

Abstract
There is an urgent need for new antibiotics which are effective against drug-resistant bacteria without contributing to resistance development. We have designed and developed antimicrobial copolymers with cationic amphiphilic structures based on the mimicry of naturally occurring antimicrobial peptides. These copolymers exhibit potent antimicrobial activity against a broad spectrum of bacteria including methicillin-resistant Staphylococcus aureus with no adverse hemolytic activity. Notably, these polymers also did not result in any measurable resistance development in E. coli. The peptide-mimetic design principle offers significant flexibility and diversity in the creation of new antimicrobial materials and their potential biomedical applications.
Keywords: antimicrobial, biomimetic, copolymer, peptides, structure-property relations
Introduction
“No action today, no cure tomorrow” The World Health Organization (WHO) recently published its global strategy to stimulate further endeavors to combat antibiotic resistant bacteria.[1, 2] The WHO advocates that antimicrobial resistance is not a new problem but one that is becoming more dangerous.[3] A high percentage of healthcare-associated or hospital-acquired infections are caused by antibiotic-resistant bacteria including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (VRE). Most recently, the emergence of “totally drug resistant” M. tuberculosis was described from clinical isolates in South Africa.[4] These resistant bacteria cause adverse infections, resulting in prolonged illness, complications to existing conditions, and can often result in mortality. The prevalence of drug resistance also rapidly diminishes the available treatment options, adding tremendous costs and burdens to the health care system worldwide.[5–10] However, the number of new antibiotics, not just new formulations of existing ones, has fallen steadily in the past few decades.[11] This is because it is a scientific challenge to develop new classes of antimicrobials with molecular mechanisms that can overcome acquired resistance and do not contribute to resistance development. Effective approaches for newer and stronger antibiotics are in urgent need to ensure the future treatment options for drug resistant infections as well as prevention of new resistance development.
Many of the clinically prevalent antibiotics are enzyme inhibitors and DNA replication inhibitors, which disrupt the essential bacterial cellular biosynthesis of nucleic acids, proteins, or cell wall components.[12] Because antibiotics target specific components in biosynthetic pathways which do not exist in humans, the antimicrobial development has been directed to identification and characterization of such molecules with high affinity for the bacterial target. However, bacteria can rapidly acquire resistance against these antibiotic drugs by numerous mechanisms including enzymes which destroy antibiotic agents, efflux pumps which excrete antibiotics from cells, or mutations which alter antibiotic target sites.[13, 14] In addition, resistance to one drug often triggers multi-drug resistance.[5, 9] The hurdle of rapid resistance development in bacteria is compounded by man-made hurdles as the number of new antibiotics in the development pipeline has declined: simply put, the conventional drug discovery approach cannot keep up with the pace of resistance development in bacteria. This is because it has been difficult to find new antimicrobial mechanisms of action which are intrinsically insensitive to the emergent and/or existing resistance mechanisms in bacteria.[8] Therefore it is critical to use creative scientific solutions for a new era of antimicrobial development to fight against resistance in bacteria.
To that end, host-defense antimicrobial peptides and their synthetic mimics have emerged as candidates for new antibiotics.[15–18] Antimicrobial peptides have been identified and isolated from many organisms including plants, insects, and animals. Theses peptides are featured by cationic, amphiphilic nature accompanied with secondary structures including α-helix, β-sheet, loop, and extended structure.[19] The proposed mechanism of action for the majority of these antimicrobial peptides is by acting on bacterial cell membranes. The peptides bind to the bacterial cell surface by electrostatic interactions between cationic side chains of the peptide and negatively charged components of the bacterial cell wall and membrane(s). The accumulation of peptides on the cell surface leads to membrane disruption, resulting in a breakdown of the membrane potential, leakage of cellular components, and bacterial cell death. Since all bacteria require an in-tact cytoplasmic membrane, the mode of action of antimicrobial peptides targeting cell membranes is effective against drug-resistant bacteria and less susceptible to the resistance mechanisms in bacteria. However, the implementation of peptides as new antibiotics have significant obstacles associated with bioavailability, high manufacturing cost, and unknown systemic toxicity.[20]
To create new antimicrobials effective to drug resistant bacteria, our laboratories have extended the molecular mimicry of antimicrobial peptides to create antimicrobials based on conventional synthetic polymers such as methacrylate (Figure 1).[18, 21–31] These polymers are designed to mimic the cationic functionality and amphiphilic nature of antimicrobial peptides, designed to act as membrane-active antimicrobial agents. The synthetic polymers such as methacrylate have been used as a molecular platform, and cationic and hydrophobic groups are randomly distributed along the polymer chains. These random copolymers do not have intrinsically programmed secondary structures such as α-helix and β-sheet and have random sequence of cationic and hydrophobic groups, which contrast the homogeneous structures of peptides. Therefore, it has been a scientific challenge for our and other laboratories in this field to provide the basis that these synthetic polymers are capable of exerting antimicrobial peptide-like activity. However, a number of studies on antimicrobial polymers with good efficacy have been documented recently,[17, 32–38] and the number of reports is increasing, supporting this new design concept of antimicrobial polymers. The availability of chemical and structural diversity of synthetic polymers has further extended this research field to include new scientific investigation of polymer architectures for use as antimicrobial macromolecules.[32, 39]
Figure 1. Antimicrobial synthetic copolymers mimicking antimicrobial peptides.

Antimicrobial copolymers are synthetic methacrylate random copolymers bearing cationic primary ammonium groups (red) and hydrophobic ethyl groups (blue) in the side chains. The methacrylate backbone was colored green. Antimicrobial peptides are α-helical cationic antimicrobial peptide LL-37. Cationic residues (blue) and hydrophobic residues (yellow) are segregated into the opposite sides of helix. The backbone structure was colored gray. (Reprinted with permission from reference.[49] Copyright 2012 American Chemical Society)
In this article, we discuss our approach to develop cationic amphiphilic polymers as antimicrobial agents and modulate their antimicrobial activity by molecular design, which has been inspired by natural antimicrobial peptides. We also discuss the structural determinants to control their antimicrobial activity and toxicity to human cells. To support these discussions, the results from biophysical experiments are described, which quantified the membrane-binding of polymers and examined the role of ammonium groups in the interactions of polymers with membranes. In addition, our recent report on the antimicrobial activity of conventional poly(ethylene imine)s, widely used as a gene carrier in drug delivery system, is discussed as a new model polymers to investigate the mode of antimicrobial action of cationic polymers.[40] The focus of this feature article is our approach with the peptide-mimetic design of antimicrobial polymers rather than an overview of reported literature on other materials. For more detailed and comprehensive discussions on preparation and efficacies of specific antimicrobial polymers, we encourage the readers to examine a number of excellent review articles referenced here.[17, 33, 34, 37, 41, 42]
Design and Activity of Cationic Amphiphilic Antimicrobial Polymers
Design and synthesis of antimicrobial methacrylate copolymers
Based on the peptide-mimetic design, we have been interested in developing antimicrobial polymers using conventional polymer platform of methacrylate, which have no intrinsic secondary structures like peptides. We synthesized poly(methacrylate)s bearing cationic and hydrophobic groups in the side chains, which are randomly distributed in a polymer chain (Figure 2).[21] These methacrylate random copolymers have primary ammonium groups as cationic sources, mimicking the cationic functionality of lysine residues which are abundant in naturally occurring and designed antimicrobial peptide sequences although quaternary ammonium groups have been traditionally used for antimicrobial polymers.[17, 37, 41] The cationic functionality of copolymers was expected to provide enhanced binding to bacterial surfaces by electrostatic attraction as in the antimicrobial peptide design template. We synthesized the copolymers in the presence of a chain transfer agent methyl 3-mercaptopropionate to prepare low molecular weight (MW) polymers (MW = 1,000 – 10,000 g·mol−1) to match with the relatively low MWs of antimicrobial peptides (a few kDa). To control the balance between net cationic charge and hydrophobicity, the mole percentages of hydrophobic groups (MPHB) were varied from 0 (cationic homopolymers) to 60 mol%. In general, the copolymers with higher MPHB of hydrophobic groups are not soluble in water which inherently limits the functionality of these compounds.
Figure 2. Synthesis of methacrylate random copolymers with cationic amphiphilic structures.
