

Abstract
Existing concepts and models for glucose‐stimulated insulin secretion (GSIS) are overviewed and a newer perspective has been formulated toward the physiological understanding of GSIS. A conventional model has been created on the basis of in vitro data on application of a square wave high glucose in the absence of any other stimulatory inputs. Glucose elicits rapid insulin release through an adenosine triphosphate‐sensitive K+ channel (KATP channel)‐dependent mechanism, which is gradually augmented in a KATP channel‐independent manner. Biphasic GSIS thus occurs. In the body, the β‐cells are constantly exposed to stimulatory signals, such as glucagon‐like peptide 1 (GLP‐1), parasympathetic inputs, free fatty acid (FFA), amino acids and slightly suprathreshold levels of glucose, even at fasting. GLP‐1 increases cellular cyclic adenosine monophosphate, parasympathetic stimulation activates protein kinase C, and FFA, amino acids and glucose generate metabolic amplification factors. Plasma glucose concentration gradually rises postprandially under such tonic stimulation. We hypothesize that these stimulatory inputs together make the β‐cells responsive to glucose independently from its action on KATP channels. Robust GSIS in patients with a loss of function mutation of the sulfonylurea receptor, a subunit of KATP channels, is compatible with this hypothesis. Furthermore, pre‐exposure of the islets to an activator of protein kinase A and/or C makes β‐cells responsive to glucose in a KATP channel‐ and Ca2+‐independent manner. We hypothesize that GSIS occurs in islet β‐cells without glucose regulation of KATP channels in vivo, for which priming with cyclic adenosine monophosphate, protein kinase C and non‐glucose nutrients are required. To understand the physiology of GSIS, comprehensive integration of accumulated knowledge is required.
Keywords: Adenosine triphosphate‐sensitive K+ channel, Modulatory signals, Physiological insulin secretion
Introduction
Insulin is the exclusive hormone that lowers plasma glucose concentration, and glucose homeostasis is maintained primarily as a result of regulated insulin secretion. Pancreatic β‐cells recognize extracellular glucose concentration and secrete insulin as required at a given time. Glucose‐stimulated insulin secretion (GSIS) is modulated by a number of factors, such as non‐glucose nutrients, hormones and neural inputs (Figure 1). Thus, the intracellular network for regulation of insulin secretion is complex and multifactorial. Although GSIS has been viewed as analogous to excitation‐contraction coupling of muscle1 and stimulus‐secretion coupling of neuron/chromaffin cells2, the regulatory system for GSIS is far more complex than these other two systems. In addition, the time frames of insulin secretion and neurotransmitter release are different: insulin secretion is tuned over minutes to hours, whereas neurotransmitter release occurs instantaneously; that is, within the subsecond range. As a counterpart for excitation‐contraction coupling and stimulus‐secretion coupling, Wollheim3 coined the term ‘metabolism‐secretion coupling’ for GSIS, which will form the basis of the present review. We present an overview of existing concepts and models for GSIS, and provide a newer perspective based on recent developments in this field toward its physiological understanding. Although evidence for a ‘glucose receptor’ has recently been reported4, this topic will not be discussed in the present review, because its physiological relevance remains to be determined.
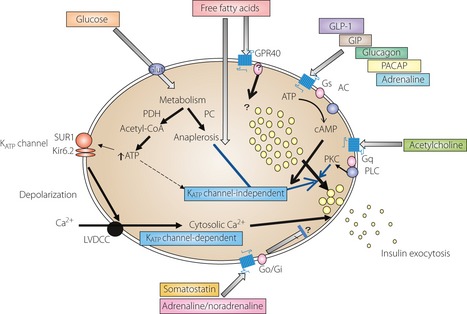
Figure 1.

A proposed signaling network of insulin exocytosis in pancreatic β‐cells. AC, adenylate cyclase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; GIP, glucose‐dependent insulinotropic peptide; GLP‐1, glucagon‐like polypeptide‐1; Glut, glucose transporter; KATP channel, adenosine triphosphate‐sensitive K+ channel; Kir6.2, K+ channel 6.2 subunits; LVDCC, L‐type voltage‐dependent calcium channel PACAP, pituitary adenylate cyclase activating peptide; PDH, pyruvate dehydrogenase; PC, pyruvate carboxylase; PKC, protein kinase C; PLC, phospholipase C; SUR1, sulfonylurea receptor 1.
