

Abstract
Aims/Introduction
Type 2 diabetes is characterized by dysregulation of immunity, oxidative stress and reduced incretin effects. Experimental studies suggest that glucagon‐like peptide (GLP‐1) might have immunomodulating effects. We hypothesize that GLP‐1 receptor agonist, exendin‐4, might reduce inflammatory response in type 2 diabetes.
Materials and Methods
Using peripheral blood mononuclear cells (PBMC) sampled from 10 type 2 diabetes and 10 sex‐ and age‐matched control subjects and supernatants from PBMC culture, the expression of phospho‐mitogen activated protein kinase (MAPK) signaling pathways in CD4+ T helper lymphocytes and monocytes was analyzed using flow cytometry. Cytokines/chemokines and superoxide anion before and after treatment with exendin‐4 were measured by cytometric bead array and chemiluminesence assay, respectively.
Results
Compared with control subjects, PBMC from type 2 diabetes patients showed activated MAPK (P38, c‐Jun NH2‐terminal protein kinase and extracellular signal‐regulated kinase) signaling pathway, elevated superoxide anion, increased pro‐inflammatory cytokines (tumor necrosis factor‐α, interleukin‐1β, interleukin‐6) and chemokines (CCL5/regulated on activation normal T‐cell expressed and secreted and CXCL10/interferon‐γ‐induced protein 10). These changes were attenuated by exendin‐4, possibly through the suppression of p38 MAPK.
Conclusions
These results suggest that exendin‐4 might downregulate pro‐inflammatory responses and reduce oxidative stress by suppressing MAPK signaling pathways in type 2 diabetes.
Keywords: Diabetes, Exendin‐4, Inflammation
Introduction
Type 2 diabetes is characterized by decreased β‐cell mass and impaired insulin secretion. Several mechanisms, such as endoplasmic reticulum (ER) stress, oxidative stress, glucotoxicity, lipotoxicity and amyloid deposition have been implicated1. Intriguingly, all these factors might induce an inflammatory response that contributes to β‐cell dysfunction and death. In patients with type 2 diabetes, insulitis characterized by islet infiltration with immune cells and increased release of cytokines/chemokines has been reported4. In the same vein, antagonists of pro‐inflammatory cytokines, such as interleukin (IL)‐1β antagonists, have been shown to improve glycemia, preserve β‐cell function and prevent diabetes development6.
Inflammation is associated with the cellular activation of mitogen‐activated protein kinase (MAPK) signaling pathways through serine or threonine phosphorylation of transcription factors, which bind to promoter regions of a gene network to mediate immune responses, resulting in pathophysiological changes and clinical consequences9. In mammalian cells, p42/p44 extracellular signal‐regulated kinase (ERK), c‐Jun NH2‐terminal protein kinase (JNK) and p38 MAPK are the three main kinases that can be activated by pro‐inflammatory cytokines (such as tumor necrosis factor (TNF)‐α, IL‐1β and IL‐6), mitogens and physical stress9. Previous studies showed abnormal ERK and p38 MAPK activities in monocytes of patients with type 2 diabetes10. In experimental models, specific inhibition of MAPK pathways can suppress pro‐inflammatory responses with delayed onset of diabetes11. Importantly, cytokines, such as TNF‐α and IL‐1β, can activate membrane‐bound nicotinamide adenine dinucleotide phosphate‐oxidase (NADPH) oxidase to generate superoxide in endothelial cells (ECs). Inhibitor of NADPH oxidase has been shown to abrogate superoxide production in response to TNF‐α in ECs. This might imply that inflammation can induce oxidative stress, which can contribute to and perpetuate the pathological process of type 2 diabetes12.
Glucagon‐like peptide‐1 (GLP‐1) is an incretin hormone secreted by enteroendocrine L cells in the gut. Apart from promoting β‐cell proliferation and inhibiting apoptosis, stimulating insulin secretion and reducing blood glucose13, GLP‐1 might possess extraglycemic activity including anti‐inflammatory effects14. In ex vivo studies, exendin‐4, a GLP‐1 analog, has been shown to improve β‐cell function by reducing pro‐inflammatory cytokines (interferon‐γ, IL‐17, IL‐1β and IL‐2) and caspase‐3 activation in human islets16. Inhibitors of dipeptidyl peptidase‐IV, known to increase circulating levels of GLP‐1, reversed new onset diabetes in non‐obese diabetic (NOD) mice by reducing insulitis, modulating inflammation and improving islet function14.
Despite these experimental findings, it remains uncertain whether exendin‐4 shows any modulating effects in the human immune system. Herein, we investigated the effects of exendin‐4 on inflammatory responses and oxidative stress in human lymphocytes and monocytes in peripheral blood mononuclear cells of type 2 diabetes patients.
Methods
Blood Samples from Patients with Type 2 diabetes and Control Subjects
A total of 10 Chinese patients with type 2 diabetes were recruited from the Department of Pediatrics and the Diabetes and Endocrine Center of the Prince of Wales Hospital, Hong Kong. Type 2 diabetes was diagnosed according to the 1985 World Health Organization criteria using diagnostic values of fasting plasma glucose (PG) ≥ 7.0 mmol/L and/or 2‐h (or random) PG ≥ 11.1 mmol/L with or without 75‐g oral glucose tolerance test, depending on the presence or absence of symptoms. All patients were non‐smokers and free from infection for 4 weeks preceding the study. Bodyweight and body height were measured for determination of body mass index (BMI). A total of 10 sex‐ and age‐matched healthy Chinese volunteers were recruited as healthy controls (CTL). From each subject, 20 mL of venous peripheral ethylenediamine tetra‐acetic acid (EDTA) blood was obtained, followed by immediate fractionation of peripheral blood mononuclear cells (PBMC) for ex vivo study. In brief, PBMCs were prepared by centrifuging 20 mL EDTA venous blood using a Ficoll–Paque plus density gradient (GE Healthcare Bio‐Sciences Corp, Piscataway, NJ, USA). Plasma and serum were stored in 300‐μL aliquots at −70°C until analysis. The aforementioned protocol was approved by the Clinical Research Ethics Committee of the Chinese University of Hong Kong‐New Territories East Cluster Hospitals. Informed consent was obtained from all participants according to the Declaration of Helsinki.