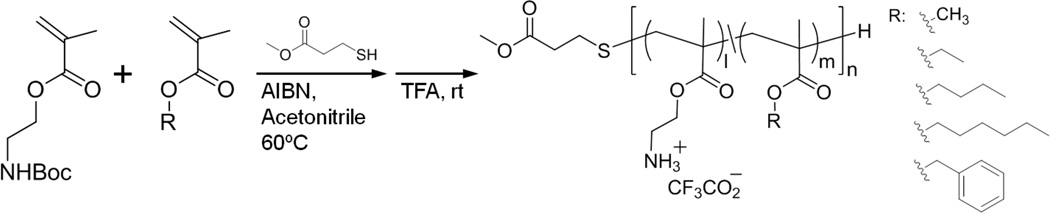
R: hydrophobic groups (methyl, ethyl, butyl, hexyl, and benzyl groups). The polymers were prepared from boc-protected amine monomer and alkyl or benzyl methacrylate monomers. The polymerization was performed using azobisisobutylonitrile (AIBN) as a radical initiator and methyl 3-mercaptopropionate as a chain transfer agent in acetonitrile. The treatment of boc-protected polymers by trifluoroacetic acid (TFA) gave the amphiphilic random copolymers with cationic ammonium groups in the side chains.
Antimicrobial and hemolytic activities of methacrylate copolymers
We first examined the antimicrobial activity of cationic copolymers with butyl methacrylate as the hydrophobic moiety (Figure 3A).[21] We synthesized a series of random copolymers with three different MW ranges (average MW = 1,600, 5,000, and 8,700 g·mol−1). We determined the minimum inhibitory concentration (MIC), which is the lowest polymer concentration to completely inhibit bacterial growth in solution, as a measure of antimicrobial activity. As a reference, a lower MIC value indicates a higher efficacy of antimicrobial activity, i.e. less of the antimicrobial is required to inhibit bacterial growth. For all the polymer series, the MIC values against E. coli decreased as the MPHB of butyl groups increased and leveled off above 30 mol%. This indicates that the hydrophobicity of polymers is one of the driving forces for the antimicrobial activity, but any further increase in hydrophobicity does not contribute to the further increases in activity (Figure 3B). Comparing the MICs of these polymers, the polymers with lowest MWs (MW = 1,300 – 1,900 g·mol−1) showed lowest MIC values (MIC = 16 µg·mL−1) above the MPHB > 30 mol%. We speculate that low MW polymer chains may be able to penetrate the cell wall structure of E. coli more effectively than high MW counterparts. We also consider that low MW of polymers yields a greater number of moles of polymer chains in a given unit of weight-based concentration compared to the high MW polymers.
Figure 3. Antimicrobial and hemolytic activities of cationic amphiphilic methacrylate random copolymers.

(A) Polymer structures. (B) Molecular weight dependence. Each series of polymers have average MW of 1,600, 5,000, and 8,700 g·mol−1. (C) Hydrophobic group dependence. Low MW polymers with different hydrophobic groups were tested for antimicrobial activity against E. coli and hemolytic activity against human red blood cells. The data are adapted from our previous reports.[21, 23]
Considering use of these copolymers as antimicrobials, it is important to evaluate their toxicity to human cells. We measured hemolytic activity against human red blood cells (RBC) as an initial assessment of toxic effect of copolymers to human cells. We determined HC50, which is a polymer concentration for 50% hemolysis. The HC50 values decreased as the MPHB of butyl groups increased, indicating hemolytic activity of polymers is also enhanced by hydrophobicity of polymers. Similar to the MICs, the HC50 values also level off at high MPHB of butyl groups. Interestingly, the HC50 values of the polymer series with lowest MWs (MW = 1,300 – 1,900 g·mol−1) were magnitudes higher than those of high MW counterparts. This result indicates that the hemolytic activity of these copolymers can be reduced by designing low MWs of copolymers, which mimic the molecular size of natural antimicrobial peptides.
The previous results suggested that polymers with small molecular sizes (low MWs) would be a good platform to prepare non-toxic antimicrobial polymers.[21] The results also indicated that the hydrophobicity of polymers enhances their antimicrobial activity, but the excess hydrophobicity renders the polymers toxic. Therefore, it is necessary to hit the right balance between hydrophobicity and cationic functionality to maximize the antimicrobial activity and minimize the hemolytic activity. To that end, we further examined the effect of side chain hydrophobicity on the antimicrobial and hemolytic activities of these copolymers (Figure 3C).[23] We synthesized low MW (1,600 to 2,000 g·mol−1) cationic methacrylate copolymers with different hydrophobic groups including methyl, ethyl, butyl, hexyl, and benzyl groups. The MIC values for E. coli decreased as the MPHB of hydrophobic groups increased and leveled off in high MPHB regions. The lowest MIC at high MPHB is almost the same value (~16 µg·mL−1) for all copolymers tested. The MPHB at the transition from high to low MIC values shifted to smaller MPHB as the alkyl chain length decreased. This result supports the notion that hydrophobicity of polymers enhances the antimicrobial activity against E. coli.
Next, the effect of hydrophobic groups on the hemolytic activity of copolymers was assessed by HC50 values. The HC50 values decreased as MPHB increased, and the curves were shifted to smaller MPHB as the alkyl chain length decreased (Figure 3B, C). For the copolymers with short alkyl chains (methyl and ethyl groups), the HC50 values are higher than MIC values at the same MPHB, indicating these copolymer series are selective to bacteria over human cells. However, the copolymers with longer alkyl chains showed lower HC50 values, resulting in no selectivity to bacteria over human cells. This is likely because the short alkyls provide small increments in the net hydrophobicity of copolymers, allowing fine-tuning of amphiphilic balance for high antimicrobial activity (low MIC) of copolymers and low hemolytic activity (high HC50). These results suggest that the antimicrobial and hemolytic activities of polymers can be controlled by careful tuning of polymer structures for optimal amphiphilic balance and molecular size. The peptide-mimetic approach has also been undertaken by the research groups, specifically investigating the structure–antimicrobial activity relationship using a variety of alternative polymer platforms including, for example, polynorbornene[35], nylon[38, 43], poly(phenyleneethynylene)s[44], polymethacrylamide[25]. These studies also reached the same conclusion that the cationic amphiphilic balance is the key determinant for the selective activity of polymers against bacteria over human cells. This indicates that the underlying design principle seems to be applicable for many synthetic polymer structures. In addition to these random copolymers, the effect of spatial separation of cationic groups and hydrophobic groups has been previously investigated using pyridinium-methacrylate copolymer[45]. A series of polymers have cationic pyridines modified with hydrophobic tails on the “same center”, which are non-hemolytic antimicrobials. On the other hand, polymers with separation of the cationic and hydrophobic tails in the different side chains onto “different centers” are antimicrobial, but also hemolytic. We have also demonstrated the segregated amphiphilic structures significantly affect their biological activities. Amphiphilic vinyl ether block copolymers with separated cationic and hydrophobic block segments showed low hemolytic activity whereas random copolymer counterparts showed high hemolytic activity.[46] Recently, Song et al. also investigated the effect of the exact distance of ammonium groups along the backbone using alternating, random, and uniform structures on antimicrobial activity.[47] The results indicated that the hydrophobic spacer distance along the polymer backbone between neighboring cationic side chains required greater than 4 Å and at least 8 – 10 Å. These reported studies indicate that the synthetic polymer platforms are able to provide a diversity of amphiphilic structures, sequences, and spatial arrangements to explore structural parameters to mimic the function and structural features of antimicrobial peptides.
Bactericidal kinetics and susceptibility to resistance
Using representative copolymers PM63 and PB27, which are methacrylate copolymers with 63 mol% methyl group and 27 mol% butyl groups respectively, we assessed the potential of these copolymers as antimicrobial agents. We first determined how quickly these polymers can reduce the number of viable E. coli cells in a solution growth phase (bactericidal kinetics) (Figure 4A).[30] At a concentration of twice the MIC, the PM63 and PB27 killed 99.99% of bacteria within 60 min for E. coli. For comparison, pexiganan, synthetic derivative of magainin was reported 99.9% killing of E. coli within one hour at the concentration of twice the MIC.[48] These results indicate that the activity of these copolymers are comparable to that of antimicrobial peptides.
Figure 4. Bactericidal kinetics and susceptibility to resistance in bacteria.