Adenosine Triphosphate‐Sensitive Potassium Channel: The Central Dogma
On elevation of plasma glucose concentration, glucose enters the pancreatic β‐cells through the glucose transporter on the plasma membrane. Glucose is then phosphorylated by glucokinase and subjected to glycolysis, by which pyruvate is generated in the cytoplasm. Pyruvate is metabolized equally by pyruvate dehydrogenase and pyruvate carboxylase (PC) in the β‐cells, and passes into the mitochondria. The former reaction leads to generation of adenosine triphosphate (ATP) in the respiratory chain and the latter is accompanied by efflux of tricarboxylic acid (TCA) cycle intermediates as anaplerosis. ATP is a signaling molecule for insulin secretion in β‐cells, because the cell is equipped with ATP‐sensitive K+ channels (KATP channels), which close on elevation of cytoplasmic ATP or ATP/adenosine diphosphate ratio. As the KATP channel is the primary determinant of the membrane potential of the β‐cells, closure of these channels causes membrane depolarization. The membrane depolarization opens L‐type voltage‐dependent Ca2+ channels (VDCC), followed by Ca2+ influx and elevation of cytosolic free Ca2+ concentration ([Ca2+]i). The elevation of [Ca2+]i rapidly increases the rate of insulin exocytosis.
This model has its origin in the pioneering work of Dean and Matthews5, and was formulated based on the nature of β‐cell electrophysiology6. Inagaki et al.8 successfully identified the KATP channel in β‐cells as a tetra‐octamer composed of four sulfonylurea receptor 1 (SUR1) and inwardly rectifying K+ channel 6.2 subunits (Kir6.2). SUR1 is the target of insulin secretagogues, such as sulfonylurea (SU) and glinide used in the treatment of type 2 diabetes, such that the channel is closed on binding of SU or glinide to the SUR1. GSIS was completely abolished in vitro by treatment with diazoxide, a KATP channel opener, or nifedipine, a dihydropyridine Ca2+ channel blocker that inhibits opening of the VDCC. On the basis of these observations, signaling through KATP channels, VDCC and [Ca2+]i elevation had been considered the exclusive mechanism underlying GSIS until 1992.
An acute elevation of [Ca2+]i facilitates fusion of insulin granules and the plasma membrane leading to an increased rate of exocytosis. This reaction is mediated by the assembly of soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor protein. However, the kinetics of exocytosis are considerably different between exocytosis of synaptic vesicles in neuronal cells and large dense‐core vesicles in β‐cells, with the former being more than five orders of magnitude faster than the latter10. In addition, glucose‐induced electrophysiological events; that is, KATP channel closure and elevation of [Ca2+]i, do not faithfully reflect the time‐course and concentration dependency of GSIS. These phenomena had been overlooked or left unanswered.
Biphasic Insulin Release and Katp Channel‐Independent Glucose Action
In the early 1970s, Grodsky11 established that GSIS is biphasic when extracellular glucose concentration is abruptly raised from sub‐ (3 mmol/L) to suprastimulatory concentrations (16.7–22.2 mmol/L). He suggested that glucose triggers insulin secretion from a threshold‐sensitive packet of insulin in the labile pool, and glucose potentiates insulin release by replenishing this labile pool by distinct mechanism(s). The first phase culminates 5–6 min after stimulation, and a gradual increase in insulin release over 60 min ensues, which is called the second phase11.
The temporal profile of the ionic events after glucose stimulation is significantly different from that of GSIS: glucose‐induced membrane depolarization and [Ca2+]i rise are continuously oscillating, and by no means biphasic. The biphasic nature of GSIS is robust in rat and human islets, weak in mouse islets, and absent in most tumor β‐cell lines. A square wave application of a depolarizing concentration of K+ sharply increases the rate of insulin exocytosis for several minutes, which temporarily resembles the first phase12. However, in contrast to the insulin response to high glucose, high K+‐induced insulin release is monophasic and followed by gradual lowering; that is, there is no second phase. Nevertheless, the time‐courses of membrane depolarization and [Ca2+]i elevation seen after K+ depolarization are similar, if not identical, to those induced by high glucose. These observations indicated that high glucose produces signal(s) for insulin secretion in addition to elevation of [Ca2+]i.