Flow Cytometric Analysis of Intracellular Activated (Phosphorylated) MAPK Signaling Molecules
Flow cytometric analysis was applied to determine the activation of MAPK signaling pathways indicated by phospho‐ERK, phospho‐p38 MAPK and phospho‐JNK in CD4+ T helper lymphocytes and monocytes in PBMC from patients and controls. Briefly, PBMC (viability > 95%) were prepared by Ficoll–Paque plus density gradient centrifugation. The PBMC culture was incubated with or without exendin‐4 (Sigma–Aldrich, St. Louis, MO, USA) at 50 nmol/L for 10 mins at 37°C in a 5% CO2 atmosphere. PBMC was then fixed/permeabilized by Cytofix/Cytoperm™ buffer (BD Biosciences, Mississauga, ON, Canada) at room temperature for 15 min. Cells were then washed with Perm/Wash™ buffer twice and resuspended in BD Pharmingen™ stain buffer (BD Pharmingen Corp, San Diego, CA, USA) at 1 × 107 cells/mL. PE‐conjugated anti‐human phospho‐ERK, phospho‐p38 MAPK, phospho‐JNK antibody or mouse immunoglobulin G (IgG) isotypic antibody (BD Pharmingen) was added to each tube and incubated at room temperature for 45 min in the dark. Cells were then washed and resuspended with stain buffer (BD Pharmingen) for flow cytometric analysis using a BD FACSCalibur flow cytometer (BD Biosciences Corp, San Diego, CA, USA). During the flow cytometric analysis of intracellular MAPK, PE‐conjugated antibodies against CD4 and CD14 cell surface markers were used for gating of CD4+ T helper lymphocyte (CD4+) and monocytes (CD14+) in PBMC. Mouse IgG isotypic antibodies were used to normalize the background signal. Results were expressed as mean fluorescence intensity (MFI) for the expression of intracellular phospho‐p38 MAPK, phospho‐ERK and phospho‐JNK in 10,000 cells using BD CellQuest™ software (BD FACSCalibur; BD Biosciences Corp).
Measurement of Clinical Biomarkers
Blood samples were collected by venepuncture after an overnight fast. Serum was centrifuged (1500 g for 15 min at 4°C) and stored at 4°C until measurement. Biochemical markers, such as insulin, plasma glucose and glycated hemoglobin (HbA1c), were measured at the Department of Chemical Pathology of the Prince of Wales Hospital, The Chinese University of Hong Kong, with an externally accredited quality assurance program.
Measurement of Concentrations of Cytokines, Chemokines, Insulin, C‐peptide and Autoantibodies (Autoantibodies to Glutamic Acid Decarboxylase and Protein Tyrosine Phosphatase)
The PBMC culture was incubated with or without exendin‐4 (50 nmol/L) for 24 h at 37°C in a 5% CO2 atmosphere. The cell‐free supernatant was harvested and stored at −80°C for subsequent assays. The supernatant concentration of inflammatory cytokines TNF‐α, IL‐1β, IL‐6, IL‐10 and IL‐12p70, and chemokines CXCL8/IL‐8, CCL5/regulated on activation normal T‐cell expressed and secreted (RANTES), CCL2/monocyte chemoattractant protein‐1 (MCP‐1), CXCL10/interferon‐γ‐induced protein 10 (IP‐10) and CXCL9/monokine induced by interferon‐γ were measured using the inflammatory cytokine and chemokine cytometric bead array (CBA) reagent kits (BD Pharmingen), respectively. Samples were analyzed using a multifluorescence BD flow cytometer (FACSCalibur™) with BD CellQuest™ software and BD™ CBA software. The coefficients of variation for all cytokine and chemokine assays were less than 10%. Serum insulin/C‐peptide concentrations were measured by enzyme‐linked immunosorbent assay (ELISA; Mercodia AB, Sylveniusgatan, Sweden). The status of diabetes was further confirmed by enzyme immunoassay for the determination of autoantibodies to glutamic acid decarboxylase (anti‐GAD) and protein tyrosine phosphatase (anti‐IA2) in serum (Medizym, Dahlewitz/Berlin, Germany).
Quantification of Superoxide Anion by Chemiluminescent Assay
The superoxide anion oxidizes luminol in a reaction that produces photons of light that are readily measured with a standard luminometer. The superoxide anion detection kit (Calbiochem Corp., San Diego, CA, USA) utilizes an enhancer that increases the sensitivity of the assay by amplifying the chemiluminescence. Cells (5 × 105) were suspended in 100 uL luminol‐enhancer assay medium and incubated for 30 mins at room temperature. oxidized luminol in a reaction that produced photons of light that were measured with a luminometer (VICTOR3 multi‐label plate reader; Perkin Elmer, Waltham, MA, USA).
Statistical Analysis
All analyses were carried out using SPSS for Windows, version 16.0, (SPSS Inc., Chicago, IL, USA). Between‐group comparison (basal and ex vivo culture supernatant concentrations) was made by Mann–Whitney U‐test, whereas within‐group comparison (before and after exendin‐4 treatment) was made by Wilcoxon signed‐rank test. A P‐value less than 0.05 (two‐tailed) was considered significant.
