

Abstract
Imaging of poly (ADP-ribose) polymerase-1 (PARP-1) expression in vivo is a potentially powerful tool for developing PARP-1 inhibitors for drug discovery and patient care. We have synthesized several derivatives of benzimidazole carboxamide as PARP-1 inhibitors, which can be 18F-labeled easily for positron emission tomographic (PET) imaging. Of the compounds synthesized, 12 had the highest inhibition potency for PARP-1 (IC50 = 6.3 nM). [18F]12 was synthesized under conventional conditions in high specific activity with 40-50 % decay-corrected yield. MicroPET studies using [18F]12 in MDA-MB-436 tumor-bearing mice demonstrated accumulation of [18F]12 in the tumor that was blocked by olaparib, suggesting that the uptake of [18F]12 in the tumor is specific to PARP-1 expression.
Keywords: PARP-1, PET, radiolabeling, imaging
1. Introduction
Poly (ADP-ribose) polymerase-1 (PARP-1) is one of the most abundant members of the PARP family of nuclear enzymes.1 Its best characterized functions are sensing DNA damage and facilitating DNA repair, but PARP-1 also participates in many other DNA-related cellular processes, such as apoptosis regulation, cell division, differentiation, transcriptional regulation, and chromosome stabilization.2-5 PARP-1, a 113 kD protein, has three unique structural domains: the N-terminal DNA binding domain with two zinc fingers that specifically bind to damaged DNA strand breaks;1, 6 the central automodification domain; and the C-terminal catalytic domain that sequentially transfers ADP-ribose subunits from nicotinamide adenine dinucleotide (NAD+) to protein acceptors.7 Due to its critical role in DNA repair, PARP-1 has been actively pursued as a drug target over the past 20 years, with tremendous efforts invested to develop several generations of PARP-1 inhibitors (Figure 1) for therapeutic purposes, especially in the area of ischemia-reperfusion injury and cancer.5, 8 More recently, PARP1 inhibition has been demonstrated as an effective method for inducing synthetic lethality in cancers that depend on PARP1 activity for survival.9 Therefore, a number of PARP inhibitors, including olaparib (AZD2281, KU-59436), veliparib (ABT-888), rucaparib (PF-01367338, AG014699), and niraparib (MK4827), are now undergoing evaluation in clinical trials as anticancer drugs10-14. These PARP inhibitors effectively inhibit PARP1 activity as well as activity of other PARP-like enzymes such as PARP2 and tankyrase115.
Figure 1. Examples of PARP-1 inhibitors.

Despite the promising results from these clinical trials related to progression-free survival, however, differences in the ability of the various PARP inhibitors to suppress tumoral PARP activity cannot be accurately determined by current assays. Additionally, the mechanism of action for iniparib, initially developed as a PARP inhibitor, was more recently discovered to be most likely unrelated to inhibition of PARP activity16. Therefore, methods that can quantify tumoral PARP activity noninvasively in vivo would be particularly useful for demonstrating tumor-specific PARP inhibition as well as assessing the duration of effective PARP inhibition to guide dosing decisions.
Imaging with positron emission tomography (PET) could be an effective approach for noninvasively determine PARP activity levels due to its high sensitivity with reasonable spatial resolution, accurate tracer uptake quantification, and minimal physiological effects from PET tracers radiolabeled with high specificity and purity. [11C]PJ-34, a PARP-1 targeted tracer, showed some potential in imaging PARP-1 expression in an animal model of diabetes.17 Recently, a 18F-labeled olaparib/AZD2281 derivative was synthesized by a two-step labeling strategy using a [4 + 2] cycloaddition between trans-cyclooctene and tetrazine.18, 19 In vitro cell studies and microPET tumor imaging using this tracer showed very promising results.19, 20 We now report a new radiolabeled PARP-1 inhibitor for measuring PARP-1 expression in vivo with PET. Based on the benzimidazole carboxamide (NU1085)21 and its derivative (AG014361),22 several analogs that could be easily labeled with 18F were synthesized, and their inhibition potency against PARP-1 was determined. 12 (IC50 = 6.3 ± 1.3 nM) was labeled with 18F in one step with high chemical and radiochemical purities. MicroPET imaging demonstrated increased uptake of [18F]12 in MDA-MB-436 tumors that was blocked by both 12 and olaparib/AZD2281.
2. Results
2.1 Chemistry
The syntheses of the new PARP-1 inhibitors are shown in Scheme 1 and 2. Methyl 2,3-diaminobenzate (5) was reacted with 4-(2-fluoroethoxy)benzoyl chloride in pyridine and dichloromethane to afford a mixture of intermediate 6a and the benzimidazole compound 6. After evaporation of the solvent, the mixture was refluxed with methanesulfonic acid in methanol to give 6. Then the methyl ester of 6 was converted to the amide compound 8 using ammonium in methanol. Similarly, the alkyne analog 9 was synthesized starting from 5 and 4-(prop-2-ynyloxy)benzoyl chloride. The triazole compound 10 was prepared by the copper(I) catalyzed click reaction of 2-fluoroethylazide and 9 using CuSO4·5H2O and sodium ascorbate in DMF.
Scheme 1. Synthesis of derivatives of NU1085 (2).

Reagents and conditions: (a) ROC6H4COCl (R = CH2CH2F for 6a and 6, R = CH2C≡CH for 7a and 7) pyridine, CH2Cl2; (b) CH3SO3H, MeOH; (c) NH3, MeOH; (d) 9, FCH2CH2N3, CuSO4, sodium ascorbate, DMF.
Scheme 2. Synthesis of derivatives of AG014361 (3).

