

Significance
We use a hybridization approach to enrich the DNA from the protein-coding fraction of the genomes of two Neandertal individuals from Spain and Croatia. By analyzing these two exomes together with the genome sequence of a Neandertal from Siberia we show that the genetic diversity of Neandertals was lower than that of present-day humans and that the pattern of coding variation suggests that Neandertal populations were small and isolated from one another. We also show that genes involved in skeletal morphology have changed more than expected on the Neandertal evolutionary lineage whereas genes involved in pigmentation and behavior have changed more on the modern human lineage.
Keywords: ancient DNA, exome capture, site frequency spectra, paleogenetics
Abstract
We present the DNA sequence of 17,367 protein-coding genes in two Neandertals from Spain and Croatia and analyze them together with the genome sequence recently determined from a Neandertal from southern Siberia. Comparisons with present-day humans from Africa, Europe, and Asia reveal that genetic diversity among Neandertals was remarkably low, and that they carried a higher proportion of amino acid-changing (nonsynonymous) alleles inferred to alter protein structure or function than present-day humans. Thus, Neandertals across Eurasia had a smaller long-term effective population than present-day humans. We also identify amino acid substitutions in Neandertals and present-day humans that may underlie phenotypic differences between the two groups. We find that genes involved in skeletal morphology have changed more in the lineage leading to Neandertals than in the ancestral lineage common to archaic and modern humans, whereas genes involved in behavior and pigmentation have changed more on the modern human lineage.
Analyses of the coding regions of multiple present-day human individuals have uncovered many amino acid-changing SNPs segregating at low frequency in present-day human populations (1–7). It also has been shown that Europeans carry a larger proportion than Africans of amino acid-changing SNPs inferred to alter the structure or function of proteins and thus to be potentially deleterious (4), a fact attributed to the bottleneck and subsequent expansion of human populations during and after the migration out of Africa (4). In contrast, little is known about the coding variation in Neandertals, an extinct hominin group closely related to present-day humans. The main reasons for this are the rarity of Neandertal remains and the fact that >99% of the DNA extracted from typical Neandertal bones is derived from microbes (8, 9), making shotgun sequencing of the endogenous DNA impractical.
Here, we use a hybridization approach to enrich and sequence the protein-coding fractions of the genomes of two Neandertals from Spain and Croatia. We analyze them together with the genome sequence recently determined from a Neandertal from southern Siberia (10) and show that the genetic diversity, pattern of coding variation, and genes that may underlie phenotypic changes in Neandertals are remarkably different from those in present-day humans.
Results and Discussion
We used densely tiled oligonucleotide probes (9) and a recently described protocol (11) to capture the protein-coding exons from 17,367 genes in a ∼49,000-y-old (12) (uncalibrated radiocarbon date) Neandertal from Spain (SD1253, El Sidrón Cave) and a ∼44,000-y-old (uncalibrated radiocarbon date) Neandertal from Croatia (Vi33.15, Vindija Cave). The DNA libraries from these two Neandertals contain 0.2% and 0.5% endogenous DNA, respectively, and the capture approach enriched the endogenous DNA 325-fold and 153-fold, resulting in an average coverage of 12.5-fold and 42.0-fold for the El Sidrón and Vindija exomes, respectively (Table 1). Present-day human mitochondrial contamination in the enriched libraries is estimated to be between 0.24% [confidence interval (CI): 0.23–0.24%] and 0.40% (CI: 0.38–0.42%) for El Sidrón and between 0.28% (CI: 0.27–0.28%) and 1.08% (CI: 1.05–1.08%) for Vindija. Before genotypes were called, we lowered the base quality scores toward the ends of the Neandertal DNA sequences to compensate for substitutions potentially due to cytosine deaminations (Methods and Fig. S2).
Table 1.
Coverage comparison of the protein-coding regions in Neandertals
| Draft genome (8) | El Sidrón | Vindija | Altai genome (10) | |
| Average coverage | 1.3-fold | 12.5-fold | 42.0-fold | 45.3-fold |
| Coding bases, n (%)* | 14,289,004 (52.3) | 25,651,281 (93.8) | 26,876,083 (98.3) | 27,313,554 (99.9) |
| Coding genes, n (%)† | ||||
| 80% | 177 (1.0) | 15,502 (89.3) | 16,944 (97.6) | 17,322 (99.7) |
| 90% | 46 (0.3) | 13,561 (78.1) | 16,294 (93.8) | 17,313 (99.7) |
| 100% | 19 (0.1) | 5,476 (31.5) | 11,696 (67.3) | 17,264 (99.4) |
Number and percentage of bases in the exome covered by at least one read.