In 1992, the present authors and another group showed that glucose augments insulin exocytosis evoked by K+ depolarization even when the KATP channels are fully open or closed by the presence of diazoxide or SU, respectively13. In the presence of a high concentration of SU with the KATP channels fully closed, glucose induces an increase, not a decrease, in 86Rb+ outflow from the islet cells, which is a surrogate index of K+ outflow15. Importantly, GSIS in the presence of a depolarizing concentration of K+ and diazoxide occurs without any further increase in [Ca2+]i16. Thus, KATP‐independent GSIS was established.
It has long been considered that there are distinct pools of insulin granules in the β‐cells, known as readily releasable pools (RRP) and reserve pools (RP)17. RRP is not necessarily physically docked to the plasma membrane. The biphasic GSIS has been understood as a product of a combination of triggering and amplification/augmentation along with this concept. That is, triggering denotes the initial rapid insulin exocytosis from the RRP by the KATP‐dependent mechanism, and amplification/augmentation indicates gradual enhancement by the KATP‐independent signal. The latter can occur with replenishment of the RRP from the RP, so we proposed viewing the biphasic insulin release as ‘fusion and replenishment’18.
The aforementioned series of secretory and ionic events are observed on sudden increase in glucose from sub‐ to suprastimulatory concentration in the absence of any other stimuli, and so the conditions are markedly different from those seen physiologically.
Metabolic Amplification Factor
Although the molecule(s) responsible for the KATP‐independent GSIS have yet to be identified, anaplerotic metabolism of pyruvate by PC and subsequent efflux of the TCA cycle intermediates are considered key events (Figure 1)19. The candidate molecules suggested to date as possible mediators of the KATP channel‐independent glucose action include ATP, guanosine 5′‐triphosphate, the reduced form of nicotinamide‐adenine dinucleotide, glutamate, malonyl‐CoA and Rab27a21. It is possible that thermosensitive transient receptor potential channel and Kv channels are involved in KATP‐independent glucose signaling23.
Priming of β‐Cells
Time‐Dependent Potentiation
Glucose causes time‐dependent potentiation (TDP) of insulin secretion; that is, exposure of islet β‐cells to high glucose enhances insulin release in response to the stimulus applied later25. Metabolizable amino acids and the glycolytic intermediate, glyceraldehyde, show similar enhancement of insulin secretion. In contrast, non‐metabolizable secretagogues, such as SU and high K+, do not prime the β‐cells in this manner. In contrast, metabolic inhibition abolishes TDP by glucose and the amino acids. Therefore, metabolic stimulus, the mitochondrial intermediates in the case of amino acids, is required for TDP. Interestingly, pharmacological activation of protein kinase C by phorbol ester also time‐dependently potentiates insulin secretion26.
The β‐cell priming by glucose occurs in a KATP channel‐independent manner, because the potentiation was totally resistant to a high concentration of diazoxide, an opener of KATP channels12. Furthermore, TDP occurs under stringent Ca2+‐free conditions and therefore [Ca2+]i elevation is not required27. Taken together, these observations suggest that the second phase observed under a square wave high‐glucose application is the triggered insulin release followed by enhancement by TDP.
Permissive Role of Hormones, Non‐Glucose Fuels and Neural Inputs
Incretins
Glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide are gastrointestinal hormones called incretins, which robustly enhance nutrient‐induced insulin secretion, and they are very important in the physiology of insulin secretion. Incretins bind to G protein‐coupled receptors on the β‐cell membrane and increase cellular 3′,5′‐cyclic adenosine monophosphate (cAMP). When the β‐cells are exposed to a stimulatory concentration of glucose, cAMP further elevates GSIS. Incretin action is resistant to diazoxide, and therefore it is independent of KATP channel closure28. cAMP enhances GSIS through protein kinase A (PKA)‐dependent and ‐independent mechanisms29. As one of the latter mechanisms, activation of the Epac2/Rap1 signaling cascade was proposed30. With regard to the insulin granule dynamics, cAMP increases the size of RRP in a glucose concentration‐dependent manner. This finding strongly indicates that incretin primes the β‐cells in the presence of stimulatory ambient glucose concentration. Incretin priming occurs in a Ca2+‐independent manner, even under stringent Ca2+‐free conditions31.