Reagents and conditions: (a) ROC6H4COCl (R = CH2CH2F for 12a and 12, R = CH2C≡CH for 13a and 13, R = CH2CH2Br for 14a and 14), pyridine, CH2Cl2; (b) CH3SO3H, MeOH; (c) 13, FCH2CH2N3, CuSO4, sodium ascorbate, DMF; (d) 14, AgOMs, acetonitrile.
The tricycle analogs were synthesized from the diamine intermediate 11. Compound 11 was reacted with 4-(2-fluoroethoxy)benzoyl chloride in pyridine and dichloromethane to afford a mixture of intermediate 12a and the benzimidazole 12. After evaporation of the solvent, the mixture was refluxed with methanesulfonic acid in methanol to give 12. Similarly, 13 and 14 were made from the corresponding benzyl chlorides. Compound 15 was prepared by the click reaction under the same condition as for 10 using 2-fluoroethylazide and 13. The mesylate precursor 16 for the labeling of 12 with 18F was synthesized by reflux of 14 and silver methanesulfonate in acetonitrile.
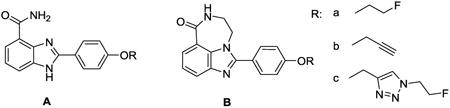
Newly synthesized PARP-1 inhibitors were assessed for their ability to inhibit active PARP-1 using the method described by Putt and Hergenrother.23 The results are shown in Table 1. The tricycle benzimidazole analogs had higher inhibition potency than their respective benzimidazole analogs (e.g., 12 vs. 8, 15 vs. 10). In both benzimidazole and tricycle benzimidazole analogs, the analogs with fluoroethoxy substituent had three times higher inhibition potencies than the respective analogs with fluoroethyl triazole group (e.g., 8 vs. 10, 12 vs. 15). Therefore, the most potent inhibitor, 12, was selected for 18F-labeling.
Table 1. Inhibition efficiency of PARP-1 inhibitors.
| |||
|---|---|---|---|
| Compound | Structure | R | IC50 (nM) |
| 1 PJ34 | / | / | 170.2 ± 8.3a |
| 8 | A | a | 10.8 ± 0.4 |
| 9 | A | b | 25.8 ± 3.3 |
| 10 | A | c | 30.3 ± 5.6 |
| 12 | B | a | 6.3 ± 1.3 |
| 13 | B | b | 18.7 ± 2.7 |
| 15 | B | c | 22.1 ± 6.3 |
2.2 Radiochemistry
[18F]12 was synthesized by the nucleophilic substitution of the mesylate precursor 16 under conventional conditions (K222/K2CO3) in DMF at 105 °C (Scheme 3), affording [18F]12 in 40-50 % yield (decay corrected) after reversed phase HPLC purification and solid phase extraction (Figure 2). The total synthesis time was 90 min. The specific activity of the final dose in 10% ethanol/saline was 5500-18000 mCi/μmol.
Scheme 3. Radiosynthesis of [18F]12.
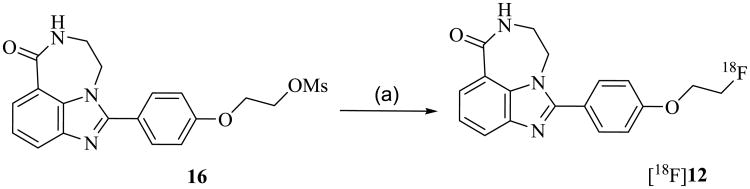
Reagents and conditions: [18F]KF, K2222, K2CO3, DMF, 105 °C, 10 min.
Figure 2.
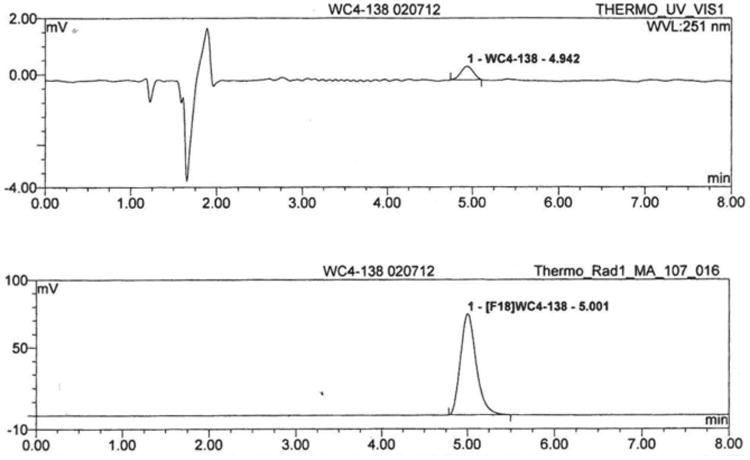
Analytical HPLC of [18F]12, showing high chemical and radiochemical purities. (Top: UV, Bottom: radioactivity; specific activity = 11500 mCi/μmol).
2.3 MicroPET studies
MDA-MB-436 human breast cancer xenograft tumors in immune-deficient mice were clearly visualized by PET using [18F]12. These tumors demonstrated increased uptake that was clearly distinguishable from background at 60 min post-tracer injection (Figure 3). The same mice treated with olaparib (50 mg/kg i.p.) or 12 (1 mg/kg i.p.) 20 min prior to tracer injection decreased [18F]12 uptake in the tumors to the background tissue activity levels (Figure 3). The time-radioactivity curves of [18F]12 in the tumors from 0-60 min of the microPET studies confirmed the visual assessment of the microPET images (Figure 4), demonstrating significantly decreased [18F]12 uptake as a result of treatment with either olaparib or 12.
Figure 3.