Results
Clinical Profile of Type 2 Diabetes Patients and Controls
None of the patients with type 2 diabetes were treated with insulin, and all had disease duration less than 5 years without clinical complications. Compared with age‐ and sex‐matched controls, type 2 diabetes patients had significantly higher fasting plasma glucose, BMI, insulin and C peptide levels than control subjects (Table 1).
Table 1. Demographic and clinical characteristics of patients with type 2 diabetes and control subjects.
| Variables | Type 2 diabetes | Controls |
|---|---|---|
| n | 10 | 10 |
| Sex (male/female) | 6/4 | 8/2 |
| Age (years) | 32 (27–33) | 26.5 (25–31.5) |
| Duration of disease (years) | 1 (1–2) | NA |
| Fasting plasma glucose (mmol/L) | 8.6 (5.9–12.1) | 4.9 (4.5–5.2) |
| Body mass index (kg/m2) | 28 (26.7–33.2) | 21.1 (19.3–22.6)* |
| Fasting serum C‐peptide (pmol/L) | 980.2 (470.5–1561.4) | 232 (170.9–275.7)* |
| Anti‐GAD | Negative | Negative |
| Anti‐IA2 | Negative | Negative |
| HOMA‐IR | 29.2 (20.1–42.6) | 7.9 (5.8–13.5)* |
| HOMA‐β% | 193.8 (123.7–505.8) | 416.4 (340.7–664.6) |
| Treatment with metformin | ||
| Patients, n (%) | 8 (80%) | NA |
| Daily dose (mg) | 2100 (1700–3000) | NA |
| Treatment with simvastatin | ||
| Patients, n (%) | 3 (30%) | NA |
| Daily dose (mg) | 26.7 (20–40) | NA |
| Treatment with gliclazide | ||
| Patients, n (%) | 2 (20%) | NA |
| Daily dose (mg) | 125 (90–160) | NA |
| Treatment with lisinopril | ||
| Patients, n (%) | 2 (20%) | NA |
| Daily dose (mg) | 12.5 (12.5–12.5) | NA |
| Cytokines/chemokines | ||
| TNF‐α (pg/mL) | 56 (7–236) | 22 (8–58)* |
| IL‐1β (pg/mlL) | 111 (6–184) | 35 (22–61)* |
| IL‐6 (pg/mL) | 114 (8–205) | 55 (20–105)* |
| RANTES (pg/mL) | 330 (53–694) | 32 (15–98)* |
| CXCL10 (pg/mL) | 76 (10–156) | 24 (16–38)* |
*P < 0.05. CXL10, interferon‐γ‐induced protein 10; GAD, glutamic acid decarboxylase; HOMA‐β, homeostatic model assessment of β‐cell finction; HOMA‐IR, homeostatic model assessment of insulin resistance; IA2, protein tyrosine phosphatase; IL, interleukin; NA, not applicable, RANTES, regulated on activation normal T‐cell expressed and secreted; TNF, tumor necrosis factor.
Exendin‐4 Suppressed TNF‐α, IL‐1β, IL‐6 Expression from PBMC in Type 2 Diabetes Patients
Previous studies reported elevated serum levels of inflammatory cytokines including TNF‐α, IL‐6, IL‐1β and IL‐6 in newly diagnosed type 2 diabetes patients17. In this experiment, after 24 h of incubation with PBMC culture, we observed significantly higher levels of inflammatory cytokines (TNF‐α, IL‐6 and IL‐1β) in PBMC supernatants from patients with type 2 diabetes than controls (Figure 1). Treatment with exendin‐4 (50 nmol/L), U0126 (specific inhibitor of ERK signaling pathway) and SB 203580 (specific inhibitor of p38 MAPK signaling pathway) for 24 h significantly reduced cytokine levels in PBMC supernatants from patients with type 2 diabetes (all P < 0.05), but not in control subjects (all P > 0.05).
Figure 1.
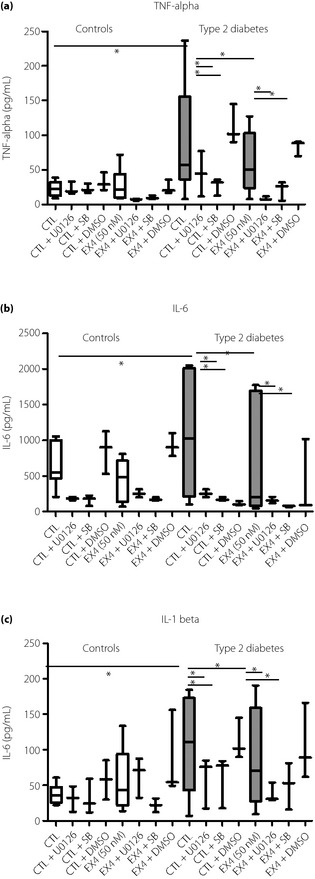
Effects of exendin‐4 on the ex vivo production of pro‐inflammatory cytokines of controls (CTL) and patients with type 2 diabetes. Peripheral blood mononuclear cells from controls and type 2 diabetes patients were pretreated with U0126 (U0126 10 μmol/L), SB203580 (SB 10 μmol/L) for 1 h followed by incubation with or without exendin‐4 (Ex4; 50 nmol/L) for 24 h. Release of (a) tumor necrosis factor (TNF)‐α, (b) interleukin (IL)‐1β and (c) IL‐6 were determined by cytometric bead array using flow cytometry. Dimethyl sulfoxide (DMSO) (0.1%) was used as the vehicle control. Results are presented with box‐and‐whisker plots. *P < 0.05.
