammatory cytokine formation. In contrast to its attenuating role in the proliferative response to vascular injury, mPGES-1 in ECs and VSMCs has no detectable impact on atherogenesis. These observations and our results with vascular injury (8) raise the possibility that the broader cardiovascular efficacies observed with global mPGES-1 deletion might be conserved by targeting macrophage mPGES-1. The contribution from LysMCre-mediated mPGES-1 deletion in dendritic cells and neutrophils is trivial, so phenotypes observed with myeloid cell deletion are largely attributable to macrophages.
Whereas augmented PGI2 formation has been observed in mPGES-1 global KO mice coincident with restraint of atherogenesis, its functional contribution to this phenotype has not been established. Here we recapitulate this atherogenic phenotype without evidence of an alteration in systemic biosynthesis of PGI2 by targeting the macrophage. This has the theoretical attraction of avoiding a perturbation that might dilute the analgesic efficacy of mPGES-1 inhibition. PGI2, like PGE2, may mediate pain and appears to dominate over PGE2 in some mouse models of analgesia (14). Although a modest rediversion to TxA2 biosynthesis was observed, thrombogenesis is not accelerated by myeloid mPGES-1 deletion. Blood pressure was also unaltered by myeloid mPGES-1 deletion, recapitulating the effects of global mPGES-1 deletion. Several strategies have emerged that favor drug delivery to activated macrophages, such as those relevant to the antiatherogenic impact of myeloid mPGES-1 deletion, including linkage to ligands for the folate receptor (40, 41) and the use of HDL-based nanoparticles (23, 42). Given that people treated with NSAIDs and mPGES-1 inhibitors are likely already to have a burden of atherosclerotic disease, the more relevant experiment will be whether a small molecule inhibitor of macrophage mPGES-1 can induce regression of established disease, rather than merely restrain atherogenesis.
Inhibition of mPGES-1 presents a considerable challenge in drug development. However, the general conservation of NSAID analgesic efficacy, the more benign cardiovascular profile in preclinical models that were predictive of NSAID-induced clinical cardiotoxicity, and the suggestion of efficacy in some inflammatory cardiovascular syndromes are encouraging. The present results strengthen the evidence that targeting macrophage mPGES-1 may be a strategy further to refine efficacy while limiting adverse effects attributable to enzyme inhibition in other tissues.
Materials and Methods
Animals.
All animal studies were performed in accordance with the guidelines approved by the Institute for Animal Care and Use Committee at the University of Pennsylvania.
For myeloid cell mPGES-1 deletion, as previously described (8), mPGES-1f/f mice, kindly provided by Mohammad Bohlooly of Astra Zeneca, were crossed with LysMCre mice to yield mice with mPGES-1 selectively deleted in myeloid cells, dominantly macrophages. These mice were further crossed with low-density lipoprotein receptor-deficient (LDLR−/−) mice to generate LysMCre, mPGES-1f/f, LDLR−/− mice (Mac-mPGES-1-KO). LysMCre-mediated recombination results in deletion of the targeted gene in the myeloid cell lineage, whereas the LysMCre transgenic mice were considered as Lysozyme-M null mice as well (43). We used mice that do or do not express LysMCre as the two wild-type controls for Mac-mPGES-1-KOs. mPGES-1f/f, LDLR−/− without LysMCre mice were used as WT1 (Mac-mPGES-1-WT1), whereas LysMCre, LDLR−/− without mPGES-1f/f mice were used as WT2 (Mac-mPGES-1-WT2).
For vascular cell mPGES-1 deletion, mPGES-1f/f mice were crossed with SM22Cre and/or Tie2Cre mice to yield mice with mPGES-1 selectively deleted in VSMCs, ECs, or both as described (8). These mice were crossed with LDLR−/− mice to generate SM22Cre, mPGES-1f/f, LDLR−/− mice (SM22 Cre); Tie2Cre, mPGES-1f/f, LDLR−/− mice (Tie2 Cre); or SM22Cre, Tie2Cre, mPGES-1f/f, LDLR−/− (double Cre) mice; the mPGES-1f/f, LDLR−/− mice (no Cre) were used as their littermate controls.