Number and percentage of genes with at least 80%, 90%, and 100% of their length covered by one read.
We also retrieved the exomes from the genome sequences of a Neandertal (10) (Table 1) and a Denisovan (13) individual, both from Denisova Cave in the Altai Mountains, Siberia, as well as from nine present-day humans (Yoruba, Mandenka, and Dinka from Africa; French, Sardinian, and Italian American from Europe; Han and Dai from Asia; and Papuan from Oceania), which have been sequenced to a quality similar to that of the two archaic genomes (13, 14) (SI Appendix and Table S7). A neighbor-joining tree based on pairwise differences (Fig. 1A) shows that the three Neandertals form a clade, whereas the Denisovan individual is a sister group to the Neandertals, in agreement with previous results (8, 13, 15).
Fig. 1.

(A) Neighbor-joining tree based on the number of pairwise differences between individuals. Phenotypic categories enriched in amino acid-changing substitutions on the lines to Neandertals and to modern humans, respectively, are indicated. The bar indicates 50 inferred substitutions per megabase. (B) Neighbor-joining tree of pairwise FST values for the same individuals. The bar indicates an FST of 0.05.
As expected for closely related groups, a substantial amount of the variation is shared between Neandertals and present-day humans. Thus, about 33% of the derived alleles seen in the Neandertals are shared with the present-day humans and about 19%, 24%, and 24% of the derived alleles seen in the Africans, Europeans, and Asians, respectively, are shared with the Neandertals (Table S14). The number of nucleotide differences within an individual (heterozygosity) is 0.143 per thousand coding bases in the El Sidrón Neandertal, 0.127 in the Vindija Neandertal, and 0.113 in the Altai Neandertal. The average heterozygosity among the three Neandertals (0.128) is 25%, 33%, and 36% of the average heterozygosity in the three African (0.507), European (0.387), and Asian (0.358) individuals, respectively (Table S12), and thus significantly smaller than that in humans today (P = 4.5 × 10−3; Mann–Whitney U test). The three Neandertals also have longer runs of homozygosity (Fig. S9), suggesting that mating among related individuals may have been more common in Neandertals than in present-day humans, compatible with the observations in the Altai Neandertal (10). Also compatible with these observations is that the genetic differentiation among individuals, as measured by the pairwise fixation index (FST) (Fig. 1B), is greater among Neandertals than among the Africans, Europeans, and Asians (P = 1.3 × 10−2; Mann–Whitney U test), suggesting that mating in small and isolated populations may have caused Neandertal populations to be more differentiated from one another than what is typical for present-day humans.
To investigate the extent to which the population history reflected in the low genetic diversity affected the evolution of coding regions, we investigated derived alleles present in Neandertals and compared them with those of the present-day Africans, Europeans, and Asians (Table 2). We found that the fraction of all derived SNPs inferred to change amino acids in Neandertals (51.1%) is larger than that in Africans (45.7%; P = 3.1 × 10−11; G-test), Europeans (46.4%; P = 6.9 × 10−10), and Asians (46.7%; P = 4.5 × 10−7) (Table 2). We used PolyPhen-2 (16) to assess whether the amino acid-changing SNPs in Neandertals are enriched in alleles inferred to affect protein function or structure and thus likely to be slightly deleterious. The proportion of such SNPs in the Neandertals (45.4%) is larger than that in Africans (36.4%; G-test; P = 1.6 × 10−14), Europeans (37.9%; P = 4.5 × 10−9), and Asians (38.1%; P = 1.3 × 10−9) (Table 2). Similarly, the proportion of such SNPs in conserved protein sites [PhastCons (17)] is larger in the Neandertals (50.1%) than in the Africans (38.1%; G-test; P < 10−20), Europeans (40.2%; P = 1.1 × 10−15), and Asians (39.7%; P < 10−20) (Table 2). Furthermore, a physicochemical classification of amino acid substitutions, the Grantham score (GS) (18), shows that the proportion of derived SNPs in the Neandertals that cause radical (GS >100) amino acid changes is larger than that in present-day humans (Table 2). Derived alleles in conserved protein sites that are homozygous in all individuals analyzed also are more frequent in Neandertals (39.6%) than in Africans (G-test; 29.4%; P = 2.1 × 10−3), Europeans (27.8%; P = 1.6 × 10−5), and Asians (26.9%; P = 5.9 × 10−6) (Table 2).