(A) Bactericidal kinetics of methacrylate copolymers against E. coli. The polymers are methacrylate copolymers with methyl group (PM63, MPHB = 63 mol%, DP = 17) and butyl groups (PB27, MPHB = 27 mol%, DP = 16) as hydrophobic side chains. The viable account below 100 CFU·mL−1 was not determined due to the detection limit of this assay condition. (B) Susceptibility to resistance in E. coli. E. coli was cultured in the presence of polymers at the half of MIC, and 21 passages did not result in resistance development while conventional antibiotics showed up to 512-fold (norfloxacin) or 256-fold (ciprofloxacin) increase in their MICs. See reference[30] for experimental details. The data are adapted from our previous report.[30]
Additionally, we also determined the susceptibility of the PM63 and PB27 copolymers to development of resistance in E. coli.[30] E. coli was sub-cultured in liquid media containing sub-inhibitory concentrations (one-half MIC) of each polymer, and the MIC of these copolymers was determined after each passage. The E. coli was sub-cultured again at the half of the MIC determined after each passage, and this procedure was repeated for up to 21 passages (Figure 4B). The MIC values of PM63 and PB27 against the cultured E. coli after each passage were constant within a single two-fold dilution through all 21 passages. In contrast, the MIC values of norfloxacin and ciprofloxacin (FDA approved drugs that act as DNA synthesis inhibitors) increased, and finally reached 512-fold or 256-fold MIC after 21 passages, respectively. This result indicates that these copolymers are not susceptible to the resistance mechanisms in E. coli under these conditions. In addition, the E. coli resistant to norfloxacin and ciprofloxacin remain susceptible to the PM63 and PB27 copolymers with the same MIC values as the original antibiotic susceptible strain. Similarly, the E. coli culture passaged with the copolymers maintains susceptibility to the norfloxacin and ciprofloxacin. These results indicate that the copolymers do not contribute to the development of cross-resistance in bacteria. It should be noted that the acquired resistance in bacteria to these antibiotics was persistent after 15 antibiotic-free passages, indicating that the antibiotic resistance was not simply physiological adaption in bacteria.
Modulating Antimicrobial Activity by Peptide-inspired Design
We have been interested in controlling the antimicrobial activity of synthetic copolymers by designing their molecular structures. As described above, our molecular design of antimicrobial copolymers is based on the mimicry of natural antimicrobial peptides. We further extended our peptide-mimetic approach to utilize the “snorkeling” effect well characterized in peptides to control the interaction of polymer chains with cell membranes.[49] The snorkeling model is one in which peptides have long spacer arms in the cationic residues (lysine and arginine), which can reach to the lipid-water interface, allowing the hydrophobic peptide helices to localize in the hydrophobic domains of lipid bilayers.[50–54] The snorkeling effect has been utilized for stabilizing hydrophobic transmembrane peptides in model and cell membranes. Inspired by this peptide design, we hypothesized that the snorkeling effect will facilitate polymer insertion to cell membranes for potent antimicrobial activity. Accordingly, we synthesized cationic amphiphilic copolymers with elongated cationic spacer arms of ethylene (E2), butylene (E4), and hexane (E6) (Figure 5A).[49] We chose ethyl methacrylate as a hydrophobic co-monomer because, as mentioned above, the short alkyl side chains can provide a means of fine-tuning overall hydrophobicity of the polymers for optimizing selective activity against bacteria over human cells. In the polymerization, the boc-protected amine monomers and ethyl methacrylate were consumed at nearly identical rates, indicating the random incorporation of these monomers into the polymer chains.[49] The representative copolymers displayed antimicrobial activity against a broad spectrum of bacteria including community-associated methicillin-resistant S. aureus.[49] The MIC values decreased as the length of spacer arms increased, indicating that the antimicrobial activity is enhanced by the elongation of spacer arms. The HC50 values of copolymers also decreased with elongation of spacer arms, indicating the hemolytic activity increased. From these results, the copolymer E428 (MPHB = 28%, DP = 12.5) showed a good balance between low MIC and high HC50 values, indicating potent activity, but low hemolytic activity. These results indicate that the copolymers can be controlled by fine-tuning of the spacer arm length to give selective activity to bacteria over human cells. To investigate the snorkeling effect on the interactions of polymers with lipid membranes, we examined the conformation of polymer chains in bacterial cell membranes by molecular dynamics simulations (Figure 5C, D). The result showed that the E2 model polymer is localized on the bilayer surface, and the polymer chain folds into a compact conformation likely due to hydrophobic collapse of polymer chains upon exposure to water. On the other hand, the polymer chains of E4 and E6 are inserted into the hydrophobic region of the bilayer, and the polymer backbone is relatively stretched. In addition, the cationic ammonium groups and ethyl side chains of ethyl methacrylate are segregated to the opposite side relative to the polymer backbone. The cationic groups appear to be interacting with phosphate from the lipid head groups, likely through an electrostatic interaction. These results indicate that the polymers can develop segregated amphiphilic structures upon binding to lipid membranes. In general, cationic amphiphilic antimicrobial peptides such as magainins and LL-37 adopt an α-helical conformation upon binding to cell membranes, in which cationic and hydrophobic residues are segregated to opposite side of the α-helix.[15, 16, 55] Although the copolymers are not likely to have any defined secondary structures such as an α-helix, the polymer chains can adopt their conformation to form similarly segregated cationic amphiphilic structures in cell membranes as peptides do. These results may indicate that such cationic amphiphilic structure is a key determinant for antimicrobial activity of peptides and polymers rather than the sequence and secondary conformation of polymers. Gellman and coworkers previously demonstrated that random nylon copolymers with cationic amphiphilic structures display high antimicrobial potency and selective activity against bacteria over human cells, which is based on the hypothesis that these copolymers are capable of forming a segregated amphiphilic conformation upon binding to bacterial cell membranes.[38, 43] Yethiraj and coworkers also previously reported that random copolymer β-peptide models also adopt the segregated amphiphilic conformation in lipid membrane.[56] These results support the design approach of antimicrobial polymers that amphiphilic copolymers can exert antimicrobial effect by mimicking the segregated amphiphilic structure of peptides, but it does not necessarily require the clearly defined facially amphiphilic secondary structures like peptides.
Figure 5. Antimicrobial polymers based on snorkeling design.

(A) Polymer structures with elongated cationic spacer arms. (B) Characteristics and properties of methacrylate copolymers with elongated cationic spacer arms. aMole percentage of ethyl group (MPethyl) and the degree of polymerization (DP) were determined by 1H NMR analysis. The number-average molecular weight (Mn) was calculated based on the MPethyl and DP. (C) Snapshot of polymer chains in lipid bilayers. (D) Polymer conformations in the XZ-plain of lipid bilayers. Reprinted with permission from reference.[49] Copyright 2012 American Chemical Society.
Mechanistic Studies by Biophysical Methods
Polymer binding to lipid membranes
We have been interested in quantifying interactions between the amphiphilic copolymers and cell membranes, which we hypothesize, determine the antimicrobial and hemolytic activities of these copolymers. To that end, we designed experiments to determine the binding properties of the amphiphilic copolymers to model lipid membranes using fluorescent dansyl-labeled polymers and large unilamellar vesicles (LUVs) or giant unilamellar vesicles (GUVs) (Figure 6).[23] The fluorescence properties of the dansyl group are sensitive to polarity of surrounding environment such that the emission spectrum of dansyl group shifts to shorter wavelengths (blue-shift) and the emission intensity increases when transferred from a polar (aqueous) to a non-polar (hydrophobic) environment. We expected that the emission from the dansyl groups conjugated to the polymer chains would increase along with blue shift when the polymers are transferred from the aqueous milieu to the hydrophobic environment of lipid membranes.[57] Thus, we prepared methacrylate copolymers modified with a dansyl group at the polymer end (Figure 6A, B). Fluorescence microscopy of GUVs composed of POPC/POPG (1:1 mol:mol) was fluorescent after addition of dansyl-labeled methacrylate copolymers, supporting the notion that the dansyl groups bind to the hydrophobic domains of lipid membranes and yielding a signal for polymer binding (Figure 6C). Likewise, the emission intensity from the dansyl groups increased with titration of liposomes, indicating that the dansyl groups inserted into the lipid bilayer when the copolymers are bound to liposomes. Accordingly, the binding isotherms of the copolymers were obtained by monitoring the emission intensity of dansyl-labeled copolymers upon titration with LUVs (Figure 6D). We used liposomes consisting of POPC or POPE/POPG (8:2) as a model bilayer composition to mimic human red blood cell (RBC) membranes or bacterial cell membranes, respectively (Figure 6D). To extend this avenue of study, we used a series of dansyl-labeled copolymers (D0, D27, and D49) with different compositions of butyl methacrylate (0, 27, and 49 mol%) to examine the effect of hydrophobic contents on their membrane binding behavior.