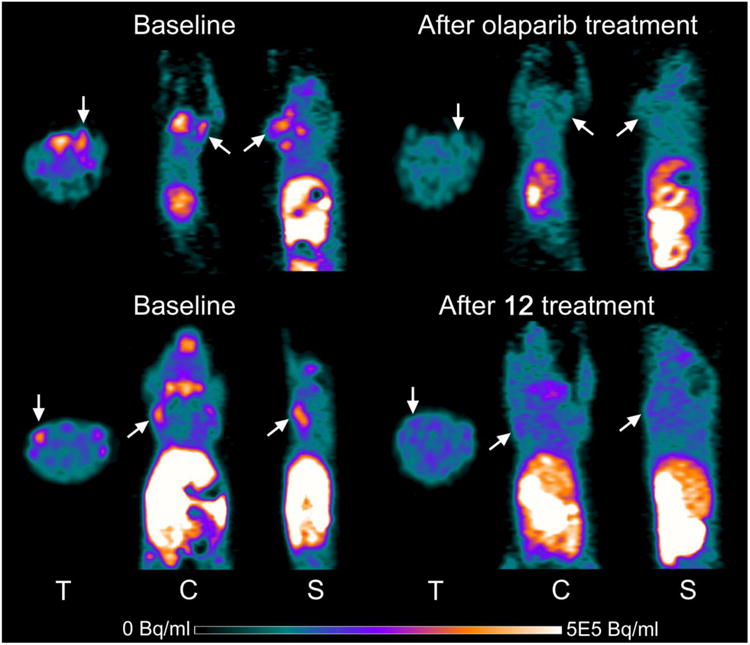
MicroPET images of MDA-MB-231 and MDA-MB-436 tumors in mice tumor at 60 min after [18F]12 injection. Top row: MDA-MB-231 tumor before and after treatment with olaparib (i.p. 50 mg/kg 20 min pretreatment. Bottom row: MDA-MB-436 (right) and MDA-MB-231 (left) tumors before and after 12 (i.p. 1 mg/kg 20 min pretreatment).
Figure 4.
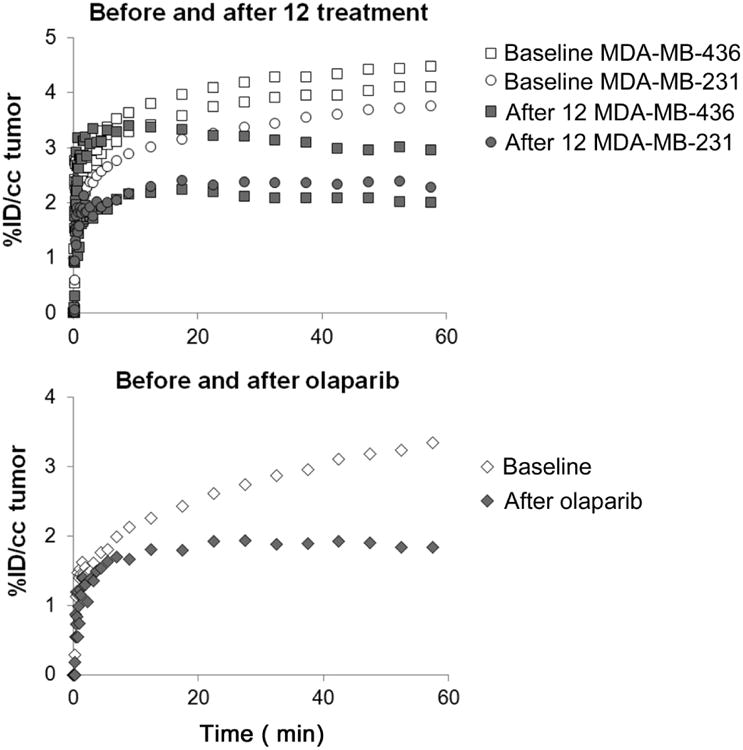
Time-radioactivity curves of [18F]12 in MDA-MB-436 and MDA-MB-231 tumors in mice under baseline conditions and blocked with 12 (i.p. 1 mg/kg 20 min pretreatment, top graph). The olaparib treated MDA-MB-231 tumor also demonstrated decreased [18F]12 uptake (bottom graph).
3. Discussion
Most PARP-1 inhibitors developed to date are NAD+ competitive inhibitors.8, 25 A large hydrophobic pocket adjacent to the nicotinamide binding site26 allows functionalization of these inhibitors to improve potency, solubility and to incorporate a radionuclide. The benzimidazole carboxamide core is one of the most potent core structures among reported PARP inhibitors.27 Therefore, we based the design of the 18F-labeled PARP-1 inhibitors for PET imaging on this core. Incorporation of the radionuclide is easily achieved either through a fluoroethoxy group via nucleophilic substitution28 with [18F]fluoride or via Cu(I) catalyzed click reaction using 2-[18F]fluoroethyl azide.29, 30 The corresponding 2-fluoroethoxy and 2-fluoroethyl triazole analogs of NU1085 and AG014361 were respectively synthesized according to Schemes 1 and 2 in only a few steps. Although the propargyl and triazole groups slightly reduced the inhibition potency of the analogs, the most potent of these were 12 (IC50 = 6.3 nM), an analog of AG014361 with a 2-fluoroethoxy group, followed by 8 (IC50 = 10.8), an analog of NU1085. Therefore, 12 was chosen to be labeled for further evaluation in animals as a PET tracer for imaging PARP-1 expression in vivo.
The one-step labeling of [18F]12 under typical conditions can be easily automated for clinical production of this tracer. The final dose for injection had very high in chemical and radiochemical purities and excellent specific activity. The injected mass of [18F]12 in the microPET studies is 0.0037-0.012 μg/dose (0.2 mCi/dose), which is much lower than the blocking dose and therapeutic doses. This amount of mass is unlikely to have a pharmacological effect, and we anticipate that [18F]12 will be a useful PET tracer for imaging PARP-1 expression.