Table 2.
Distribution of derived alleles by functional class
| Derived alleles, n (%) | ||||||||
| Polymorphic positions (SNPs) | Homozygous positions (“fixed”) | |||||||
| Category | Neandertals* | Africans† | Europeans† | Asians† | Neandertals* | Africans† | Europeans† | Asians† |
| Coding | ||||||||
| Synonymous | 2,501 (48.9) | 8,250 (54.3) | 5,571 (53.6) | 5,518 (53.3) | 1,273 (56.1) | 383 (59.3) | 624 (57.2) | 603 (57.8) |
| Nonsynonymous | 2,611 (51.1) | 6,947 (45.7) | 4,820 (46.4) | 4,839 (46.7) | 996 (43.9) | 263 (40.7) | 467 (42.8) | 441 (42.2) |
| PP-2 | ||||||||
| Benign | 1,354 (54.6) | 3,596 (63.6) | 2,266 (62.1) | 2,292 (61.9) | 600 (63.8) | 16 (55.2) | 53 (66.3) | 59 (64.1) |
| Deleterious | 1,128 (45.4) | 2,057 (36.4) | 1,385 (37.9) | 1,409 (38.1) | 340 (36.2) | 13 (44.8) | 31 (36.9) | 33 (35.9) |
| PC | ||||||||
| Benign | 1,296 (49.9) | 4,280 (61.9) | 2,872 (59.8) | 2,903 (60.3) | 601 (60.4) | 185 (70.6) | 335 (72.2) | 321 (73.1) |
| Deleterious | 1,303 (50.1) | 2,635 (38.1) | 1,931 (40.2) | 1,913 (39.7) | 394 (39.6) | 77 (29.4) | 129 (27.8) | 118 (26.9) |
| GS | ||||||||
| Benign | 2,112 (80.9) | 5,673 (81.7) | 3,936 (81.7) | 3,954 (81.7) | 833 (83.6) | 231 (87.8) | 413 (88.4) | 374 (84.8) |
| Deleterious | 499 (19.1) | 1,274 (18.3) | 884 (18.3) | 885 (18.3) | 163 (16.4) | 32 (12.2) | 54 (11.6) | 67 (15.2) |
For the coding category, the number and proportion of coding alleles that are either synonymous or nonsynonymous are shown. For the PolyPhen-2 (PP-2), PhastCons (PC) and Grantham score (GS) categories, the number and proportion of nonsynonymous alleles that are predicted to be either benign or deleterious are shown.
Derived in Neandertals and homozygous ancestral in Africans. See Table S18 for additional details.
Derived in Africans, Europeans, or Asians and homozygous ancestral in Neandertals.
Although only six Neandertal chromosomes are available, we note that among derived amino acid-changing alleles likely to have been at low frequency in Neandertals because they are seen once among the six chromosomes (Fig. 2), a higher proportion is inferred to alter protein structure or function in Neandertals (49.1% by PolyPhen-2; similar results with PhastCons) than in Africans (G-test; 38.3%; P = 2.2 × 10−12), Europeans (40.6%; P = 8.8 × 10−7), and Asians (41.8%; P = 5.1 × 10−6). Furthermore, we note that these low-frequency, putatively deleterious alleles have a stronger inferred impact on proteins in Neandertals than in Africans (P < 2.2 × 10−16), Europeans (P = 6.7 × 10−9), or Asians (P = 2.0 × 10−7) (Fig. 2 and SI Appendix).
Fig. 2.

Frequency spectra of nonsynonymous derived alleles classified by PolyPhen-2 as either benign or deleterious in Neandertals and present-day humans. The ratio of deleterious to benign derived alleles is higher in Neandertals than in the present-day humans. See Table S19 for the actual allele counts and Fig. S10 for the frequency spectra of nonsynonymous derived alleles assessed by PhastCons and GS.