Exendin‐4 Suppressed CXCL10 and CXCL5 Production from PBMC in Type 2 Diabetes Patients
These findings together with the anti‐inflammatory effects of GLP‐1 prompted us to further investigate the immunoregulatory role of exendin‐4 on production of chemokines from PBMC in type 2 diabetes patients. The normal physiological plasma concentration of GLP‐1 is approximately 10 pg/mL, but the local concentration of GLP‐1 can be 100–1000 fold higher than that of the circulating level20. In this ex vivo experiment, the exendin‐4, used as the GLP‐1 mimic, is expected to produce a concentration similar to that in the local tissues. Here in, chemokines recruit activated immune cells to sites of inflammation and are important mediators of insulitis. A suppressor of cytokine‐induced chemokine and Fas expression has been shown to inhibit apoptosis‐mediated β‐cell death21. As shown in Figure 2, exendin‐4 significantly suppressed the release of CCL5/RANTES and CXCL10/IP‐10 from PBMC in the type 2 diabetes group compared with the control group (all P < 0.05).
Figure 2.
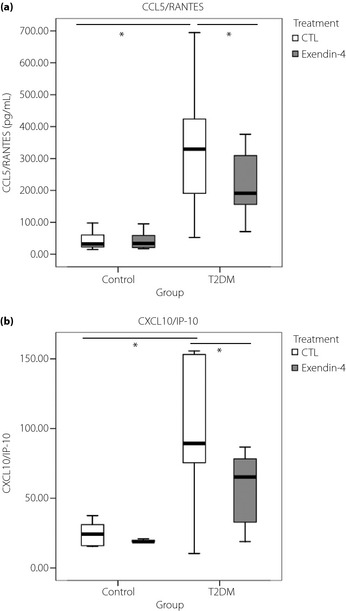
(a) CCL5/regulated on activation normal T‐cell expressed and secreted (RANTES) and (b) CXCL10/interferon‐γ‐induced protein 10 (IP‐10) levels in culture supernatants of peripheral blood mononuclear cell from patients with type 2 diabetes (T2DM) and normal controls (CTL) with or without treatment of exendin‐4 (50 nmol/L) for 24 h at 37°C. Results are presented with box‐and‐whisker plots. *P < 0.05.
Effects of Exendin‐4 on the Activation of Intracellular ERK and p38 MAPK but not JNK in CD4+ T Helper Lymphocytes and Monocytes
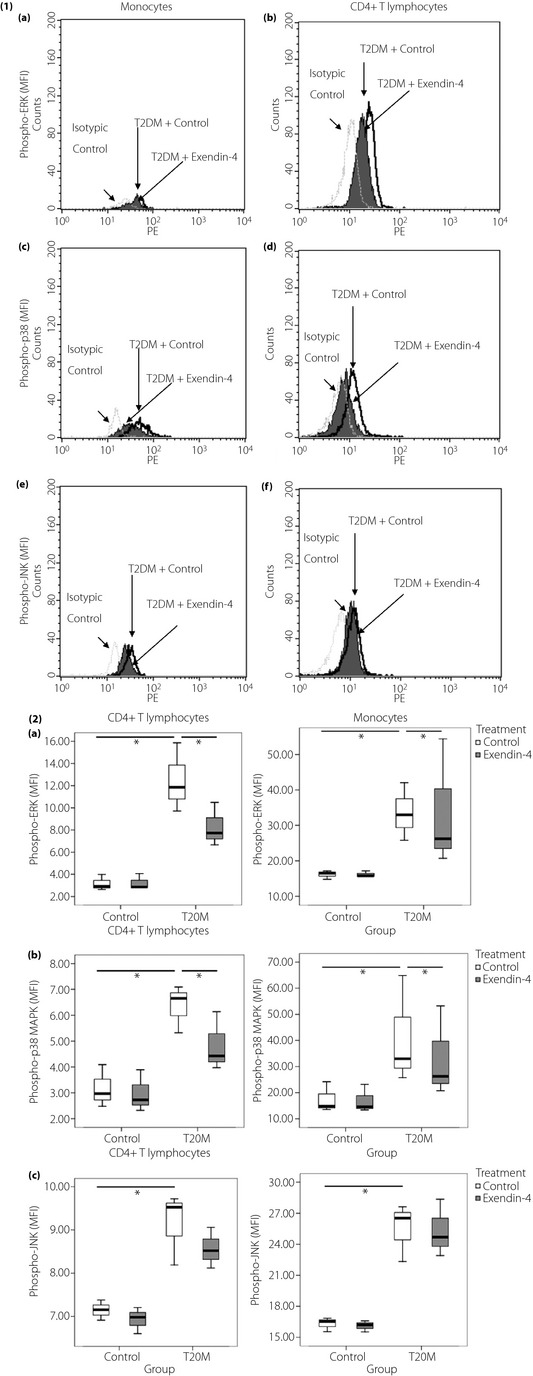
To characterize the intracellular mechanism for the release of cytokines and chemokines from PBMC, we investigated the phosphorylation of intracellular ERK and p38 MAPK in CD4+ T lymphocytes and monocytes. As shown in Figure 3, the representative histograms of flow cytometric analysis illustrated increased levels of phospho‐ERK, phospho‐p38, and phospho‐JNK in CD4+ T lymphocytes and monocytes in type 2 diabetes patients compared with control subjects. Figure 4 showed higher phosphorylation of ERK and p38 at baseline in type 2 diabetes patients than control subjects (all P < 0.05). Exendin‐4 suppressed phosphorylation of ERK and p38 MAPK (all P < 0.05) in CD4+ T lymphocytes and monocytes from type 2 diabetes patients (P > 0.05).