MDA-MB-436 is a human breast cancer cell line with innately high levels of PARP-1 activity19 and has been used in a mouse models to assess the efficacy of the 18F-labeled olaparib derivative for imaging PARP-1 activity with microPET. [18F]12 progressively accumulated in the tumor during the 1 hour microPET acquisition, and [18F]12 uptake was blocked in animals pretreated with either olaparib or 12. Both olaparib and 12 are competitive PARP-1 inhibitors with high inhibition potencies (IC50 = 5 nM31 and 6.3 nM, respectively). Thus, our data indicate that [18F]12 uptake in the mouse model is due to PARP-1 expression, and that [18F]12 is a promising PET tracer for in vivo imaging of PARP-1 expression specifically.
4. Conclusion
A highly potent PARP-1 inhibitor 12 (IC50 = 6.3 nM) has been developed based on the tricycle benzimidazole core. [18F]12 was synthesized using conventional labeling method with high purities and specific activity. MicroPET studies of [18F]12 in MBA-MD-436 tumor-bearing mice demonstrated accumulation of the radioactivity in these tumors with known PARP-1 overexpression; the uptake in the tumor was blocked by the highly potent PARP-1 inhibitor olaparib and by 12, supporting the specificity of [18F]12 uptake for PARP-1 activity. Therefore, [18F]12 is a promising PET tracer for imaging PARP-1 expression in vivo that should be evaluated for clinical utility.
5. Experimental
5.1 General methods and materials
All chemicals were obtained from standard commercial sources and used without further purification. All reactions were carried out by standard air-free and moisture-free techniques under an inert nitrogen atmosphere with dry solvents unless otherwise stated. Flash column chromatography was conducted using Scientific Adsorbents, Inc. silica gel, 60A, “40 Micron Flash” (32-63 μm). Melting points were determined using MEL-TEMP 3.0 apparatus and are uncorrected. Routine 1H and 13C NMR spectra were recorded at 300 MHz on a Varian Mercury-VX spectrometer. All chemical shifts were reported as parts per million (ppm) downfield from tetramethylsilane (TMS). All coupling constants (J) are given in Hertz (Hz). Splitting patterns are typically described as follows: s, singlet; d, doublet; t, triplet; m, multiplet. Elemental analysis (C, H, N) were determined by Atlantic Microlab, Inc., Norcross, GA. High performance liquid chromatography (HPLC) was performed with an ultraviolet detector and a well-scintillation NaI (Tl) detector and associated electronics for radioactivity detection. A Grace Altima C18 250 × 10 mm 10 μ semi-preparative column (A) and an Altima C18 250 × 4.6 mm 10 μ analytical column (B) were used for preparation and analysis respectively. [18F]Fluoride was produced at Washington University by the 18O(p,n)18F reaction through proton irradiation of enriched (95%) [18O] water in the RDS111 cyclotron. Radio-TLC was accomplished using a Bioscan AR-2000 imaging scanner (Bioscan, Inc., Washington, DC). Published methods were used for the synthesis of compound 527 and 1122. All animal experiments were conducted under Washington University Animal Studies Committee IACUC-approved protocols in accordance with the recommendations of the National Research Council's ‘Guide for the Care and Use of Laboratory Animals’.
5.2 Synthesis
5.2.1 Methyl 2-(4-(2-fluoroethoxy)phenyl)-1H-benzo[d]imidazole-4-carboxylate (6)
A mixture of methyl 2,3-diaminobenzoate 5 (500 mg, 3 mmol) and 4-(2-fluoroethoxy)benzoyl chloride (638 mg, 3.15 mmol) in CH2Cl2 (10 mL) and pyridine (10 mL) was stirred overnight at 23 °C. After removal of the solvent under reduced pressure, the residue was dissolved in methanol (50 mL), and followed by addition of CH3SO3H (1 mL). After the mixture was refluxed for 3 h, methanol was removed under reduced pressure, and the residue was dissolved in ethyl acetate (75 mL). The solution was washed with saturated Na2CO3 (50 mL), water (50 mL) and saturated NaCl (50 mL), and dried over Na2SO4. After evaporation of the solvent, the crude product was purified by silica gel column chromatography eluting with hexane-ethyl acetate (1 : 1) to afford 6 as white solid (686 mg, 73 %), mp 134.2-134.6 °C. 1H NMR (300 MHz, CDCl3) δ 10.58 (s, 1H), 7.98 (d, J = 8.7 Hz, 2H), 7.95 (d, J = 8.7 Hz, 1H), 7.83 (d, J = 7.2 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 6.98 (d, J = 9.0 Hz, 2H), 4.75 (dt, J = 47.1 Hz, 4.2 Hz, 2H), 4.22 (dt, J = 27.6 Hz, 4.2 Hz, 2H), 3.96 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 167.0, 160.2, 152.3, 144.8, 135.0, 128.2, 124.4, 124.2, 122.3, 121.7, 114.9, 113.0, 81.6 (d, J = 169.7 Hz), 67.1 (d, J = 20.6 Hz), 52.0.
5.2.2 Methyl 2-(4-(prop-2-ynyloxy)phenyl)-1H-benzo[d]imidazole-4-carboxylate (7)
Compound 7 was prepared according to the same procedure for compound 6, except using compound 5 (500 mg, 3 mmol) and 4-(prop-2-ynyloxy)benzoyl chloride (613 mg, 3.15 mmol) as starting materials. The crude product was purified by silica gel column chromatography eluting with hexane-ethyl acetate (1 : 1) to afford 7 as white solid (724 mg, 79 %), mp 176.0-176.8 °C. 1H NMR (300 MHz, CDCl3) δ 10.58, 8.03 (d, J = 9.0 Hz, 2H), 7.98 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.29 (t, J = 8.1 Hz, 1H), 7.10 (d, J = 9.0 Hz, 2H), 4.76 (d, J = 2.4 Hz, 2H), 4.00 (s, 3H), 2.57 (t, J = 2.4 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 167.1, 159.4, 152.4, 144.9, 135.1, 128.2, 124.6, 124.3, 122.8, 121.8, 115.4, 113.0, 77.9, 76.0, 55.8, 52.1.