When analyzed individually, each of the three Neandertal individuals carries a larger fraction of derived amino acid-changing alleles (P = 7.6 × 10−3 for SNPs; P = 7.0 × 10−3 for homozygous; Mann–Whitney U test) as well as alleles inferred to change protein function (PhastCons; P = 8.0 × 10−3 for heterozygous, P = 8.0 × 10−3 for homozygous) than any of the present-day individuals analyzed (Tables S15 and S16). Thus, not only Neandertals as a group, but also each Neandertal individual analyzed, carried a larger fraction of putatively deleterious alleles than present-day humans. This also is the case for the Denisovan individual, who represents a little-known Asian population related to Neandertals.
We conclude that the patterns of coding variation in Neandertals differ strongly from those in present-day humans, a fact likely to be the result of differences in their demographic histories. Inferences from the complete Neandertal genome suggest that sometime after 0.5–1.0 Mya, the ancestral Neandertal population decreased in size, whereas the ancestors of present-day humans increased in number (10). A low population size over a long time would reduce the efficacy of purifying selection and contribute to a larger fraction of slightly deleterious alleles, particularly at low frequency, but not necessarily to the absolute number of slightly deleterious alleles per individual (19) (SI Appendix). The fact that the three Neandertals carry longer tracts of homozygosity and differ more from one another than present-day humans within continents further suggests that Neandertals may have been subdivided in small local populations. However, we note that the three Neandertal individuals analyzed are not contemporaneous but differ in time by perhaps as much as 20,000 y. It therefore is crucial for DNA sequences from more Neandertals to be generated when suitable specimens become available. It is also crucial for more complex demographic scenarios, for example involving recurrent population reductions and expansions, to be explored to fully understand the differences in population history between Neandertals and modern humans.
We previously used the Neandertal and Denisova genomes (8, 10, 13) to identify amino acid changes that are shared by all humans today but do not occur in the archaic genomes and therefore are inferred to have risen to high frequency on the modern human lineage. The exomes of the three Neandertals and the Denisovan individual allow us, for the first time, to identify derived amino acid changes shared by the three Neandertals as well as the Denisovan individual that are not seen, or occur at low frequency, in present-day humans. Such changes are of interest because they may underlie phenotypes specific to the archaic populations. We calculated the fraction of all amino acid changes specific to either the archaic or present-day human lineages for each phenotype category of genes in the Human Phenotype Ontology (20), and compared it with the fraction of all amino acid changes in the same phenotype categories that occurred on the human lineage after its divergence from the chimpanzee lineage but before the divergence of modern and archaic humans (SI Appendix). Thus, unlike in previous approaches (8, 10, 13), we assessed the enrichment of amino acid changes in phenotypes across multiple archaic individuals while correcting for the size and base composition of genes across the genome (Methods and SI Appendix).
For the archaic lineage before the divergence of Neandertals and Denisovans, we find an overrepresentation of changes in genes related to metabolism, the cardiovascular system, hair distribution, and morphology (genitals, palate, face, extremities, joints, digits, thorax, orbital, and occipital skull regions and general mobility) [Table S28; binomial test; P values and false discovery rates (FDRs) in the table]. Thus, it may be that future functional investigations of the changes in the 22 genes identified in enriched categories (Table S29) will reveal the genetic basis for some features shared by Neandertals and Denisovans, although almost no morphologic information currently is available for the latter (15).
In the Neandertal lineage after the split from Denisovans, only the phenotype category “hyperlordosis” is overrepresented (Fig. 1A; binomial test; P = 4.5 × 10−5; FDR = 2.1 × 10−2). This is interesting because Neandertals had a reduced lordotic curvature relative to other archaic hominins as well as to modern humans (21). Changes in the six genes associated with this phenotype (CUL7, GLB1, NEB, COL2A1, HSPG2, and VPS13B) thus may have contributed to this feature of the Neandertal morphology.