Free Fatty Acid
Free fatty acid (FFA) participates in the regulation of GSIS. Insulin secretion is suppressed with long‐term exposure of β‐cells to excessively high concentration of FFA, which is called lipotoxicity32. However, short‐term (hours) exposure to the physiological concentration of FFA causes TDP33. The action of FFA appears to be mediated by FFA oxidation per se or accumulation of long chain acyl‐CoA in the cytoplasm. The insulinotropic action of FFA might be partially mediated by membrane receptor(s) for FFA, which is called GPR40 (Figure 1)35. An agonist of GPR40 that is currently in clinical trials36 elevates [Ca2+]i through activation of phospholipase C and protein kinase D137. At any rate, FFA augmentation of GSIS is also independent of KATP channel33.
Parasympathetic Nerves and Neuropeptides
Activation of the parasympathetic nervous system causes release of acetylcholine in the islets, and binding of acetylcholine to the membrane receptor leads to hydrolysis of phospholipids, accumulation of cellular inositol 1, 4, 5‐triphosphate and activation of PKC38. As a result, GSIS is strongly enhanced. This pathway might play a role in the cephalic phase of insulin release39 and TDP (as aforementioned). Pituitary adenylate cyclase‐activating polypeptide (PACAP), which is located in the nerve endings around pancreatic islets, binds to its receptor on β‐cells and enhances insulin secretion at the picomolar range by increasing cellular cAMP40. Although the physiological significance of PACAP in controlling insulin secretion is still controversial, it is interesting that PACAP is one of the substrates for dipeptidyl‐peptidase 4.
Inhibitory Signals for Insulin Secretion
An important aspect of the physiological control of insulin secretion is the presence of inhibitory signals41. Heightened sympathetic tone suppresses insulin secretion through an increase in noradrenaline42. Noradrenaline binds to the α2‐adrenergic receptor and activates the heterotrimeric G protein. Somatostatin, which is secreted from pancreatic δ‐cells, suppresses insulin secretion by binding to the specific heterotrimeric G protein‐coupled receptor as well. These inhibitory hormones activate Gi and/or Go, and inhibit insulin exocytosis at multiple sites. It is likely that changes in the inhibitory signals are intimately involved in the physiology of insulin secretion in vivo.
Toward a Physiological Understanding
In the body, the β‐cells are primed by glucose, incretins, FFA and parasympathetic neural input, even under fasting conditions, because fasting plasma concentrations of glucose, active GLP‐1, and FFA are 5 mmol/L, 10 pmol/L and 500 μEq/L, respectively, which are within the stimulatory ranges for β‐cells. On intake of a meal, gradual elevation of glucose and amino acids occurs, and incretins and the parasympathetic input further increase. Concomitant suppression of sympathetic nerve input might influence to increase in insulin secretion.
Role of KATP Channels
SU and diazoxide, a closer and opener of KATP channels, enhance and reduce meal‐induced insulin secretion, respectively. The absence of KATP channels; that is, persistent closure, causes persistent hyperinsulinemic hypoglycemia, and activating mutation of the channel causes impaired insulin secretion and diabetes. Taken together, these observations imply that GSIS is affected by KATP channel closing and opening in vivo.
KATP‐Independent GSIS
Robust insulin secretion in response to the intravenous bolus injection of glucose occurs in patients with loss of function SUR1 mutation, provided euglycemia has been maintained for hours by continuous glucose infusion43. This in vivo observation was supported by experimental data showing rapid insulin secretion in response to a square wave application of glucose by the islets from such patients or KATP channel knockout mice44. It is also important to recognize that the β‐cells from patients with persistent hyperinsulinemic hypoglycemia of infancy (PHHI) or KATP channel knockout mice do not secrete excess insulin in vitro with substimulatory concentration of glucose despite persistent elevation of [Ca2+]i46. Under these conditions, β‐cell [Ca2+]i is elevated, but there are no other stimuli/priming, such as incretin/cAMP elevation, FFA and parasympathetic input/PKC activation. We hypothesize that GSIS occurs in the islet β‐cells, even in the absence of glucose regulation of KATP channels in vivo under priming with cAMP, non‐glucose nutrients and PKC. GSIS in PHHI patients after partial pancreatectomy provides further support for this hypothesis. The patients maintain non‐diabetic glycemia for years, and oral GSIS and meal‐stimulated insulin secretion do occur in them48. Post‐challenge incretin secretion would significantly assist GSIS on oral glucose or meal intake.