5.2.3 2-(4-(2-Fluoroethoxy)phenyl)-1H-benzo[d]imidazole-4-carboxamide (8)
A solution of 6 (315 mg, 1 mmol) in 7 N NH3 in methanol (10 mL) was stirred for 3 days at 23 °C. After evaporation of the solvent, the crude product was purified by silica gel column chromatography eluting with hexane-ethyl acetate (1 : 2) to afford 8 as white solid (245 mg, 82 %), mp 195.8-197.4 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.23 (d, J = 9.0 Hz, 2H), 7.89 (d, J = 7.5 Hz, 1H), 7.80 (s, 2H), 7.74 (d, J = 7.8 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 7.20 (d, J = 8.4 Hz, 2H), 4.81 (dt, J = 47.7 Hz, 3.6 Hz, 2H), 4.37 (dt, J = 30.0 Hz, 3.9 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 166.4, 160.1, 152.0, 135.5, 128.6, 122.7, 122.0, 115.1, 82.1 (d, J = 166.2 Hz), 67.3 (d, J = 19.4 Hz). Anal. Calcd for C16H14FN3O2.0.5H2O: C, 62.33; H, 4.90; N, 13.63. Found: C, 62.54; H, 4.87; N, 13.67.
5.2.4 2-(4-(Prop-2-ynyloxy)phenyl)-1H-benzo[d]imidazole-4-carboxamide (9)
Compound 9 was prepared according to the same procedure for compound 8, except using compound 7 (460 mg, 1.5 mmol) as starting material. The crude product was purified by silica gel column chromatography eluting with hexane-ethyl acetate (1: 2) to afford 9 as white solid (378 mg, 86 %), mp 183.4-183.9 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.21 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 7.8 Hz, 1H), 7.78 (s, 2H), 7.72 (d, J = 7.8 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.20 (d, J = 9.0 Hz, 2H), 4.92 (d, J = 1.8 Hz, 2H), 3.63 (s, 1H). 13C NMR (75 MHz, DMSO-d6) δ 166.4, 159.0, 152.0, 128.5, 122.7, 122.3, 121.9, 115.4, 78.9, 78.6, 55.7. Anal. Calcd for C17H18N3O2.0.5H2O: C, 67.99; H, 4.70; N, 13.99. Found: C, 67.97; H, 4.72; N, 13.71.
5.2.5 2-(4-((1-(2-Fluoroethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-1H-benzo[d]imidazole-4-carboxamide (10)
A mixture of 9 (291 mg, 1.0 mmol), 1-azido-2-fluoroethane (1.68 mmol), sodium ascorbate (990 mg, 5.0 mmol), and CuSO4.5H2O (125 mg, 0.5 mmol) in DMF (10 mL) was stirred overnight at 23 °C. The reaction mixture was diluted with ethyl acetate (75 mL), and washed with water (2 × 50 mL), and saturated NaCl (50 mL), dried over Na2SO4. After evaporation of the solvent, the crude product was purified by silica gel column chromatography eluting with ethyl acetate to afford 10 as white solid (255 mg, 67 %), mp 256.4-257.3 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.33 (s, 1H), 8.21 (d, J = 8.7 Hz, 2H), 7.87 (d, J = 7.5 Hz, 1H), 7.78 (s, 2H), 7.71 (d, J = 7.8 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.27 (d, J = 8.7 Hz, 2H), 5.28 (s, 2H), 4.85 (dt, J = 46.8 Hz, 4.5 Hz, 2H), 4.76 (dt, J = 27.6 Hz, 4.2 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 166.3, 159.9, 152.0, 142.5, 141.6, 135.3, 128.6, 125.1, 122.7, 122.1, 121.9, 115.3, 114.7, 99.5, 81.9 (d, J = 167.3 Hz), 61.3, 50.1 (d, J = 20.5 Hz). Anal. Calcd for C19H17FN6O2: C, 59.99; H, 4.50; N, 22.09. Found: C, 60.10; H, 4.67; N, 21.49.
5.2.6 5,6-Dihydro-2-(4-(2-fluoroethoxy)phenyl)-imidazo[4,5,1-jk][1,4]benzodiazepin-7(4H)-ones(12)
Compound 12 was prepared according to the same procedure for compound 6, except using compound 11 (177 mg, 1 mmol) and 4-(2-fluoroethoxy)benzoyl chloride (213 mg, 1.05 mmol) as starting materials. The crude product was purified by silica gel column chromatography eluting with ethyl acetate-methanol (10: 1) to afford 12 as white solid (247 mg, 76 %), mp 236.0-237.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.44 (t, J = 5.1 Hz, 1H), 7.89-7.80 (m, 4H), 7.34 (t, J = 7.8 Hz, 1H), 7.17 (d, J = 8.7 Hz, 2H), 4.79 (dt, J = 48.9 Hz, 3.6 Hz, 2H), 4.44 (m, 2H), 4.35 (dt, J = 31.2 Hz, 3.9 Hz, 2H), 3.53 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 167.8, 159.9, 154.1, 143.7, 132.9, 131.7, 125.5, 123.1, 122.5, 121.9, 118.1, 115.1, 82.5 (d, J = 165.0 Hz), 67.7 (d, J = 19.3 Hz), 50.9, 40.8. Anal. Calcd for C18H16FN3O2: C, 66.45; H, 4.96; N, 12.92. Found: C, 66.43; H, 5.03; N, 12.92.