Figure 3.
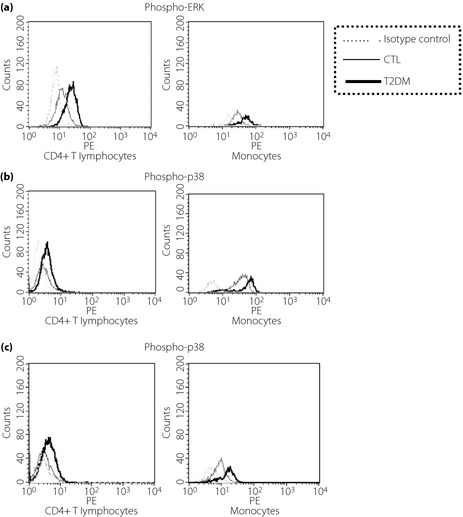
Representative histograms of flow cytometry analysis of phospho‐mitogen‐activated protein kinases including (a) phospho‐extracellular signal‐regulated kinase (ERK), (b) phospho‐p38, and (c) phospho‐c‐Jun NH2‐terminal protein kinase (JNK) in CD4+ T lymphocytes and monocytes from peripheral blood mononuclear cells in patients with type 2 diabetes (T2DM) and the control group (CTL). Triplicate experiments were carried out with essentially identical results and representative figures are shown. Immunoglobulin G1 isotypic control antibodies, which have no specificity for target cells within a particular experiment yet retain all the non‐specific characteristics of the antibodies used in the experiment. Inclusion of this antibody is to confirm the specificity of primary antibody binding and exclude non‐specific fragment crystallizable receptor binding to cells or other cellular protein interactions.
Figure 4.
Basal and ex vivo expression of phospho‐mitogen‐activated protein kinases (MAPK) in exendin‐4 treated CD4+ T lymphocytes and monocytes from peripheral blood mononuclear cells in patients with type 2 diabetes (T2DM) and healthy subjects. (4‐1) Representative histograms show the intracellular expression of (a,b) phospho‐extracellular signal‐regulated kinase (ERK), (c,d) phosphor‐p38 and (e,f) phosphor‐c‐Jun NH2‐terminal protein kinase (JNK) in peripheral blood mononuclear cells incubated without or with exendin‐4 (50 nmol/L) for 10 min. (4‐2) Results are expressed in bar charts. (a) Phosphorylation of ERK, (b), phosphorylation of p38 MAPK and (c) phosphorylation of JNK in monocytes and CD4+ T lymphocytes from patients with type2 diabetes and control subjects were analyzed by flow cytometry. Results are presented with box‐and‐whisker plots.
Exendin‐4 Suppressed Oxidative Stress by Decreasing Superoxide Anion Production
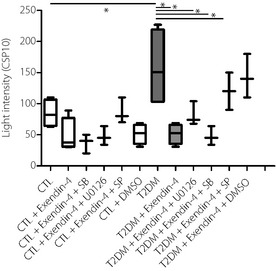
Using the chemiluminescence assay, we detected higher levels of superoxide ions in supernatant of PBMC cultures from type 2 diabetes patients than control subjects, suggestive of oxidative stress, which could be inhibited with 24‐h exendin‐4 treatment, and U0126 (specific inhibitor of ERK signaling pathway) and SB 203580 (specific inhibitor of p38 MAPK signaling pathway; Figure 5).
Figure 5.
Effect of exendin‐4 on the ex vivo production of superoxide anion in culture supernatant of peripheral blood mononuclear cells (PBMC) from type 2 diabetes patients and control subjects. PBMC culture was incubated with or without exendin‐4 at 50 nmol/L for 24 h, with or without pretreatment with U0126 (U0126 10 μmol/L), SP600125 (SP 10 μmol/K) and SB203580 (SB 10 μmol/L) for 1 h. Culture supernatants were then harvested for the assay of superoxides. Results are presented with box‐and‐whisker plots. *P < 0.05. CTL, control; DMSO, dimethyl sulfoxide; T2DM, type 2 diabetes.
Discussion
In the present study, for the first time we have confirmed the activation of monocytes and CD4+ T cells with increased activation of MAPK signaling molecules including ERK, JNK and p38, as well as elevated production of pro‐inflammatory cytokines, chemokines and superoxide anions in PBMC from patients with type 2 diabetes compared with healthy subjects. Interestingly, most of these immunological changes were reversed or attenuated by ex vivo treatment with exendin‐4. These findings lend support to the dysregulation of immunity in type 2 diabetes, which might be ameliorated by direct immunomodulating effects of exendin‐4 treatment.
There is now emerging evidence showing the importance of immune dysregulation in type 2 diabetes17. Chemokines belong to a family of more than 50 proteins that play key roles in recruitment of leukocytes at the site of inflammation22. In diabetes, CD4+ T lymphocytes, once recruited by chemokines such as CCL5/RANTES to the islets, will differentiate into T helper1 (Th1) to secret Th1 and pro‐inflammatory cytokines that activate other immune cells to cause islet inflammation and β‐cell disorder22. In mammalian cells, the MAPK molecules are serine and threonine protein kinases, which can be activated by pro‐inflammatory cytokines to regulate a wide range of cellular responses23. Although ERK signaling is involved in cell proliferation, differentiation, transformation and cytokine production25, activation of p38 MAPK in immune cells can lead to apoptosis, stress responses and inflammation27. Phosphorylation of p38 MAPK also participates in the production of inflammatory cytokines and chemokines, including CCL2, IL‐1β, IL‐6 and TNF‐α, from human mesangial cells23, endothelial cells24, memory T cells27 and eosinophils28.