Additional experimental evidence for the hypothesis is as follows. Glucose increases insulin secretion by the islets, even under stringently Ca2+‐free conditions, given pre‐exposure of the islets to forskolin and phorbol ester, activators of PKA and PKC, respectively49. Recently, we showed rapid insulin secretion in islets pre‐exposed to forskolin on exposure to square wave high glucose in the presence of diazoxide and nifedipine50.
cAMP enhancement of GSIS has attracted renewed interest with the development of incretin mimetics for clinical use. Although detailed discussion of this topic is beyond the scope of the present review, it occurs only when ambient glucose concentration exceeds a threshold and is resistant to diazoxide, and so is a manifestation of KATP‐independent GSIS28.
Summary
Insulin secretion in vivo is finely tuned by a variety of stimulatory and inhibitory signals. It is important to appreciate that the β‐cells are being tonically primed with nutrients, and hormonal and neural inputs, even in the fasting state (Figure 2). Meal intake produces gradual rather than instantaneous elevation of glucose. Thus, to gain a comprehensive understanding of the physiology of GSIS, careful experimentation and integration of accumulated knowledge are required. Further studies are needed to fully understand the complex and multifactorial mechanisms involved in GSIS.
Figure 2.
Distinction of glucose‐stimulated insulin release in (a) in vitro and (b) in vivo. (a) Square‐wave application of high glucose produces biphasic insulin release in vitro. Adenosine triphosphate‐sensitive K+ channel (KATP channel) closure is mandatory for this response. Nevertheless, glucose gradually enhances the Ca2+‐stimulated secretion in a KATP channel‐independent manner. (b) Under the physiological condition, the β‐cells are being continuously primed. Namely, the plasma level of glucose and other nutrients is at the weakly stimulatory range, and hormonal and neural stimuli are also present, even at fasting. A gradual elevation of plasma glucose after a meal robustly raises the rate of insulin exocytosis adenosine triphosphate‐sensitive K+ channel‐independently under this condition.  , Constitutive secretion;
, Constitutive secretion;  , K+ channel‐dependent secretion;
, K+ channel‐dependent secretion;  , K+ channel‐independent secretion.
, K+ channel‐independent secretion.
Acknowledgements
The authors declare no conflict of interest. We express our gratitude to Professor Toru Aizawa for constructive discussion.
(J Diabetes Invest, doi: 10.1111/jdi.12094, 2013)
References
- 1.Sandow A. Excitation‐contraction coupling in muscular response. Yale J Biol Med 1952; 25: 176–201 [PMC free article] [PubMed] [Google Scholar]
- 2.Douglas WW. Stimulus‐secretion coupling: the concept and clues from chromaffin and other cells. Br J Pharmacol 1968; 34: 451–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maechler P, Kennedy ED, Pozzan T, et al Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic β‐cells. EMBO J 1997; 16: 3833–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakagawa Y, Nagasawa M, Yamada S, et al Sweet taste receptor expressed in pancreatic beta‐cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS ONE 2009; 4: e5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dean PM, Matthews EK. Electrical activity in pancreatic islet cells. Nature 1968; 219: 389–390 [DOI] [PubMed] [Google Scholar]
- 6.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic β‐cells. Nature 1984; 312: 446–448 [DOI] [PubMed] [Google Scholar]
- 7.Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B‐cells. Nature 1984; 311: 271–273 [DOI] [PubMed] [Google Scholar]
- 8.Inagaki N, Gonoi T, Clement JP, et al Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 1995; 270: 1166–1170 [DOI] [PubMed] [Google Scholar]
- 9.Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic beta‐cell ATP‐sensitive K+ channel. FEBS Lett 1997; 409: 232–236 [DOI] [PubMed] [Google Scholar]
- 10.Kasai H, Takahashi N, Tokumaru H. Distinct initial SNARE configurations underlying the diversity of exocytosis. Physiol Rev 2012; 92: 1915–1964 [DOI] [PubMed] [Google Scholar]