5.2.7 5,6-Dihydro-2-(4-(prop-2-ynyloxy)phenyl)-imidazo[4,5,1-jk][1,4]benzodiazepin-7(4H)-ones(13)
Compound 13 was prepared according to the same procedure for compound 6, except using compound 11 (177 mg, 1 mmol) and 4-(prop-2-ynyloxy)benzoyl chloride (204 mg, 1.05 mmol) as starting materials. The crude product was purified by silica gel column chromatography eluting with ethyl acetate-methanol (10: 1) to afford 13 as white solid (268 mg, 84 %), mp 258.0-259.1 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.47 (s, 1H), 7.85 (m, 4H), 7.34 (t, J = 7.8 Hz, 1H), 7.18 (d, J = 6.9 Hz, 2H), 4.93 (s, 2H), 4.46 (m, 2H), 3.64 (s, 1H), 3.54 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 167.4, 158.5, 153.6, 143.3, 132.5, 131.1, 125.1, 122.7, 122.4, 121.5, 117.7, 115.0, 79.0, 78.5, 55.6, 50.5. Anal. Calcd for C19H15N3O2: C, 71.91; H, 4.76; N, 13.24. Found: C, 71.71; H, 4.82; N, 12.98.
5.2.8 2-(4-(2-Bromoethoxy)phenyl)-5,6-dihydro-imidazo[4,5,1-jk][1,4]benzodiazepin-7(4H-ones(14)
Compound 14 was prepared according to the same procedure for compound 6, except using compound 11 (177 mg, 1 mmol) and 4-(2-bromoethoxy)benzoyl chloride (277 mg, 1.05 mmol) as starting materials. The crude product was purified by silica gel column chromatography eluting with ethyl acetate-methanol (10: 1) to afford 14 as white solid (255 mg, 66 %), mp decomposed 280 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.44 (t, J = 5.6 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.34 (t, J = 7.6 Hz, 1H), 7.16 (d, J = 8.4 Hz, 2H), 4.44 (m, 4H), 3.86 (t, J = 5.2 Hz, 2H), 3.53 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.8, 159.7, 154.1, 143.7, 132.9, 131.7, 125.5, 123.1, 122.6, 122.0, 118.1, 115.2, 68.4, 50.5, 40.8.
5.2.9 5,6-Dihydro-2-(4-((1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)-imidazo[4,5,1-jk][1,4]benzodiazepin-7(4H)-ones (15)
Compound 15 was prepared according to the same procedure for compound 10, except using compound 13 (159 mg, 0.5 mmol) as starting material. The crude product was purified by silica gel column chromatography eluting with ethyl acetate-methanol (10: 1) to afford 15 as white solid (147 mg, 72 %), mp 226.5-227.6 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.46 (t, J = 5.7 Hz, 1H), 8.33 (s, 1H), 7.89-7.81 (m, 4H), 7.34 (t, J = 7.8 Hz, 1H), 7.25 (d, J = 8.4 Hz, 2H), 5.28 (s, 2H), 4.85 (dt, J = 47.1 Hz, 4.2 Hz, 2H), 4.76 (dt, J = 27.6 Hz, 4.2 Hz, 2H), 4.45 (m, 2H), 3.54 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 167.4, 159.4, 143.3, 142.6, 131.2, 125.1, 125.0, 122.7, 122.0, 121.5, 117.7, 114.8, 81.9 (d, J = 167.3 Hz), 61.2, 50.5, 50.1 (d, J = 20.5 Hz), 40.4. Anal. Calcd for C21H19FN6O2.1.5H2O: C, 58.19; H, 5.12; N, 19.39. Found: C, 57.89; H, 4.54; N, 18.92.
5.2.10 5,6-Dihydro-2-(4-(2-(methylsulfonyloxy)ethoxy)phenyl)-imidazo[4,5,1-jk][1,4]benzodiazepin-7(4H)-ones (16)
A mixture of 14 (193 mg, 0.5 mmol) and AgOMs (508 mg, 2.5 mmol) was refluxed for 8 h. After evaporation of the solvent, the crude product was purified by silica gel column chromatography eluting with ethyl acetate-methanol (10 : 1) to afford 16 as white solid (129 mg, 64 %), mp 253.2-254.1 °C. 1H NMR (400 MHz, CD3OD) δ 7.97 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 8.8 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 7.17 (d, J = 8.8 Hz, 2H), 4.59 (t, J = 4.0 Hz, 2H), 4.49 (t, J = 4.0 Hz, 2H), 4.35 (t, J = 4.0 Hz, 2H), 3.65 (m, 2H), 3.12 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 160.2, 148.1, 142.7, 135.7, 132.2, 131.1, 131.0, 125.8, 122.6, 122.1, 121.4, 116.9, 114.6, 68.3, 66.0, 50.5, 40.6, 36.0.