Hitherto, it remains uncertain whether MAPK pathways are implicated in the production of pro‐inflammatory cytokines/chemokines from human PBMC in type 2 diabetes. In the present study, we were able to confirm the mechanisms of activation of MAPK signaling in both monocytes and CD4+ T helper lymphocytes in type 2 diabetes.
Type 2 diabetes is characterized by reduced incretin effect and GLP‐1 hyposecretion30. However, it is now widely accepted that GLP‐1 secretion does not differ between patients with type 2 diabetes and normal controls31. For the first time, using human PBMC, we have shown that exendin‐4 treatment suppressed the secretion of IL‐1β, TNF‐α and IL‐6, possibly through the suppression of phosphorylation of p38 MAPK and ERK. Apart from augmenting insulin release during meal time, GLP‐1/exendin‐4 exerts extrapancreatic effects including suppressing inflammatory cytokines and chemokines release from human islets and β‐cell lines15. In support of this notion, exendin‐4 has been shown to prevent encephalomyocarditis virus‐induced β‐cell destruction by reducing inflammatory immune response of macrophages15.
In type 2 diabetes, exendin‐4 has been consistently shown to reduce serum inflammatory markers, such as high sensitivity C‐reactive protein, and cytokines such as CCL2/MCP‐1, compared with the placebo treatment33. In obese mice, treatment with dipeptidyl peptidase‐4 inhibitor, sitagliptin, reduced local inflammation in adipose tissue and pancreatic islets34. In the same vein, in the colon of mice with T‐cell‐induced inflammatory bowel disease resembling Crohn's disease and ulcerative colitis, there was marked reduction in proglucagon‐derived intestinal peptides including GLP‐1, GLP‐2 and glicentin35. Apart from the detection of messenger ribonucleic acid transcripts of GLP‐1 receptor in ribonucleic acid isolated from murine spleen, thymus and lymph nodes36, other researchers have reported the expression of GLP‐1 receptor on natural killer T (NKT) cells with GLP‐1, inducing a dose‐dependent inhibition of cytokine secretion from NKT cells in a case report of two patients with type 2 diabetes and psoriasis37. In streptozotocin‐induced rats, GLP‐1 receptor agonist protected against renal damage and polyneuropathy through its anti‐inflammatory action38.
Given the circulating and ubiquitous nature of monocytes, our results suggested that the anti‐inflammatory effects of exendin‐4 might be mediated partly through its direct effects on immune cells. In experimental studies, treatment with exendin‐4 reduced monocyte/macrophage accumulation in the arterial wall by inhibiting the inflammatory response in macrophages39. In the present study, we confirmed the signaling pathways associated with immune activation of PBMC. In this regard, our group also showed that exendin‐4 protected human islet amyloid polypeptide‐induced apoptosis of rat β insulinoma cell line, INS1‐E, through similar pathways although p‐JNK was not affected40. Taken together, the immunomodulating effects of exendin‐4 on multiple immune effectors were in part mediated by its inhibition on the activation of p38 MAPK, cytokines and chemokine release.
Activation of cytokine release can lead to increased oxidative stress as a result of membrane‐bound NADPH oxidase activation12. In pancreatic islets, activation of immune cells and cytokine release increased oxidative stress with the loss of β‐cells, which was prevented by anti‐oxidant compounds41. With the onset of hyperglycemia, which promotes oxidative stress42, a vicious cycle of inflammation, oxidative stress, β‐cell loss and hyperglycemia can be set up. In support of this notion43, in the present ex vivo study, we confirmed increased production of superoxide ions in PBMC from patients with type 2 diabetes, which could be attenuated by the treatment with exendin‐4.
Several studies have reported the ameliorating effects of exendin‐4 on diabetic vascular complications, such as nephropathy, by decreasing oxidative stress44. In primary neuronal cultures and human SH‐SY5Y cells, exendin‐4 reduced amyloid β toxicity and oxidative stress46. Interestingly, superoxide dismutase activity was enhanced in recombinant human GLP‐1‐pretreated streptozotocin‐induced diabetic mice with improved hyperglycemia47. In the present experiment, exendin‐4 treatment inhibited superoxide anion in PBMC in type 2 diabetes, which might add to the anti‐inflammatory effects of exendin‐4 and break the vicious cycle.
Using PBMC from patients with type 2 diabetes, we have shown, for the first time, the activation of monocytes and CD4+ T cells indicated by increased MAPK signaling pathways, cytokines/chemokines release and oxidative stress. Apart from adipokines and cytokines, metabolic triggers such as blood glucose and lipids, notably free fatty acids17, might trigger an inflammatory cascade to induce cellular dysfunction. In this connection, our novel findings regarding the direct immunomodulating effects of exendin‐4 in PBMC though suppression of phosphorylation of p38 MAPK and ERK, and production of superoxide anion, suggest that the beneficial effects of exentin‐4 might extend beyond diabetes to include other autoimmune or inflammatory diseases.
Acknowledgements
We thank our nurses, Ms Kitman Loo, Harriet Chung, Cherry Chiu and Lindy Chan for their assistance; our technologists, Mr Raymond Wong, Dr Sharon Lee, Miss Ma Shihong and Miss Lik Kwan for their assistance in flow cytometry. This work was supported in part by the Hong Kong Government Research Grants Council (CUHK4462/06M) and Hong Kong Jockey Club Charities Trust (JCICM‐P2‐05 (CUHK). All authors declare no conflict of interest.