Figure 6. Polymer-binding to lipid bilayers.

(A) Polymer synthesis of dansyl-labeled methacrylate copolymers. (B) Characteristics and of dansyl-labeled methacrylate copolymers. aMole percentage of butyl group (MPbutyl) and the degree of polymerization (DP), were determined by 1H NMR analysis. The number-average molecular weight (Mn) was calculated based on the MPbutyl and DP. (C) Confocal image of GUVs of POPC/POPG (1:1) incubated with dansyl-labeled methacrylate copolymers (unpublished data). (D) Binding isotherms of dansyl-labeled methacrylate copolymers to liposomes. The data are adapted from our previous report.[23]
Using this approach, we were able to determine the dissociation constant (Kd) of each copolymer by curve fitting analysis. In general, the dissociation constant Kd values of copolymers decreased as the MPHB of hydrophobic butyl groups of copolymers increases for both of neutral RBC-mimetic and the anionic bacterial-mimetic membrane compositions. This indicates the net hydrophobicity of the polymers enhances binding to membrane regardless of anionic charge carried on the lipid headgroup. On the other hand, this series of methacrylate copolymers (D0, D27, and D49) showed lower Kd values for POPC liposomes than those of POPE/POPG liposomes, indicating that these copolymers preferentially bind RBC-mimetic lipid compositions over bacterial-mimetic ones. This result reflects the relatively high hemolytic activity of these copolymers with butyl side chains against human RBCs. It is interesting that the Kd values displayed only maximum 4-fold increase as the MPHB of butyl groups increased from 0 to 49 mol% while the HC50 values of corresponding polymers without dansyl groups decreased up to 100 fold. This result suggests that the activity of polymers against cell membranes is determined by not only binding, but also the subsequent mechanism of membrane disruption or pore formation upon binding the membrane. We have previously demonstrated that the hemolysis induced by methacrylate copolymers is caused by osmotic lysis due to the formation of nano-sized pores in the RBC membranes.[29] It will be necessary to further study the inter-relationship among polymer hydrophobicity, hemolytic activity, and underlying mechanism of bilayer disruption for the de novo polymer engineering principles to design non-toxic antimicrobial polymers.
Role of amine functionality in antimicrobial mechanism
In our polymer design, primary ammonium groups serve as the source of cationic charge. We initially selected these primary amines in order to mimic the structural features of the host defense peptides, which typically contain multiple lysine residues.[15] The cationic ammonium groups of peptide side chains are expected to bind to the highly negatively-charged bacterial cell surface, thus which provides a high affinity mechanism for the polymers to exert their antibacterial effects. This also facilitates the selective electrostatic attraction to bacteria cells over human cells, which have a significantly lower net negative charge on the extracellular surface. Using this rationale, if the electrostatic binding is a dominant factor in their antimicrobial mechanism, polymers with quaternary ammonium groups could yield molecules with the highest affinity for bacteria and most potent antimicrobial activity compared to polymers with primary or tertiary ammonium groups. To test this hypothesis, we prepared copolymers with primary, tertiary, or quaternary ammonium groups (Figure 7A).[24, 28] The replacement of these primary amine groups with tertiary amines or quaternary ammonium moieties resulted in diminished antimicrobial activity (Figure 7B). Based on these initial structure-activity data, we further hypothesized that the chemical structure of the cationic groups in the polymer side chain plays a key role in not only electrostatic binding of polymers to bacterial membranes, but also the molecular mechanism of membrane insertion and disruption.
Figure 7. Role of amine functionality in antimicrobial mechanism.
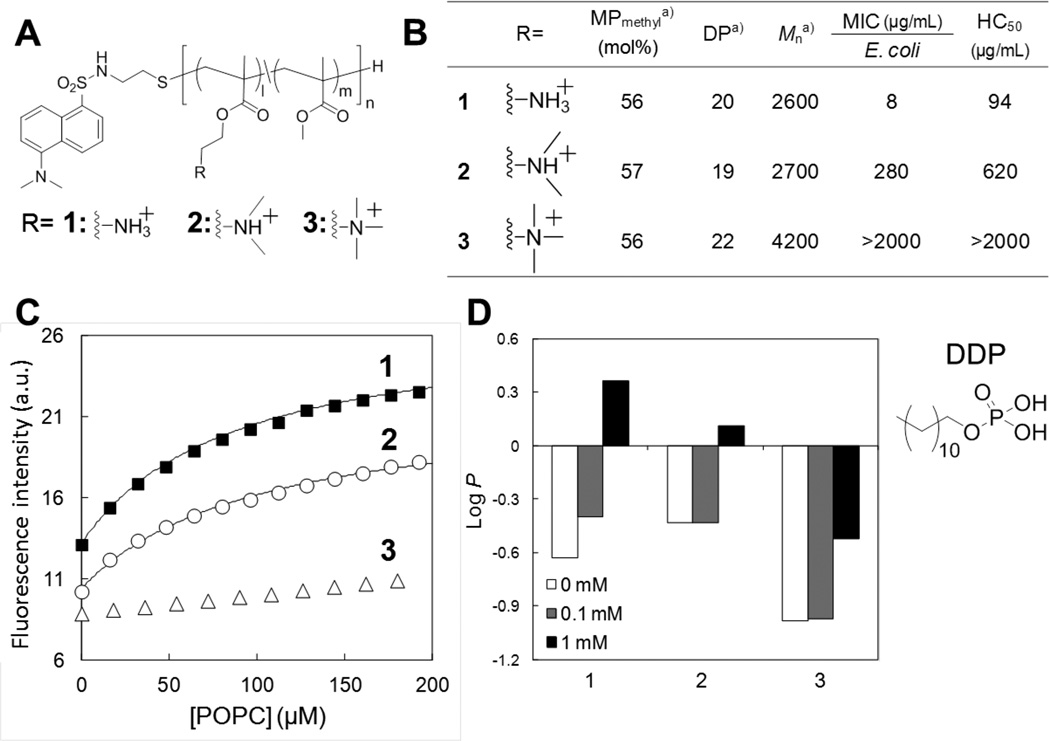
(A) Polymer structures with (1) primary, (2) tertiary, or (3) quaternary ammonium groups and a dansyl end group. (B) Characteristics and biological properties of methacrylate copolymers with primary, tertiary, or quaternary ammonium groups. aMole percentage of methyl group (MPmethyl) and the degree of polymerization (DP), were determined by 1H NMR analysis. The number-average weight (Mn) was calculated based on the MPmethyl and DP. These copolymers have similar MPmethyl and DP. (C) Fluorescence emission from dansyl groups upon binding to lipid bilayers of POPC. (D) Partitioning of polymers between octanol and water in the absence and presence of phosphate surfactant DDP. The partition coefficient P is defined as P = [Polymer]octanol/[Polymer]water. The data are adapted from our previous report.[28]
What is the role of the primary ammonium group in the mechanism of membrane disruption?
To address this question, we first investigated the affinity of polymers for lipid membrane using a representative set of polymers with different ammonium groups.[28] Again, each polymer end group was covalently attached to the membrane-sensitive dye dansyl for determining the binding behavior of these polymers to lipid membranes as described above (Figure 6). We used liposomes consisting of POPC as model lipids for initial testing. This zwitterionic lipid is a representative model for human RBC membranes, and results can be qualitatively compared with the lytic activity of the polymers against human RBCs (HC50 values). The experimental results of the binding of dye-labeled copolymers to liposomes supported our hypothesis: the fluorescence intensity of copolymers bearing primary ammonium cationic groups increased significantly compared to the other copolymers bearing tertiary or quaternary ammonium groups (Figure 7C). This suggests that the primary-ammonium containing copolymers can insert into the hydrophobic bilayer core more readily than other copolymers, whereas the copolymer with quaternary ammonium groups largely remains partitioned in the aqueous milieu.