Finally, the modern human lineage has an overrepresentation of amino acid-changing substitutions in genes related to pigmentation (Fig. 1A; P = 3.2 × 10−5; FDR = 5.9 × 10−2) and behavioral traits (Fig. 1A; P < 5 × 10−4; FDR < 7.8 × 10−2). The overrepresentation in the pigmentation category is the result of amino acid substitutions in two genes involved in melanosome function (GPR143 and LYST), for which the derived alleles are at high frequency in Africa and fixed outside Africa. These mutations may contribute to differences in pigmentation among present-day humans (22, 23). In contrast, the behavioral category is the result of substitutions that seem to be fixed in all present-day humans and occur in the three genes adenylosuccinate lyase (ADSL), glycine dehydrogenase (GLDC), and transmembrane protein SLITRK1. These genes are associated with de novo purine biosynthesis, mitochondrial glycine degradation, and neurite outgrowth, respectively. In the case of ADSL, mutations known to impair the gene result in varying degrees of psychomotor retardation, autism, and muscle wasting, whereas in the case of GLDC, mutations result in glycine encephalopathy and in the case of SLITRK1, in Tourette syndrome, characterized by motoric and vocal ticks. In the Human Phenotype Ontology (20), all these genes are annotated as being involved in “hyperactivity” and “aggressive behavior.” Thus, it is possible that the mutations in these genes may have affected some component of behavior unique to modern humans. However, we note that even if they did, the way in which they occurred is unknown. For example, if they affected activity or aggression levels, it is unclear whether they increased or decreased such traits. In any event, these changes clearly warrant further investigation.
In conclusion, we show that the genetic diversity of late Neandertals across their entire geographical range was lower than that of present-day humans and that there is evidence suggesting that Neandertal populations may have been small and isolated from one another. We also show that genes involved in morphology may have changed more on the Neandertal and Denisova lineages than on the preceding lineage from the common ancestor shared with chimpanzees, whereas genes involved in behavior may have changed more on the modern human lineage.
Methods
DNA Extraction and Library Preparation.
We prepared DNA extracts from two Neandertal bones, SD1253 from El Sidrón Cave and Vi33.15 from Vindija Cave, as described in Rohland and Hofreiter (24) (Table S1), and prepared DNA sequencing libraries containing a special 4-bp clean-room tag sequence to avoid contamination in later steps (25). During library preparation, we used a uracil-DNA glycosylase (UDG) and endonuclease VIII mix to remove uracils resulting from cytosine deaminations (26).
Gene Annotation and Array Design.
We designed 22 arrays as described in Burbano et al. (9) to capture 173,086 coding exons annotated in the human reference genome (GRCh37/1000 Genome Project release). These exons encompass ∼27 Mb and belong to 17,367 genes annotated in consensus coding sequences (CCDS) (27), RefSeq (28), and GENCODE (29). A total of 20,005,501 probes 60 bases long were generated, at 3-base tile density, to cover up to 100 bases upstream and downstream of each coding exon. Probes containing repetitive elements were discarded (Fig. S1).
Exome Capture Experiment.
We used 40 libraries from the El Sidrón and 19 from the Vindija Neandertals for capture as described in Fu et al. (11). The pools of exome-enriched libraries from El Sidrón and Vindija were sequenced from both sides on 11 and 15 lanes of the Genome Analyzer IIx (Illumina), respectively. After sequences from the same DNA fragments were merged, sequences of at least 35 bases were aligned to the human reference genome with Burrows-Wheeler aligner (BWA) (30). Removal of PCR duplicates resulted in ∼1.0 and ∼3.4 Gb of sequence aligned to the human genome in El Sidrón and Vindija exomes, respectively (Table S4). The percentage of DNA sequences with a mapping quality of at least one that map on the tiled regions is 64.7% for the El Sidrón and 64.0% for the Vindija capture. Coverage is higher in longer exons with intermediate GC content (Figs. S5 and S6). Reference allele frequencies in El Sidrón and Vindija exomes are within the range of reference allele frequencies in present-day human DNA captures (SI Appendix). Note that a capture bias toward alleles present on the probes should not affect the nonsynonymous-to-synonymous and deleterious-to-benign ratios or the variance of the allele frequency distribution in Neandertals; that is, capture bias should not affect the test of proportions or the frequency spectra of Neandertals presented in this work.
Contamination Estimates.