Quantification of Atherosclerosis.
All mice were fed a HFD (0.2% cholesterol/21% saturated fat; formula TD 88137, Harlan Teklad) from 2 mo of age for 3 mo or 6 mo. Mouse aortic trees were prepared and stained and atherosclerotic lesions were quantified as described (17). Briefly, the entire aorta from the aortic root to the iliac bifurcation was collected and fixed in formalin-free fixative Prefer (Anatech LTD). Adventitial fat was removed. The aorta was opened longitudinally, stained with Sudan IV (Sigma), and pinned down on black wax to expose the intima. The extent of atherosclerosis was determined by the en face lesion area percentage to the entire intimal area.
Immunohistochemical Analysis of Lesion Morphology.
Mouse hearts were embedded in optimal cutting temperature compound, and 10-μm serial sections of the aortic root were cut and mounted on Superfrost Plus slides (Fisher Scientific). Samples were fixed in acetone for 15 min at −20 °C. Before treatment with the first antibody, sections were consecutively treated to block endogenous peroxidase [3% (vol/vol) H2O2 for 15 min], with 10% (vol/vol) normal serum blocking solution (dependent on host of secondary antibody, in 1% BSA/PBS for 15 min), and for endogenous biotin blocking (streptavidin–biotin blocking kit; Vector Laboratories). Sections were then incubated with the desired primary antibody in blocking solution overnight at 4 °C. Samples were individually stained for collagen type I (1 μg/mL; 1310-01, Southern Biotech), fibronectin (0.3 μg/mL; F3648, Sigma), α-SMA (12.3μg/mL; F3777, Sigma), VCAM-1 (10 μg/mL; 553331, BD Bioscience), CD11b (5 μg/mL; 557395, BD Bioscience), CD206 (1 μg/mL; MCA2235GA, Serotech), and iNOS (0.5 μg/mL; 15323, Abcam), all with isotype-matched controls. Where required, sections were then incubated with biotinylated-IgG secondary antibody (specific to host of primary antibody, all 1 μg/mL; Vector Laboratories) diluted in 1% BSA/PBS for 1 h at room temperature. Sections were then incubated with streptavidin-horseradish peroxidase (1 μg/mL; 016-030-084, Jackson Immunoresearch) diluted in 1% BSA/PBS for 30 min at room temperature. Slides were equilibrated in sterile H2O for 5 min and then developed using the DAB substrate kit (K3468; Dako) per the manufacturer’s protocol. Samples were then counterstained with hematoxylin, dehydrated, and mounted in Cytoseal-60 (12-547; Fisher Scientific). Images were captured on a Nikon Eclipse 80i upright microscope at 10× magnification, using Nikon NIS-Elements software.
DiR-HDL Nanoparticle Experiments.
To assess macrophage accumulation in whole aortas, Mac-mPGES-1-WT and Mac-mPGES-1-KO mice received an i.v. (tail vein) injection of high-density lipoprotein nanoparticles labeled with fluorophore DiR-HDL (D-12731; Invitrogen) in 0.9% sterile saline at 1 µmol DiR/kg. Twenty-four hours later, the mice were killed and whole aortas were dissected and subjected to the near infrared fluorescence immediately, using an IVIS Spectrum fluorescence imaging system, as described in ref. 23.
Cell Culture Experiments.
Thioglycollate-elicited peritoneal macrophages from Mac-mPGES-1-WT and Mac-mPGES-1-KO mice were harvested and cultured as previously described (17). For the Boyden chamber assay, cells were added to the top wells of Costar Transwell modified Boyden chambers (6.5-mm diameter tissue culture-treated polycarbonate membranes containing 8-µm pores; Corning). Media with or without 10 ng/mL MCP-1 were placed in the lower chambers. After allowing cell migration for 24 h, cells that remained in the top wells were scraped off, using cotton swabs, and all remaining cells were stained with 0.1% crystal violet (Sigma) for 20 min. Cells were then washed off by 1% deoxycholic acid (Sigma) solution and absorbance was measured at 595 nm. For the 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyane perchlorate (DiI)-labeled acetylated LDL (Ac-LDL) uptake assay, cells were incubated with 10 µg/mL DiI-Ac-LDL for 4 h at 37°. Then cells were washed with PBS twice and visualized under a fluorescence microscope. DAPI was used for nuclei staining. For real-time RT-PCR analysis, cells were treated with or without 50 μg/mL oxidized LDL (ox-LDL) for 24 h. Total RNA was prepared using the RNeasy Mini kit (Qiagen) and was reverse transcribed into cDNA, using M-MLV reverse transcriptase (Promega). Taqman universal PCR Master Mix and probes (Applied Biosystems) were used for real-time PCR.