We speculated that the polymer with primary ammonium groups binds more effectively to liposomes because of the nature of the interaction with the phosphate lipid headgroups, specifically a combination of electrostatic and hydrogen-bonding effects. This is because the affinity of polymers for lipid membranes appears to be correlated to the ability of ammonium groups for hydrogen bonding. In comparison to primary amines, tertiary amines are weaker hydrogen bond donors and their interaction with phosphates may be inhibited by the presence of two bulky methyl groups. Furthermore, the quaternary ammonium salt groups are incapable of hydrogen-bonding. To test this hypothesis, we evaluated partitioning of copolymers into water-octanol phases in the presence and absence of a phosphate surfactant (dodecyl phosphate, DDP), which mimics the phospholipid functionality (Figure 7D).[28] The copolymer with primary ammonium groups was partitioned from the aqueous to the organic layer upon addition of the anionic phosphate DDP, whereas the copolymers with tertiary and quaternary ammonium groups are less sensitive to the DDP. This result supports the hypothesis of specific complexation of primary ammonium groups with phosphate groups. This complexation also facilitates polymer partitioning to the octanol phase, which indicates that the complexation of primary ammonium groups to phosphate headgroups likely enhances the polymer insertion into the hydrophobic bilayer core, which ultimately enhances the membrane disruption. The insertion of polymers into lipid bilayers was further examined by sum frequency generation (SFG) vibrational spectroscopy.[27] This technique allows for the determination of molecular orientation within the bilayer. The SFG spectra of copolymers with 33 mol% of butyl side chains showed that the butyl groups are oriented parallel to the surface normal of lipid bilayer. This result suggests that the hydrophobic groups of polymers are likely insert into the hydrophobic core of lipid membrane. These results are consistent with the results of computational studies on the amphiphilic conformations of polymers in lipid membranes (Figure 5).
Cationic Polymer Functionality for Anti-S. aureus Activity
Recently, we have expanded the types of polymer backbones for antibiotic development and investigated the antimicrobial activity and toxicity of cationic poly(ethylene imine)s (PEIs).[40] PEIs have been extensively used as drug and gene carriers in biomedical applications because of their ability to enter cells through endosomal escape.[58–61] In the development of antimicrobial polymers, a number of studies reported antimicrobial activity of PEI derivatives modified with hydrophobic long alkyl groups[62–64] or quaternized by alkyl groups to give water-soluble antimicrobials or water insoluble antimicrobial coatings.[65–67] All of these studies focused on the activity of modified PEIs, but the PEI backbone was not considered as a contributing, and possibly functionally active, antimicrobial. As such, we have been interested in the intrinsic antimicrobial activity of un-modified PEIs as molecular models to investigate the antimicrobial mode of action and molecular mechanism driven by the cationic functionality and structures of synthetic polymers.
We tested a series of commercially available PEIs with different molecular weights (500 – 12,000 g·mol−1) and polymer structures (branched and linear) (Figure 8A, B).[40] In general, the branched PEIs (B-PEIs) showed potent antimicrobial activity against S. aureus (MIC = 16 – 32 µg·mL−1), while these B-PEIs are not active against E. coli (MIC > 250 µg·mL−1). On the other hand, linear PEIs (L-PEIs) are relatively active against both E. coli (MIC = 31 µg·mL−1) and S. aureus (MIC = 8 µg·mL−1). In addition, the B-PEIs showed no hemolytic activity up to 4,000 µg·mL−1 while the L-PEIs are relatively hemolytic. These results indicate that the B-PEIs appear to be selective to S. aureus over E. coli and human RBCs. Such selective activity has been reported previously for cationic compounds including polynorbornene derivatives[35] and acyl-Lys oligomers[68]. Dissection of the structure-activity relationship yielding bacterial selectivity will be of great interest for future studies.
Figure 8. Antimicrobial activity of poly(ethylene imine)s.

(A) Chemical structures and schematic presentations of branched and liner PEIs. (B) Characteristics and biological properties of PEIs. aNumber-average molecular weight (Mn) was determined by aqueous GPC using PEGs as standards. (C) S. aureus membrane depolarization induced by PEIs in HEPES buffer. A membrane-potential sensitive dye DiSC3(5) was added to S. aureus suspension at 20 seconds. PEI or melittin was added at 100 seconds. At 200 seconds, melittin was added to the bacterial solution containing PEIs. The concentrations of PEIs are the MIC of each PEI determined in Muller Hinton broth. The data are adapted from our previous report.[40]
Intrigued by the bacterial selectivity, we further examined the antimicrobial mechanism of these PEIs. We tested the ability of PEIs to permeabilize cell membranes of S. aureus using the membrane-potential depolarization assay using the fluorescent dye DiSC3(5) which is sensitive to membrane potential.[68, 69] This dye accumulates in the S. aureus cell membrane and the fluorescence emission is self-quenched due to high local dye concentrations. If PEIs damage the membrane, it would cause membrane depolarization and subsequent alleviation of the self-quenching, i.e. a resultant increases in emission intensity. Interestingly, the B-PEIs did not cause any significant membrane depolarization up to concentrations 5 times greater than the MIC (Figure 8C). This result indicates that membrane permeabilization is not likely to be the primary mechanism of antimicrobial activity of B-PEIs against S. aureus. Although the detailed mechanism is not clear at this point, cationic B-PEIs could bind to anionic components in the cell wall including cell-wall and lipoteichoic acids and further interact with proteins and membranes in the cell, disrupting cellular biological functions as speculated for natural antimicrobial polymer chitosan[70].
On the other hand, the L-PEIs caused substantial depolarization of the S. aureus membrane, which is comparable to lytic peptide melittin. This indicates that the L-PEIs may exert their antimicrobial effect, at least in part, by membrane disruption. This mode of action of L-PEIs may reflect the potent activity against both E. coli and S. aureus as membrane-active antimicrobial peptides show a broad spectrum of activity in general. Notably, 1H NMR analysis indicated that the L-PEIs have 4 – 11 mol% of N-propionyl groups still remaining in the final preparations, after acid hydrolysis in the PEI synthesis. It is likely because of these hydrophobic propionyl groups that the L-PEIs can disrupt bacterial cell membranes, resulting in inhibition of bacterial growth as shown for other amphiphilic copolymers.
In addition to antimicrobial activity, PEIs are also capable of inducing fusion of lipid vesicles.[71] Anionic vesicles consisting of 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) in the presence of PEI with MW of 600 – 10,000 g·mol−1 undergo fusion to form larger vesicles. Interestingly, the ability of PEIs to induce fusion of vesicles depends on the PEI concentration (Figure 9). Dynamic light scattering (DLS) measurement revealed that, in the case of PEI with MW of 10,000 g·mol−1, the diameter of fused vesicles showed the maximum value of 600 nm around 1 µM of PEI, while the higher or lower PEI concentrations did not affect the vesicle size (Figure 9A). PEIs-induced lipid mixing between vesicles through membrane fusion was further evaluated by means of Förster resonance energy transfer (FRET)-based lipid mixing assay.[72] We used a FRET pair consisting of N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phoshoethanolamine, triethylammonium salt (NBD-PE, donor) and N-(Lissamine rhodamine B)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt (Rh-PE, acceptor).[73] Dye-labeled vesicles and unlabeled vesicles were independently prepared and pre-mixed with each other prior to the addition of the PEIs. The intervesicular lipid mixing was detected as recovery of the donor fluorescence observed at 530 nm (I530) due to the dilution of donor/acceptor pair in the membrane originated in the fusion between dye-labeled and unlabeled vesicles (Figure 9B). We observed that addition of PEI showed apparent increase of I530 in a specific concentration range (0.1 – 1 µM), in which an apparent increase of vesicular size was also observed. These results indicate that the PEIs have specific concentration regions for enhancing membrane fusion. Our results show that PEIs bound to liposomal surfaces only induce the membrane fusion at the intermediate concentrations tested but NOT at the highest concentrations. This can be thought of as a “sweet spot” of balance between bilayer destabilization induced by PEI binding and electrostatic repulsions between PEIs associated with different vesicles (Figure 9C). At the intermediate concentrations, PEIs create enough disruption of bilayer packing order, allowing for vesicles to fuse however do NOT impart enough surface charge to prevent close approach of fusion partner vesicles. However, at the high PEI concentrations, the high density of PEIs on the liposomal surface inhibits the interactions between vesicles due to electrostatic repulsions between high net positive charges present on the liposome surfaces, as confirmed by the zeta-potential measurement (Figure 9B). These results indicate that PEIs are capable of modulating interactions between anionic cell membranes which represent the properties of bacterial cell membranes. PEIs have been extensively used in biomedical applications, however, to the best of our knowledge, the interactions of PEIs with bacterial cell wall and cell membranes have not been investigated systematically, and the understanding of biological function of PEIs in bacteria cells is limited. Synthetic polymers such PEIs are very cost-effective compared to other drugs and peptides and will serve as models for the design and mechanism of future antimicrobial polymers.