- 11.Grodsky GM. A threshold distribution hypothesis for packet storage of insulin and its mathematical modeling. J Clin Invest 1972; 51: 2047–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komatsu M, Yokokawa N, Takeda T, et al Pharmacological characterization of the voltage‐dependent calcium channel of pancreatic B‐cell. Endocrinology 1989; 125: 2008–2014 [DOI] [PubMed] [Google Scholar]
- 13.Sato Y, Aizawa T, Komatsu M, et al Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic B‐cell. Diabetes 1992; 41: 438–443 [DOI] [PubMed] [Google Scholar]
- 14.Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP‐sensitive K+ channels in mouse B cells. J Clin Invest 1992; 89: 1288–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Best L, Yates AP, Tomlinson S. Stimulation of insulin secretion by glucose in the absence of diminished potassium (86Rb+) permeability. Biochem Pharmacol 1992; 43: 2483–2485 [DOI] [PubMed] [Google Scholar]
- 16.Gembal M, Detimary P, Gilon P, et al Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate‐sensitive K+ channels in mouse B cells. J Clin Invest 1993; 91: 871–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bratanova‐Tochkova TK, Cheng H, Daniel S, et al Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes 2002; 51(Suppl 1): S83–S90 [DOI] [PubMed] [Google Scholar]
- 18.Aizawa T, Komatsu M. Rab27a: a new face in beta cell metabolism‐secretion coupling. J Clin Invest 2005; 115: 227–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brun T, Roche E, Assimacopoulos‐Jeannet F, et al Evidence for an anaplerotic/malonyl‐CoA pathway in pancreatic β‐cell nutrient signaling. Diabetes 1996; 45: 190–198 [DOI] [PubMed] [Google Scholar]
- 20.Sugden MC, Holness MJ. The pyruvate carboxylase‐pyruvate dehydrogenase axis in islet pyruvate metabolism: going round in circles? Islets 2011; 3: 302–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasai K, Ohara‐Imaizumi M, Takahashi N, et al Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J Clin Invest 2005; 115: 388–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komatsu M, Sato Y, Aizawa T, et al KATP channel‐independent glucose action: an elusive pathway in stimulus‐secretion coupling of pancreatic β‐cell. Endocr J 2001; 48: 275–288 [DOI] [PubMed] [Google Scholar]
- 23.Uchida K, Tominaga M. The role of thermosensitive TRP (transient receptor potential) channels in insulin secretion. Endocr J 2011; 58: 1021–1028 [DOI] [PubMed] [Google Scholar]
- 24.Yoshida M, Nakata M, Yamato S, et al Voltage‐dependent metabolic regulation of Kv2.1 channels in pancreatic β‐cells. Biochem Biophys Res Commun 2010; 396: 304–309 [DOI] [PubMed] [Google Scholar]
- 25.Grill V, Adamson U, Cerasi E. Immediate and time‐dependent effects of glucose on insulin release from rat pancreatic tissue. Evidence for different mechanisms of action. J Clin Invest 1978; 61: 1034–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niki I, Tamagawa T, Niki H, et al Possible involvement of diacylglycerol‐activated, Ca2+‐dependent protein kinase in glucose memory of the rat pancreatic B‐cell. Acta Endocrinol (Copenh) 1988; 118: 204–208 [DOI] [PubMed] [Google Scholar]
- 27.Yamada S, Komatsu M, Aizawa T, et al Time‐dependent potentiation of the β‐cell is a Ca2+‐independent phenomenon. J Endocrinol 2002; 172: 345–354 [DOI] [PubMed] [Google Scholar]
- 28.Yajima H, Komatsu M, Schermerhorn T, et al cAMP enhances insulin secretion by an action on the ATP‐sensitive K+ channel‐independent pathway of glucose signaling in rat pancreatic islets. Diabetes 1999; 48: 1006–1012 [DOI] [PubMed] [Google Scholar]
- 29.Seino S, Shibasaki T. PKA‐dependent and PKA‐independent pathways for cAMP‐regulated exocytosis. Physiol Rev 2005; 85: 1303–1342 [DOI] [PubMed] [Google Scholar]
- 30.Shibasaki T. Elucidation of the function and role of cAMP sensor Epac2A in insulin secretion. Diabetol Int 2012; 3: 187–196 [Google Scholar]