Neutrophils were harvested by lavage of the peritoneal cavity with 5 mL of cold PBS 4 h after injection of 1 mL 3% (wt/vol) thioglycollate broth into the peritoneal cavity of the mice as described in ref. 44. The cells were pelleted after lysis of the residual red blood cells (ACK lysing buffer; Life Technologies). Resuspended cells were then incubated in RPMI 1640 supplemented with 10% (vol/vol) FBS for 2 h. Nonadherent cells were then stimulated with 5 μg/mL LPS for 24 h and supernatants were collected for measurement of prostanoids and leukotrienes.
Dendritic cells were isolated as described in ref. 45. Briefly, spleens were injected with Liberase TL (Roche Diagnostics GmbH) and DNase I (Roche Diagnostics GmbH) before being teased apart and incubated at 37 °C for 60 min. They were then mashed through a 70-µm nylon strainer and red blood cells were lysed with the ACK lysing buffer. Cells were enriched with CD11c microbeads (130-052-001; Miltenyi Biotec) on a magnetic column (130-042-401; Miltenyi Biotec). The resulting population was 90% CD11c+ by flow cytometry. Cells were then cultured and stimulated with 5 μg/mL LPS for 24 h and supernatants were collected for measurement of prostanoids and leukotrienes.
Mass Spectrometric Analysis of Eicosanoids and Their Metabolites.
Eicosanoids were quantified in cell culture supernatants as previously described (17). Systemic production of PGE2, PGI2, PGD2, and TxA2 was determined by mass spectrometric quantitation of their major urinary metabolites: PGE-M, PGI-M, PGD-M, and Tx-M, respectively. The nonenzymatic lipid peroxidation product, 8,12-iso-iPF2α-VI, was measured as previously described (46). Data were corrected for urinary creatinine (Oxford Biomedical Research).
Lipid Analysis in Plasma.
Blood was collected from the vena cava of CO2-euthanized mice. Plasma total cholesterol and triglyceride levels were measured by commercial kits from Wako Chemicals.
Blood Pressure and Photochemical Injury-Induced Thrombosis.
Systolic blood pressure was measured in conscious mice by using a computerized noninvasive tail cuff system as previously described (32). Blood pressure was recorded once each day for 7 consecutive days, and the average blood pressure was calculated. Photochemical injury-induced thrombosis was performed as previously described (8).
Statistical Analysis.
Statistical analyses were performed with two-sample unpaired t tests with or without Welch’s correction, depending on whether the variances were significantly different. All data were expressed as means ± SEM. A total of 76 two-sample tests were performed, with a significance threshold of 0.05. A total of 29 of the 76 P values are significant at the 0.05 level. We therefore would conservatively expect 0.05 × 76 = 3.8 false positives from 76 tests if all null hypotheses were true. Thus, we expect ∼25 of our rejected null hypotheses to be correctly rejected. With an expectation of 25 true positives, we can recalculate the expected number of falsely rejected null hypotheses to conservatively be 0.05 × 51 = 2.55. So, we can (conservatively) expect less than 3 false positives from the 25 rejected null hypotheses. This number is safely conservative because many of the observed P values are very small. The overall conclusions of this article are robust to such a small number of false positives, in particular, among the hypotheses with marginal P values.
Supplementary Material
Acknowledgments
We thank Dr. Mohammad Bohlooly and Dr. Xiufeng Xu from Astra Zeneca for kindly providing the mPGES-1f/f mice. We thank Weili Yan, Helen Zou, Wenxuan Li, and Ashmita Saigal for their technical support. This work was supported by a National Institutes of Health Grant HL062250 (to G.A.F., the McNeil Professor in Translational Medicine and Therapeutics).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1401797111/-/DCSupplemental.