Figure 9. Membrane fusion induced by PEI (MW = 10,000 g·mol−1).

(A) Effect of the PEI on the size distribution of DOPS vesicles as evaluated by DLS. (B) Concentration dependence of the PEI on fluorescence intensity at 530 nm (I530) as an index of lipid mixing in FRET-based assay and zeta potential of DOPS vesicles. (C) Cryo-TEM images of the DOPS vesicles in the presence of the PEI at various concentrations (Bar = 100 nm). The data are adapted from our previous report.[71]
Conclusion and Future Perspectives
In summary, we have developed a number of antimicrobial copolymers based on the mimicry of naturally occurring antimicrobial peptides. The copolymers showed antimicrobial activity against a broad spectrum of bacteria including drug-resistant S. aureus. Importantly, these copolymers did not result in the resistance development in E. coli after over 21 passages in the presence of the copolymers while the MICs of conventional antibiotic norfloxacin increased up to 512-fold under the same conditions. The activity of copolymers was modulated using the snorkeling effect exerted by cationic elongated side chains. These activity profiles represent the hallmarks of antimicrobial peptides, and these peptide-inspired design approaches have been successfully translated to improve the antimicrobial activity of synthetic polymers.
From these results, antimicrobial polymers offer a great deal of promise for development of new antimicrobials which are less susceptible to the emergent and current resistance mechanisms in bacteria. The development of these new polymers and compounds provides the basis to further investigate their potential clinical and industrial applications. While this promise is evident, it does not come without the need for significantly enhanced studies into the mechanism of action of the compounds and their physiological relevance. One of the complicating factors in this line of study is that the parent model, the antimicrobial peptides, is not fully understood in terms of molecular mechanism.[74–77] This is partially because of the number and sequence variability in naturally occurring host defense peptides. In addition, it has been recently reported that the antimicrobial peptides have low affinity multiple targets in bacteria, contrasting to conventional antibiotics which have high affinity specific targets.[15, 16, 78, 79] Therefore, the antimicrobial activity of peptides may rise from the results from assembles of each component of mechanism or their synergistic effects. The antimicrobial peptides-mimetic polymers may inherit this complexity of mechanism, which would require new methodology to elucidate the molecular mechanism of antimicrobial polymers. Alternatively, use of polymers may be able to dissect these mechanisms by targeting different functions and thus isolating molecular properties relevant for specific aspects of molecular action.
As future perspectives to the research filed of antimicrobial polymers, continuing research interest is the development of novel polymers and macromolecules. The myriad of possibilities afforded by the variety of copolymer building blocks expands the possibilities for new active compounds with variable or tailored activity. In this regard, we have recently developed cationic block copolymers which displayed selective activity against E. coli over human RBCs while the counterpart random copolymers showed high hemolytic activity.[46] The macromolecular approach will offer new design of antimicrobials from nano-scale particles to possibly macroscopic gels,[32, 39, 80] which are not readily accessible by single peptides. For example, Nederberg et al. have recently prepared a biodegradable cationic nanoparticle by self-assembly of block copolymers, which is likely to exert antimicrobial effect by attacking bacterial cell membranes.[39] The nanoparticle showed a broad spectrum of activity and is effective against bacterial infections in vivo. These new approaches will open new avenues to designing polymers and macromolecules with new and tunable antimicrobial activity profiles while adding functionalities such as biodegradability and additional payload delivery.
In addition to the development of new materials, there will be a continual need for biophysical, cellular, and microbiological characterization of novel compounds to elucidate mechanism of action. As seen from the experiences with development of antimicrobial polymers based on antimicrobial peptides, retaining specific molecular signatures does not guarantee identical mechanism when applied to bacteria. This also gives rise a need for further development of new methodology to answer biophysical questions for these systems. New methods for characterizing cellular binding mechanisms, mechanism of membrane permeability, and intracellular interactions are necessary as the activity of antimicrobial polymers is tuned to be more specific and targeted in nature. These novel methods may also be directed at better mimicry of the physiological environment, such that biophysical measurements can more accurately reflect the behavior of antimicrobials in the in vivo conditions.
Finally, of specific interest to the field is the deeper understanding of the complex relationship between bacteria, host and antimicrobial polymers.[77, 81–84] As noted, the antimicrobial polymers mimic many aspects of the antimicrobial peptide mechanism of action when studied from a biophysical perspective as well as with respect to in vitro cellular, microbiological, and biochemical assays. That said, there are still many unanswered questions regarding the specific, mechanistic details of the interactions between antimicrobial polymers and the bacteria they are targeting. This includes not only the detailed mechanism of action, which is likely variable between polymer classes, but also the bacterial responses to these compounds which relate to resistance mechanisms.[85–88] This lack of understanding is equally evident regarding the systemic host response to the polymers and to any metabolic or reaction side-products created through the bacteriolytic functions. In particular, the bacterial responses to antimicrobial exposure have recently garnered significant interest with regard to development of antimicrobials[85–89] although there is a dearth of information regarding bacterial responses to antimicrobial polymers. Numerous studies have been performed to investigate the bacterial transcriptional responses to antimicrobial peptide exposure. The results provided insight into the relationships of antimicrobial mechanism with bacterial resistance as well as the activation mechanism of virulence factors upon antimicrobial exposure. Antimicrobial peptide exposure has also been shown to modulate bacterial motility and the ability to form biofilms, an important consideration when dosing antimicrobials.[85–90] These studies will provide a starting point for investigating antimicrobial polymers and their action.
Overall the future is promising for the development of antimicrobial polymers that can transition from the laboratory to the clinic or to applied materials. The ease and cost effective synthesis of polymers combined with variety of building blocks presents tremendous opportunities for the development of new, tunable, highly effective antimicrobials.
Acknowledgements
This work was supported in part by a JSPS research fellowship grant (No. 22-5165) (H.T.), a Grant-in-Aid for Young Scientists B (No. 22700494) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (K.Y.), and NIH award (1R15GM094330) (G.C.).
Biographies

Dr. Haruko Takahashi
Haruko Takahashi received her PhD degree from the Tokyo Medical and Dental University, Japan in 2012. In 2012 she was a postdoctoral fellow at the Kyoto University, Japan on a fellowship supported by the Japan Society for the Promotion of Science. Starting in 2013 she has been a visiting scholar at the University of Michigan School of Dentistry. Her research focuses on the design of biologically functional materials based on synthetic polymers for biomedical applications in the fields of drug delivery, protein engineering, and antimicrobial development.

Prof. Gregory A. Caputo
Gregory A. Caputo received his PhD degree from Stony Brook University in 2003. During 2004 and 2005, he was a postdoctoral researcher at Texas A&M Health Sciences center followed by a postdoctoral fellowship at the University of Pennsylvania School of Medicine from 2005–2007. He is currently an Associate Professor in the Department of Chemistry and Biochemistry at Rowan University. The focus of research in his laboratory is on the characterization and development of peptides and peptide mimetics for use in antimicrobial applications, green energy applications, and as models for fundamental biophysical studies.

Prof. Kenichi Kuroda
Kenichi Kuroda received his B.S. in 1995 and M. Eng. in 1997 from Kyoto University, Japan and his PhD in 2003 from the Massachusetts Institute of Technology. After his postdoctoral training at the University of Pennsylvania School of Medicine, he joined the faculty at the University of Michigan School of Dentistry as Assistant Professor in 2006. His research interests include the design and development of peptide-mimetic polymers and oligomers as antimicrobial materials effective against drug-resistant bacteria and as membrane-active agents for use in biomedical applications. His laboratory also focuses on the development of new dental materials including biomimetic polymer adhesives.
Footnotes
Conflict of interest:
One of the authors (K. Kuroda) is a co-inventor on a patent application filed by the University of Pennsylvania covering “Antimicrobial Copolymers and Uses Thereof”. The patent application has been licensed to PolyMedix Inc. (Radnor, PA). PolyMedix did not play a role in the design and conduct of this study, in the collection, analysis, or interpretation of the data, or in the preparation, review, or approval of the article.