To measure the present-day human mtDNA contamination in the El Sidrón and Vindija exomes, we used the sequences aligned by BWA (30) to the human reference mitochondrial genome (31) and realigned them to the El Sidrón and Vindija mitochondrial genomes (32), using the mapping iterative assembler (33). We then computed a contamination upper estimate using 81 and 84 diagnostic positions at which El Sidrón or Vindija mitochondrial genomes, respectively, differed from all sequences in a worldwide panel of 311 present-day human mitochondrial genomes. Sequences overlapping at least one of the diagnostic positions were classified as contaminants if they showed the present-day human allele (Table S8). We computed a lower estimate using 483 and 631 diagnostic positions at which El Sidrón and Vindija mitochondrial genomes, respectively, differed from at least one sequence in the worldwide panel of 311 present-day human mitochondrial genomes. Sequences overlapping at least one of the diagnostic positions were classified as contaminants if they showed the differing present-day human allele (Table S8).
Computational Correction of Cytosine Deaminations.
Sequences may carry residual cytosine deaminations in the first positions of the 5′ end and the last positions in the 3′ end despite the UDG treatment (26) (Fig. S2A). These bases are read as thymine and adenosine, respectively. We decreased the quality score to 2 of any “T” base occurring within the first five bases or “A” base within the last five positions in the El Sidrón and Vindija sequences. Because a 2 on the phred scale (34) represents a 63% chance of sequencing error, any downstream analysis will consider these bases as sequencing artifacts. This approach resulted in comparable C-to-T and G-to-A substitution patterns in ancient and present-day individuals after genotype calling (Fig. S2B), suggesting that cytosine deaminations do not significantly contribute SNPs at low frequency in Neandertals (Fig. 2) or to their proportion of putatively deleterious alleles (Table S17).
Variation Discovery.
We called Neandertal genotypes with GATK (35) and obtained high-quality calls for 18,811,848 and 23,283,112 sites in El Sidrón and Vindija exomes, respectively (Table S10). Genotypes in the Altai Neandertal, Denisovan, and present-day human individuals in Fig. 1 were obtained similarly (Tables S10 and S11), and a combined file for all individuals was created and annotated as in Meyer et al. (13) (Table S9). High-quality genotype calls for the longest protein-coding transcript in each gene were used for analysis (SI Appendix).
Genetic Differentiation.
We computed the pairwise FST among individuals using the pairwise number of differences between individuals (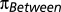 ) and the number of heterozygous sites in each individual (
) and the number of heterozygous sites in each individual (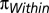 ). The FST value for individuals A and B is then
). The FST value for individuals A and B is then
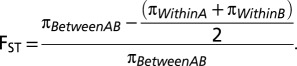 |
Derived Alleles.
Neandertals were compared with each of the three Africans, Europeans, and Asians separately. In each pairwise comparison, derived alleles present in either Neandertals or the three present-day humans were used (Table 2, Fig. 2, and Table S18). Derived states for alleles were determined from the inferred ancestral base in the Enredo Pecan Ortheus (EPO) six-primate genome alignments (36) and were classified as synonymous or nonsynonymous based on the gene annotation used in the array design (SI Appendix). When counting alleles in heterozygous and homozygous positions for each archaic or present-day individual (Tables S15 and S16), we used derived alleles that are ancestral in the three Africans or ancestral in the three Neandertals, respectively.
Prediction of Functional Consequences.
Nonsynonymous derived alleles classified in the “possibly” and “probably” damaging categories in PolyPhen-2 (16), in a position with a PhastCons (17) posterior probability larger than 0.9 or with a GS (18) of 101 or more, were considered to alter protein structure or function (Table 2 and Tables S15–S18). These alleles likely are deleterious. We used the HumDiv model of PolyPhen-2, which is based on the differences between human proteins and closely related mammalian homologs but does not incorporate present-day human polymorphism. Thus, this model is meant for the analysis of patterns of natural selection in which even slightly deleterious alleles must be considered. Furthermore, when counting putatively deleterious alleles per individual, we used only derived alleles that did not match the human reference sequence. This minimizes the bias in the counts of benign and deleterious alleles in polymorphic positions (Table 2) caused by the presence of present-day human, but not Neandertal, sequences in PolyPhen-2 alignments. It also results in the few counts of benign and deleterious alleles in homozygous positions in present-day humans using PolyPhen-2 (Table 2). The PhastCons conservation scores were computed on alignments of mammalian but not human proteins.
Phenotype Enrichment Analysis.