References
- 1.Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50(Suppl):S423–S428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu Y, et al. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4(132):32ra54. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannon CP, et al. MEDAL Steering Committee Cardiovascular outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) programme: A randomised comparison. Lancet. 2006;368(9549):1771–1781. doi: 10.1016/S0140-6736(06)69666-9. [DOI] [PubMed] [Google Scholar]
- 4.Struthmann L, et al. Prothrombotic effects of diclofenac on arteriolar platelet activation and thrombosis in vivo. J Thromb Haemost. 2009;7(10):1727–1735. doi: 10.1111/j.1538-7836.2009.03582.x. [DOI] [PubMed] [Google Scholar]
- 5.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: A novel therapeutic target. Pharmacol Rev. 2007;59(3):207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 6.Kudo I, Murakami M. Prostaglandin E synthase, a terminal enzyme for prostaglandin E2 biosynthesis. J Biochem Mol Biol. 2005;38(6):633–638. doi: 10.5483/bmbrep.2005.38.6.633. [DOI] [PubMed] [Google Scholar]
- 7.Wang M, Song WL, Cheng Y, Fitzgerald GA. Microsomal prostaglandin E synthase-1 inhibition in cardiovascular inflammatory disease. J Intern Med. 2008;263(5):500–505. doi: 10.1111/j.1365-2796.2008.01938.x. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, et al. Cell selective cardiovascular biology of microsomal prostaglandin E synthase-1. Circulation. 2013;127(2):233–243. doi: 10.1161/CIRCULATIONAHA.112.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang M, et al. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci USA. 2006;103(39):14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, et al. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011;123(6):631–639. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang M, et al. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117(10):1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- 12.Trebino CE, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA. 2003;100(15):9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamei D, et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004;279(32):33684–33695. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- 14.Chen M, et al. Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum-transfer arthritis. Arthritis Rheum. 2008;58(5):1354–1365. doi: 10.1002/art.23453. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki J, et al. Roles of prostaglandin E2 in cardiovascular diseases. Int Heart J. 2011;52(5):266–269. doi: 10.1536/ihj.52.266. [DOI] [PubMed] [Google Scholar]
- 16.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: Structures, properties, and functions. Physiol Rev. 1999;79(4):1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 17.Yu Z, et al. Disruption of the 5-lipoxygenase pathway attenuates atherogenesis consequent to COX-2 deletion in mice. Proc Natl Acad Sci USA. 2012;109(17):6727–6732. doi: 10.1073/pnas.1115313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egan KM, et al. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306(5703):1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi T, et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114(6):784–794. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fries S, Grosser T. The cardiovascular pharmacology of COX-2 inhibition. Hematology (Am Soc Hematol Educ Program) 2005;2005:445–451. doi: 10.1182/asheducation-2005.1.445. [DOI] [PubMed] [Google Scholar]
- 21.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 22.Hallemeesch MM, et al. NO production by cNOS and iNOS reflects blood pressure changes in LPS-challenged mice. Am J Physiol Endocrinol Metab. 2003;285(4):E871–E875. doi: 10.1152/ajpendo.00004.2002. [DOI] [PubMed] [Google Scholar]
- 23.Duivenvoorden R, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. doi: 10.1038/ncomms4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawson JA, et al. Identification of two major F2 isoprostanes, 8,12-iso- and 5-epi-8, 12-iso-isoprostane F2alpha-VI, in human urine. J Biol Chem. 1998;273(45):29295–29301. doi: 10.1074/jbc.273.45.29295. [DOI] [PubMed] [Google Scholar]