Contributor Information
Haruko Takahashi, Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Kyoto, Japan.
Edmund F. Palermo, Macromolecular Science and Engineering Center, University of Michigan, Ann Arbor, MI, USA
Kazuma Yasuhara, Graduate School of Materials Science, Nara Institute of Science and Technology, Nara, Japan.
Gregory A. Caputo, Department of Chemistry and Biochemistry, Rowan University, Glassboro, NJ, USA
Kenichi Kuroda, Email: kkuroda@umich.edu, Department of Biologic and Materials Sciences, School of Dentistry, University of Michigan, Ann Arbor, MI, USA.
References
- 1.World Health Organization. WHO Global Strategy for Containment of Antimicrobial Resistance. 2013 Apr; http://www.who.int.proxy.lib.umich.edu/csr/resources/publications/drugresist/en/EGlobal_Strat.pdf.
- 2.World Health Organization. The evolving threat of antimicrobial resistance Options for action. 2013 Apr; http://whqlibdoc.who.int/publications/2012/9789241503181_eng.pdf.
- 3.World Health Organization. World Health Day — 7 April 2011. 2013 Apr; http://www.who.int.proxy.lib.umich.edu/world-health-day/2011/en/
- 4.Klopper M, Warren RM, Hayes C, van Pittius NCG, Streicher EM, Müller B, Sirgel FA, Chabula-Nxiweni M, Hoosain E, Coetzee G, van Helden PD, Victor TC, Trollip AP. Emerg. Infect. Dis. 2013;19:449. doi: 10.3201//EID1903.120246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy SB. The Antibiotic Paradox. How Miracle Drugs Are Destroying the Miracle. New York: Plenum Press; 1992. [Google Scholar]
- 6.Levy SB, Marshall B. Nat. Med. 2004;10:S122. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 7.Nathan C. Nature. 2004;431:899. doi: 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- 8.Fernandes P. Nat. Biotechnol. 2006;24:1497. doi: 10.1038/nbt1206-1497. [DOI] [PubMed] [Google Scholar]
- 9.Fischbach MA, Walsh CT. Science. 2009;325:1089. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambers HF, Deleo FR. Nat. Rev. Microbiol. 2009;7:629. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Castro FR, Naranjo OR, Marco JA, Violan JS. Semin. Respir. Crit. Care Med. 2009;30:161. doi: 10.1055/s-0029-1202935. [DOI] [PubMed] [Google Scholar]
- 12.Kohanski MA, Dwyer DJ, Collins JJ. Nat. Rev. Microbiol. 2010;8:423. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert PA. Adv. Drug Deliver. Rev. 2005;57:1471. doi: 10.1016/j.addr.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Tenover FC. Am. J. Infect. Control. 2006;34:S3. doi: 10.1016/j.ajic.2006.05.219. [DOI] [PubMed] [Google Scholar]
- 15.Zasloff M. Nature. 2002;415:389. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 16.Brogden KA. Nat. Rev. Microbiol. 2005;3:238. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 17.Munoz-Bonilla A, Fernandez-Garcia M. Prog. Polym. Sci. 2012;37:281. [Google Scholar]
- 18.Kuroda K, Caputo GA. Wires Nanomed. Nanobi. 2013;5:49. doi: 10.1002/wnan.1199. [DOI] [PubMed] [Google Scholar]
- 19.Powers JPS, Hancock REW. Peptides. 2003;24:1681. doi: 10.1016/j.peptides.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 20.Marr AK, Gooderham WJ, Hancock REW. Curr. Opin. Pharmacol. 2006;6:468. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Kuroda K, DeGrado WF. J. Am. Chem. Soc. 2005;127:4128. doi: 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]
- 22.Ivanov I, Vemparala S, Pophristic V, Kuroda K, DeGrado WF, McCammon JA, Klein ML. J. Am. Chem. Soc. 2006;128:1778. doi: 10.1021/ja0564665. [DOI] [PubMed] [Google Scholar]
- 23.Kuroda K, Caputo GA, DeGrado WF. Chem.-Eur. J. 2009;15:1123. doi: 10.1002/chem.200801523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palermo EF, Kuroda K. Biomacromolecules. 2009;10:1416. doi: 10.1021/bm900044x. [DOI] [PubMed] [Google Scholar]
- 25.Palermo EF, Sovadinova I, Kuroda K. Biomacromolecules. 2009;10:3098. doi: 10.1021/bm900784x. [DOI] [PubMed] [Google Scholar]
- 26.Palermo EF, Kuroda K. Appl. Microbiol. Biot. 2010;87:1605. doi: 10.1007/s00253-010-2687-z. [DOI] [PubMed] [Google Scholar]
- 27.Avery CW, Palermo EF, McLaughin A, Kuroda K, Chen Z. Anal. Chem. 2011;83:1342. doi: 10.1021/ac1025804. [DOI] [PubMed] [Google Scholar]
- 28.Palermo EF, Lee DK, Ramamoorthy A, Kuroda K. J. Phys. Chem. B. 2011;115:366. doi: 10.1021/jp1083357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sovadinova I, Palermo EF, Huang R, Thoma LM, Kuroda K. Biomacromolecules. 2011;12:260. doi: 10.1021/bm1011739. [DOI] [PubMed] [Google Scholar]
- 30.Sovadinova, EF IP, Urban M, Mpiga P, Caputo GA, Kuroda K. Polymers. 2011;3:1512. [Google Scholar]
- 31.Mizutani M, Palermo EF, Thoma LM, Satoh K, Kamigaito M, Kuroda K. Biomacromolecules. 2012;13:1554. doi: 10.1021/bm300254s. [DOI] [PubMed] [Google Scholar]
- 32.Engler AC, Wiradharma N, Ong ZY, Coady DJ, Hedrick JL, Yang YY. Nano Today. 2012;7:201. [Google Scholar]
- 33.Gabriel GJ, Som A, Madkour AE, Eren T, Tew GN. Mat. Sci. Eng. R. 2007;57:28. doi: 10.1016/j.mser.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kenawy ER, Worley SD, Broughton R. Biomacromolecules. 2007;8:1359. doi: 10.1021/bm061150q. [DOI] [PubMed] [Google Scholar]
- 35.Lienkamp K, Madkour AE, Musante A, Nelson CF, Nusslein K, Tew GN. J. Am. Chem. Soc. 2008;130:9836. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tew GN, Scott RW, Klein ML, Degrado WF. Accounts Chem. Res. 2009;43:30. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timofeeva L, Kleshcheva N. Appl. Microbiol. Biot. 2011;89:475. doi: 10.1007/s00253-010-2920-9. [DOI] [PubMed] [Google Scholar]
- 38.Mowery BP, Lindner AH, Weisblum B, Stahl SS, Gellman SH. J. Am. Chem. Soc. 2009;131:9735. doi: 10.1021/ja901613g. [DOI] [PubMed] [Google Scholar]
- 39.Nederberg F, Zhang Y, Tan JPK, Xu KJ, Wang HY, Yang C, Gao SJ, Guo XD, Fukushima K, Li LJ, Hedrick JL, Yang YY. Nat. Chem. 2011;3:409. doi: 10.1038/nchem.1012. [DOI] [PubMed] [Google Scholar]
- 40.Gibney KA, Sovadinova I, Lopez AI, Urban M, Ridgway Z, Caputo GA, Kuroda K. Macromol. Biosci. 2012;12:1279. doi: 10.1002/mabi.201200052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tashiro T. Macromol. Mater. Eng. 2001;286:63. [Google Scholar]
- 42.Siedenbiedel F, Tiller JC. Polymers. 2012;4:46. [Google Scholar]
- 43.Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. J. Am. Chem. Soc. 2007;129:15474. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 44.Ishitsuka Y, Arnt L, Majewski J, Frey S, Ratajczek M, Kjaer K, Tew GN, Lee KYC. J. Am. Chem. Soc. 2006;128:13123. doi: 10.1021/ja061186q. [DOI] [PubMed] [Google Scholar]
- 45.Sambhy V, Peterson BR, Sen A. Angew. Chem. Int. Edit. 2008;47:1250. doi: 10.1002/anie.200702287. [DOI] [PubMed] [Google Scholar]