- 31.Yamada S, Komatsu M, Sato Y, et al Time‐dependent stimulation of insulin exocytosis by 3′,5′‐cyclic adenosine monophosphate in the rat islet beta‐cell. Endocrinology 2002; 143: 4203–4209 [DOI] [PubMed] [Google Scholar]
- 32.Liu YQ, Tornheim K, Leahy JL. Shared biochemical properties of glucotoxicity and lipotoxicity in islets decrease citrate synthase activity and increase phosphofructokinase activity. Diabetes 1998; 47: 1889–1893 [DOI] [PubMed] [Google Scholar]
- 33.Komatsu M, Sharp GW. Palmitate and myristate selectively mimic the effect of glucose in augmenting insulin release in the absence of extracellular Ca2+. Diabetes 1998; 47: 352–357 [DOI] [PubMed] [Google Scholar]
- 34.Komatsu M, Yajima H, Yamada S, et al Augmentation of Ca2+‐stimulated insulin release by glucose and long‐chain fatty acids in rat pancreatic islets: free fatty acids mimic ATP‐sensitive K+ channel‐independent insulinotropic action of glucose. Diabetes 1999; 48: 1543–1549 [DOI] [PubMed] [Google Scholar]
- 35.Itoh Y, Kawamata Y, Harada M, et al Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003; 422: 173–176 [DOI] [PubMed] [Google Scholar]
- 36.Ferdaoussi M, Bergeron V, Zarrouki B, et al G protein‐coupled receptor (GPR)40‐dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 2012; 55: 2682–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaku K, Araki T, Yoshinaka R. Randomized, double‐blind, dose‐ranging study of TAK‐875, a novel GPR40 agonist, in Japanese patients with inadequately controlled Type 2 diabetes. Diabetes Care 2013; 36: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta‐cell function. Endocr Rev 2001; 22: 565–604 [DOI] [PubMed] [Google Scholar]
- 39.Ahren B. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia 2000; 43: 393–410 [DOI] [PubMed] [Google Scholar]
- 40.Yada T, Sakurada M, Ishihara H, et al Pituitary adenylate cyclase‐activating polypeptide (PACAP) is an islet substance serving as an intra‐islet amplifier of glucose‐induced insulin secretion in rats. J Physiol 1997; 505: 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharp GW. Mechanisms of inhibition of insulin release. Am J Physiol 1996; 271: C1781–C1799 [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Fang Q, Straub SG, et al Noradrenaline inhibits exocytosis via the G protein betagamma subunit and refilling of the readily releasable granule pool via the alphai1/2 subunit. J Physiol 2010; 588: 3485–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grimberg A, Ferry RJ Jr, Kelly A, et al Dysregulation of insulin secretion in children with congenital hyperinsulinism due to sulfonylurea receptor mutations. Diabetes 2001; 50: 322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Straub SG, Cosgrove KE, Ammala C, et al Hyperinsulinism of infancy: the regulated release of insulin by KATP channel‐independent pathways. Diabetes 2001; 50: 329–339 [DOI] [PubMed] [Google Scholar]
- 45.Seghers V, Nakazaki M, DeMayo F, et al Sur1 knockout mice. A model for K(ATP) channel‐independent regulation of insulin secretion. J Biol Chem 2000; 275: 9270–9277 [DOI] [PubMed] [Google Scholar]
- 46.Aguilar‐Bryan L, Bryan J. Molecular biology of adenosine triphosphate‐sensitive potassium channels. Endocr Rev 1999; 20: 101–135 [DOI] [PubMed] [Google Scholar]
- 47.Aguilar‐Bryan L, Bryan J, Nakazaki M. Of mice and men: K(ATP) channels and insulin secretion. Recent Prog Horm Res 2001; 56: 47–68 [DOI] [PubMed] [Google Scholar]
- 48.Beltrand J, Caquard M, Arnoux JB, et al Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care 2012; 35: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Komatsu M, Schermerhorn T, Aizawa T, et al Glucose stimulation of insulin release in the absence of extracellular Ca2+ and in the absence of any increase in intracellular Ca2+ in rat pancreatic islets. Proc Natl Acad Sci USA 1995; 92: 10728–10732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takei M, Dezaki K, Ishii H, et al A new experimental model of ATP‐sensitive K+ channel‐independent insulinotropic action of glucose: a permissive role of cAMP for triggering of insulin release from rat pancreatic β‐cells. Endocr J 2013; 60: 599–607 [DOI] [PubMed] [Google Scholar]