(J Diabetes Invest, doi: 10.1111/jdi.12063, 2013)
References
- 1.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 2002; 51(Suppl 3): S455–S461 [DOI] [PubMed] [Google Scholar]
- 2.Masters SL, Dunne A, Subramanian SL, et al Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL‐1beta in type 2 diabetes. Nat Immunol 2010; 11: 897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson RP, Harmon J, Tran PO, et al Beta‐cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004; 53(Suppl 1): S119–S124 [DOI] [PubMed] [Google Scholar]
- 4.Richardson SJ, Willcox A, Bone AJ, et al Islet‐associated macrophages in type 2 diabetes. Diabetologia 2009; 52: 1686–1688 [DOI] [PubMed] [Google Scholar]
- 5.Donath MY, Boni‐Schnetzler M, Ellingsgaard H, et al Islet inflammation impairs the pancreatic beta‐cell in type 2 diabetes. Physiology (Bethesda) 2009; 24: 325–331 [DOI] [PubMed] [Google Scholar]
- 6.Pickersgill LM, Mandrup‐Poulsen TR. The anti‐interleukin‐1 in type 1 diabetes action trial–background and rationale. Diabetes Metab Res Rev 2009; 25: 321–324 [DOI] [PubMed] [Google Scholar]
- 7.Mandrup‐Poulsen T. The role of interleukin‐1 in the pathogenesis of IDDM. Diabetologia 1996; 39: 1005–1029 [DOI] [PubMed] [Google Scholar]
- 8.Eizirik DL, Mandrup‐Poulsen T. A choice of death–the signal‐transduction of immune‐mediated beta‐cell apoptosis. Diabetologia 2001; 44: 2115–2133 [DOI] [PubMed] [Google Scholar]
- 9.Schett G, Tohidast‐Akrad M, Smolen JS, et al Activation, differential localization, and regulation of the stress‐activated protein kinases, extracellular signal‐regulated kinase, c‐JUN N‐terminal kinase, and p38 mitogen‐activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum 2000; 43: 2501–2512 [DOI] [PubMed] [Google Scholar]
- 10.Tchaikovski V, Olieslagers S, Bohmer FD, et al Diabetes mellitus activates signal transduction pathways resulting in vascular endothelial growth factor resistance of human monocytes. Circulation 2009; 120: 150–159 [DOI] [PubMed] [Google Scholar]
- 11.Jeong HW, Hsu KC, Lee JW, et al Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab 2009; 296: E955–E964 [DOI] [PubMed] [Google Scholar]
- 12.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 1999; 85: 753–766 [DOI] [PubMed] [Google Scholar]
- 13.Drucker DJ. The biology of incretin hormones. Cell Metab 2006; 3: 153–165 [DOI] [PubMed] [Google Scholar]
- 14.Tian L, Gao J, Hao J, et al Reversal of new‐onset diabetes through modulating inflammation and stimulating beta‐cell replication in nonobese diabetic mice by a dipeptidyl peptidase IV inhibitor. Endocrinology 2010; 151: 3049–3060 [DOI] [PubMed] [Google Scholar]
- 15.Sano H, Terasaki J, Mishiba Y, et al Exendin‐4, a glucagon‐like peptide‐1 receptor agonist, suppresses pancreatic beta‐cell destruction induced by encephalomyocarditis virus. Biochem Biophys Res Commun 2010; 404: 756–761 [DOI] [PubMed] [Google Scholar]
- 16.Li L, El‐Kholy W, Rhodes CJ, et al Glucagon‐like peptide‐1 protects beta cells from cytokine‐induced apoptosis and necrosis: role of protein kinase B. Diabetologia 2005; 48: 1339–1349 [DOI] [PubMed] [Google Scholar]
- 17.Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 2004; 27: 813–823 [DOI] [PubMed] [Google Scholar]
- 18.Hussain MJ, Peakman M, Gallati H, et al Elevated serum levels of macrophage‐derived cytokines precede and accompany the onset of IDDM. Diabetologia 1996; 39: 60–69 [DOI] [PubMed] [Google Scholar]
- 19.Pickup JC, Chusney GD, Thomas SM, et al Plasma interleukin‐6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci 2000; 67: 291–300 [DOI] [PubMed] [Google Scholar]
- 20.Orskov C, Rabenhoj L, Wettergren A, et al Tissue and plasma concentrations of amidated and glycine‐extended glucagon‐like peptide I in humans. Diabetes 1994; 43: 535–539 [DOI] [PubMed] [Google Scholar]
- 21.Jacobsen ML, Ronn SG, Bruun C, et al IL‐1beta‐induced chemokine and Fas expression are inhibited by suppressor of cytokine signalling‐3 in insulin‐producing cells. Diabetologia 2009; 52: 281–288 [DOI] [PubMed] [Google Scholar]
- 22.Baggiolini M. Chemokines in pathology and medicine. J Intern Med 2001; 250: 91–104 [DOI] [PubMed] [Google Scholar]
- 23.Rovin BH, Wilmer WA, Danne M, et al The mitogen‐activated protein kinase p38 is necessary for interleukin 1beta‐induced monocyte chemoattractant protein 1 expression by human mesangial cells. Cytokine 1999; 11: 118–126 [DOI] [PubMed] [Google Scholar]
- 24.Goebeler M, Kilian K, Gillitzer R, et al The MKK6/p38 stress kinase cascade is critical for tumor necrosis factor‐alpha‐induced expression of monocyte‐chemoattractant protein‐1 in endothelial cells. Blood 1999; 93: 857–865 [PubMed] [Google Scholar]
- 25.Chambard JC, Lefloch R, Pouyssegur J, et al ERK implication in cell cycle regulation. Biochim Biophys Acta 2007; 1773: 1299–1310 [DOI] [PubMed] [Google Scholar]