- 25.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: Therapeutic challenges and opportunities. J Clin Invest. 2006;116(1):4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudic RD, et al. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96(12):1240–1247. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- 27.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Claveau D, et al. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003;170(9):4738–4744. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- 29.Barbieri SS, et al. Cyclooxygenase-2-derived prostacyclin regulates arterial thrombus formation by suppressing tissue factor in a sirtuin-1-dependent-manner. Circulation. 2012;126(11):1373–1384. doi: 10.1161/CIRCULATIONAHA.112.097295. [DOI] [PubMed] [Google Scholar]
- 30.Nagai N, Hoylaerts MF, Gallacher DJ, Lu HR, Lijnen HR. Prothrombotic effect of Rofecoxib in a murine venous thrombosis model. Thromb Res. 2008;122(5):668–673. doi: 10.1016/j.thromres.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 31.Murata T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388(6643):678–682. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Y, et al. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116(5):1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia Z, Aoyagi T, Yang T. mPGES-1 protects against DOCA-salt hypertension via inhibition of oxidative stress or stimulation of NO/cGMP. Hypertension. 2010;55(2):539–546. doi: 10.1161/HYPERTENSIONAHA.109.144840. [DOI] [PubMed] [Google Scholar]
- 34.Jia Z, et al. Microsomal prostaglandin synthase-1-derived prostaglandin E2 protects against angiotensin II-induced hypertension via inhibition of oxidative stress. Hypertension. 2008;52(5):952–959. doi: 10.1161/HYPERTENSIONAHA.108.111229. [DOI] [PubMed] [Google Scholar]
- 35.Francois H, et al. Role of microsomal prostaglandin E synthase 1 in the kidney. J Am Soc Nephrol. 2007;18(5):1466–1475. doi: 10.1681/ASN.2006040343. [DOI] [PubMed] [Google Scholar]
- 36.Facemire CS, Griffiths R, Audoly LP, Koller BH, Coffman TM. The impact of microsomal prostaglandin e synthase 1 on blood pressure is determined by genetic background. Hypertension. 2010;55(2):531–538. doi: 10.1161/HYPERTENSIONAHA.109.145631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Degousee N, et al. Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation. 2008;117(13):1701–1710. doi: 10.1161/CIRCULATIONAHA.107.749739. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, et al. Cyclooxygenase-2-derived prostaglandin E2 promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res. 2013;113(2):104–114. doi: 10.1161/CIRCRESAHA.113.301033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghoshal S, Loftin CD. Cyclooxygenase-2 inhibition attenuates abdominal aortic aneurysm progression in hyperlipidemic mice. PLoS ONE. 2012;7(11):e44369. doi: 10.1371/journal.pone.0044369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Y, et al. Treatment of experimental adjuvant arthritis with a novel folate receptor-targeted folic acid-aminopterin conjugate. Arthritis Res Ther. 2011;13(2):R56. doi: 10.1186/ar3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayala-López W, Xia W, Varghese B, Low PS. Imaging of atherosclerosis in apoliprotein e knockout mice: Targeting of a folate-conjugated radiopharmaceutical to activated macrophages. J Nucl Med. 2010;51(5):768–774. doi: 10.2967/jnumed.109.071324. [DOI] [PubMed] [Google Scholar]
- 42.Cormode DP, et al. Atherosclerotic plaque composition: Analysis with multicolor CT and targeted gold nanoparticles. Radiology. 2010;256(3):774–782. doi: 10.1148/radiol.10092473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sinha P, et al. Mouse lysozyme-M knockout mice reveal how the self-determinant hierarchy shapes the T cell repertoire against this circulating self antigen in wild-type mice. J Immunol. 2004;173(3):1763–1771. doi: 10.4049/jimmunol.173.3.1763. [DOI] [PubMed] [Google Scholar]
- 44.Hu Y. Isolation of human and mouse neutrophils ex vivo and in vitro. Methods Mol Biol. 2012;844:101–113. doi: 10.1007/978-1-61779-527-5_7. [DOI] [PubMed] [Google Scholar]
- 45.Inaba K, et al. Isolation of dendritic cells. Curr Protoc Immunol. 2009;Chapter 3:Unit 3.7. doi: 10.1002/0471142735.im0307s86. [DOI] [PubMed] [Google Scholar]
- 46.Song WL, Lawson JA, Wang M, Zou H, FitzGerald GA. Noninvasive assessment of the role of cyclooxygenases in cardiovascular health: A detailed HPLC/MS/MS method. Methods Enzymol. 2007;433:51–72. doi: 10.1016/S0076-6879(07)33003-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.