5.3 PARP-1 enzymatic assay
This assay is based on the chemical quantification of NAD+, i.e. the amount of NAD+ consumed when the active PARP-1 C-terminal catalytic domain sequentially transfers ADP-ribose subunits from nicotinamide adenine dinucleotide (NAD+) to protein acceptors.7
High-specific-activity PARP-1 and activated DNA were purchased from Trevigen (Gaithersburg, MD). All other reagents required for this assay including NAD+ were purchased from Sigma-Aldrich (St. Louis, MO). Known PARP-1 inhibitor PJ-34, used as a control in these experiments, was synthesized in-house. To test the compounds for PARP-1 inhibition, a solution of 250 nM NAD+ was first made in 50 mM Tris-HCl, 2 mM MgCl2, at pH 8.0 (PARP assay buffer) and 20 μL transferred to each well of a 96-well black fluorescence plate. A solution of 50 μg/mL of activated DNA was made in PARP assay buffer and 10 μL was added to each well. Stock solutions of test compounds were first prepared in DMSO, diluted to varying concentrations in PARP assay buffer, and 10 μL was added to each well. To initiate the reaction, 10 μL of 10 μg/mL PARP-1 enzyme in PARP assay buffer was added to each well. The total volume was 50 μL, bringing the final concentrations to 2 μg/mL PARP-1 enzyme, 10 μg/mL activated DNA, and 100 nM NAD+ per well. The plate was then incubated at room temperature for 20 min. The amount of NAD+ present was then determined by first adding 20 μL 2 M KOH, followed by 20 μL of 20 % acetophenone (in ethanol) to each well. The plate was allowed to incubate at 4 °C for 10 min. Then 90 μL of 88 % formic acid was added, resulting in a final concentration of 222 mM KOH, 2.2% acetophenone, 44% formic acid, and varying concentrations of NAD+. The plate was incubated at 100 °C for 5 min., allowed to cool, and then read on a Perkin Elmer Victor microplate fluorometer (Waltham, MA) using 360 nm excitation and 450 nm emission filters. Dose-response curves were generated using GraphPad Prism version 5.04 for Windows (San Diego, CA) where control wells containing NAD+ only were set at 0% PARP activity and control wells containing PARP-1 only were set at 100% PARP activity. IC50 values were calculated from the dose-response curves generated from at least three independent experiments and reported in Table 1 as mean ± standard deviation (SD).
5.4 Radiosynthesis of [18F]12
[18F]fluoride (up to 50 mCi in 100-500 μL [18O]water) was transferred to a BD vacutainer (13 × 75 mm, 5 mL, glass, no additives) containing K222 (2.2 mg, 5.8 μmol) and K2CO3 (0.3 mg, 2.2 μmol), then the mixture was dried by azeotropic distillation at 105 °C using acetonitrile (3 × 1 mL) under a gentle flow of N2 gas. When the drying was nearly finished, the vacutainer was removed from the oil bath and the solvent residue (< 100 μL) was removed by a flow of N2 at room temperature. A solution of 16 (0.65 mg, 1.6 μmol) in DMF (300 μL) was added to the vacutainer and then shaken and heated at 105 °C for 10 min. At room temperature, the reaction mixture was diluted with water (2 mL) and then loaded onto a semi-preparative column (A) for purification (18% acetonitrile/82% water/0.1% TFA, 4 mL/min, 251 nm). The HPLC fraction containing [18F]12 was collected, and [18F]12 was obtained in ethanol using standard solid phase extraction method. The dose was diluted to 10% ethanol in saline. An analytical column (B) was used to analyze the dose (32% acetonitrile/68% 0.1 M ammonium formate buffer pH = 4.5, 2 mL/min, 251 nm). The total synthesis time was 90 min, the decay corrected yields 40-50%, the radiochemical purity 100%, and the specific activity ranged from 5500 to 18000 mCi/μmol at the end of synthesis.
5.5 MicroPET studies
MDA-MB-436 human breast cancer cells were maintained in cell culture under standard conditions with 5% CO2 atmosphere using Leibovitz's L-15 medium supplemented with 10% FBS, 10μg/ml bovine insulin and 16μg/ml Glutathione. MDA-MB-231 human breast cancer cells were also maintained in cell culture under standard conditions with 5% CO2 atmosphere using minimum essential medium (MEM) supplemented with 1% 200mM L-glutamine, 1% 100mM sodium pyruvate, 1% 10mM NEAA, 2% vitamins for MEM and 5% FBS. Cells in exponential growth were trypsinized and harvested for tumor implantation. After counting, cells were re-suspended in ice-cold 1:1 Matrigel and PBS to give the desired concentration and held on ice.
Mature female athymic nu/nu mice from Charles River Laboratories are allowed to acclimate in an AALAC accredited housing facility for at least 1 week prior to tumor implantation for these serial imaging studies. Female nu/nu mice were implanted in the mammary fat pads (near the axillary lymph nodes) with 1 × 107 MDA-MB-436 breast cancer cells or either 1 × 107 or 5 × 106 MDA-MB-231 breast cancer cells in ∼100 μL of 1:1 Matrigel:PBS. Imaging studies were conducted 18-19 days post implantation.
Tumor-bearing mice were placed in an induction chamber containing ∼2% isoflurane/oxygen and then secured to a custom double bed for placement of tail vein catheters; anesthesia was maintained via nose-cone at ∼1% isoflurane/oxygen for the dynamic imaging procedure. The mice were injected with 150-200 μCi of [18F]12 and scanned for 0-60 min using Focus 220 and Inveon PET/CT scanners. The standard uptake values (SUVs) were generated from manually drawn regions of interests for tumors and surrounding ‘background’ tissue. Visualization of specific uptake was determined by comparison of baseline scans with images acquired by tracer injection 20 minutes after pre-treatment with the blocking agents olaparib (50 mg/kg, IP) or 12 (1 mg/kg, IP).
Acknowledgments
NIH P01 HL13851 (PI: Gropler, Welch) supported this study. The Doris Duke Foundation Clinical Investigator Award (PI: Chen) and NIH R01 HL116389 (PI: Chen) supported DLC's effort. We thank Robert Dennett and Brian Wingbermuehle for the production of [18F]fluoride, Justin Rothfuss for conducting the binding affinity assays, and the staff of the Washington University Small Animal Imaging Facility for their technical assistance.