We identified Human Phenotype Ontology (20) categories that are enriched for genes with amino acid-changing derived alleles in the archaic (Neandertal and Denisovan), the Neandertal, or the modern human lineages compared with amino acid-changing derived alleles shared among archaic and present-day humans in the same genes and phenotype categories using the program FUNC (37). Comparing the genes associated with a phenotype category with themselves at two different periods controls for differences in the number of genes in each category, as well as the length, sequence composition, and distribution across the genome of the genes associated with it. We restricted the analysis to phenotype ontology terms to which at least two genes are mapped and performed four separate comparisons for ontology enrichment (Tables S24, S26, and S28) that focus on different evolutionary time frames in the archaic, the Neandertal, and the modern human lineages. Our significance criteria are a P value <0.01 and an FDR ≤0.1 within each comparison in each lineage, but we also highlight categories that have a family-wise error rate ≤0.1 as a more conservative cutoff (Tables S24, S26, and S28). Derived alleles in the modern human lineage were required to be fixed or at high frequency (>90%) in the 1000 Genomes Project (14). Note that the high number of present-day humans makes the test more conservative in the modern than in the archaic human lineage.
Supplementary Material
Acknowledgments
We thank David Reich and Montgomery Slatkin for comments on the manuscript, Oscar Lao for clarifying the ancestry of one present-day individual, Cesare de Filippo for help with the principal components analysis plots, Agilent Technologies for the capture arrays, and the Presidential Innovation Fund of the Max Planck Society for support. C.L.-F. and F.A.S.-Q. are supported by the Ministerio de Economía y Competitividad (Grant BFU2012-34157).
Footnotes
The authors declare no conflict of interest.
Data deposition: The exome sequence capture data reported in this paper have been deposited in the European Nucleotide Archive (accession no. ERP002457). These data are also available at http://cdna.eva.mpg.de/neandertal/exomes/.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1405138111/-/DCSupplemental.
References
- 1.Cargill M, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22(3):231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 2.Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158(3):1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bustamante CD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437(7062):1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 4.Lohmueller KE, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451(7181):994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42(11):969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 7.Tennessen JA, et al. Broad GO Seattle GO NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burbano HA, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328(5979):723–725. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prüfer K, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505(7481):43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Q, et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci USA. 2013;110(6):2223–2227. doi: 10.1073/pnas.1221359110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wood RE, et al. A new date for the Neanderthals of El Sidrón Cave (Asturias, Northern Spain) Archaeometry. 2013;55(1):148–158. [Google Scholar]
- 13.Meyer M, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338(6104):222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abecasis GR, et al. 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468(7327):1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 19.Simons YB, Turchin MC, Pritchard JK, Sella G. The deleterious mutation load is insensitive to recent population history. Nat Genet. 2014;46(3):220–224. doi: 10.1038/ng.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson PN, Mundlos S. The human phenotype ontology. Clin Genet. 2010;77(6):525–534. doi: 10.1111/j.1399-0004.2010.01436.x. [DOI] [PubMed] [Google Scholar]
- 21.Been E, Gómez-Olivencia A, Kramer PA. Lumbar lordosis of extinct hominins. Am J Phys Anthropol. 2012;147(1):64–77. doi: 10.1002/ajpa.21633. [DOI] [PubMed] [Google Scholar]
- 22.Hider JL, et al. Exploring signatures of positive selection in pigmentation candidate genes in populations of East Asian ancestry. BMC Evol Biol. 2013;13:150. doi: 10.1186/1471-2148-13-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu F, et al. Digital quantification of human eye color highlights genetic association of three new loci. PLoS Genet. 2010;6(5):e1000934. doi: 10.1371/journal.pgen.1000934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rohland N, Hofreiter M. Comparison and optimization of ancient DNA extraction. Biotechniques. 2007;42(3):343–352. doi: 10.2144/000112383. [DOI] [PubMed] [Google Scholar]
- 25.Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40(1):e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Briggs AW, et al. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010;38(6):e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pruitt KD, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19(7):1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37(Database issue):D32–D36. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coffey AJ, et al. The GENCODE exome: Sequencing the complete human exome. Eur J Hum Genet. 2011;19(7):827–831. doi: 10.1038/ejhg.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews RM, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23(2):147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 32.Briggs AW, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325(5938):318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 33.Green RE, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134(3):416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8(3):186–194. [PubMed] [Google Scholar]
- 35.McKenna A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paten B, et al. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 2008;18(11):1829–1843. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prüfer K, et al. FUNC: A package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics. 2007;8:41. doi: 10.1186/1471-2105-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.