- 26.Wong CK, Wang CB, Ip WK, et al Role of p38 MAPK and NF‐kB for chemokine release in coculture of human eosinophils and bronchial epithelial cells. Clin Exp Immunol 2005; 139: 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong CK, Li PW, Intracellular Lam CW, et al p38 MAPK and NF‐kappaB regulate IL‐25 induced release of cytokines and chemokines from costimulated T helper lymphocytes. Immunol Lett 2007; 112: 82–91 [DOI] [PubMed] [Google Scholar]
- 28.Wong CK, Cheung PF, Ip WK, et al Interleukin‐25‐induced chemokines and interleukin‐6 release from eosinophils is mediated by p38 mitogen‐activated protein kinase, c‐Jun N‐terminal kinase, and nuclear factor‐kappaB. Am J Respir Cell Mol Biol 2005; 33: 186–194 [DOI] [PubMed] [Google Scholar]
- 29.Cheung PF, Wong CK, Lam CW. Molecular mechanisms of cytokine and chemokine release from eosinophils activated by IL‐17A, IL‐17F, and IL‐23: implication for Th17 lymphocytes‐mediated allergic inflammation. J Immunol 2008; 180: 5625–5635 [DOI] [PubMed] [Google Scholar]
- 30.Nauck MA, Baller B, Meier JJ. Gastric inhibitory polypeptide and glucagon‐like peptide‐1 in the pathogenesis of type 2 diabetes. Diabetes 2004; 53(Suppl 3): S190–S196 [DOI] [PubMed] [Google Scholar]
- 31.Nauck MA, Vardarli I, Deacon CF, et al Secretion of glucagon‐like peptide‐1 (GLP‐1) in type 2 diabetes: what is up, what is down? Diabetologia 2010; 54: 10–18 [DOI] [PubMed] [Google Scholar]
- 32.Pugazhenthi U, Velmurugan K, Tran A, et al Anti‐inflammatory action of exendin‐4 in human islets is enhanced by phosphodiesterase inhibitors: potential therapeutic benefits in diabetic patients. Diabetologia 2010; 53: 2357–2368 [DOI] [PubMed] [Google Scholar]
- 33.Wu JD, Xu XH, Zhu J, et al Effect of exenatide on inflammatory and oxidative stress markers in patients with type 2 diabetes mellitus. Diabetes Technol Ther 2011; 13: 143–148 [DOI] [PubMed] [Google Scholar]
- 34.Dobrian AD, Ma Q, Lindsay JW, et al Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. Am J Physiol Endocrinol Metab 2010; 300: E410–E421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt PT, Hartmann B, Bregenholt S, et al Deficiency of the intestinal growth factor, glucagon‐like peptide 2, in the colon of SCID mice with inflammatory bowel disease induced by transplantation of CD4+ T cells. Scand J Gastroenterol 2000; 35: 522–527 [DOI] [PubMed] [Google Scholar]
- 36.Hadjiyanni I, Baggio LL, Poussier P, et al Exendin‐4 modulates diabetes onset in nonobese diabetic mice. Endocrinology 2008; 149: 1338–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hogan AE, Tobin AM, Ahern T, et al Glucagon‐like peptide‐1 (GLP‐1) and the regulation of human invariant natural killer T cells: lessons from obesity, diabetes and psoriasis. Diabetologia 2011; 54: 2745–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Himeno T, Kamiya H, Naruse K, et al Beneficial effects of exendin‐4 on experimental polyneuropathy in diabetic mice. Diabetes 2011; 60: 2397–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arakawa M, Mita T, Azuma K, et al Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon‐like peptide‐1 receptor agonist, exendin‐4. Diabetes 2010; 59: 1030–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan R, Li X, Gu X, et al Exendin‐4 protects pancreatic beta cells from human islet amyloid polypeptide‐induced cell damage: potential involvement of AKT and mitochondria biogenesis. Diabetes Obes Metab 2010; 12: 815–824 [DOI] [PubMed] [Google Scholar]
- 41.Kanitkar M, Gokhale K, Galande S, et al Novel role of curcumin in the prevention of cytokine‐induced islet death in vitro and diabetogenesis in vivo. Br J Pharmacol 2008; 155: 702–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alhaider AA, Korashy HM, Sayed‐Ahmed MM, et al Metformin attenuates streptozotocin‐induced diabetic nephropathy in rats through modulation of oxidative stress genes expression. Chem Biol Interact 2011; 192: 233–242 [DOI] [PubMed] [Google Scholar]
- 43.Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes 1991; 40: 405–412 [DOI] [PubMed] [Google Scholar]
- 44.Kodera R, Shikata K, Kataoka HU, et al Glucagon‐like peptide‐1 receptor agonist ameliorates renal injury through its anti‐inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia 2011; 54: 965–978 [DOI] [PubMed] [Google Scholar]
- 45.Park CW, Kim HW, Ko SH, et al Long‐term treatment of glucagon‐like peptide‐1 analog exendin‐4 ameliorates diabetic nephropathy through improving metabolic anomalies in db/db mice. J Am Soc Nephrol 2007; 18: 1227–1238 [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Duffy KB, Ottinger MA, et al GLP‐1 receptor stimulation reduces amyloid‐beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. J Alzheimers Dis 2010; 19: 1205–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu YL, Huang J, Liu J, et al Protective effect of recombinant human glucagon‐like peptide‐1 (rhGLP‐1) pretreatment in STZ‐induced diabetic mice. J Pept Sci 2010; 17: 499–504 [DOI] [PubMed] [Google Scholar]