- 46.Oda Y, Kanaoka S, Sato T, Aoshima S, Kuroda K. Biomacromolecules. 2011;12:3581. doi: 10.1021/bm200780r. [DOI] [PubMed] [Google Scholar]
- 47.Song AR, Walker SG, Parker KA, Sampson NS. ACS Chem. Biol. 2011;6:590. doi: 10.1021/cb100413w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge YG, MacDonald DL, Holroyd KJ, Thornsberry C, Wexler H, Zasloff M. Antimicrob. Agents Chemother. 1999;43:782. doi: 10.1128/aac.43.4.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palermo EF, Vemparala S, Kuroda K. Biomacromolecules. 2012;13:1632. doi: 10.1021/bm300342u. [DOI] [PubMed] [Google Scholar]
- 50.Strandberg E, Morein S, Rijkers DTS, Liskamp RMJ, van der Wel PCA, Killian JA. Biochemistry. 2002;41:7190. doi: 10.1021/bi012047i. [DOI] [PubMed] [Google Scholar]
- 51.Caputo GA, London E. Biochemistry. 2003;42:3275. doi: 10.1021/bi026697d. [DOI] [PubMed] [Google Scholar]
- 52.Strandberg E, Killian JA. FEBS Lett. 2003;544:69. doi: 10.1016/s0014-5793(03)00475-7. [DOI] [PubMed] [Google Scholar]
- 53.Jaud S, Fernandez-Vidal M, Nilsson I, Meindi-Beinker NM, Hubner NC, Tobias DJ, von Heijne G, White SH. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11588. doi: 10.1073/pnas.0900638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schow EV, Freites JA, Cheng P, Bernsel A, von Heijne G, White SH, Tobias DJ. J. Membrane Biol. 2011;239:35. doi: 10.1007/s00232-010-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hancock REW, Sahl HG. Nat. Biotechnol. 2006;24:1551. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 56.Mondal J, Zhu X, Cui QA, Yethiraj A. J. Phys. Chem. B. 2010;114:13585. doi: 10.1021/jp1070242. [DOI] [PubMed] [Google Scholar]
- 57.Li YH, Chan LM, Tyer L, Moody RT, Himel CM, Hercules DM. J. Am. Chem. Soc. 1975;97:3118. [Google Scholar]
- 58.Neu M, Fischer D, Kissel T. J. Gene. Med. 2005;7:992. doi: 10.1002/jgm.773. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen DN, Green JJ, Chan JM, Longer R, Anderson DG. Adv. Mater. 2009;21:847. doi: 10.1002/adma.200801478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia T, Kovochich M, Liong M, Meng H, Kabehie S, George S, Zink JI, Nel AE. ACS Nano. 2009;3:3273. doi: 10.1021/nn900918w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miyata K, Nishiyama N, Kataoka K. Chem. Soc. Rev. 2012;41:2562. doi: 10.1039/c1cs15258k. [DOI] [PubMed] [Google Scholar]
- 62.Chen CZS, Cooper SL. Biomaterials. 2002;23:3359. doi: 10.1016/s0142-9612(02)00036-4. [DOI] [PubMed] [Google Scholar]
- 63.Gao BJ, Zhang X, Zhu Y. J. Biomater. Sci.-Polym. Ed. 2007;18:531. doi: 10.1163/156856207780852523. [DOI] [PubMed] [Google Scholar]
- 64.Pasquier N, Keul H, Heine E, Moeller M, Angelov B, Linser S, Willumeit R. Macromol. Biosci. 2008;8:903. doi: 10.1002/mabi.200800121. [DOI] [PubMed] [Google Scholar]
- 65.Lin J, Qiu SY, Lewis K, Klibanov AM. Biotechnol. Bioeng. 2003;83:168. doi: 10.1002/bit.10651. [DOI] [PubMed] [Google Scholar]
- 66.Haldar J, Weight AK, Klibanov AM. Nat. Protoc. 2007;2:2412. doi: 10.1038/nprot.2007.353. [DOI] [PubMed] [Google Scholar]
- 67.Dhende VP, Samanta S, Jones DM, Hardin IR, Locklin J. ACS Appl. Mater. Interfaces. 2011;3:2830. doi: 10.1021/am200324f. [DOI] [PubMed] [Google Scholar]
- 68.Epand RF, Sarig H, Mor A, Epand RM. Biophys. J. 2009;97:2250. doi: 10.1016/j.bpj.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6968. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raafat D, von Bargen K, Haas A, Sahl HG. Appl. Environ. Microbiol. 2008;74:3764. doi: 10.1128/AEM.00453-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yasuhara K, Tsukamoto M, Tsuji Y, Kikuchi J. Colloid. Surface. A. 2012;415:461. [Google Scholar]
- 72.Hoekstra D, Düzgüneş N. Methods in Enzymology. San Diego, CA, USA: Academic Press; 1993. Lipid mixing assays to determine fusion in liposome systems; p. 115. [DOI] [PubMed] [Google Scholar]
- 73.Struck DK, Hoekstra D, Pagano RE. Biochemistry. 1981;20:4093. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 74.Rahnamaeian M. Plant signaling & behavior. 2011;6:1325. doi: 10.4161/psb.6.9.16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hancock REW, Nijnik A, Philpott DJ. Nat. Rev. Microbiol. 2012;10:243. doi: 10.1038/nrmicro2745. [DOI] [PubMed] [Google Scholar]
- 76.Bowie JH, Separovic F, Tyler MJ. Peptides. 2012;37:174. doi: 10.1016/j.peptides.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 77.Afacan NJ, Yeung ATY, Pena OM, Hancock REW. Curr. Pharm. Design. 2012;18:807. doi: 10.2174/138161212799277617. [DOI] [PubMed] [Google Scholar]
- 78.Franco OL. FEBS Lett. 2011;585:995. doi: 10.1016/j.febslet.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 79.Peschel A, Sahl H-G. Nat. Rev. Microbiol. 2006;4:529. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- 80.Liu SQ, Yang C, Huang Y, Ding X, Li Y, Fan WM, Hedrick JL, Yang Y-Y. Adv. Mater. 2012;24:6484. doi: 10.1002/adma.201202225. [DOI] [PubMed] [Google Scholar]
- 81.Shai Y, Makovitzky A, Avrahami D. Curr. Protein Pept. Sci. 2006;7:479. doi: 10.2174/138920306779025620. [DOI] [PubMed] [Google Scholar]
- 82.Karavolos MH, Spencer H, Bulmer DM, Thompson A, Winzer K, Williams P, Hinton JCD, Khan CMA. Bmc Genomics. 2008;9 doi: 10.1186/1471-2164-9-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ahmad A, Ahmad E, Rabbani G, Haque S, Arshad M, Khan RH. Curr. Protein Pept. Sci. 2012;13:211. doi: 10.2174/138920312800785076. [DOI] [PubMed] [Google Scholar]
- 84.Madi A, Alnabhani Z, Leneveu C, Mijouin L, Feuilloley M, Connil N. Arch. microbiol. 2013;195:189. doi: 10.1007/s00203-012-0865-3. [DOI] [PubMed] [Google Scholar]
- 85.Hirakawa H, Nishino K, Hirata T, Yamaguchi A. J. Bacteriol. 2003;185:1851. doi: 10.1128/JB.185.6.1851-1856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bader MW, Navarre WW, Shiau W, Nikaido H, Frye JG, McClelland M, Fang FC, Miller SI. Mol. Microbiol. 2003;50:219. doi: 10.1046/j.1365-2958.2003.03675.x. [DOI] [PubMed] [Google Scholar]
- 87.Groisman EA, Mouslim C. Nat. Rev. Microbiol. 2006;4:705. doi: 10.1038/nrmicro1478. [DOI] [PubMed] [Google Scholar]
- 88.Mensa B, Kim YH, Choi S, Scott R, Caputo GA, DeGrado WF. Antimicrob. Agents Chemother. 2011;55:5043. doi: 10.1128/AAC.05009-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Costechareyre D, Chich J-F, Strub J-M, Rahbe Y, Condemine G. PloS one. 2013;8:e54118. doi: 10.1371/journal.pone.0054118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hunke S, Keller R, Muller VS. Fems Microbiol. Lett. 2012;326:12. doi: 10.1111/j.1574-6968.2011.02436.x. [DOI] [PubMed] [Google Scholar]