Abbreviations
- PARP
poly (ADP-ribose) polymerase
- NAD+
nicotinamide adenine dinucleotide
- PET
positron emission tomography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Hassa PO, Hottiger MO. Frontiers in bioscience : a journal and virtual library. 2008;13:3046. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 2.d'Adda di Fagagna F, Hande MP, Tong WM, Lansdorp PM, Wang ZQ, Jackson SP. Nature genetics. 1999;23:76. doi: 10.1038/12680. [DOI] [PubMed] [Google Scholar]
- 3.Jagtap P, Szabo C. Nature reviews Drug discovery. 2005;4:421. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber V, Dantzer F, Ame JC, de Murcia G. Nature reviews Molecular cell biology. 2006;7:517. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 5.Virag L, Szabo C. Pharmacological reviews. 2002;54:375. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 6.Gradwohl G, Menissi de Murciaer JM, Molinete M, Simonin F, Koken M, Hoeijmakers JH, de Murcia G. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:2990. doi: 10.1073/pnas.87.8.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. The Journal of biological chemistry. 1984;259:4770. [PubMed] [Google Scholar]
- 8.Ferraris DV. Journal of medicinal chemistry. 2010;53:4561. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- 9.Basu B, Sandhu SK, de Bono JS. Drugs. 2012;72:1579. doi: 10.2165/11635510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, Toniatti C, Carpenter CL, Iannone R, Kaye SB, de Bono JS, Wenham RM. The lancet oncology. 2013;14:882. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 11.Plummer R, Lorigan P, Steven N, Scott L, Middleton MR, Wilson RH, Mulligan E, Curtin N, Wang D, Dewji R, Abbattista A, Gallo J, Calvert H. Cancer chemotherapy and pharmacology. 2013;71:1191. doi: 10.1007/s00280-013-2113-1. [DOI] [PubMed] [Google Scholar]
- 12.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A. The lancet oncology. 2011;12:852. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 13.Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, Jia L, Weil M, Speranza G, Murgo AJ, Kinders R, Wang L, Parchment RE, Carter J, Stotler H, Rubinstein L, Hollingshead M, Melillo G, Pommier Y, Bonner W, Tomaszewski JE, Doroshow JH. Cancer research. 2011;71:5626. doi: 10.1158/0008-5472.CAN-11-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Javle M, Curtin NJ. Therapeutic advances in medical oncology. 2011;3:257. doi: 10.1177/1758834011417039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, Pol E, Frostell A, Ekblad T, Oncu D, Kull B, Robertson GM, Pellicciari R, Schuler H, Weigelt J. Nature biotechnology. 2012;30:283. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Shi Y, Maag DX, Palma JP, Patterson MJ, Ellis PA, Surber BW, Ready DB, Soni NB, Ladror US, Xu AJ, Iyer R, Harlan JE, Solomon LR, Donawho CK, Penning TD, Johnson EF, Shoemaker AR. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:510. doi: 10.1158/1078-0432.CCR-11-1973. [DOI] [PubMed] [Google Scholar]
- 17.Tu Z, Chu W, Zhang J, Dence CS, Welch MJ, Mach RH. Nuclear medicine and biology. 2005;32:437. doi: 10.1016/j.nucmedbio.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 18.Keliher EJ, Reiner T, Turetsky A, Hilderbrand SA, Weissleder R. ChemMedChem. 2011;6:424. doi: 10.1002/cmdc.201000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiner T, Keliher EJ, Earley S, Marinelli B, Weissleder R. Angew Chem Int Ed Engl. 2011;50:1922. doi: 10.1002/anie.201006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiner T, Lacy J, Keliher EJ, Yang KS, Ullal A, Kohler RH, Vinegoni C, Weissleder R. Neoplasia. 2012;14:169. doi: 10.1593/neo.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, Durkacz BW, Hostomsky Z, Newell DR. Clinical cancer research : an official journal of the American Association for Cancer Research. 2000;6:2860. [PubMed] [Google Scholar]
- 22.Skalitzky DJ, Marakovits JT, Maegley KA, Ekker A, Yu XH, Hostomsky Z, Webber SE, Eastman BW, Almassy R, Li J, Curtin NJ, Newell DR, Calvert AH, Griffin RJ, Golding BT. Journal of medicinal chemistry. 2003;46:210. doi: 10.1021/jm0255769. [DOI] [PubMed] [Google Scholar]
- 23.Putt KS, Hergenrother PJ. Analytical biochemistry. 2004;326:78. doi: 10.1016/j.ab.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 24.Jagtap P, Soriano FG, Virag L, Liaudet L, Mabley J, Szabo E, Hasko G, Marton A, Lorigados CB, Gallyas F, Jr, Sumegi B, Hoyt DG, Baloglu E, VanDuzer J, Salzman A, Southan GJ, Szabo C. Critical care medicine. 2002;30:1071. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 25.Ruf A, de Murcia G, Schulz GE. Biochemistry. 1998;37:3893. doi: 10.1021/bi972383s. [DOI] [PubMed] [Google Scholar]
- 26.Kinoshita T, Nakanishi I, Warizaya M, Iwashita A, Kido Y, Hattori K, Fujii T. FEBS letters. 2004;556:43. doi: 10.1016/s0014-5793(03)01362-0. [DOI] [PubMed] [Google Scholar]
- 27.White AW, Almassy R, Calvert AH, Curtin NJ, Griffin RJ, Hostomsky Z, Maegley K, Newell DR, Srinivasan S, Golding BT. Journal of medicinal chemistry. 2000;43:4084. doi: 10.1021/jm000950v. [DOI] [PubMed] [Google Scholar]
- 28.Lasne MC, Perrio C, Rouden J, Barre L, Roeda D, Dolle F, Crouzel C. Top Curr Chem. 2002;222:201. [Google Scholar]
- 29.Glaser M, Arstad E. Bioconjugate chemistry. 2007;18:989. doi: 10.1021/bc060301j. [DOI] [PubMed] [Google Scholar]
- 30.Zhou D, Chu W, Dence CS, Mach RH, Welch MJ. Nuclear medicine and biology. 2012 doi: 10.1016/j.nucmedbio.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VM, Jr, Matthews IT, Moore S, O'Connor MJ, Smith GC, Martin NM. Journal of medicinal chemistry. 2008;51:6581. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]